

Abstract
Background and Purpose
Results regarding protective effects of dipeptidyl peptidase 4 (DPP4) inhibitors in renal ischaemia–reperfusion injury (IRI) are conflicting. Here we have compared structurally unrelated DPP4 inhibitors in a model of renal IRI.
Experimental Approach
IRI was induced in uninephrectomized male rats by renal artery clamping for 30 min. The sham group was uninephrectomized but not subjected to IRI. DPP4 inhibitors or vehicle were given p.o. once daily on three consecutive days prior to IRI: linagliptin (1.5 mg·kg−1·day−1), vildagliptin (8 mg·kg−1·day−1) and sitagliptin (30 mg·kg−1·day−1). An additional group received sitagliptin until study end (before IRI: 30 mg·kg−1·day−1; after IRI: 15 mg·kg−1·day−1).
Key Results
Plasma‐active glucagon‐like peptide type 1 (GLP‐1) increased threefold to fourfold in all DPP4 inhibitor groups 24 h after IRI. Plasma cystatin C, a marker of GFR, peaked 48 h after IRI. Compared with the placebo group, DPP4 inhibition did not reduce increased plasma cystatin C levels. DPP4 inhibitors ameliorated histopathologically assessed tubular damage with varying degrees of drug‐specific efficacies. Renal osteopontin expression was uniformly reduced by all DPP4 inhibitors. IRI‐related increased renal cytokine expression was not decreased by DPP4 inhibition. Renal DPP4 activity at study end was significantly inhibited in the linagliptin group, but only numerically reduced in the prolonged/dose‐adjusted sitagliptin group. Active GLP‐1 plasma levels at study end were increased only in the prolonged/dose‐adjusted sitagliptin treatment group.
Conclusions and Implications
In rats with renal IRI, DPP4 inhibition did not alter plasma cystatin C, a marker of glomerular function, but may protect against tubular damage.
Abbreviations
- AKI
acute kidney injury
- CKD
chronic kidney disease
- DPP4
dipeptidyl peptidase 4
- ESRD
end‐stage renal disease
- GIP
gastric inhibitory polypeptide
- GLP‐1
glucagon‐like peptide type 1
- IRI
ischaemia reperfusion injury
- KC
keratinocyte chemoattractant
- MCP‐1
monocyte chemotactic protein 1
- UniNX
uni‐nephrectomy
Introduction
Acute kidney injury (AKI) is a frequent and increasingly prevalent syndrome, defined by a rapid deterioration of kidney function (Kam Tao Li et al., 2013; Kramann et al., 2015), which is associated with high morbidity and mortality (Lafrance and Miller, 2010; Kam Tao Li et al., 2013). The incidence of AKI is increasing and expected to double over the next 10 years (Silver et al., 2015). Data from recent observational studies suggest an association between AKI and chronic kidney disease (CKD) and end‐stage renal disease (ESRD) (Coca et al., 2012). About 14% of patients with AKI progress to stage 4 CKD, and more severe cases of AKI are associated with a higher risk for progression into CKD (Chawla et al., 2011; Coca et al., 2012). The high incidence of AKI is discussed as one possible explanation for the increasing burden of CKD and ESRD (Coca et al., 2012; Belayev and Palevsky, 2014). There are many risk factors and causes for AKI (Hsu et al., 2008; Basile et al., 2012). Among these, the most prominent aetiology of AKI is acute tubular necrosis as a consequence of an ischaemic or nephrotoxic insult (Basile et al., 2012; Kramann et al., 2015). In a clinical setting, kidney hypoxia or ischaemia–reperfusion injury (IRI) inevitably occurs during surgery involving renal or aortic vascular occlusion. Of all cases of hospital‐acquired AKI, approximately 30–40% occur during surgery, making the perioperative period one of the leading causes of AKI (Uchino et al., 2005; Thakar et al., 2009). The most vulnerable renal tissue with regards to both ischaemic and toxic insults are tubular epithelial cells (Bonventre and Yang, 2011; Kramann et al., 2015). These cells rely on aerobic respiration and have a high metabolic demand due to their involvement in fluid and electrolyte reabsorption, making them uniquely susceptible to IRI (Uchida and Endou, 1988; Kramann et al., 2015).
Dipeptidyl peptidase 4 (DPP4) inhibitors are a new drug class approved for the treatment of type 2 diabetes. Its anti‐diabetic effects are elicited by an inhibition of the ubiquitously expressed serine exopeptidase DPP4, which mediates the degradation of incretins such as glucagon‐like peptide 1 (GLP‐1) and gastric inhibitory polypeptide (GIP) (Mentlein, 1999). Moreover, DPP4 also has several other substrates, including growth factors, neuro‐ and vasoactive peptides and chemokines (Mentlein, 1999). Based on the structure, DPP4 inhibitors can be broadly divided into peptidomimetics (e.g. vildagliptin) and non‐peptidomimetics (e.g. linagliptin, sitagliptin) (Baetta and Corsini, 2011; Ceriello et al., 2014). Although their chemical structures are different, all DPP4 inhibitors are substrate competitive active site binders but show different binding characteristics on the DPP4 protein (Schnapp et al., 2016).
Preclinical evidence suggests that DPP4 inhibitors have protective effects on IRI of the heart and lungs (Sauvé et al., 2010; Jungraithmayr et al., 2012; Hocher et al., 2013). As DPP4 is strongly expressed in the kidney (Mentlein, 1999), DPP4 inhibition might also elicit protective effects in renal IRI. Several studies already investigated putative protective effects in renal IRI, but with conflicting results. Therefore, we studied the effects of three structurally different DPP4 inhibitors ‐ linagliptin, vildagliptin and sitagliptin ‐ on the outcome of IR‐induced AKI in uni‐nephrectomized rats. One additional arm (sitagliptin) was treated throughout the entire experiment and dose adjusted.
Methods
Animals
All animal care and experimental procedures were approved by the Committee on the Ethics of Animal Experiments (Landesamt fuer Gesundheit und Soziales), Berlin, Germany. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; Kilkenny et al., 2012; McGrath & Lilley, 2015). Male Wistar rats (10 sham animals, 14 placebo‐treated animals and 14 per each treatment group), aged 5–6 weeks, weighing 200–250 g, were obtained from Charles River, Germany, and housed in groups under standardized specific pathogen‐free conditions (12 h light/dark cycle, 23°C, humidity of 50–60%, rat type IV cages) with food and water ad libitum. After an acclimatization period of 1 week, all animals were subjected to uni‐nephrectomy. Two weeks later, the remaining kidney was exposed to IRI by clamping the renal artery for 30 min, and sham surgery was performed without clamping. Animals were randomly allocated to six different groups: sham, placebo, linagliptin (1.5 mg·kg−1·day−1), vildagliptin (8 mg·kg−1·day−1), sitagliptin (30 mg·kg−1·day−1) and prolonged/dose‐adjusted sitagliptin, a group which was treated with sitagliptin until the end of study in order to block the DPP4 activity during the entire experiment. In this group, treatment started with 30 mg·kg−1 once daily on two consecutive days prior to IRI; after that, the dose was adjusted to 15 mg·kg−1·day−1 because of renal failure. All groups received DPP4 inhibitor treatment or vehicle via gavage once daily on two consecutive days prior to IRI and on the day of surgery 2 h before IRI. Doses in the current study were selected based on previous DPP4 inhibitor studies in similar settings and calculated on AUC (Chaykovska et al., 2011; Connelly et al., 2014).
Baseline blood samples were taken from the tail vein. Blood samples were collected at baseline 24 h before IRI, and 24, 48, 72 h and 1 week (168 h) after IRI. Afterwards, animals were killed under deep isoflurane anaesthesia and organs were harvested. Two sham animals were excluded from the study because of critical weight loss occurring shortly after the surgical sham intervention (probably due to infection), reducing the n‐value of the sham group to eight. The n‐value variation of measured plasma parameters among the analysed experimental groups originated from either limited plasma sample volume or from plasma samples that gave a NaN (not a number) result in the respective assay. The n‐value variation of histological readouts among the experimental groups occurred because of suboptimal tissue processing in one batch of samples due to technical difficulties of the automated tissue processor. The whole animal experiment was performed in a randomized fashion using six arms. Rats were randomly allocated to six different arms, with each arm containing about the same number of animals from every treatment group. The animal study was performed by different investigators than the following experiments, ensuring that all analyses were performed in a blinded fashion.
Biomarkers of renal function and injury
Plasma cystatin C, a biomarker of glomerular filtration rate and osteopontin, a biomarker of tubular damage, were measured by automated immunoassays (Myriad RBM, Inc., Austin, Texas, USA).
