

Abstract
Hepatocyte nuclear factor 4α (HNF4α) controls the expression of liver-specific protein-coding genes. However, some microRNAs are also modulated by HNF4α, and it is not known whether they are direct targets of HNF4α and whether they influence hepatic function. In this study, we found that HNF4α regulates microRNAs, indicated by marked down-regulation of miR-194 and miR-192 (miR-194/192) in liver-specific Hnf4a-null (Hnf4aΔH) mice. Transactivation of the shared miR-194/192 promoter was dependent on HNF4α expression, indicating that miR-194/192 is a target gene of HNF4α. Screening of potential mRNAs targeted by miR-194/192 revealed that expression of genes involved in glucose metabolism (glycogenin 1 (Gyg1)), cell adhesion and migration (activated leukocyte cell adhesion molecule (Alcam)), tumorigenesis and tumor progression (Rap2b and epiregulin (Ereg)), protein SUMOylation (Sumo2), epigenetic regulation (Setd5 and Cullin 4B (Cln4b)), and the epithelial-mesenchymal transition (moesin (Msn)) was up-regulated in Hnf4aΔH mice. Moreover, we also found that miR-194/192 binds the 3′-UTR of these mRNAs. siRNA knockdown of HNF4α suppressed miR-194/192 expression in human hepatocellular carcinoma (HCC) cells and resulted in up-regulation of their mRNA targets. Inhibition and overexpression experiments with miR-194/192 revealed that Gyg1, Setd5, Sumo2, Cln4b, and Rap2b are miR-194 targets, whereas Ereg, Alcam, and Msn are miR-192 targets. These findings reveal a novel HNF4α network controlled by miR-194/192 that may play a critical role in maintaining the hepatocyte-differentiated state by inhibiting expression of genes involved in dedifferentiation and tumorigenesis. These insights may contribute to the development of diagnostic markers for early HCC detection, and targeting of the miR-194/192 pathway could be useful for managing HCC.
Keywords: gene regulation, hepatocyte nuclear factor 4 (HNF-4), liver, microRNA (miRNA), transcription
Introduction
Liver has diverse functions such as metabolism of carbohydrates, lipids, and proteins; detoxification of ammonia and xenobiotics; and synthesis of bile acids and blood coagulation factors. Various liver-enriched transcription factors such as hepatocyte nuclear factor 1 (HNF1),2 HNF3, HNF4, and HNF6 are needed to maintain differentiated liver-specific functions (1). Of these, HNF4α is a member of the nuclear receptor superfamily and a key regulator involved in the transcription regulatory network in the liver (2, 3), and re-expression of HNF4α in dedifferentiated hepatoma cells has the potential to revert cells back to the differentiated state (4). As Hnf4a-null mice are embryonic lethal during gastrulation (5), two types of liver-specific Hnf4a-null mice (Hnf4aΔH) were established (6, 7). Disruption of HNF4α in the fetal livers showed that hepatic HNF4α is essential for formation of a hepatic epithelium, liver morphogenesis, and liver homeostasis by positive regulation of a large number of genes during development into the mature liver (6). Disruption of HNF4α in the Hnf4aΔH mice led to diverse phenotypes such as fatty livers and elevation of serum bile acids and ammonia (7–9). Hnf4aΔH mice also exhibit increased spontaneous hepatocyte proliferation and increased susceptibility to diethylnitrosamine-induced liver cancer, indicating that HNF4α has tumor suppressor activity, although the mechanism has not been established (10, 11). Because HNF4α regulates many liver-enriched genes via HNF4α-binding sites in the promoter regions of the target genes and HNF4α is bound to about 12% of the gene promoters expressed in human hepatocytes (12), the HNF4α-derived transcriptional network in the liver is expected to be considerably complicated.
The gene expression pattern in the livers of Hnf4aΔH mice is markedly changed largely due to loss of expression of direct HNF4α target genes that are down-regulated, giving rise to the altered phenotypes of these mice (13). For instance, reduced expression of ornithine transcarbamylase (Otc), encoded by a HNF4α target gene, caused elevated serum ammonia due to the inability to degrade ammonia (8). In contrast, many mRNAs are up-regulated in Hnf4aΔH mice. Although direct transcriptional repression by HNF4α itself could be considered as a mechanism by which these mRNAs are reduced in Hnf4aΔH mice, another possibility is the involvement of microRNAs (miRNAs) that could be under direct transcriptional control of HNF4α. miRNAs are small non-coding RNAs involved in various physiological functions such as development, differentiation, inflammation, and tumorigenesis through their ability to down-regulate protein-coding gene expression by binding to target mRNAs and either promoting mRNA degradation or inhibiting mRNA translation (14, 15). miRNAs have important roles in liver diseases such as viral hepatitis, steatohepatitis, liver fibrosis, polycystic liver disease, and hepatocellular carcinoma (HCC) (16, 17).
Among the miRNAs, miR-122 is the most abundant in adult liver (18). Expression of miR-122 is reduced in HCC, and overexpression of miR-122 suppresses cell proliferation (19, 20). Thus, miR-122 could function as a tumor suppressor gene against HCC. Expression of miR-122 positively correlates with HNF4α expression in HCC cell lines, and miR-122 is directly regulated by HNF4α, indicating that the HNF4α-directed miR-122 pathway could be essential for inhibition of hepatocyte proliferation and HCC development (21, 22). Likewise, HNF4α directly regulates the expression of miR-134 and miR-134-suppressed oncogenic KRAS expression, revealing that miR-134 has a suppressive effect on HCC progression (23). Moreover, the following HNF4α–miRNA inflammatory feedback circuits in human HCC have been proposed. HNF4α positively regulates miR-7 and miR-124 followed by suppression of IL6R, STAT3, and RelA-mediated NF-κB pathways, eventually resulting in the up-regulation of HNF4α through down-regulation of miR-24, miR-629, or miR-21 (24, 25). These results also indicate that lack of HNF4α-controlled miRNAs could result in HCC development by activation of inflammatory signaling. These miRNAs were identified from studies using immortalized hepatocytes or HCC cell lines and thus could be biomarkers for HCC diagnosis and candidates for new target genes for HCC therapy. However, miRNAs regulated by HNF4α in normal hepatocytes but not in immortalized or transformed hepatocytes remain to be identified.
In this study, the miRNAs regulated by HNF4α were examined using Hnf4aΔH mice. miR-194 and miR-192, controlled by the same promoter, were down-regulated in Hnf4aΔH mice, and HNF4α directly bound to the promoter region of the miR-194–2 and miR-192 (miR-194/192) gene cluster. Furthermore, miR-194 and miR-192 directly targeted many mRNAs including frizzled-6 (Fzd6), Gyg1, Setd5, Sumo2, Cln4b, Rap2b, Ereg, Alcam, and Msn. The HNF4α–miR-194/192 cascade was also conserved in the human HCC cell line. Consequently, the HNF4α–miR-194/192 pathway could be a novel HNF4α cascade and a candidate for early detection of HCC and development of anti-HCC drugs.
Results
Down-regulation of hepatic miR-194 and miR-192 in Hnf4aΔH mice
HNF4α positively regulates the expression of many genes through HNF4α-binding sites in promoter regions (1, 3). A marked change of the mRNA expression pattern was observed in the livers of Hnf4aΔH mice (13), including many down-regulated mRNAs identified as encoded by direct HNF4α target genes (8, 9, 26). In addition, several up-regulated genes were also detected in Hnf4aΔH mice, but their mechanism of increase is not known. Although transcriptional repression of genes such as Snail, Slug, and HMGA2 by HNF4α was demonstrated (27), the mechanism of negative regulation of other genes by HNF4α has not been established. Another possibility for negatively regulated mRNAs is indirectly through miRNA. Indeed, HNF4α is known to regulate the expression of miR-7, miR-122, miR-124, and miR-134 (22–25). Thus, expression profiling of hepatic miRNAs in Hnf4a-floxed (Hnf4af/f) and Hnf4aΔH mice was investigated by microarray analysis (supplemental Table S1). Of 610 miRNAs, expression of 24 and 19 miRNAs was down-regulated or up-regulated more than 2-fold in Hnf4aΔH mice, respectively (Table 1). In general, because HNF4α positively regulates the target genes, down-regulated miRNAs in Hnf4aΔH mice could be direct target genes for HNF4α. Although other miRNAs, including miR-34a and miR-301a, were increased in Hnf4aΔH mice, they were not investigated in the current study. They could be either directly repressed by HNF4α or the result of downstream events mediated by loss of expression of other direct HNF4α target genes.
Table 1.
Expression profiling of hepatic miRNAs in Hnf4aΔH mice
miRNAs down-regulated or up-regulated more than 2-fold in Hnf4aΔH mice are listed. No significant change of miR-215 and miR-122 was detected. Raw data of each miRNA are given in parentheses.
| miRNA | Fold change (Hnf4aΔH/Hnf4af/f) |
|---|---|
| Down-regulated | |
| miR-194 | 0.11 (77.4/698.7) |
| miR-455 | 0.11 (1.4/12.5) |
| miR-805 | 0.16 (21.8/137.6) |
| miR-192 | 0.17 (96.5/559.5) |
| miR-193 | 0.25 (22.3/89.5) |
| miR-365 | 0.27 (4.4/16.1) |
| miR-193b | 0.31 (19.8/64.1) |
| miR-203 | 0.31 (6.9/22.1) |
| miR-130a | 0.32 (55.3/171.8) |
| miR-467a*, -d* | 0.32 (1.3/4.1) |
| miR-377 | 0.33 (1.3/4.0) |
| miR-220 | 0.37 (1.1/3.0) |
| miR-466b-3-3p | 0.38 (1.0/2.6) |
| miR-101b | 0.39 (29.6/75.5) |
| miR-323-5p | 0.43 (1.6/3.7) |
| miR-299* | 0.43 (1.3/3.0) |
| miR-574-3p | 0.44 (8.3/19.0) |
| miR-669e | 0.46 (1.7/3.7) |
| miR-21 | 0.47 (127.8/270.0) |
| miR-680 | 0.47 (3.7/7.8) |
| miR-467f | 0.47 (2.8/6.0) |
| miR-802 | 0.47 (2.1/4.5) |
| miR-425* | 0.48 (1.2/2.5) |
| miR-290-3p | 0.48 (1.0/2.1) |
| Up-regulated | |
| miR-34a | 5.99 (53.9/9.0) |
| miR-301a | 4.59 (7.8/1.7) |
| miR-28 | 3.34 (9.7/2.9) |
| miR-497 | 3.06 (19.9/6.5) |
| miR-484 | 2.97 (11.3/3.8) |
| miR-181a | 2.92 (11.1/3.8) |
| miR-689 | 2.56 (4.6/1.8) |
| miR-350 | 2.41 (14.0/5.8) |
| miR-500 | 2.41 (7.0/2.9) |
| miR-152 | 2.32 (24.6/10.6) |
| miR-125a-5p | 2.31 (65.1/28.2) |
| miR-31 | 2.29 (151.7/66.2) |
| miR-195 | 2.28 (50.9/22.3) |
| miR-335-5p | 2.27 (11.8/5.2) |
| miR-31* | 2.26 (4.3/1.9) |
| miR-142-5p | 2.23 (13.6/6.1) |
| miR-140* | 2.16 (14.9/6.9) |
| miR-151-5p | 2.10 (26.1/12.4) |
| miR-200b | 2.05 (19.5/9.5) |
| miR-215 | 1.38 (2.5/1.8) |
| miR-122 | 0.93 (7459/8046) |
The expression of miR-194 was decreased by 90% in Hnf4aΔH mice compared with Hnf4af/f mice. miR-194 was also expressed at considerably higher levels in Hnf4af/f mice when compared with other miRNAs. Similarly, the expression of miR-192 was also higher in Hnf4af/f mice and markedly lower in Hnf4aΔH mice. Indeed, qRT-PCR quantification showed that hepatic expression of miR-194 and miR-192 was suppressed by about one-tenth in Hnf4aΔH mice (Fig. 1A). Expression of other liver-enriched miRNAs such as miR-21, -101b, -130a, -193, -193b, -455, and -805 was also decreased in Hnf4aΔH mice (Fig. 1A). Others have reported that the expression of miR-194 and miR-192 is regulated under the same promoter, and tissue distribution of miR-194 and miR-192 is almost equivalent to that of HNF4α (28, 29), suggesting that HNF4α could positively regulate the expression of miR-194 and miR-192. Therefore, further studies were performed on miR-194 and miR-192. No significant difference in hepatic expression of miR-7, miR-122, miR-124, and miR-134, which were reported as the target miRNAs for HNF4α (22–25), was observed in Hnf4aΔH mice (supplemental Table S1).
Figure 1.
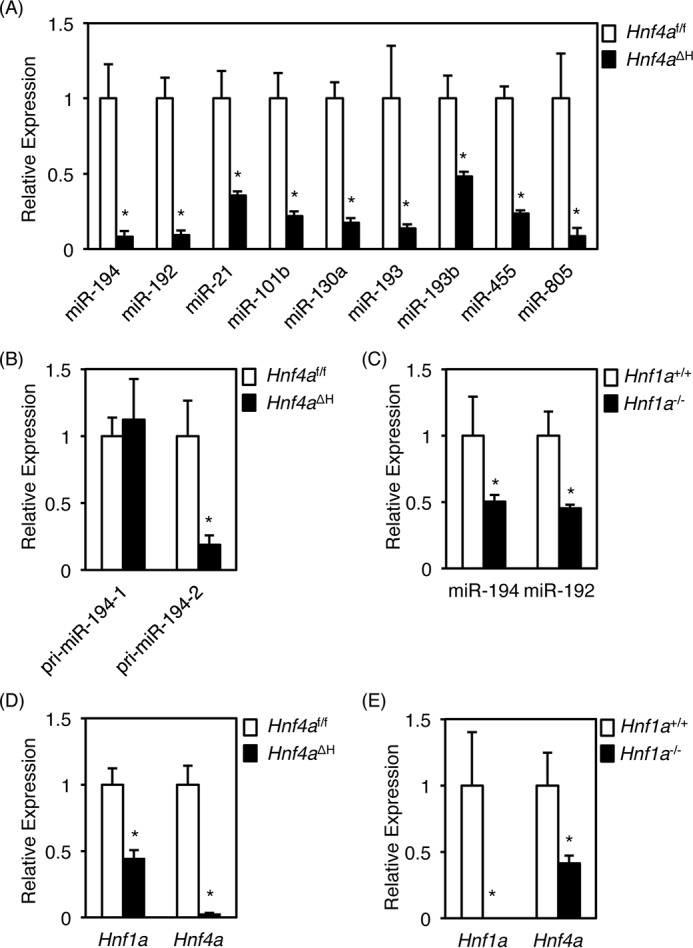
Down-regulation of hepatic miRNAs in Hnf4aΔH mice. Real-time PCR data for miR-194, miR-192, miR-21, miR-101b, miR-130a, miR-193, miR-193b, miR-455, and miR-805 (A), pri-miR-194–1 and pri-miR-194–2 (B), and Hnf1a and Hnf4a (D) from total liver RNA of Hnf4aΔH and Hnf4af/f mice and for miR-194 and miR-192 (C) and Hnf1a and Hnf4a (E) from total liver RNA of Hnf1a−/− and Hnf1a+/+ mice are shown. Error bars represent S.D. Data are mean ± S.D. of four independent mice. *, p < 0.05.
In mice, mature miR-194 is located on two chromosomes with miR-194-1 and miR-194-2 clustered with miR-215 and miR-192, respectively. Expression of miR-215 was unchanged in Hnf4aΔH mice, and the expression level was very low as compared with miR-194 and miR-192 (Table 1). Thus, miR-194-2, but not miR-194-1, could be highly expressed in normal liver and preferentially suppressed in Hnf4aΔH mice. As expected, hepatic expression of pri-miR-194-1 was unchanged, whereas expression of pri-miR-194-2 was markedly suppressed in Hnf4aΔH mice (Fig. 1B). These data indicate that HNF4α could directly activate the miR-194/192 gene via a single promoter.
Regulation of hepatic miR-194/192 by HNF4α and HNF1α
HNF1α, another liver-enriched transcription factor, directly regulates the miR-194/192 gene through a binding site in the miR-194/192 promoter region (28). Accordingly, qRT-PCR analysis using the livers of Hnf4aΔH mice and Hnf1a-null mice was performed to determine which factor, HNF1α or HNF4α, predominates in controlling expression of the miR-194/192 gene. Hepatic expression of both miRNAs in Hnf1a-null mice was reduced to 50%, and the remnant levels were much higher when compared with Hnf4aΔH mice (Fig. 1C). Because HNF4α positively regulates Hnf1a in the liver (2), decreased expression of miR-194/192 in Hnf4aΔH mice could be due to decreased expression of both Hnf1a and Hnf1a. Therefore, the expression of Hnf4a and Hnf1a mRNAs in Hnf1a-null mice was also investigated. The expression of Hnf4a was reduced to 3%, and the expression of Hnf1a mRNA was reduced to 45% in Hnf4aΔH mice (Fig. 1D). Conversely, expression of Hnf1a mRNA was not detected, and expression of Hnf4a mRNA was reduced to 42% in Hnf1a-null mice (Fig. 1E), revealing that the remaining expression of Hnf1a in Hnf4aΔH mice and Hnf4a in Hnf1a-null mice is similar. Hence, HNF4α could be a more essential factor for controlling expression of miR-194/192 as compared with HNF1α in the liver.
p53 also has the potential to transcriptionally control the miR-194/192 gene (29, 30). However, p53 was up-regulated in Hnf4aΔH mice, whereas it was unchanged in Hnf1a-null mice (Fig. 2, A and C). Similarly, the expression of p21 (Cdkn1a) mRNA, encoded by a direct target gene for p53, was also up-regulated in Hnf4aΔH mice and unchanged in Hnf1a-null mice (Fig. 2, B and D), indicating that up-regulation of p53 in Hnf4aΔH mice could not overcome repression of miR-194/192 expression caused by loss of HNF4α.
Figure 2.
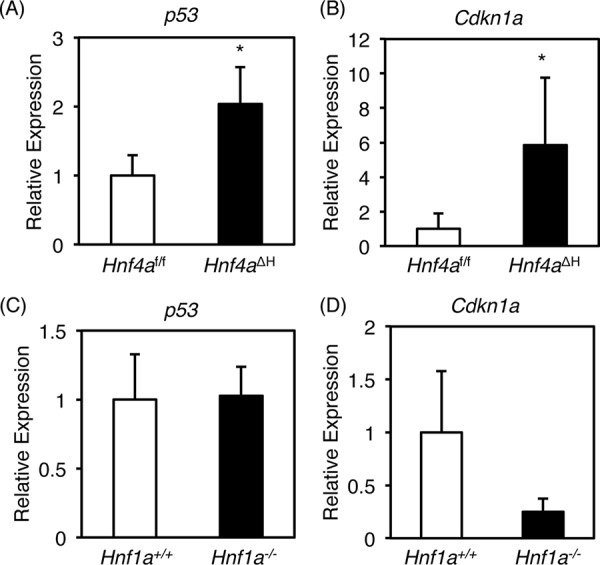
Expression of hepatic p53 and Cdkn1a in Hnf4aΔH and Hnf1a-null mice. Real-time PCR data for p53 (A and C) and Cdkn1a (B and D) from total liver RNA of liver-specific Hnf4aΔH and Hnf4af/f mice (A and B) and Hnf1a-null (Hnf1a−/−) and control (Hnf1a+/+) mice (C and D) are shown. Error bars represent S.D. Data are mean ± S.D. of four independent mice. *, p < 0.05.