DPP4 activity and GLP‐1 measurements
Whole blood was collected into lithium heparin‐treated tubes and plasma was separated by centrifugation. Activity of plasma DPP4 was assessed using a previously reported method (Chaykovska et al., 2011). A 20 μL volume of plasma was diluted with 30 μL of DPP4 assay buffer (100 mmol·L−1 Tris, 100 mmol·L−1 NaCl, adjusted to pH 7.8 with HCl) and mixed with 50 μL substrate (final concentration 100 mmol·L−1 H‐Ala‐Pro‐7‐amido‐4‐trifluoromethylcoumarin). After incubation at room temperature for 10 min, fluorescence of the wells was determined using a Wallac Victor 1420 Multilabel Counter at an excitation wavelength of 405 nm and an emission wavelength of 535 nm. Active (uncleaved, 7–36 amide or 7–37) GLP‐1 was detected in plasma samples using a commercially available multiarray assay system (K150JWC from Meso Scale Discovery, Rockwell, Maryland, USA) following the instructions provided by the supplier. This antibody only detects active GLP‐1 (7–36 amide or 7–37 GLP‐1) but not cleaved and inactive GLP‐1 (9–36 amide or 9–37 GLP‐1).
Cytokine detection from plasma and kidney homogenates
For preparation of kidney extracts, 50 mg of tissue was lysed in 500 μL of Meso Scale Discovery (Gaithersburg, MD, USA) lysis buffer (MSD R60TX‐2) with protease inhibitor cocktails from Thermo Fisher, Germany (1861278), and Sigma, Germany (P5726), using FastPrep devices (MP Biomedicals) at 4°C, 30 s, 6000 × g. Homogenates were centrifuged for 10 min, 4°C at 10 000 × g. Supernatants were adjusted to protein concentration of 20 mg·mL−1 and used for cytokine analysis and DPP4 activity measurements.
Cytokine concentrations of kidney tissue (50 μL of lysate) and plasma samples (undiluted sample) were analysed by Meso Scale Discovery (Gaithersburg, MD, USA) immunoassays according to the manufacturer's protocols using a MSD Custom Rat Cytokine kit (K153AOH‐2). Concentrations of CCL2 were analysed in 25 μL aliquots by the Rat Mcp‐1 Ultra Sensitive Kit (K153AYC‐2). Absolute concentrations were calculated by comparison with a series of standard dilutions of the respective cytokine measured on the same plate. In both plasma and renal homogenates, concentrations of CCL2, IFN‐γ, IL‐10, IL‐13, IL‐1β, IL‐4, IL‐5, IL‐6, keratinocyte chemoattractant (KC) and TNF‐α were analysed.
Renal DPP4 activity
For DPP4 activity, 2 μL of kidney homogenate extracts were diluted with 130 mL of HEPES buffer (10 mM) pH 7.6 and incubated for 10 min at 37°C in 96‐well plates. A total of 100 μL of Gly‐Pro‐pNA (4‐nitroaniline, Sigma Aldrich, Germany) was added as DPP4 substrate (1 mM solution) and kinetics (20 min, every 60 s) were detected at 405 nm. The reaction was observed to be linear up until 11 min; thus, this timeframe was used to assess renal DPP4 activity. Because Gly‐Pro‐pNA is an artificial DPP4 substrate and high protease activity is present in the kidney, we added 10 μM sitagliptin to a parallel set of extracts, to define DPP4‐specific activity baseline. Values were given as specific DPP4 activity in (δ OD405 nm·min−1) by subtracting sitagliptin‐inhibited baseline. For graphic display and statistical analysis, treatment group‐specific AUC of the linear kinetic period (0–11 min) were calculated (minutes were converted to seconds).
Histological assessment of kidney tubular necrosis and tubular dilatation
For pathohistological evaluation, all samples were embedded in paraffin, cut in 3 μm sections and stained with haematoxylin–eosin. Tubular dilatation and tubular necrosis were analysed using a semiquantitative grading score on haematoxylin–eosin stained kidney samples by two blinded independent investigators. At least 25 viewing fields of the outer medulla were randomly chosen with 200× magnification per each kidney sample. Tubular dilatation was assessed by a three‐point scoring system, where 1 indicates <25% tubules show dilation and thinned epithelium, 2 indicates 25 to 50% tubules show dilation and thinned epithelium, and 3 indicates >50% tubules show dilation and thinned epithelium (Blydt‐Hansen et al., 2003). Tubular necrosis was classified according to a 1–5 score as follows: grade 1 (no to mild damage) = less than 10% tubular necrosis, grade 2 (mild to moderate damage) = 10 to 25% tubular cell necrosis, grade 3 (moderate to severe damage) = 25 to 50% tubular cell necrosis, grade 4 (severe damage) = 50 to 75% and grade 5 (severe to very severe) = greater than 75% tubular cell necrosis (Park et al., 2008).
Western blot analysis of renal protein expression
Western blots were performed as previously described (Putra et al., 2014). Briefly, renal samples containing both cortex and medulla were crushed in a metal mortar after cooling in liquid nitrogen. Protein was extracted using a urea/thiourea buffer [2 M thiourea, 7 M urea, 2% SDS, 1% DTT and protease inhibitor (Complete Mini, Cat. no.: 11 697 498 001, Roche)]. Protein extracts were separated by SDS‐PAGE employing a 10% polyacrylamide gel. Following SDS‐PAGE gels were blotted to nitrocellulose membrane (AmershamTM HybondTM ECL, GE Healthcare) using a Biorad Trans‐Blot semidry blotter and transfer buffer (184 mM glycine, 24 mM Tris, 20% methanol). The anti‐osteopontin antibody (sc‐21742, Santa Cruz Biotechnology) was used at a dilution of 1:1000 and the anti‐housekeeping protein actin antibody (A5060, Sigma‐Aldrich) at a dilution of 1:10 000. The signals were developed using enhanced chemiluminescence. The developed membranes were digitalized using a 600 dpi scanner resolution and analysed with AlphaEaseFCTM.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analyses were performed using SPSS Version 20 and Graph Pad Prism 6. Quantitative data in text and tables are expressed as mean ± SD. In the figures, data are presented as means ± SEM. Data were analysed for normal distribution using the Kolmogorov–Smirnov test. Not normally distributed data were analysed using the Kruskal–Wallis test followed by a Dunn post hoc test. For normally distributed data, group comparisons were performed employing one‐way ANOVA. Heterogeneity of variance was assessed using Levene's test. If results of the test were significant (P < 0.05), Welch's F was used. According to heterogeneity of variances, either a Bonferroni (equal variances assumed) or a Games–Howell (unequal variances assumed) post hoc test was used, as recommended (Field, 2013; Muth, 2014). Post hoc tests were only run if F achieved P < 0.05. For the comparison of parameters measured over time, two‐way ANOVA was used followed by a Bonferroni post hoc test. To analyse if two parameters are correlated, Pearson bivariate correlation analysis was employed. P values lower than 0.05 were considered statistically significant.
Materials
Linagliptin [BI1356; 8‐[(3R)‐aminopiperidin‐1‐yl]‐7‐(but‐2‐yn‐1‐yl)‐3‐methyl‐1‐[(4‐methyl‐quinazolin‐2‐yl)methyl]‐3,7‐dihydro‐1H‐purine‐2,6‐dione] was developed and synthesized by Boehringer Ingelheim Pharma GmbH and Co. KG (Biberach an der Riss, Germany), and vildagliptin [(2S)‐1‐[2‐[(3‐hydroxy‐1‐adamantyl)amino]acetyl]pyrrolidine‐2‐carbonitrile] and sitagliptin [(3R)‐3‐amino‐1‐[3‐(trifluoromethyl)‐6,8‐dihydro‐5H‐[1,2,4]triazolo[4,3‐a]pyrazin‐7‐yl]‐4‐(2,4,5‐trifluorophenyl)butan‐1‐one] were from Sequoia, Oxford, UK. The compounds were dissolved in 0.5% Natrosol, and were administered p.o.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Basic characteristics of the treatment groups are displayed in Table 1. There were no statistical significant differences regarding baseline and final body weight.
Table 1.
Basic animal characteristics and summary of kidney parameters
| Sham (8) | Placebo (14) | Linagliptin (14) | Vildagliptin (14) | Sitagliptin (14) | Prolonged/dose‐adjusted sitagliptin (14) | |
|---|---|---|---|---|---|---|
| Baseline weight (g) | 335.1 ± 10.7 | 333.1 ± 32.8 | 327.3 ± 24.1 | 327.1 ± 30.8 | 337.1 ± 21.6 | 321.3 ± 27.3 |
| Final Weight (g) | 362.4 ± 15.9 | 345.3 ± 26.4 | 344.9 ± 21.9 | 343.7 ± 28.0 | 351.6 ± 21.6 | 334.0 ± 28.8 |
| Kidney Weight (g) | 1.7 ± 0.2* | 2.5 ± 0.7 | 2.3 ± 0.2 | 2.5 ± 0.5 | 2.4 ± 0.5 | 2.2 ± 0.3 |
| Rel. Kidney Weight (g·g−1 BW × 100) | 0.46 ± 0.04* | 0.72 ± 0.17 | 0.66 ± 0.10 | 0.74 ± 0.12 | 0.68 ± 0.12 | 0.66 ± 0.12 |
| Tubular dilatation score | 1.1 ± 0.16* | 2.8 ± 0.17 | 2.4 ± 0.41 | 2.8 ± 0.26 | 2.3 ± 0.46* | 2.0 ± 0.52* |
| Tubular necrosis score | 1.2 ± 0.15* | 4.5 ± 0.48 | 4.0 ± 0.78 | 4.5 ± 0.62 | 3.6 ± 0.94 | 3.0 ± 1.03* |
| Renal osteopontin expression | 0.75 ± 0.41* | 1.17 ± 0.43 | 0.68 ± 0.22* | 0.70 ± 0.34* | 0.76 ± 0.34* | 0.67 ± 0.29* |
P < 0.05, significantly different from placebo; one‐way ANOVA. Values displayed are means ± SD.