The miR-194/192 gene promoter is transactivated by HNF4α
To investigate the mechanism by which HNF4α regulates the miR-194/192 gene, the promoter region of the miR-194/192 gene was analyzed. As described above, HNF1α was already found to transactivate the miR-194/192 gene through an HNF1α-binding site in colorectal adenocarcinoma-derived Caco-2 cells, and two regions located at −130 to −118 and −110 to −98 upstream of the HNF1α-binding site were highly conserved among species (28), but the binding proteins were not identified. Because HNF4α binding to both regions was predicted by JASPAR, an open access database (Fig. 3A), promoter analysis was performed to determine whether HNF4α has the potential to transactivate the miR-194/192 promoter. In HCC-derived and endogenous HNF4α-expressing HepG2 cells, the −70/+21 fragment containing an HNF1α-binding site was transactivated by 50-fold as compared with the promoterless vector pGL4.11 (Fig. 3B). The promoter activity of the −115/+21 fragment containing the HNF1α-binding site and a proximal HNF4α-binding site was much higher, and that of the −162/+21 fragment was highest in the presence of both HNF4α-binding sites, indicating that the minimum promoter of the miR-194/192 gene is located between −162 and +21. Introduction of mutations into the proximal (M1) or distal (M2) HNF4α-binding site resulted in significant reduction of the activities, and mutations in both binding sites (M1 + M2) did not transactivate the −162 and −2032/+21 regions (Fig. 3C). By co-transfection with the HNF4α expression vector in HEK293T cells, the promoter activity was slightly induced in the −115/+21 fragment containing the distal HNF4α-binding site and strongly activated in the −162/+21 fragment in the presence of both HNF4α-binding sites (Fig. 3D). Furthermore, the induced activities in the −162 and −2032/+21 promoters were markedly suppressed by introduction of mutations into one or both HNF4α-binding sites (Fig. 3E). These results indicate that both HNF4α-binding sites cooperatively transactivate the miR-194/192 gene.
Figure 3.
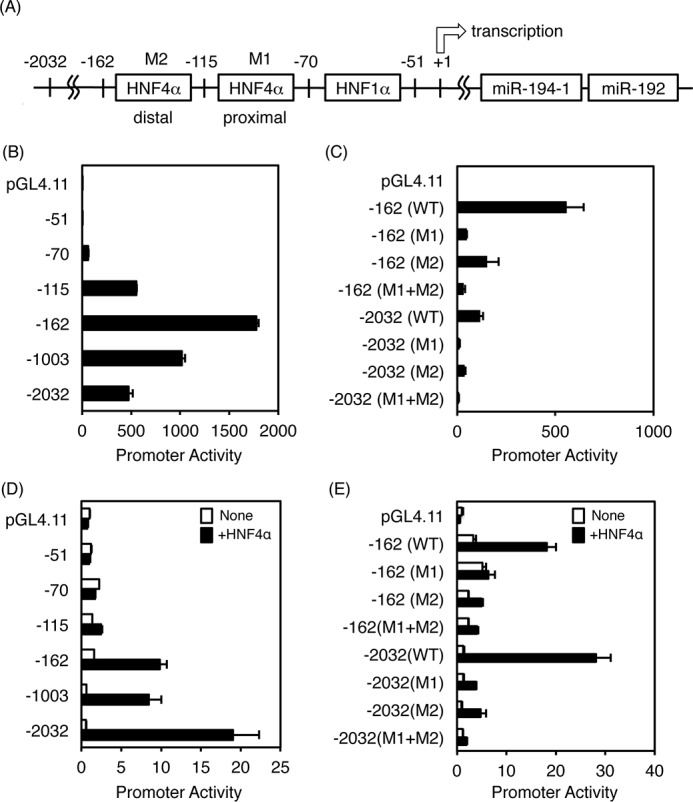
Promoter analysis of the human miR-194/192 gene. A, schematic diagram of the promoter region of the human miR-194/192 gene. B, promoter constructs of the miR-194/192 gene (WT) were transfected into HepG2 cells. C, mutations were introduced into the HNF4α-binding sites (M1 and M2), and the constructs were transfected into HepG2 cells. D and E, promoter constructs of the WT and mutants (M1 and M2) of the miR-194/192 gene were transfected into HEK293T cells with empty vector (white bar) or HNF4α expression vector (black bar). The normalized activity is presented as relative activity based on the promoterless vector pGL4.11. Error bars represent S.D. Data are mean ± S.D. of three independent experiments.
To determine whether HNF4α can directly bind to both HNF4α-binding sites in the miR-194/192 promoter, gel mobility shift analysis was performed (Fig. 4A). Nuclear extracts from HepG2 cells bound to the distal and proximal HNF4α-binding sites (Fig. 4A, lane 2, lower arrows). This complex was diminished by the addition of excess amounts of the unlabeled competitor and OTC competitor that contains an HNF4α-binding site (Fig. 4A, lanes 3 and 5) but not the competitor that contains mutations in the HNF4α-binding site (Fig. 4A, lane 4). Moreover, the complex was supershifted by anti-HNF4α antibody (Fig. 4A, lane 6, upper arrow) but not the anti-C/EBPα antibody (lane 7). These results indicate that HNF4α binds to both HNF4α-binding sites of the miR-194/192 promoter. Next, chromatin immunoprecipitation (ChIP) was used to determine whether HNF4α directly binds to the miR-194/192 promoter in vivo. We found that HNF4α was bound to the HNF4α-binding sites in HepG2 cells (Fig. 4B). In addition, ChIP analysis using the livers of Hnf4af/f and Hnf4aΔH mice indicated that hepatic HNF4α in Hnf4af/f mice bound to the promoter region, but the binding was decreased in Hnf4aΔH mice (Fig. 4C). Similar results were obtained in the mouse Otc promoter that is dependent on an HNF4α-binding site (8), suggesting that HNF4α physiologically binds to the promoter region of the miR-194/192 gene in humans and mice.
Figure 4.
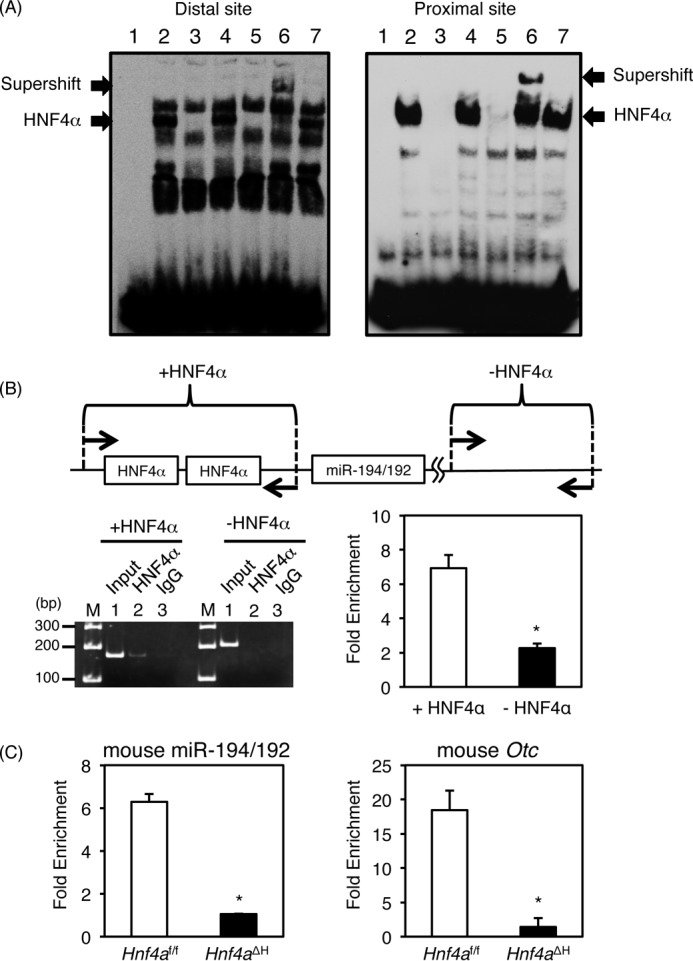
Identification of HNF4α-binding sites in the miR-194/192 promoter. A, gel shift analysis. Nuclear extracts from HepG2 cells were incubated with biotin-labeled probe carrying the distal (left lane) and proximal (right lane) HNF4α-binding sites in the miR-194/192 promoter in the absence (lane 2) or presence of a 50-fold excess of the unlabeled miR-194/192 probe (lane 3), the mutated miR-194/192 probe (lane 4), and the OTC probe (lane 5). For supershift analysis, anti-HNF4α and anti-C/EBPα antibodies were added (lanes 6 and 7). The complex between HNF4α and the probe and the supershifted complex are indicated by the lower and upper arrows, respectively. B, chromatin immunoprecipitation using HepG2 cells was performed with anti-HNF4α antibody (lane 2) and normal goat IgG (lane 3) (left). Input DNA was used as a positive control (lane 1). The regions between −201 and −41 containing both HNF4α-binding sites (+HNF4α) and between +16667 and +16874 without HNF4α-binding site (−HNF4α) in the human miR-194/192 gene were amplified. The data from qPCR were normalized relative to the input and expressed as -fold enrichment (right). C, chromatin immunoprecipitation using the livers of Hnf4aΔH and Hnf4af/f mice with anti-HNF4α antibody and normal goat IgG. The regions between −164 and −70 containing both HNF4α-binding sites in the mouse miR-194/192 promoter (left), between −264 and −165 containing two HNF4α-binding sites in the mouse Otc promoter (right), and between +45820 and +45893 without an HNF4α-binding site in the mouse Hmgcs2 gene were amplified, respectively. The data from qPCR was normalized relative to the input and expressed as -fold enrichment over data from IgG control. Error bars represent S.D. Data are mean ± S.D. of three independent experiments. *, p < 0.05.
Identification of miR-194 and miR-192 target mRNAs
It was reported that miR-194 is involved in epithelial-mesenchymal transition (EMT) and HCC metastasis (31). In addition, miR-194 regulates mRNAs encoded by many genes including N-cadherin (Cdh2), Rac1, heparin-binding epidermal growth factor (Hbegf), tyrosine protein phosphatase non-receptor type 2 (Ptpn2), integrin α9 (Itga9), suppressor of cytokine signaling 2 (Socs2), DNA methyltransferase 3A (Dnmt3a), Fzd6, and talin 2 (Tln2) that play important roles in tumorigenesis, tumor progression, cell adhesion, cell migration, and EMT (31–33). miR-192 is also known to negatively regulate mRNAs encoding IGF-1, IGF-1R, and ZEB2 that are also involved in EMT and tumor progression and invasion (30, 34). Therefore, qRT-PCR was performed to determine whether hepatic expression of these mRNAs was increased in Hnf4aΔH mice. Expression of Fzd6, Hbegf, Ptpn2, Dnma3a, Itga9, and Rac1 mRNAs was significantly increased, whereas expression of Socs2, Cdh2, Tln2, and Zeb2 mRNAs was unchanged in Hnf4aΔH mice (Fig. 5, A and C). Next, novel target genes for miR-194 and miR-192 were investigated using TargetScan, DIANA-microT, and miRDB databases, and the resulting high-scoring hits that overlapped such as Hnf1b, Gyg1, hook microtubule-tethering protein 3 (Hook3), Setd5, selenophosphate synthetase 1 (Sephs1), Sumo2, Cln4b, and Rap2b were selected as the target candidates for miR-194 (supplemental Fig. S1, B–F). Regarding the candidates of miR-192 target gene mRNAs, Ereg, ADP-ribosylation factor guanine nucleotide-exchange factor 1 (Arfgef1), radical S-adenosyl methionine domain-containing 2 (Rsad2), Alcam, and Msn were selected (supplemental Fig. S2, G–I). As expected, the expression of all candidate mRNAs was significantly increased in Hnf4aΔH mice (Fig. 5, B and C). The expression of CLN4B protein, a candidate for miR-194 targeting, and ALCAM protein, a candidate for miR-192 targeting, was also increased more than 2-fold in Hnf4aΔH mice compared with Hnf4af/f mice (Fig. 5D). Therefore, studies were done to determine whether miR-194 and miR-192 could directly interact with the 3′-UTR of these candidate mRNAs and inhibit the protein expression (Fig. 6). miR-194 and miR-192 markedly repressed the 3′-UTR activities in the presence of the complementary sequences of miR-194- and miR-192-binding sites (c-miR-194 and c-miR-192) (supplemental Fig. S2). The 3′-UTR activities of Fzd6, Gyg1, Setd5, Sumo2, Cln4B, and Rap2b using a luciferase reporter system were significantly inhibited by miR-194 (Fig. 6A, wild type (WT)). Because two miR-194-binding sites were predicted within the 3′-UTR of Fzd6 and Rap2b, mutations were introduced to disrupt the binding of miR-194 in both binding sites. Each mutant (M1 and M2) partially rescued the 3′-UTR activity, and both mutants (M1 + M2) completely recovered the activities (Fig. 6A). The same results were also obtained in the mutated 3′-UTR regions of the Gyg1, Setd5, Sumo2, and Cln4B (Fig. 6B). Similarly, 3′-UTR activities of Ereg, Alcam, and Msn were suppressed in a miR-192-dependent manner, and the activities were recovered by introduction of mutations into the miR-192-binding sites (Fig. 6B). These results indicate that Fzd6, Gyg1, Setd5, Sumo2, Cln4B, and Rap2b are the target mRNAs for miR-194 and that Ereg, Alcam, and Msn mRNAs are targets for miR-192.
Figure 5.
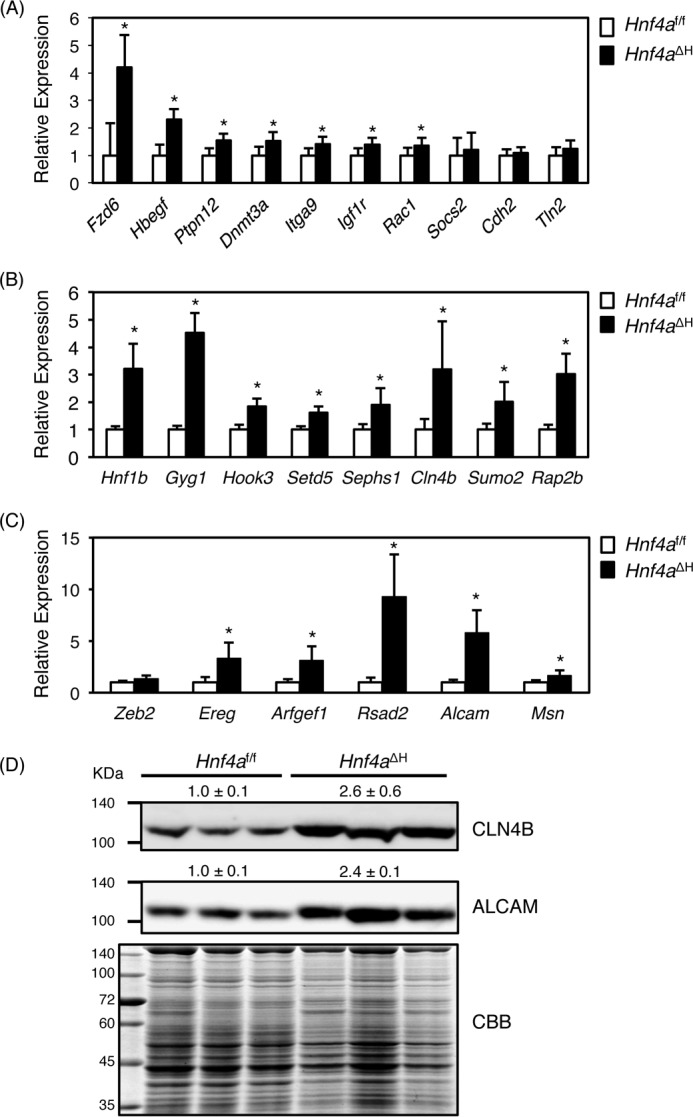
Hepatic expression of the target genes for miR-194 and miR-192 in Hnf4aΔH mice. A, real-time PCR from total liver RNA of Hnf4aΔH and Hnf4af/f mice. Known (A) and new (B) mRNA targets for miR-194 and known and new targets (C) for miR-192. Error bars represent S.D. Data are mean ± S.D. of five independent mice. *, p < 0.05. D, Western blot of CLN4B and ALCAM protein from livers of Hnf4aΔH and Hnf4af/f mice (n = 3 for each genotype). CLN4B and ALCAM expression normalized by total protein in Hnf4aΔH was presented as relative to Hnf4af/f mice. CBB, Coomassie Brilliant Blue.
Figure 6.
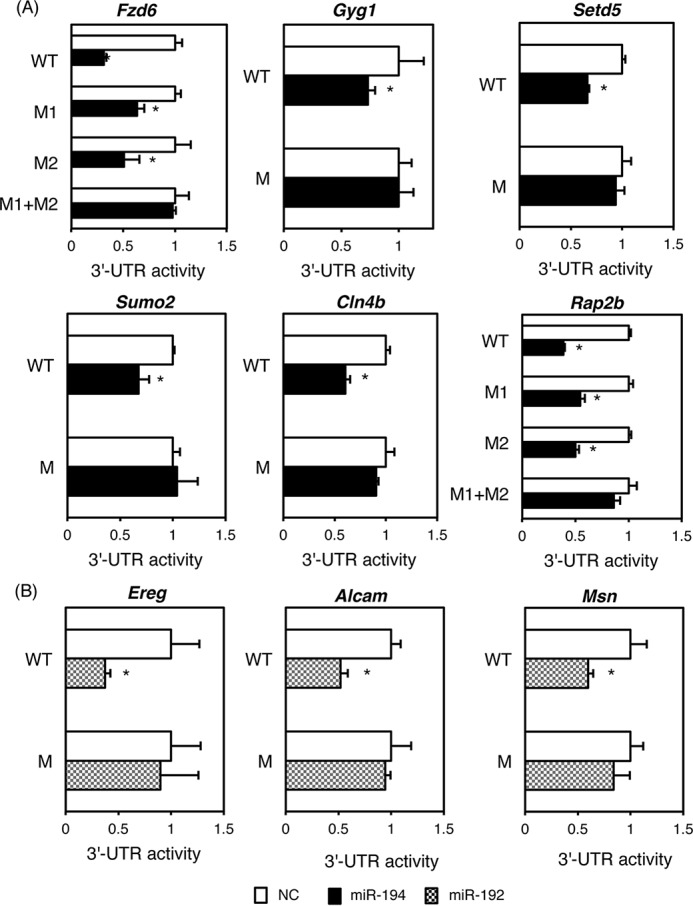
3′-UTR activities of the target mRNAs for miR-194 and miR-192. Luciferase vectors harboring the WT 3′-UTR of the target mRNAs for miR-194 (A) and miR-192 (B) were transfected into HEK293T cells with miR-194 (black bar), miR-192 (striped bar), or negative control (NC; white bar). Mutations were introduced into miR-194-binding sites (M). Error bars represent S.D. Data are mean ± S.D. of three independent experiments. *, p < 0.05.
To determine whether the HNF4α-directed miR-194/192 pathway is also conserved in human HCC-derived cell lines, HNF4α was knocked down by siRNA. HNF4α siRNA inhibited mRNA and protein expression of HNF4α in HepG2 cells, and expression of both miR-194 and miR-192 was largely repressed (Fig. 7, A and B), whereas expression of target mRNAs for miR-194 and miR-192 such as Fzd6, Gyg1, Cln4b, Rap2b, Ereg, Alcam, and Msn was increased by HNF4α knockdown (Fig. 7C). Inhibition of miR-194 and miR-192 also enhanced expression of target mRNAs such as Fzd6, Setd5, Sumo2, Cln4b, Ereg, and Alcam. (Fig. 7D). Conversely, miR-194 and miR-192 mimics suppressed expression of all target genes in human HCC-derived HLE cells that hardly express endogenous HNF4α (Fig. 7E). These results indicate that regulation of miR-194/192 by HNF4α and the target mRNAs by miR-194/192 may also be conserved in human HCC cell lines.
Figure 7.