Kidney function
The impact of IRI on kidney function was assessed by plasma cystatin C and osteopontin levels. IRI induced steadily increasing plasma cystatin C concentrations, which peaked 48 h after clamping. The placebo group displayed significantly higher cystatin C levels 48 h after IRI compared with the sham group (Figure 1A). Treatment with DPP4 inhibitors did not significantly affect plasma cystatin C concentrations compared with placebo at any point (Figure 1A). Furthermore, IRI induced an increase in plasma osteopontin levels, which peaked 72 h after clamping (Figure 1B). Compared with the sham group, the placebo group displayed significantly elevated plasma osteopontin levels at 24 h, 48 h and 72 h after clamping (Figure 1B). Treatment with DPP4 inhibitors resulted in numerically lower plasma osteopontin concentrations compared with the placebo group, but these differences did not reach statistical significance at any point (Figure 1B).
Figure 1.
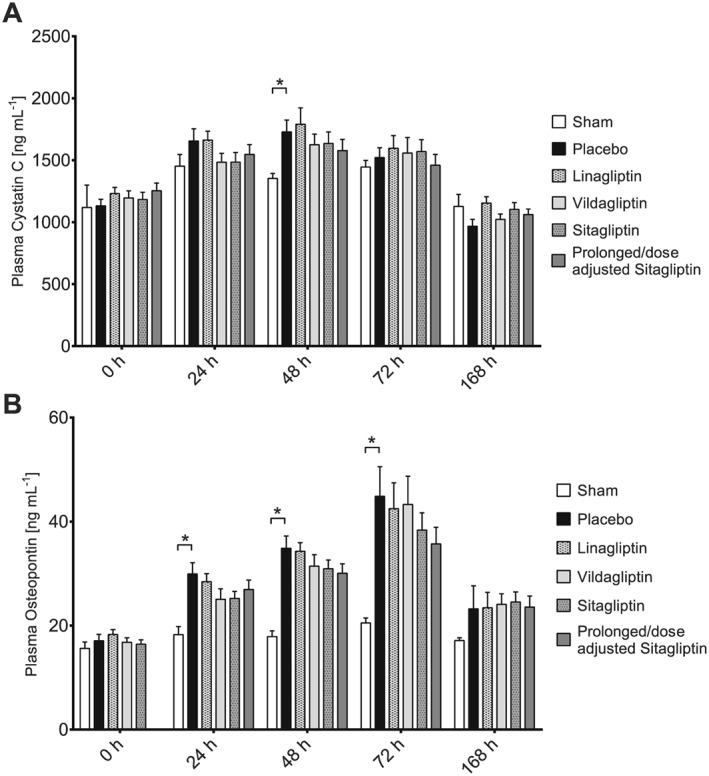
Concentrations of plasma cystatin C and osteopontin over the course of the study. (A) Plasma cystatin C concentrations; n‐values: sham (0 h: n = 8; 24 h: n = 8; 48 h: n = 8; 72 h: n = 8; 168 h: n = 8), placebo (0 h: n = 12; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14), linagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 13; 168 h: n = 14), vildagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14), sitagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 13), prolonged/dose‐adjusted sitagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14). Values shown are means ± SEM. *P < 0.05, significantly different as indicated; two‐way ANOVA. (B) Plasma osteopontin concentrations; n‐values: sham (0 h: n = 8; 24 h: n = 8; 48 h: n = 8; 72 h: n = 8; 168 h: n = 8), placebo (0 h: n = 12; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14), linagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 13; 168 h: n = 14), vildagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14), sitagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 13), prolonged/dose‐adjusted sitagliptin (0 h: n = 14; 24 h: n = 14; 48 h: n = 14; 72 h: n = 14; 168 h: n = 14). Values shown are means ± SEM. *P < 0.05, significantly different as indicated; two‐way ANOVA.
DPP4 activity and active GLP‐1 levels in plasma
All groups with DPP4 inhibitor treatment displayed a significantly reduced DPP4 activity 24 h after induction of IRI compared with the placebo group (Figure 2A). Plasma concentrations of active GLP‐1 were significantly increased in all DPP4 inhibitor treatment groups versus the placebo group, up to 24 h after IRI (Figure 2B). At the end of the study, 168 h after IRI, active GLP‐1 plasma concentrations of all treatment groups except the prolonged/dose‐adjusted sitagliptin group returned to baseline. The prolonged/dose‐adjusted sitagliptin group, as expected, still displayed significantly elevated active GLP‐1 plasma levels (Figure 2C).
Figure 2.
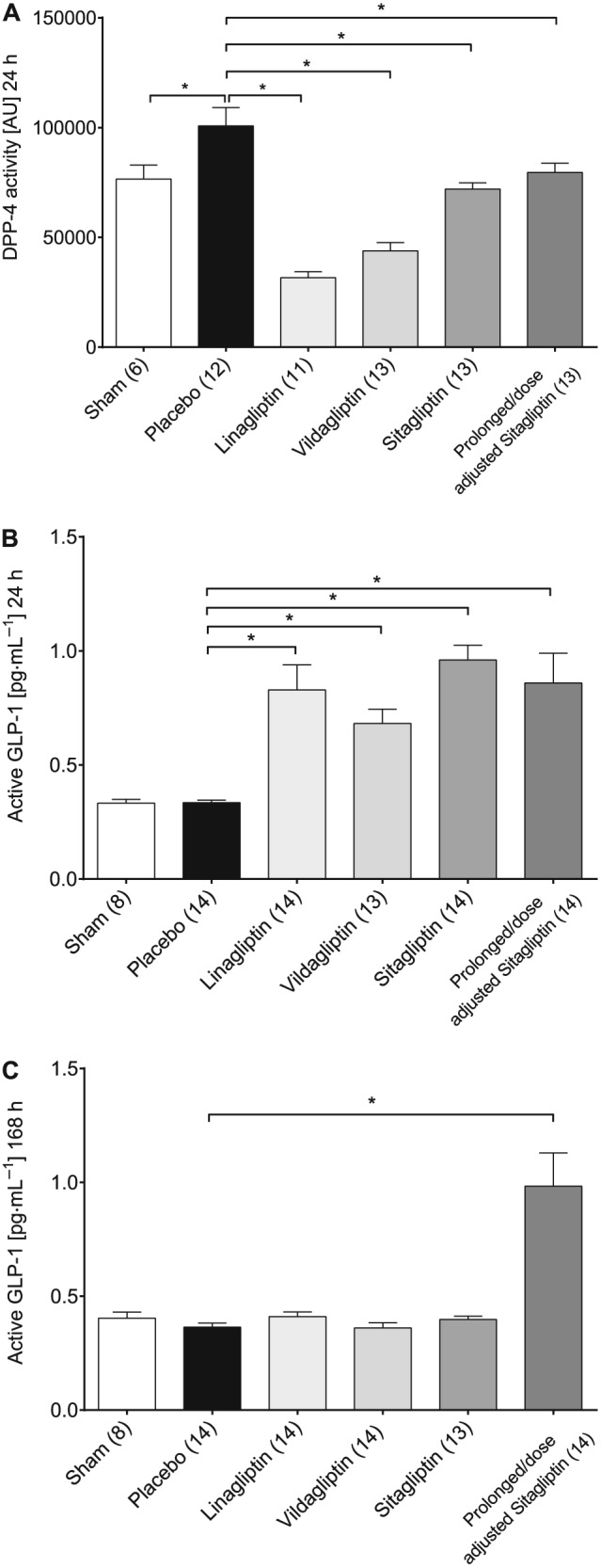
Plasma activity of DPP4 (A), plasma concentration of active GLP‐1 24 h (B) and 168 h post IRI (C). Values shown are means ± SEM. *P < 0.05, significantly different as indicated; Kruskal–Wallis test or one‐way ANOVA.
Renal DPP4 activity
The AUC of renal DPP4 activity, assessed 168 h after IRI, was not significantly different between the sham and placebo group. Kidney homogenates of linagliptin‐treated animals still displayed a significantly reduced DPP4 activity compared with placebo‐treated animals. Also renal DPP4 activity of prolonged/dose‐adjusted sitagliptin‐treated animals was numerically reduced, but did not reach statistical significance. Vildagliptin‐ and sitagliptin‐treated animals displayed no reduction of renal DPP4 activity (Figure 3).
Figure 3.