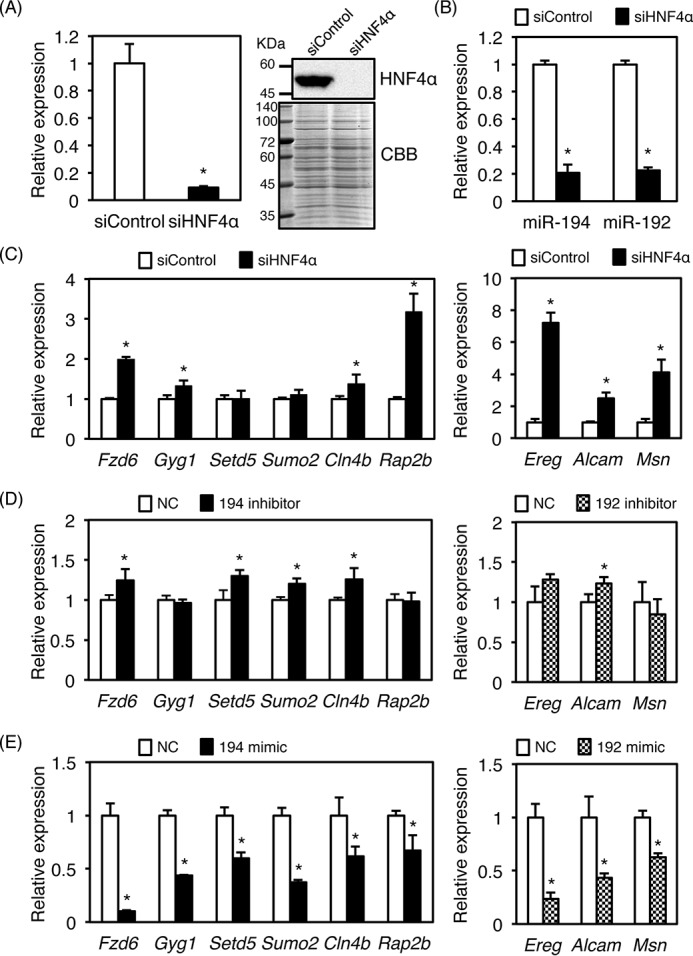
Conservation of HNF4α–miR-194/192 cascade in human HCC cell lines. siRNA (10 nm) was transfected into HepG2 cells. Real-time PCR data for HNF4α (A), miR-194 and miR-192 (B), and target mRNAs for miR-194 and miR-192 (C) are shown. A Western blot of HNF4α is shown in A. D, miR-194 and miR-192 inhibitors (100 nm) were transfected into HepG2 cells, and relative expression of the target genes was analyzed by real-time PCR. E, miR-194 and miR-192 mimics (10 nm) were transfected into HLE cells, and relative expression of the target genes was analyzed by real-time PCR. Error bars represent S.D. Data are mean ± S.D. of three independent experiments. *, p < 0.05. NC, negative control; CBB, Coomassie Brilliant Blue.
Discussion
In this study, HNF4α was found to directly activate the miR-194/192 gene promoter. Down-regulation of HNF4α target genes partially could explain the onset of phenotypes in Hnf4aΔH mice (7, 8, 26). From microarray analysis using livers of Hnf4aΔH mice (13), most mRNAs were markedly down-regulated, probably due to the loss of direct positive regulation of the respective genes by HNF4α. However, up-regulation of many mRNAs was also observed in Hnf4aΔH mice. Thus, HNF4α might negatively regulate these genes via direct positive regulation of miRNAs that in turn could bind to destabilize mRNAs. HNF4α was shown to bind to the promoter region of miR-122, the most abundant miRNA in the liver, and transactivate the miR-122 in an HNF4α-dependent manner (22). However, hepatic expression of miR-122 was unchanged in Hnf4aΔH mice. HNF4α knockdown also inhibited miR-122 expression in mouse liver (22), suggesting that transactivation of miR-122 by HNF4α would be transient in Hnf4aΔH mice as observed, probably due to secondary effects. Likewise, HNF4α positively regulates miR-7, miR-124, and miR-134 in human HCC and tumor-derived cell lines (23–25). However, unexpectedly, the hepatic expression of these miRNAs was also unchanged in Hnf4aΔH mice. Because the expression levels of miR-7, miR-124, and miR-134 were very low or undetectable in normal livers of Hnf4af/f mice, HNF4α may not directly regulate these miRNAs as found in normal liver. Thus, HNF4α might regulate these miRNAs only during the later stages of HCC progression but not the early stages of HCC development because Hnf4aΔH mice do not exhibit early spontaneous HCC development.
Unlike miR-7, miR-122, and miR-134, miR-194 and miR-192 were not reported to be down-regulated in human HCC specimens used in these studies, indicating that down-regulation of miR-194 and miR-192 in the liver could be an early event in the onset of liver inflammation and pretumorigenesis. miR-194 is involved in the differentiation of intestinal epithelial cells, and HNF1α was also reported to transactivate miR-194/192 via an HNF1α-binding site (28). HNF1α is a direct target for HNF4α (2), and hepatic expression of HNF1α was actually down-regulated in Hnf4aΔH mice as expected. Thus, both HNF4α and HNF1α might cooperatively control the miR-194/192 gene promoter. However, down-regulation of miR-194 and miR-192 in Hnf1a-null mice was not as pronounced when compared with Hnf4aΔH mice because sufficient HNF4α still remains in Hnf1a-null mice, indicating that HNF4α might be a predominant factor in regulation of the miR-194/192 gene as compared with HNF1α.
p53 was also reported to control expression of the miR-194/192 gene via a p53-binding site (29, 30). The p53-binding site is located upstream of the distal HNF4α-binding site, and the promoter of −162/+21 fragment including the p53- and HNF4α-binding sites was activated in an HNF4α-dependent manner, suggesting that p53 and HNF4α might synergistically transactivate the miR-194/192 gene. Expression of miR-194 and miR-192 was markedly down-regulated in a p53-dependent manner in B cell neoplasm multiple myeloma, which rarely expresses endogenous HNF4α (30). Conversely, the expression of miR-194 and miR-192 was slightly down-regulated in human HCC including mutated or deleted p53 without statistical significance as compared with wild-type p53 (34), indicating that HNF4α and HNF1α mainly transactivate the miR-194/192 gene despite p53 status.
EMT is an important process for cells to acquire tumor metastatic potential. Expression of miR-194 was also down-regulated in liver mesenchymal-like cancer cell lines as compared with liver epithelial cancer cell lines, and overexpression of miR-194 suppressed invasion and migration of hepatoma cells (31). Thus, miR-194 and miR-192 expression in liver might retain the differentiated status of hepatocytes and suppress proliferation, migration, and malignant transformation. Re-expression of HNF4α in dedifferentiated hepatoma cells resulted in reversion to differentiated phenotypes (4). Moreover, reduced expression of HNF4α correlated with human HCC progression (25), and forced expression of HNF4α reduced the proliferation and induced epithelial formation (25, 35, 36). These results strongly suggest that HNF4α is one of the most important factors to induce hepatocyte differentiation by direct transactivation of liver-specific genes and to inhibit dedifferentiation of hepatocytes by indirect negative regulation of oncogenes and EMT-related genes via direct up-regulation of miR-194 and miR-192. However, hepatic expression of EMT-activating genes including Cdh2 as an miR-194 target and Zeb2 as an miR-192 target mRNA was not increased in mice, showing that down-regulation of miR-194 and miR-192 in inflammatory and non-/pretransformed hepatocytes in Hnf4aΔH mice could not yet activate EMT-related genes because Hnf4aΔH mice die at about 8 weeks of age before showing tumorigenesis (7).
Others have reported that HNF1α-regulated miR-194 repressed the 3′-UTR activity of FZD6 (32). In this study, two functional miR-194-binding sites were identified in the 3′-UTR of Fzd6 mRNA. FZD6 is a membrane receptor for Wnt ligand signaling and controls cell differentiation (37). Because overexpression of FZD6 was confirmed in human HCC (38) and hepatic expression of FZD6 was not significantly increased in HNF1α-null mice (32), repression of FDZ6 expression through negative regulation by miR-194 via HNF4α could be important for suppression of tumorigenesis.
There are no reports that GYG1 in involved in HCC and metastasis. Glycogen synthesis starts from self-glucosylation of GYG1 followed by elongation of glucose chains by glycogen synthase (39). Hepatic expression of glycogen synthase 2, a limited enzyme for glycogen synthesis in the liver and a direct target for HNF4α, was markedly decreased in Hnf4aΔH mice (40). Eventually, expression of GYG1 might be increased to compensate for reduced production of de novo glycogen synthesis in Hnf4aΔH mice.
CUL4B is the main component of Cullin4B-Ring ubiquitin ligase complex and promotes tumorigenesis (41) and SETD5 is a histone methyltransferase on the histone H4 tail at lysines 5, 8, and 12 in yeast (42). Loss of function mutations in the SETD5 gene were found in patients with intellectual disability (43); however, the physiological significance of H4 methylation by SETD5 remains unsolved. Histone H4 lysines 5, 8, and 12 are also acetylated by histone acetyltransferases (44), and methylation of these sites by SETD5 might change chromatin structure and influence transcription. HNF4α-dependent suppression of Cul4B and Setd5 could partially control the epigenetic program and be important to retain the differentiated status in normal hepatocytes.
SUMO2 has an important role to regulate a variety of biological functions such as transcription, DNA repair, cell cycle, and subcellular transport by SUMOylation to the target proteins (45). Similar to an ES cell-based model of hepatocyte differentiation in which SUMO2-modified proteins were decreased during hepatocyte differentiation (46), increased activity of SUMOylation by SUMO2 could induce hepatocyte dedifferentiation in Hnf4aΔH mice.
RAP2B functions as a p53-mediated prosurvival factor. RAP2B was also found to be up-regulated in many types of tumors, but RAP2B had very weak transformation activity, indicating that RAP2B itself is not an oncogene but maintains the tumorigenic status (47). Because hepatic expression of p53 and Cdkn1a was increased in Hnf4aΔH mice, induced p53 might be responsible for additional induction of Rap2b mRNA.