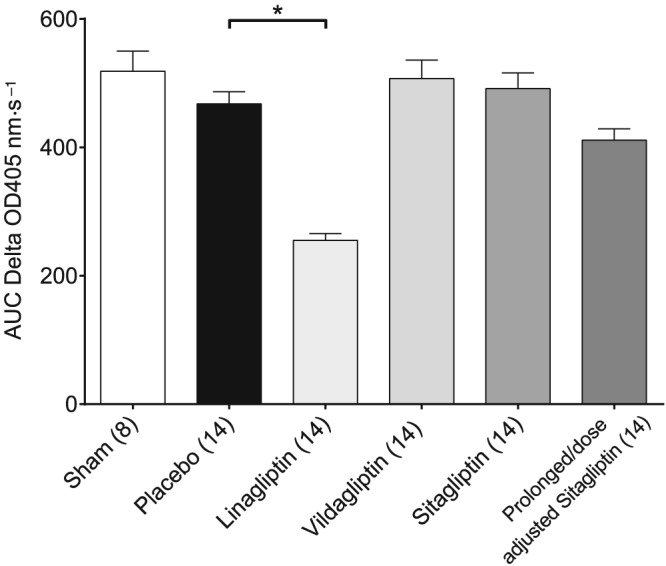
Renal DPP4 activity. Mean area AUC was calculated for each treatment group‐specific δ OD 405 nm·s−1 curve, and resulting mean values were analysed. Values shown are means ± SEM. *P < 0.05, significantly different as indicated; one‐way ANOVA.
Kidney morphology
Kidney weight, both absolute and in relation to body weight, was significantly increased in the placebo group compared with the sham group (Table 1). IRI caused a significant increase of the tubular necrosis and tubular dilatation score in the placebo group compared with the sham group (Table 1; Figure 4B). The prolonged/dose‐adjusted treatment with sitagliptin led to a significant decrease of both tubular necrosis and dilatation. Regular sitagliptin treatment significantly reduced tubular dilatation, but only numerically affected tubular necrosis score. Also linagliptin treatment resulted in a non‐significant, numerically decreased tubular necrosis and dilatation score, whereas vildagliptin treatment was without effect (Table 1; Figure 4C).
Figure 4.
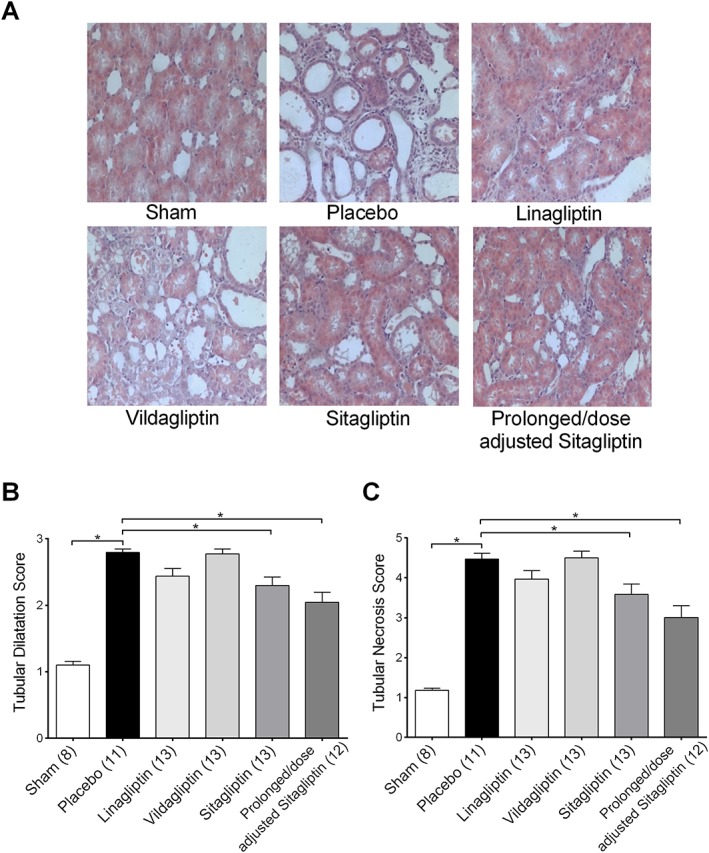
Tubular dilatation and tubular necrosis score. (A) Representative microphotographs from kidney sections of each treatment group. (B) Tubular dilatation score. (C) Tubular necrosis score. Values shown are means ± SEM. *P < 0.05, significantly different as indicated; one‐way ANOVA.
Furthermore, to assess the validity of the histological data, a correlation analysis was performed between peak plasma cystatin C (48 h) and peak plasma osteopontin (72 h) concentrations and tubular necrosis score. Peak plasma cystatin C concentrations showed only a marginal correlation (Figure 5A), while peak plasma osteopontin levels were strongly and significantly correlated with the extent of tubular necrosis and dilatation (Figure 5B). Based on the observation of a strong positive correlation between plasma osteopontin concentrations and the extent of tubular damage, together with a trend towards a reduction of plasma osteopontin by DPP4 inhibition, renal expression of osteopontin was analysed by Western blot (Figure 6A, B). Compared with the sham group, renal osteopontin expression was significantly increased in the placebo group. DPP4 inhibition prevented any increases in renal osteopontin levels, leading to significantly reduced osteopontin levels compared with the placebo group in all DPP4 inhibitor treatment groups (Figure 6A, B). Contrary to plasma osteopontin, renal osteopontin levels were not correlated to the extent of tubular damage (data not shown).
Figure 5.
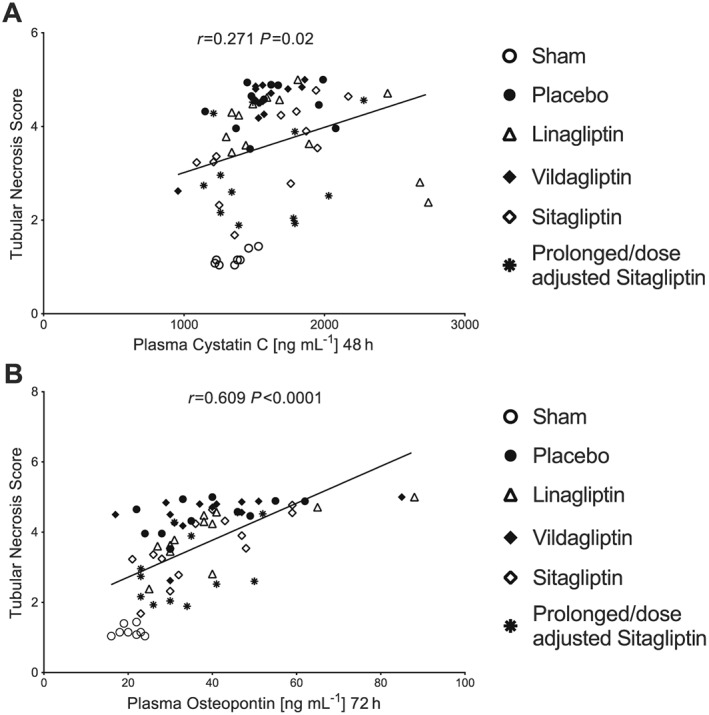
Correlation between plasma cystatin C/osteopontin and tubular necrosis score.
Figure 6.
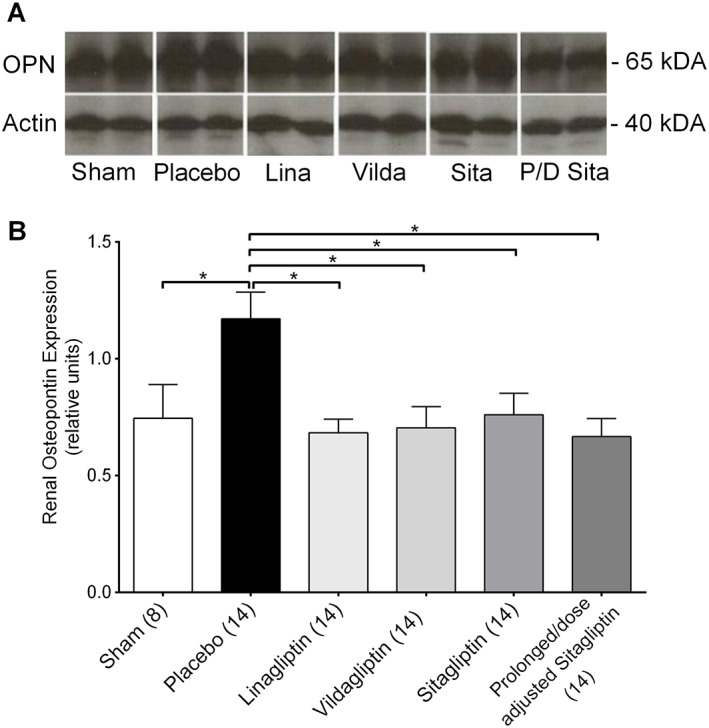
Renal expression of osteopontin. (A) Representative Western blot bands showing the expression of osteopontin (OPN, upper band; around 65 kDA) and the expression of actin (lower band; around 40 kDa). (B) Protein expression of renal osteopontin (random units). Values shown are means ± SEM. *P < 0.05, significantly different as indicated; one‐way ANOVA.
Plasma and renal cytokine levels
Analysis of cytokine levels in plasma collected at study end showed no significant alterations among all groups (Table 2). However, the renal expression of several cytokines was differently regulated. IRI lead to a significant up‐regulation of CCL2, IL‐1β, IL‐5 and KC in placebo‐treated animals compared with sham animals. Linagliptin, vildagliptin and sitagliptin treatment did not result in any significant differences compared with the placebo group. However, prolonged/dose‐adjusted sitagliptin‐treated animals showed significantly higher concentrations of IL‐6 compared with the placebo group (Table 2).
Table 2.
Plasma and renal cytokine levels
| Sham | Placebo | Linagliptin | Vildagliptin | Sitagliptin | Prolonged/dose‐adjusted sitagliptin | |
|---|---|---|---|---|---|---|
| Plasma | ||||||
| CCL2 pg·mL−1 | 2864.7 ± 684.6 [8] | 4477.9 ± 2262.9 [12] | 3293.2 ± 2675.1 [13] | 3420.1 ± 1277.0 [13] | 2841.0 ± 944.0 [14] | 2829.9 ± 832.6 [13] |
| IFN‐γ pg·mL−1 | 0.8 ± 0.6 [8] | 1.1 ± 0.8 [13] | 1.4 ± 0.8 [14] | 1.0 ± 0.6 [13] | 1.2 ± 0.6 [14] | 1.4 ± 0.7 [13] |
| IL‐10 pg·mL−1 | 5.3 ± 2.1 [8] | 8.8 ± 5.4 [13] | 8.4 ± 3.7 [14] | 8.4 ± 3.1 [13] | 7.1 ± 2.9 [14] | 10 ± 4.1 [13] |
| IL‐13 pg·mL−1 | 1.3 ± 0.5 [8] | 1.4 ± 0.6 [13] | 1.6 ± 0.7 [14] | 1.2 ± 0.6 [13] | 1.4 ± 0.5 [14] | 1.7 ± 0.8 [13] |
| IL‐1β plasma pg·mL−1 | 3.1 ± 2.3 [7] | 3.4 ± 2.2 [9] | 3.7 ± 2.5 [12] | 3.0 ± 2.8 [10] | 5.6 ± 4.6 [11] | 4.8 ± 3.9 [12] |
| IL‐4 pg·mL−1 | 0.3 ± 0.1 [8] | 0.4 ± 0.3 [13] | 0.5 ± 0.2 [14] | 0.4 ± 0.2 [13] | 0.4 ± 0.1 [14] | 0.5 ± 0.2 [13] |
| IL‐5 pg·mL−1 | 5.2 ± 3.7 [5] | 4.3 ± 4.0 [11] | 8.9 ± 4.7 [13] | 6.1 ± 5.0 [11] | 4.1 ± 3.4 [14] | 5.9 ± 4.6 [11] |
| IL‐6 pg·mL−1 | 14.3 ± 11.5 [8] | 17.0 ± 16.9 [13] | 27.4 ± 16.5 [13] | 18.3 ± 18.3 [13] | 17.5 ± 11.1 [14] | 23.4 ± 16.4 [13] |
| KC pg·mL−1 | 53.3 ± 22.1 [8] | 75.1 ± 47.5 [13] | 50.4 ± 37.6 [14] | 54.9 ± 29.1 [13] | 38.6 ± 8.5 [14] | 49.9 ± 16.2 [13] |
| TNF‐α pg·mL−1 | 2.6 ± 0.7 [7] | 2.6 ± 1.1 [14] | 2.1 ± 0.7 [14] | 2.1 ± 0.7 [13] | 2.6 ± 0.9 [14] | 3.0 ± 0.8 [13] |
| Kidney | ||||||
| CCL2 pg·mL−1 | 59.1 ± 24.7 [8]* | 192.2 ± 98.5 [14] | 247.6 ± 164.6 [14] | 221.2 ± 172.9 [14] | 165.7 ± 75.7 [14] | 266.0 ± 187.4 [14] |
| IFN‐γ pg·mL−1 | 4.0 ± 1.2 [8] | 3.0 ± 1.0 [14] | 3.0 ± 1.0 [14] | 2.9 ± 1.3 [14] | 3.1 ± 0.9 [14] | 3.6 ± 1.4 [14] |
| IL‐10 pg·mL−1 | 1.4 ± 0.5 [6] | 2.9 ± 2.3 [14] | 3.1 ± 2.0 [14] | 3.4 ± 1.7 [14] | 3.8 ± 3.6 [14] | 3.5 ± 2.0 [14] |
| IL‐13 pg·mL−1 | 2.4 ± 1.7 [8] | 2.6 ± 0.6 [14] | 2.7 ± 0.6 [14] | 3.1 ± 0.9 [14] | 2.8 ± 0.8 [14] | 3.2 ± 0.9 [14] |
| IL‐1β plasma pg·mL−1 | 44.2 ± 8.7 [8]* | 76.4 ± 19.7 [14] | 81.7 ± 17.6 [14] | 74.5 ± 24.1 [14] | 60.4 ± 16.8 [14] | 66.6 ± 20.7 [14] |
| IL‐4 pg·mL−1 | 0.4 ± 0.3 [8] | 0.5 ± 0.2 [14] | 0.5 ± 0.1 [14] | 0.5 ± 0.1 [14] | 0.5 ± 0.3 [14] | 0.6 ± 0.2 [14] |
| IL‐5 pg·mL−1 | 17.3 ± 5.6 [8]* | 34.0 ± 14.3 [14] | 35.1 ± 10.6 [14] | 36.6 ± 16.2 [14] | 30.3 ± 8.7 [14] | 37.1 ± 14.4 [14] |
| IL‐6 pg·mL−1 | 139.7 ± 40.9 [8] | 149.7 ± 41.9 [14] | 150.8 ± 33.1 [14] | 181.6 ± 65.0 [14] | 172.9 ± 44.0 [14] | 200.2 ± 57.5 [14]* |
| KC pg·mL−1 | 12.3 ± 2.9 [8]* | 48.6 ± 41.9 [14] | 45.8 ± 15.6 [14] | 49.5 ± 29.9 [14] | 31.1 ± 23.3 [14] | 44.5 ± 40.6 [14] |
| TNF‐α pg·mL−1 | 8.5 ± 1.5 [8] | 11.9 ± 2.9 [14] | 11.3 ± 2.2 [14] | 11.9 ± 4.4 [14] | 9.4 ± 2.5 [14] | 11.0 ± 3.5 [14] |
Values displayed are means ± SD.
P < 0.05, significantly different from placebo; Kruskal–Wallis test or one‐way ANOVA.
Discussion
In the current study, the effects of the DPP4 inhibitors linagliptin, vildagliptin and sitagliptin on renal IRI were analysed. DPP4 inhibition induced a reduced plasma DPP4 activity in all treatment groups, paralleled by an increase of active GLP‐1 concentrations, 24 h after IRI. Renal DPP4 activity measured at study end was significantly inhibited in the linagliptin group, but only numerically reduced in the prolonged/dose‐adjusted sitagliptin group. Plasma cystatin C concentrations peaked 48 h after IRI and were higher in the placebo group compared with the sham group. Plasma osteopontin concentrations showed a peak at 72 h, with higher plasma levels in the placebo compared with the sham group. DPP4 inhibitor treatment did not affect plasma cystatin C concentrations at any point but showed a trend towards a reduction of plasma osteopontin levels. Histological evaluation of the kidney samples revealed tubular necrosis and tubular dilatation in all groups subjected to IRI. DPP4 inhibition by prolonged/dose‐adjusted sitagliptin treatment (and partly by regular sitagliptin treatment) reduced both tubular necrosis and dilatation. Peak plasma cystatin C concentrations at 48 h were weakly correlated, and peak plasma osteopontin concentrations at 72 h were strongly correlated with the extent of tubular necrosis and dilatation. Analysis of renal osteopontin expression revealed a significant reduction in all DPP4 inhibitor groups, compared with placebo. Measurement of renal cytokine expression demonstrated an IRI‐related up‐regulation of CCL2, IL‐1β, IL‐5 and KC. Furthermore, a significant up‐regulation of renal IL‐6 expression, which was absent in the placebo group, was observed in prolonged/dose‐adjusted sitagliptin treated animals. Taken together, DPP4 inhibition did not affect the impairment of glomerular function due to IRI, measured by assessing plasma cystatin C concentrations, but did provide beneficial effects, in terms of tubular damage. One limitation of the current study, similar to the majority of previous studies (Vaghasiya et al., 2011; Glorie et al., 2012; Nuransoy et al., 2015; Youssef et al., 2015), is that no urinary parameters were investigated, which could have improved assessments of the effects of DPP4 inhibition in renal IRI on glomerular function.