In addition, Ereg, Alcam, and Msn mRNAs are targets for miR-192 in the liver. EREG is one of the ligands for epidermal growth factor receptor and is involved in inflammation and tumorigenesis (48). Knockdown of EREG suppressed cell growth in human hepatoma cell lines (49), and Ereg-null mice exhibited a significant reduction of tumor number and size in HCC model experiments (50), indicating that EREG may contribute to HCC progression. Unlike other target genes for miR-194 and miR-192 examined in this study, EREG is more highly expressed in hepatic stellate cells than in hepatocytes (50), suggesting that miR-192 synthesized in hepatocytes might be secreted into hepatic stellate cells followed by repression of EREG in normal liver. ALCAM, a cell adhesion molecule, is a marker of colorectal cancer stem cells (51). Although ALCAM expression was inhibited by miR-192 in a gastric cell line, the 3′-UTR of Alcam mRNA failed to respond to miR-192 (52). By contrast, the 3′-UTR activity of Alcam mRNA was dependent on a miR-192-binding site. ALCAM was overexpressed in human hepatoma cell lines, and silencing of ALCAM suppressed cell growth and promoted apoptosis (53), suggesting that ALCAM could accelerate tumor cell survival by an antiapoptotic role in HCC.
MSN functions as a cross-linker between the plasma membrane and actin filament. High expression of moesin was observed in breast cancers and implicated in EMT (54, 55). However, expression of Msn mRNA was induced only 1.6-fold in Hnf4aΔH mice. Because other EMT-related mRNAs such as Cdh2 and Zeb2 were not induced in Hnf4aΔH mice, these EMT-related mRNAs might not be induced sufficiently in inflammatory and non-transformed hepatocytes of Hnf4aΔH mice.
In conclusion, HNF4α was found to repress the expression of a variety of mRNAs through positive regulation of miR-194 and miR-192 in non-transformed liver. Most of these mRNAs encode proteins involved in tumorigenesis, prosurvival, epigenetic change, and EMT. These observations revealed an HNF4α cascade that inhibits hepatocyte dedifferentiation. Further studies may contribute to the development of novel diagnostic markers for the early detection of HCC using the HNF4α–miR-194/192 axis, and inhibition of the miR-194/192 targets by induced expression and/or activation of miR-194/192 could be useful for HCC therapy.
Experimental procedures
Animals
Hnf4aΔH mice carrying a mixed SvJ129, FVB, and C57BL/6 background were generated as described previously (7). All experiments were performed with 45-day-old male Hnf4af/f and Hnf4aΔH mice. Hnf1a-null mice were also used (56). The mice were housed in a pathogen-free animal facility under a standard 12-h light/12-h dark cycle with ad libitum water and chow. All experiments with mice were carried out under Association for Assessment and Accreditation of Laboratory Animal Care guidelines with approval of the National Cancer Institute Animal Care and Use Committee and Gunma University Animal Care and Experimentation Committee.
miRNA microarrays
Total RNA from the livers of Hnf4af/f and Hnf4aΔH mice was purified using the miRNeasy mini kit (Qiagen, Tokyo, Japan). Extracted RNA was labeled with Cy5 using the Label IT miRNA labeling kit (Takara Bio, Otsu, Japan). Labeled RNAs were hybridized onto 3D-Gene Mouse miRNA Oligo chips (v.11.1; Toray Industries, Tokyo, Japan). The annotation and oligonucleotide sequences of the probes conformed to the miRBase miRNA database (http://www.mirbase.org/).3 After stringent washes, fluorescence signals were scanned with the ScanArray Express Scanner (PerkinElmer Life Sciences) and analyzed using GenePix Pro version 5.0 (Molecular Devices, Sunnyvale, CA). The raw data of each spot were normalized by subtraction of the mean intensity of the background signal determined by all blank spots' signal intensities of 95% confidence intervals. Measurements of both duplicate spots with signal intensities greater than two standard deviations (S.D.) of the background signal intensity were considered to be valid. A relative expression level of a given miRNA was calculated by comparing the signal intensities of the averaged valid spots with their mean value throughout the microarray experiments after normalization by their median values adjusted equivalently. miRNA microarray data are described in supplemental Table S1. The data are also available at the Gene Expression Omnibus under accession number GSE70516.
Reverse transcription and real-time PCR
Total RNA was extracted using TriPure Isolation Reagent (Roche Applied Science). Reverse transcription for mature miRNA was performed using a Mir-X miRNA First-Strand Synthesis kit (Clontech). Real-time PCR was performed on a LightCycler 480 system II (Roche Applied Science). cDNAs encoding mature miRNA and U6 RNA were amplified using a Mir-X miRNA qRT-PCR SYBR kit (Clontech). The levels of mature miRNA were expressed relative to U6 RNA as an internal control using the ΔΔCt method. For detection of two transcripts for pri-miR-194 and mRNAs, cDNA from the livers of Hnf4af/f and Hnf4aΔH mice was transcribed using ReverTraAce qPCR RT Master with gDNA Remover (Toyobo, Osaka, Japan), and real-time PCR was performed using FastStart SYBR Green Master (Roche Applied Science). The levels of expression were normalized relative to Gapdh mRNA as an internal control using the ΔΔCt method. Sequences for the primers are shown in supplemental Table S2.
Cloning of the human miR-194/192 gene promoter region
The −2032, −1003, −162, −115, −70, and −51/+21 fragments of the human miR-194/192 promoter containing KpnI and XhoI sites were amplified with genomic DNA from HepG2 cells by PCR and cloned into the luciferase reporter vector pGL4.11 (Promega, Madison, WI). Mutations were introduced into the HNF4α-binding sites in the miR-194/192 reporter vector by PCR-based site-directed mutagenesis. Sequences for the primers are shown in supplemental Table S3.
Transient transfection and luciferase assay
HepG2 and HEK293T cells were cultured at 37 °C in Dulbecco's modified Eagle's medium (Wako, Osaka, Japan) containing 10% fetal bovine serum (HyClone, Logan, UT) and 1,000 units of penicillin and 0.1 mg of streptomycin/ml (Sigma-Aldrich). For suspension transfection, miR-194 promoters cloned into pGL4.11 and pGL4.74 encoding Renilla luciferase regulated under herpes simplex virus thymidine kinase promoter (Promega) were transfected into HepG2 cells with polyethylenimine Max (Polyscience, Warrington, PA) as a transfection reagent. For co-transfection using HEK293T cells, HNF4α expression plasmid was used. After 48 h of transfection, the cells were washed with phosphate-buffered saline (PBS), and promoter activities were measured using the Dual-Luciferase Reporter Assay System (Promega).
Transfection of siRNA and miRNA mimics and miRNA inhibitors
siRNA (10 nm) against Hnf4a mRNA (Sigma-Aldrich), 10 nm miR-192 and miR-194 mimics (miRIDIAN microRNA mimics, GE Healthcare), and 100 nm miR-192 and 194 inhibitors (miScript miRNA inhibitors, Qiagen) were transfected into HepG2 and HLE cells with Lipofectamine RNAiMAX (Life Technologies). After 48 h of transfection, total RNA and protein were extracted from the transfected cells.
Western blotting
Liver samples from Hnf4af/f and Hnf4aΔH mice and HepG2 cells treated with siRNA against Hnf4a mRNA were homogenized in lysis buffer (7 m urea, 2 m thiourea, and 1% Triton X-100) and allowed to sit on ice for 30 min. The homogenate was centrifuged at 12,000 × g for 30 min at 4 °C, and the supernatants were used as whole-cell lysates. Total protein (40 μg) with Laemmli sample buffer was incubated at 65 °C for 15 min and fractionated by 10% SDS-polyacrylamide gel electrophoresis. The gels were stained with Coomassie Brilliant Blue R-250 or blotted onto a PVDF membrane (GE Healthcare). The membrane was incubated for 1 h with mouse monoclonal antibodies against human HNF4α (Perceus Proteomics, Tokyo, Japan) and rabbit polyclonal antibodies against human CLN4B (GeneTex, Irvine, CA) and human ALCAM (Sigma-Aldrich). After washing, the membrane was incubated for 1 h with horseradish peroxidase-conjugated anti-mouse IgG (Cell Signaling Technology, Tokyo, Japan), and the reaction product was visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce) on an ImageQuant LAS4000 (GE Healthcare). Expression of CLN4B and ALCAM proteins was quantified by densitometric analysis using ImageJ software, and the expression normalized by total protein in Hnf4aΔH was presented as expression differences relative to the Hnf4af/f mice.
Gel mobility shift analysis
Nuclear extracts from HepG2 cells were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), and gel shift analysis carried out using the LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific). The following double-stranded probes were used: wild-type and mutants of distal and proximal HNF4α-binding sites for the human miR-194/192 promoter and the HNF4α-binding site for the mouse Otc promoter as a positive control. Nuclear extracts and the 5′-biotin-labeled probe of the HNF4α-binding sites for the miR-194/192 promoter (wild type) were added, and the reaction mixture was incubated on ice for 10 min. For competition experiments, a 50-fold excess of unlabeled probe was added to the reaction mixture, and the mixture was incubated on ice for 10 min prior to the addition of the 5′-biotin-labeled probe. For supershift analysis, anti-HNF4α or anti-C/EBPα antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were added to the mixture, and the mixture was incubated on ice for 10 min after the addition of the 5′-biotin-labeled probe. DNA-protein complexes were fractionated by 7% PAGE and blotted onto a Biodyne B nylon membrane (Pall, Tokyo, Japan). After washing, complexes were visualized using the detection module in the kit on an ImageQuant LAS4000. Sequences for the primers are shown in supplemental Table S4.
Chromatin immunoprecipitation
HepG2 cells were fixed in 0.5% formaldehyde and quenched by 125 mm glycine at room temperature. After washing, cells were resuspended in 3 ml of lysis buffer 1 (50 mm HEPES-KOH (pH 7.5), 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100, and protease inhibitor (Roche Applied Science)) on ice for 10 min and then centrifuged at 1,400 × g for 5 min. The cell pellet was resuspended in 3 ml of lysis buffer 2 (10 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm EDTA, and 5 mm EGTA) for 10 min at room temperature and then centrifuged at 1,400 × g for 5 min. The cell pellet was resuspended in 1 ml of lysis buffer 3 (10 mm Tris-HCl (pH 8.0), 300 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, and 0.1% sodium deoxycholate). Liver samples were ground into pieces with a pestle and mortar under liquid nitrogen and fixed in PBS containing 20 mm sodium butyrate, 1% formaldehyde, and protease inhibitor mixture for 10 min at room temperature. After centrifugation, the pellet was resuspended in lysis buffer (50 mm Tris-HCl (pH 8.0), 10 mm EDTA, 1% sodium dodecylsulfate, 20 mm sodium butyrate, and protease inhibitor). The cell lysate from HepG2 cells and liver samples was disrupted by sonication (UR-20P, Tomy, Tokyo, Japan) for 5 min on ice and then diluted 10-fold with RIPA ChIP buffer (10 mm Tris-HCl (pH 8.0), 140 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 20 mm sodium butyrate, and protease inhibitor) followed by centrifugation at 20,000 × g for 10 min. A small volume of the supernatant was stored at 4 °C as the input sample. The remaining supernatant was precleared by adding of 15 μl of a 50% slurry of protein G-Sepharose 4 Fast Flow (GE Healthcare) with sonicated salmon sperm DNA and rotated for 4 h at 4 °C followed by centrifugation at 1,900 × g for 5 min. The supernatant was divided into two aliquots and incubated with 4 μg of anti-HNF4α antibody or control normal goat IgG (Santa Cruz Biotechnology) for 16 h at 4 °C and incubated with 40 μl of a 50% slurry of protein G-Sepharose 4 Fast Flow. The reaction mixture was centrifuged at 1,900 × g for 5 min at 4 °C, and the pellet was collected. The pellet was washed for 5 min at 4 °C with 1 ml of RIPA-1 buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, and 0.1% sodium deoxycholate), RIPA-1 buffer containing 300 mm NaCl, LiCl wash solution (10 mm Tris-HCl (pH 8.0), 0.25 m LiCl, 1 mm EDTA, 0.5% Nonidet P-40, and 0.5% sodium deoxycholate), and Tris-EDTA. Then ChIP direct elution buffer (10 mm Tris-HCl (pH 8.0), 300 mm NaCl, 5 mm EDTA, and 0.5% SDS) was added to the pellet and incubated for 16 h at 65 °C for decross-linking. After treatment with RNase A for 30 min at 37 °C and proteinase K for 2 h at 55 °C, DNA was purified and amplified by PCR and real-time PCR using the ΔΔCt method. Enrichment of HNF4α binding was normalized to the input samples and expressed as -fold enrichment as compared with the control normal IgG antibody. Sequences for the primers are shown in supplemental Table S4.
3′-UTR assays
3′-UTRs of the mouse candidate genes for the miR-194 and miR-192 were amplified by PCR and cloned downstream of firefly luciferase gene in pGL3-control (Promega). As positive controls, complementary sequences for miR-194 and miR-192 (c-miR-194 and c-miR-192) were self-annealed and then cloned into the XbaI site of the pGL3-control. For 3′-UTR assay, pGL3-control/3′-UTR plasmids and pGL4.74 with 20 nm miR-194 or miR-192 mimic were co-transfected into HEK293T cells using HilyMax (Dojindo, Kumamoto, Japan) as a transfection reagent and assayed by the Dual-Glo Luciferase Assay System (Promega). Sequences for the primers of the 3′-UTRs are shown in supplemental Table S5.
Statistical analysis
All values are expressed as the mean ± S.D. All data were analyzed by unpaired Student's t test for significant differences between the mean values of each group.
Author contributions
A. M., M. K., and Y. T. conducted most of the experiments and analyzed the results. T. N. conducted experiments of microarray analysis. S. S. and T. Maeda conducted experiments of promoter analysis. C. S., T. Matsuta, M. S., M. A., and N. N. conducted experiments of real-time PCR and ChIP analysis. Y. I. conceived the idea for the project and wrote the paper with F. J. G.
Supplementary Material
Acknowledgment
We acknowledge members of the Inoue Laboratory for discussions and comments on the manuscript.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grant-in-aid for Scientific Research 25460490) (to Y. I.), the JGC-S scholarship Foundation (to Y. I.), Gunma University, Akita University, Nagoya University, Collaborative Investigation Project (to Y. I.), and Gunma University Medical Innovation Project (to Y. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2 and Tables S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HNF
- hepatocyte nuclear factor
- miRNA
- microRNA
- miR-194/192
- miR-194 and miR-192
- Hnf4aΔH mice
- liver-specific Hnf4a-null mice, Hnf4af/f mice, Hnf4a-floxed mice
- HCC
- hepatocellular carcinoma
- EMT
- epithelial-mesenchymal transition
- Otc
- ornithine transcarbamylase
- Gyg1
- glycogenin 1
- Fzd6
- frizzled-6
- Alcam
- activated leukocyte cell adhesion molecule
- Ereg
- epiregulin
- Cln4b
- Cullin 4B
- Msn
- moesin
- Cdh2
- N-cadherin
- Hbegf
- heparin-binding epidermal growth factor
- Ptpn2
- tyrosine protein phosphatase non-receptor type 2
- Itga9
- integrin α9
- Socs2
- suppressor of cytokine signaling 2
- Dnmt3a
- DNA methyltransferase 3A
- Tln2
- talin 2
- Hook3
- hook microtubule-tethering protein 3
- Sephs1
- selenophosphate synthetase 1
- SUMO
- small ubiquitin-like modifier
- Arfgef1
- ADP-ribosylation factor guanine nucleotide-exchange factor 1
- Rsad2
- radical S-adenosylmethionine domain-containing 2
- qRT-PCR
- quantitative RT-PCR
- pri-miR
- primary miRNA
- C/EBPα
- CCAAT-enhancer-binding protein α
- RIPA
- radioimmune precipitation assay
- qPCR
- quantitative PCR.
References
- 1. Schrem H., Klempnauer J., and Borlak J. (2002) Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol. Rev. 54, 129–158 [DOI] [PubMed] [Google Scholar]
- 2. Kuo C. J., Conley P. B., Chen L., Sladek F. M., Darnell J. E. Jr., and Crabtree G. R. (1992) A transcriptional hierarchy involved in mammalian cell-type specification. Nature 355, 457–461 [DOI] [PubMed] [Google Scholar]
- 3. Sladek F. M., and Seidel S. D. (2001) Hepatocyte nuclear factor 4α, in Nuclear Receptor and Genetic Disease (Burris T. P., and McCabe E., eds) pp. 309–361, Academic Press, San Diego, CA [Google Scholar]
- 4. Späth G. F., and Weiss M. C. (1998) Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J. Cell Biol. 140, 935–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen W. S., Manova K., Weinstein D. C., Duncan S. A., Plump A. S., Prezioso V. R., Bachvarova R. F., and Darnell J. E. Jr. (1994) Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 8, 2466–2477 [DOI] [PubMed] [Google Scholar]
- 6. Parviz F., Matullo C., Garrison W. D., Savatski L., Adamson J. W., Ning G., Kaestner K. H., Rossi J. M., Zaret K. S., and Duncan S. A. (2003) Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 34, 292–296 [DOI] [PubMed] [Google Scholar]
- 7. Hayhurst G. P., Lee Y. H., Lambert G., Ward J. M., and Gonzalez F. J. (2001) Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Inoue Y., Hayhurst G. P., Inoue J., Mori M., and Gonzalez F. J. (2002) Defective ureagenesis in mice carrying a liver-specific disruption of hepatocyte nuclear factor 4α (HNF4α). HNF4α regulates ornithine transcarbamylase in vivo. J. Biol. Chem. 277, 25257–25265 [DOI] [PubMed] [Google Scholar]
- 9. Inoue Y., Yu A. M., Inoue J., and Gonzalez F. J. (2004) Hepatocyte nuclear factor 4α is a central regulator of bile acid conjugation. J. Biol. Chem. 279, 2480–2489 [DOI] [PubMed] [Google Scholar]
- 10. Bonzo J. A., Ferry C. H., Matsubara T., Kim J. H., and Gonzalez F. J. (2012) Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J. Biol. Chem. 287, 7345–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walesky C., Edwards G., Borude P., Gunewardena S., O'Neil M., Yoo B., and Apte U. (2013) Hepatocyte nuclear factor 4α deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 57, 2480–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Odom D. T., Zizlsperger N., Gordon D. B., Bell G. W., Rinaldi N. J., Murray H. L., Volkert T. L., Schreiber J., Rolfe P. A., Gifford D. K., Fraenkel E., Bell G. I., and Young R. A. (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303, 1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wiwi C. A., Gupte M., and Waxman D. J. (2004) Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4α-deficient mice. Mol. Endocrinol. 18, 1975–1987 [DOI] [PubMed] [Google Scholar]
- 14. Ambros V. (2004) The functions of animal microRNAs. Nature 431, 350–355 [DOI] [PubMed] [Google Scholar]
- 15. Taganov K. D., Boldin M. P., and Baltimore D. (2007) MicroRNAs and immunity: tiny players in a big field. Immunity 26, 133–137 [DOI] [PubMed] [Google Scholar]
- 16. Chen Y., and Verfaillie C. M. (2014) MicroRNAs: the fine modulators of liver development and function. Liver Int. 34, 976–990 [DOI] [PubMed] [Google Scholar]
- 17. Wang X. W., Heegaard N. H., and Orum H. (2012) MicroRNAs in liver disease. Gastroenterology 142, 1431–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., and Tuschl T. (2002) Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 [DOI] [PubMed] [Google Scholar]