To the best of our knowledge, there are six published studies and one conference abstract that investigated DPP4 inhibition in renal IRI (Vaghasiya et al., 2011; Daniel et al., 2012; Glorie et al., 2012; Chen et al., 2013; Chang et al., 2015; Nuransoy et al., 2015; Youssef et al., 2015). Details of these studies are summarized in Table 3. Albeit employing a different assessment, quantitative and qualitative scoring systems, six of these studies were able to demonstrate a beneficial effect of DPP4 inhibition in renal IRI on tubular damage (Vaghasiya et al., 2011; Glorie et al., 2012; Chen et al., 2013; Chang et al., 2015; Nuransoy et al., 2015; Youssef et al., 2015). Results of the current study substantiate these previous findings. In regard to beneficial effects of DPP4 inhibition on glomerular function, two studies, both investigating sitagliptin in renal IRI, also did not demonstrate any significant results (Chang et al., 2015; Nuransoy et al., 2015). Four other studies did observe significant effects of DPP4 inhibition on glomerular function (Vaghasiya et al., 2011; Glorie et al., 2012; Chen et al., 2013; Youssef et al., 2015). However, both conflicting and supporting data have to be interpreted carefully, as the designs of all the available studies are very heterogeneous (Table 3). In a study of Vaghasiya et al. (2011), the authors did show protective effects of DPP4 inhibition on glomerular function, but only in a diabetic rat model of renal IRI. Glorie et al. (2012), using a non‐diabetic rat model (left renal pedicle clamping for 30 min followed by right nephrectomy), induced mild renal impairment with peak glomerular dysfunction 12 h after IRI. The authors demonstrated a significant reduction of serum creatinine at 12 h and to a minor extent at 48 h after IRI. Contrary to all other available studies that used oral drug administration, vildagliptin was administered intravenously 15 min prior to IRI (Glorie et al., 2012). Chen et al. (2013) did observe positive effects of DPP4 inhibition on parameters of glomerular function, yet excessive supratherapeutic doses of sitagliptin (600 mg·kg−1·day−1) were used, which might elicit a positive effect on renal function in this animal model, but could possibly pose a risk and would be hard to translate to humans (Bloomfield et al., 2009). Setting results of the current study into context with results from comparable studies (Glorie et al., 2012; Youssef et al., 2015), it has to be taken into account that in the current study, different to all previous studies, IRI was induced 2 weeks after uni‐nephrectomy, in a state of mild renal impairment (Arsenijevic et al., 2015). It has been shown that this additional insult results in a more pronounced post‐ischaemic functional impairment, which could have masked minor positive effects of DPP4 inhibition (Vercauteren et al., 1999). However, inducing renal IRI on top of mild renal impairment might represent the clinical situation better, as impaired renal function is a strong risk factor for acute renal failure, and the prevalence for acute renal failure in healthy individuals is low (Hsu et al., 2008). Furthermore, the different DPP4 inhibitor dosing, treatment timing and pharmacokinetic profiles must also be considered as other factors influencing the study results (Table 3). Vildagliptin, for example, has a considerably lower IC50 towards DPP‐8 and DPP‐9 than other DPP4 inhibitors, as has linagliptin towards fibroblast activating protein (Thomas et al., 2008; Huan et al., 2015). Depending on the drug doses used, together with putative disease‐related expression changes, different outcomes in animal studies might be observed. Furthermore, there are differences in the pharmacokinetics of DPP4 inhibitors. In the current study, renal DPP4 activity was measured at study end 168 h after induction of IRI. Although linagliptin treatment was discontinued on the day of IRI induction, renal DPP4 activity was still significantly inhibited 1 week later. This reduction of renal DPP4 activity was not accompanied by increased plasma active GLP‐1 levels, suggesting a minor role of renal DPP4 in inactivating circulating GLP‐1 levels. Contrary to data from the linagliptin group, no significant inhibition could be observed in the vildagliptin and sitagliptin group. The prolonged and dose‐adjusted sitagliptin group only showed a trend towards reduced renal DPP4 activity, but displayed significantly increased active GLP‐1 plasma levels. A possible explanation of the different renal DPP4 inhibition can be based on pharmacokinetic properties. Linagliptin exhibits a 10–15 times greater apparent volume of distribution, with a higher tissue‐to‐blood ratio and more than 1000‐fold lower koff rate from the DPP4 protein compared with sitagliptin (Filippatos et al., 2014; Huan et al., 2015; Nakamaru et al., 2016; Schnapp et al., 2016). Regardless of renal DPP4 activity, the strongest effects on tubular damage were observed in the prolonged/dose‐adjusted sitagliptin group. In this group, contrary to all other treatment groups, active GLP‐1 levels were increased until the end of the study, which, in context with previous findings, could be one underlying mechanism (Chang et al., 2015). In summary, different models of IRI, different study durations and different drug dosing, employing structurally different DPP4 inhibitors, could have been influential factors underlying the different results in AKI settings with regards to DPP4 inhibition, IRI and glomerular function. Another important reason for the observed heterogeneity in study results might be that current clinical definitions of AKI are focused on changes of glomerular function, by measuring suitable surrogate parameters, such as creatinine or cystatin C (Kellum, 2015). These definitions ignore the actual morphological consequences of renal IRI, which is an impairment of tubular function due to tubular alterations such as tubular necrosis and tubular dilatation (Racusen and Solez, 1986; Endre et al., 2013). In the current study, IRI had a modest effect on increasing plasma cystatin C, although tubular damage was very pronounced. Furthermore, correlation analysis displayed only a weak positive association between plasma cystatin C and tubular necrosis but a very strong positive correlation between plasma osteopontin, a marker of tubular damage and tubular necrosis (Xie et al., 2001; Zhang et al., 2010). A previous study showed that DPP4 inhibition leads to a reduction of plasma osteopontin levels (Chaykovska et al., 2011). Also in the current study, DPP4 inhibition lowered plasma osteopontin levels, but this effect did not reach statistical significance. However, the most pronounced effect in this regard was observed for the prolonged/dose‐adjusted sitagliptin treatment, which also was associated with the strongest amelioration of histomorphological tubular damage and the only group with significantly elevated active GLP‐1 plasma levels until study end. This might indicate an advantage of a prolonged treatment regimen. Analysis of renal osteopontin expression showed that osteopontin was significantly up‐regulated in the placebo group and uniformly down‐regulated to levels of the sham group in all DPP4 inhibitor groups. To the best of our knowledge, this is the first observation of a down‐regulating effect elicited by DPP4 inhibition on renal osteopontin expression. Osteopontin, a secreted 44 kDa glycoprotein, is up‐regulated in injured kidney cells and is involved in immune and anti‐apoptotic processes next to a variety of other functions (Xie et al., 2001). Previous studies have demonstrated that osteopontin expression is increased in renal IRI and that its modulation can alter the course of disease (Padanilam et al., 1996; Wagner et al., 2003; Zhang et al., 2010). However, results are conflicting, attributing to osteopontin both a positive and a negative impact on renal IRI (Padanilam et al., 1996; Nambi et al., 1997; Noiri et al., 1999; Persy et al., 1999, 2003; Zhang et al., 2010). Interpretation of results on osteopontin from the current study indicate a mild positive impact of reduced osteopontin levels in renal IRI, but leave room for speculation. Other differently designed studies with kidney assessment at various earlier time points are needed to properly investigate the seemingly complex relationship between DPP4 inhibition, renal and plasma osteopontin levels and renal damage. Given the role of osteopontin in inflammation, cytokine levels in plasma (obtained at study end) and cytokine levels in kidney homogenates were measured. No significant changes in plasma cytokine levels could be observed, which might be due to the long interval between IRI and cytokine measurements (168 h post IRI). Cytokine levels in renal homogenates of placebo‐treated animals displayed a significant up‐regulation of CCL2, IL‐1β, IL‐5 and KC levels. DPP4 inhibitor treatment did not significantly affect these IRI‐related changes in renal cytokine expression. The prolonged/dose‐adjusted sitagliptin group displayed a weak but significant up‐regulation of renal IL‐6 expression. As an up‐regulation of IL‐6 was absent from the placebo group, the observed increase of IL‐6 in the prolonged/dose‐adjusted sitagliptin group, which displayed significantly elevated active GLP‐1 plasma levels, might have been due to an GIP‐mediated effect on IL‐6 expression (Ellingsgaard et al., 2011; Timper et al., 2013; Kahles et al., 2014; Timper et al., 2016).
Table 3.