- 19. Kutay H., Bai S., Datta J., Motiwala T., Pogribny I., Frankel W., Jacob S. T., and Ghoshal K. (2006) Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell Biochem. 99, 671–678 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Bai S., Nasser M. W., Wang B., Hsu S. H., Datta J., Kutay H., Yadav A., Nuovo G., Kumar P., and Ghoshal K. (2009) MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J. Biol. Chem. 284, 32015–32027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coulouarn C., Factor V. M., Andersen J. B., Durkin M. E., and Thorgeirsson S. S. (2009) Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 28, 3526–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Z. Y., Xi Y., Zhu W. N., Zeng C., Zhang Z. Q., Guo Z. C., Hao D. L., Liu G., Feng L., Chen H. Z., Chen F., Lv X., Liu D. P., and Liang C. C. (2011) Positive regulation of hepatic miR-122 expression by HNF4α. J. Hepatol. 55, 602–611 [DOI] [PubMed] [Google Scholar]
- 23. Yin C., Wang P. Q., Xu W. P., Yang Y., Zhang Q., Ning B. F., Zhang P. P., Zhou W. P., Xie W. F., Chen W. S., and Zhang X. (2013) Hepatocyte nuclear factor-4α reverses malignancy of hepatocellular carcinoma through regulating miR-134 in the DLK1-DIO3 region. Hepatology 58, 1964–1976 [DOI] [PubMed] [Google Scholar]
- 24. Hatziapostolou M., Polytarchou C., Aggelidou E., Drakaki A., Poultsides G. A., Jaeger S. A., Ogata H., Karin M., Struhl K., Hadzopoulou-Cladaras M., and Iliopoulos D. (2011) An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147, 1233–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ning B. F., Ding J., Liu J., Yin C., Xu W. P., Cong W. M., Zhang Q., Chen F., Han T., Deng X., Wang P. Q., Jiang C. F., Zhang J. P., Zhang X., Wang H. Y., et al. (2014) Hepatocyte nuclear factor 4α-nuclear factor-κB feedback circuit modulates liver cancer progression. Hepatology 60, 1607–1619 [DOI] [PubMed] [Google Scholar]
- 26. Inoue Y., Peters L. L., Yim S. H., Inoue J., and Gonzalez F. J. (2006) Role of hepatocyte nuclear factor 4α in control of blood coagulation factor gene expression. J. Mol. Med. 84, 334–344 [DOI] [PubMed] [Google Scholar]
- 27. Santangelo L., Marchetti A., Cicchini C., Conigliaro A., Conti B., Mancone C., Bonzo J. A., Gonzalez F. J., Alonzi T., Amicone L., and Tripodi M. (2011) The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4α. Hepatology 53, 2063–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hino K., Tsuchiya K., Fukao T., Kiga K., Okamoto R., Kanai T., and Watanabe M. (2008) Inducible expression of microRNA-194 is regulated by HNF-1α during intestinal epithelial cell differentiation. RNA 14, 1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jenkins R. H., Martin J., Phillips A. O., Bowen T., and Fraser D. J. (2012) Transforming growth factor beta1 represses proximal tubular cell microRNA-192 expression through decreased hepatocyte nuclear factor DNA binding. Biochem. J. 443, 407–416 [DOI] [PubMed] [Google Scholar]
- 30. Pichiorri F., Suh S. S., Rocci A., De Luca L., Taccioli C., Santhanam R., Zhou W., Benson D. M. Jr., Hofmainster C., Alder H., Garofalo M., Di Leva G., Volinia S., Lin H. J., Perrotti D., et al. (2010) Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 18, 367–381 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Meng Z., Fu X., Chen X., Zeng S., Tian Y., Jove R., Xu R., and Huang W. (2010) miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 52, 2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krützfeldt J., Rösch N., Hausser J., Manoharan M., Zavolan M., and Stoffel M. (2012) MicroRNA-194 is a target of transcription factor 1 (Tcf1, HNF1α) in adult liver and controls expression of frizzled-6. Hepatology 55, 98–107 [DOI] [PubMed] [Google Scholar]
- 33. Le X. F., Almeida M. I., Mao W., Spizzo R., Rossi S., Nicoloso M. S., Zhang S., Wu Y., Calin G. A., and Bast R. C. Jr. (2012) Modulation of microRNA-194 and cell migration by HER2-targeting trastuzumab in breast cancer. PLoS One 7, e41170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim T., Veronese A., Pichiorri F., Lee T. J., Jeon Y. J., Volinia S., Pineau P., Marchio A., Palatini J., Suh S. S., Alder H., Liu C. G., Dejean A., and Croce C. M. (2011) p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 208, 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazarevich N. L., Cheremnova O. A., Varga E. V., Ovchinnikov D. A., Kudrjavtseva E. I., Morozova O. V., Fleishman D. I., Engelhardt N. V., and Duncan S. A. (2004) Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 39, 1038–1047 [DOI] [PubMed] [Google Scholar]
- 36. Yin C., Lin Y., Zhang X., Chen Y. X., Zeng X., Yue H. Y., Hou J. L., Deng X., Zhang J. P., Han Z. G., and Xie W. F. (2008) Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4α gene. Hepatology 48, 1528–1539 [DOI] [PubMed] [Google Scholar]
- 37. Cui C. Y., Klar J., Georgii-Heming P., Fröjmark A. S., Baig S. M., Schlessinger D., and Dahl N. (2013) Frizzled6 deficiency disrupts the differentiation process of nail development. J. Invest. Dermatol. 133, 1990–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bengochea A., de Souza M. M., Lefrançois L., Le Roux E., Galy O., Chemin I., Kim M., Wands J. R., Trepo C., Hainaut P., Scoazec J. Y., Vitvitski L., and Merle P. (2008) Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br. J. Cancer 99, 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S. (2012) Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mandard S., Stienstra R., Escher P., Tan N. S., Kim I., Gonzalez F. J., Wahli W., Desvergne B., Müller M., and Kersten S. (2007) Glycogen synthase 2 is a novel target gene of peroxisome proliferator-activated receptors. Cell. Mol. Life Sci. 64, 1145–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu H., Yang Y., Ji Q., Zhao W., Jiang B., Liu R., Yuan J., Liu Q., Li X., Zou Y., Shao C., Shang Y., Wang Y., and Gong Y. (2012) CRL4B catalyzes H2AK119 monoubiquitination and coordinates with PRC2 to promote tumorigenesis. Cancer Cell 22, 781–795 [DOI] [PubMed] [Google Scholar]
- 42. Green E. M., Mas G., Young N. L., Garcia B. A., and Gozani O. (2012) Methylation of H4 lysines 5, 8 and 12 by yeast Set5 calibrates chromatin stress responses. Nat. Struct. Mol. Biol. 19, 361–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grozeva D., Carss K., Spasic-Boskovic O., Parker M. J., Archer H., Firth H. V., Park S. M., Canham N., Holder S. E., Wilson M., Hackett A., Field M., Floyd J. A., UK10K Consortium, Hurles M., et al. (2014) De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 94, 618–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shahbazian M. D., and Grunstein M. (2007) Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100 [DOI] [PubMed] [Google Scholar]
- 45. Geiss-Friedlander R., and Melchior F. (2007) Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 46. Zhou W., Hannoun Z., Jaffray E., Medine C. N., Black J. R., Greenhough S., Zhu L., Ross J. A., Forbes S., Wilmut I., Iredale J. P., Hay R. T., and Hay D. C. (2012) SUMOylation of HNF4α regulates protein stability and hepatocyte function. J. Cell Sci. 125, 3630–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang X., He Y., Lee K. H., Dubois W., Li Z., Wu X., Kovalchuk A., Zhang W., and Huang J. (2013) Rap2b, a novel p53 target, regulates p53-mediated pro-survival function. Cell Cycle 12, 1279–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riese D. J. 2nd, and Cullum R. L. (2014) Epiregulin: roles in normal physiology and cancer. Semin. Cell Dev. Biol. 28, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao M., He H. W., Sun H. X., Ren K. H., and Shao R. G. (2009) Dual knockdown of N-ras and epiregulin synergistically suppressed the growth of human hepatoma cells. Biochem. Biophys. Res. Commun. 387, 239–244 [DOI] [PubMed] [Google Scholar]
- 50. Dapito D. H., Mencin A., Gwak G. Y., Pradere J. P., Jang M. K., Mederacke I., Caviglia J. M., Khiabanian H., Adeyemi A., Bataller R., Lefkowitch J. H., Bower M., Friedman R., Sartor R. B., Rabadan R., et al. (2012) Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levin T. G., Powell A. E., Davies P. S., Silk A. D., Dismuke A. D., Anderson E. C., Swain J. R., and Wong M. H. (2010) Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 139, 2072–2082.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin Z., Selaru F. M., Cheng Y., Kan T., Agarwal R., Mori Y., Olaru A. V., Yang J., David S., Hamilton J. P., Abraham J. M., Harmon J., Duncan M., Montgomery E. A., and Meltzer S. J. (2011) MicroRNA-192 and -215 are upregulated in human gastric cancer in vivo and suppress ALCAM expression in vitro. Oncogene 30, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma L., Wang J., Lin J., Pan Q., Yu Y., and Sun F. (2014) Cluster of differentiation 166 (CD166) regulated by phosphatidylinositide 3-Kinase (PI3K)/AKT signaling to exert its anti-apoptotic role via yes-associated protein (YAP) in liver cancer. J. Biol. Chem. 289, 6921–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Charafe-Jauffret E., Monville F., Bertucci F., Esterni B., Ginestier C., Finetti P., Cervera N., Geneix J., Hassanein M., Rabayrol L., Sobol H., Taranger-Charpin C., Xerri L., Viens P., Birnbaum D., et al. (2007) Moesin expression is a marker of basal breast carcinomas. Int. J. Cancer 121, 1779–1785 [DOI] [PubMed] [Google Scholar]
- 55. Wang C. C., Liau J. Y., Lu Y. S., Chen J. W., Yao Y. T., and Lien H. C. (2012) Differential expression of moesin in breast cancers and its implication in epithelial-mesenchymal transition. Histopathology 61, 78–87 [DOI] [PubMed] [Google Scholar]
- 56. Lee Y. H., Sauer B., and Gonzalez F. J. (1998) Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1α knockout mouse. Mol. Cell. Biol. 18, 3059–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.