Studies investigating DPP4 inhibition in models of renal IRI
| Study | Design | IRI | Outcome | Overall |
|---|---|---|---|---|
|
Sitagliptin Nuransoy et al. (2015) |
Healthy, female rats, 14 days, 5 mg·kg−1·day−1
pre IRI |
60 min IRI + UniNX |
↓ Oxidative stress ↑ Antioxidative enzymes ↓ Tubular damage ↔ Serum creatinine 24 h post IRI |
Tubular effects |
|
Sitagliptin Vaghasiya et al. (2011) |
Diabetic, male/female rats, 14 days, 5 mg·kg−1·day−1
pre IRI |
30 min bilateral IRI |
↓ Serum AST, creatinine, urea nitrogen 24 h post IRI ↑ Antioxidative enzymes ↓ Tubular damage |
Tubular and glomerular effects |
|
Sitagliptin Youssef et al. (2015) |
Healthy, male rats, 5 mg·kg−1
5/22 h post IRI |
30 min bilateral IRI |
↓ Serum urea, creatinine, cystatin C 24 h post IRI ↓ Tubular damage 24 h |
Tubular and glomerular effects |
|
Sitagliptin Chang et al. (2015) |
Healthy, male rats, 3 days, 300/600 mg·kg−1·day−1 post IRI |
60 min bilateral IRI |
↑ GLP‐1 and GLP‐1R ↓ Tubular damage 72 h post IRI Numerical reduction of serum creatinine 72 h post IRI |
Tubular effects |
|
Sitagliptin Chen et al. (2013) |
Healthy, male rats, 3 days, 600 mg·kg−1·day−1 post IRI | 60 min bilateral IRI |
↓ Serum creatinine 24 h and 72 h post IRI ↓ Tubular damage 24 and 72 h post IRI ↓ Inflammation/apoptosis/oxidative stress |
Tubular and glomerular effects |
|
Vildagliptin Glorie et al., 2012 |
Healthy, male rats, 1/10 mg·kg−1
i.v. 15 pre IRI |
30 min IRI + UniNX |
↓ Serum creatinine 12 and 48 h post IRI ↓ Tubular necrosis 12 h post IRI, ↔ at 48 h post IRI ↓ Inflammation/apoptosis/oxidative stress |
Tubular and glomerular effects |
| DPP4−/− rats Daniel et al. (2012) (Abstract) | DPP4−/− rats | 45 min IRI + UniNX |
↑ Serum creatinine 24 h post IRI versus wildtype ↑ Tubular damage versus wildtype ↑ Macrophage infiltration versus wildtype |
Detrimental tubular and glomerular effects |
Taking into account that the design of the available studies of DPP4 inhibition in renal IRI is very heterogenous (Table 3), results of the current study are in agreement with previous studies, especially regarding beneficial effects of DPP4 inhibition on IRI‐mediated tubular damage. Although only some studies employed a quantitative assessment of histopathological kidney changes, all of the previously mentioned studies observed positive effects of DPP4 inhibition on histological readouts of kidney injury (Vaghasiya et al., 2011; Glorie et al., 2012; Chen et al., 2013; Chang et al., 2015; Nuransoy et al., 2015; Youssef et al., 2015). One strength of the current study is the head‐to‐head comparison of several structurally unrelated DPP4 inhibitors, also in a clinical relevant regimen of dose adjustment. Taken together, our data suggest DPP4 inhibition is safe in models of IRI and might elicit beneficial effects. In this respect, a continuous treatment with DPP4 inhibitors or having patients on such drugs, when expecting an increased risk for AKI (e.g. during cardiac and transplantation surgeries) could be of benefit. Detrimental effects of DPP4 deficiency, which were observed in DPP4‐deficient rats subjected to IRI (Daniel et al., 2012), were not observed by any other study including the current one. This might be related to differences of a pharmacological inhibition of DPP4 in comparison with a full genetic knockout, probably due to missing crucial protein/protein interaction of DPP4 (Mulvihill and Drucker, 2014). Results of the current study support earlier evidence that DPP4 inhibition exerts beneficial effects in renal tubules in models of IRI. Regarding favourable effects of DPP4 inhibition on glomerular function, more homogenously designed studies, measuring the same parameters of glomerular function, are needed.
In conclusion, this study adds substantial evidence to previous findings, showing that DPP4 inhibition in IRI is safe and might exert beneficial effects on the renal tubules. Clinical evaluation of the current findings is still missing, but results of this and previous studies in regard to drug safety could be of clinical importance. Current evidence implies that no drug discontinuation would be needed in clinical situations with an elevated risk for renal IRI, such as cardiac surgery. However, due to the impairment of renal function, dose adjustment would be necessary (Scheen, 2015). From a practical point of view, this would favour linagliptin in a clinical setting, as it is the only DPP4 inhibitor, which does not need to be dose‐adjusted in renal impairment (Scheen, 2015).
Author contributions
B.H., C.R. and K.v.W. designed the experiment. C.R., K.v.W. and O.T. performed the animal experiment. A.M.S., L.G.F., V.A., C.C., S.E.D.P. and A.A.H. performed the molecular biological and histological analyses and statistical evaluation of the acquired data. C.R. analysed the final data set and wrote the initial manuscript. T.K. performed analysis of renal and plasma DPP4 activity and contributed to the discussion. J.R. analysed cytokine levels in plasma and renal homogenates. All authors contributed to revising and editing of the manuscript and gave their approval to publishing the final manuscript.
Conflict of interest
T.K. and J.R. are research employees of Boehringer Ingelheim Pharma. The other authors declare no conflicts interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
B.H. received a research grant from Boehringer Ingelheim Pharma for this project.
Reichetzeder, C. , von Websky, K. , Tsuprykov, O. , Mohagheghi Samarin, A. , Falke, L. G. , Dwi Putra, S. E. , Hasan, A. A. , Antonenko, V. , Curato, C. , Rippmann, J. , Klein, T. , and Hocher, B. (2017) Head‐to‐head comparison of structurally unrelated dipeptidyl peptidase 4 inhibitors in the setting of renal ischemia reperfusion injury. British Journal of Pharmacology, 174: 2273–2286. doi: 10.1111/bph.13822.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Cajot J‐F, Dulloo AG, Montani J‐P (2015). Uninephrectomy in rats on a fixed food intake results in adipose tissue lipolysis implicating spleen cytokines. Front Physiol 6: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetta R, Corsini A (2011). Pharmacology of dipeptidyl peptidase‐4 inhibitors: similarities and differences. Drugs 71: 1441–1467. [DOI] [PubMed] [Google Scholar]
- Basile DP, Anderson MD, Sutton TA (2012). Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev LY, Palevsky PM (2014). The link between acute kidney injury and chronic kidney disease. Curr Opin Nephrol Hypertens 23: 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield DM, Krishna R, Hreniuk D, Hickey L, Ghosh K, Bergman AJ et al. (2009). A thorough QTc study to assess the effect of sitagliptin, a DPP4 inhibitor, on ventricular repolarization in healthy subjects. J Clin Pharmacol 49: 937–946. [DOI] [PubMed] [Google Scholar]
- Blydt‐Hansen TD, Katori M, Lassman C, Ke B, Coito AJ, Iyer S et al. (2003). Gene transfer‐induced local heme oxygenase‐1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J Am Soc Nephrol 14: 745–754. [DOI] [PubMed] [Google Scholar]
- Bonventre JV, Yang L (2011). Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriello A, Sportiello L, Rafaniello C, Rossi F (2014). DPP‐4 inhibitors: pharmacological differences and their clinical implications. Expert Opin Drug Saf 13 (Suppl 1): S57–S68. [DOI] [PubMed] [Google Scholar]
- Chang M, Chen C, Chen Y, Wu Y, Zhen Y, Leu S et al. (2015). Sitagliptin protects rat kidneys from acute ischemia‐reperfusion injury via upregulation of GLP‐1 and GLP‐1 receptors. Acta Pharmacol Sin 36: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE (2011). The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaykovska L, von Websky K, Rahnenführer J, Alter M, Heiden S, Fuchs H et al. (2011). Effects of DPP‐4 inhibitors on the heart in a rat model of uremic cardiomyopathy. PLoS One 6: e27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐T, Tsai T‐H, Yang C‐C, Sun C‐K, Chang L‐T, Chen H‐H et al. (2013). Exendin‐4 and sitagliptin protect kidney from ischemia‐reperfusion injury through suppressing oxidative stress and inflammatory reaction. J Transl Med 11: 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coca SG, Singanamala S, Parikh CR (2012). Chronic kidney disease after acute kidney injury: a systematic review and meta‐analysis. Kidney Int 81: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly KA, Bowskill BB, Advani SL, Thai K, Chen L‐H, Kabir MG et al. (2014). Dipeptidyl peptidase‐4 inhibition improves left ventricular function in chronic kidney disease. Clin Invest Med 37: E172. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Grigo C, Wang Z, von Hörsten S, Amann K (2012). CD26/DPP4 deficiency impairs kidney function in the ischemia/reperfusion model in the rat. Poster (P351) presented at: 4. Jahrestagung der Deutschen Gesellschaft für Nephrologie. 08.10.2012. Hamburg. Germany. Available at: http://www.aey-congresse.com/nephrokongress2012/Sitzung.aspx?PSID=1921&PAID=4866 [Google Scholar]
- Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT et al. (2011). Interleukin‐6 enhances insulin secretion by increasing glucagon‐like peptide‐1 secretion from L cells and alpha cells. Nat Med 17: 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endre ZH, Kellum JA, Di Somma S, Doi K, Goldstein SL, Koyner JL et al. (2013). Differential diagnosis of AKI in clinical practice by functional and damage biomarkers: workgroup statements from the tenth Acute Dialysis Quality Initiative Consensus Conference. Contrib Nephrol 182: 30–44. [DOI] [PubMed] [Google Scholar]
- Field A (2013). Discovering Statistics Using IBM SPSS Statistics. SAGE: London, UK. [Google Scholar]
- Filippatos TD, Athyros VG, Elisaf MS (2014). The pharmacokinetic considerations and adverse effects of DPP‐4 inhibitors [corrected]. Expert Opin Drug Metab Toxicol 10: 787–812. [DOI] [PubMed] [Google Scholar]
- Glorie LLF, Verhulst A, Matheeussen V, Baerts L, Magielse J, Hermans N et al. (2012). DPP4 inhibition improves functional outcome after renal ischemia‐reperfusion injury. Am J Physiol Renal Physiol 303: F681–F688. [DOI] [PubMed] [Google Scholar]
- Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T (2013). The novel DPP‐4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol 167: 87–93. [DOI] [PubMed] [Google Scholar]
- Hsu CY, Ordoñez JD, Chertow GM, Fan D, McCulloch CE, Go AS (2008). The risk of acute renal failure in patients with chronic kidney disease. Kidney Int 74: 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan Y, Jiang Q, Liu J, Shen Z (2015). Establishment of a dipeptidyl peptidases (DPP) 8/9 expressing cell model for evaluating the selectivity of DPP4 inhibitors. J Pharmacol Toxicol Methods 71: 8–12. [DOI] [PubMed] [Google Scholar]
- Jungraithmayr W, De Meester I, Matheeussen V, Baerts L, Arni S, Weder W (2012). CD26/DPP‐4 inhibition recruits regenerative stem cells via stromal cell‐derived factor‐1 and beneficially influences ischaemia‐reperfusion injury in mouse lung transplantation. Eur J Cardiothorac Surg 41: 1166–1173. [DOI] [PubMed] [Google Scholar]
- Kahles F, Meyer C, Möllmann J, Diebold S, Findeisen HM, Lebherz C et al. (2014). GLP‐1 secretion is increased by inflammatory stimuli in an IL‐6‐dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes 63: 3221–3229. [DOI] [PubMed] [Google Scholar]
- Kam Tao Li P, Burdmann EA, Mehta RL (2013). Acute kidney injury: global health alert. J Nephropathol 2: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellum JA (2015). Diagnostic criteria for acute kidney injury: present and future. Crit Care Clin 31: 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2012). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage 20: 256–260. [DOI] [PubMed] [Google Scholar]
- Kramann R, Kusaba T, Humphreys BD (2015). Who regenerates the kidney tubule? Nephrol Dial Transplant gfu281 30: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrance J‐P, Miller DR (2010). Acute kidney injury associates with increased long‐term mortality. JASN 21: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentlein R (1999). Dipeptidyl‐peptidase IV (CD26) – role in the inactivation of regulatory peptides. Regul Pept 85: 9–24. [DOI] [PubMed] [Google Scholar]
- Mulvihill EE, Drucker DJ (2014). Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase‐4 inhibitors. Endocr Rev 35: 992–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth JED (2014). Basic Statistics and Pharmaceutical Statistical Applications, Third edn. CRC Press: Boca Raton, FL, USA. [Google Scholar]
- Nakamaru Y, Akahoshi F, Iijima H, Hisanaga N, Kume T (2016). Tissue distribution of teneligliptin in rats and comparisons with data reported for other dipeptidyl peptidase‐4 inhibitors. Biopharm Drug Dispos 37: 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambi P, Gellai M, Wu HL, Prabhakar U (1997). Upregulation of osteopontin in ischemia‐induced renal failure in rats: a role for ET‐1? Biochem Biophys Res Commun 241: 212–214. [DOI] [PubMed] [Google Scholar]
- Noiri E, Dickman K, Miller F, Romanov G, Romanov VI, Shaw R et al. (1999). Reduced tolerance to acute renal ischemia in mice with a targeted disruption of the osteopontin gene. Kidney Int 56: 74–82. [DOI] [PubMed] [Google Scholar]
- Nuransoy A, Beytur A, Polat A, Samdanci E, Sagir M, Parlakpinar H (2015). Protective effect of sitagliptin against renal ischemia reperfusion injury in rats. Ren Fail 37: 687–693. [DOI] [PubMed] [Google Scholar]
- Padanilam BJ, Martin DR, Hammerman MR (1996). Insulin‐like growth factor I‐enhanced renal expression of osteopontin after acute ischemic injury in rats. Endocrinology 137: 2133–2140. [DOI] [PubMed] [Google Scholar]
- Park Y, Hirose R, Dang K, Xu F, Behrends M, Tan V et al. (2008). Increased severity of renal ischemia‐reperfusion injury with venous clamping compared to arterial clamping in a rat model. Surgery 143: 243–251. [DOI] [PubMed] [Google Scholar]
- Persy VP, Verstrepen WA, Ysebaert DK, De Greef KE, De Broe ME (1999). Differences in osteopontin up‐regulation between proximal and distal tubules after renal ischemia/reperfusion. Kidney Int 56: 601–611. [DOI] [PubMed] [Google Scholar]
- Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME (2003). Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int 63: 543–553. [DOI] [PubMed] [Google Scholar]
- Putra SED, Tsuprykov O, Von Websky K, Ritter T, Reichetzeder C, Hocher B (2014). Dealing with large sample sizes: comparison of a new one spot dot blot method to western blot. Clin Lab 60: 1871–1877. [DOI] [PubMed] [Google Scholar]
- Racusen LC, Solez K (1986). Nephrotoxic tubular and interstitial lesions: morphology and classification. Toxicol Pathol 14: 45–57. [DOI] [PubMed] [Google Scholar]
- Sauvé M, Ban K, Momen MA, Zhou Y‐Q, Henkelman RM, Husain M et al. (2010). Genetic deletion or pharmacological inhibition of dipeptidyl peptidase‐4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 59: 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen AJ (2015). Pharmacokinetics and clinical use of incretin‐based therapies in patients with chronic kidney disease and type 2 diabetes. Clin Pharmacokinet 54: 1–21. [DOI] [PubMed] [Google Scholar]
- Schnapp G, Klein T, Hoevels Y, Bakker RA, Nar H (2016). Comparative analysis of binding kinetics and thermodynamics of dipeptidyl peptidase‐4 inhibitors and their relationship to structure. J Med Chem 59: 7466–7477. [DOI] [PubMed] [Google Scholar]
- Silver SA, Cardinal H, Colwell K, Burger D, Dickhout JG (2015). Acute kidney injury: preclinical innovations, challenges, and opportunities for translation. Can J Kidney Health Dis 2: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakar CV, Christianson A, Freyberg R, Almenoff P, Render ML (2009). Incidence and outcomes of acute kidney injury in intensive care units: a veteran's administration study. Crit Care Med 37: 2552–2558. [DOI] [PubMed] [Google Scholar]
- Thomas L, Eckhardt M, Langkopf E, Tadayyon M, Himmelsbach F, Mark M (2008). (R)‐8‐(3‐Amino‐piperidin‐1‐yl)‐7‐but‐2‐ynyl‐3‐methyl‐1‐(4‐methyl‐quinazolin‐2‐ylmethyl)‐3,7‐dihydro‐purine‐2,6‐dione (BI 1356), a novel xanthine‐based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase‐4 inhibitors. J Pharmacol Exp Ther 325: 175–182. [DOI] [PubMed] [Google Scholar]
- Timper K, Grisouard J, Sauter NS, Herzog‐Radimerski T, Dembinski K, Peterli R et al. (2013). Glucose‐dependent insulinotropic polypeptide induces cytokine expression, lipolysis, and insulin resistance in human adipocytes. Am J Physiol Endocrinol Metab 304: E1–13. [DOI] [PubMed] [Google Scholar]
- Timper K, Dalmas E, Dror E, Rütti S, Thienel C, Sauter NS et al. (2016). Glucose‐dependent insulinotropic peptide stimulates glucagon‐like peptide 1 production by pancreatic islets via interleukin 6, produced by α cells. Gastroenterology 151: 165–179. [DOI] [PubMed] [Google Scholar]
- Uchida S, Endou H (1988). Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol 255: F977–F983. [DOI] [PubMed] [Google Scholar]
- Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S et al. (2005). Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818. [DOI] [PubMed] [Google Scholar]
- Vaghasiya J, Sheth N, Bhalodia Y, Manek R (2011). Sitagliptin protects renal ischemia reperfusion induced renal damage in diabetes. Regul Pept 166: 48–54. [DOI] [PubMed] [Google Scholar]
- Vercauteren SR, Ysebaert DK, De Greef KE, Eyskens EJ, De Broe ME (1999). Chronic reduction in renal mass in the rat attenuates ischemia/reperfusion injury and does not impair tubular regeneration. J Am Soc Nephrol 10: 2551–2561. [DOI] [PubMed] [Google Scholar]
- Wagner M, Cadetg P, Ruf R, Mazzucchelli L, Ferrari P, Redaelli CA (2003). Heme oxygenase‐1 attenuates ischemia/reperfusion‐induced apoptosis and improves survival in rat renal allografts. Kidney Int 63: 1564–1573. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sakatsume M, Nishi S, Narita I, Arakawa M, Gejyo F (2001). Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int 60: 1645–1657. [DOI] [PubMed] [Google Scholar]
- Youssef MI, Mahmoud AAA, Abdelghany RH (2015). A new combination of sitagliptin and furosemide protects against remote myocardial injury induced by renal ischemia/reperfusion in rats. Biochem Pharmacol 96: 20–29. [DOI] [PubMed] [Google Scholar]
- Zhang Z‐X, Shek K, Wang S, Huang X, Lau A, Yin Z et al. (2010). Osteopontin expressed in tubular epithelial cells regulates NK cell‐mediated kidney ischemia reperfusion injury. J Immunol 185: 967–973. [DOI] [PubMed] [Google Scholar]