

Abstract
Background and Purpose
N6‐Isopentenyladenosine (i6A) is a modified nucleoside exerting in vitro and in vivo antiproliferative effects. We previously demonstrated that the actions of i6A correlate with the expression and activity of farnesyl pyrophosphate synthase (FPPS), a key enzyme involved in the mevalonate (MVA) pathway, which is aberrant in brain cancer. To develop new anti‐glioma strategies, we tested related compounds exhibiting greater activity than i6A.
Experimental Approach
We designed and synthesized i6A derivatives characterized by the introduction of diverse chemical moieties in the N6 position of adenosine and tested for their efficacy in U87 cells and in primary glioma cultures, derived from patients. NMR‐based structural analysis, molecular docking calculations and siRNA mediated knockdown were used to clarify the molecular basis of their action, targeting FPPS protein.
Key Results
CM223, the i6A derivative including a benzyl moiety in N6 position of adenine, showed marked activity in selectively targeting glioma cells, but not normal human astrocytes. This was due to induction of intrinsic pathways of apoptosis and inhibition of proliferation, along with blockade of FPPS‐dependent protein prenylation, which counteracted oncogenic signalling mediated by EGF receptors.
Conclusion and Implications
The biological effects together with structural data on interaction of CM223 with FPPS, provided additional evidence for the correlation of the i6A/CM223 antitumor activity with FPPS modulation. Because the MVA pathway is an important promising target, CM223 and its derivatives should be considered interesting active molecules in antiglioma research.
Abbreviations
- FPPS
farnesyl pyrophosphate synthase
- i6A
N6‐isopentenyladenosine
- PI
propidium iodide
- STD
saturation transfer difference
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| Akt (protein kinase B) |
| ERK |
| FPPS, farnesyl pyrophosphate synthase |
| Rap: RAS subfamily |
| Catalytic receptors b |
| EGFR, epidermal growth factor receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
N6‐Isopentenyladenosine (i6A) is a modified nucleoside, consisting of adenosine with an isopentenyl chain derived from dimethylallyl pyrophosphate in the N6 position. It belongs to the cytokinin family, involved in control of many processes in plants (Kersten, 1984; Laten and Zahareasdoktor, 1985; Bifulco et al., 2008). In humans, many biological actions, both in vitro and in vivo, including antitumour effects, have been attributed to i6A (Woo et al., 2010; Castiglioni et al., 2013; Laezza et al., 2014; Pisanti et al., 2014; Laezza et al., 2015; Ciaglia et al., 2017). Interest in the study of this compound has been reinforced by experimental observations that i6A interferes with the growth of k‐ras transformed thyroid cells (KiMol) xenograft tumours by inhibiting the expression and activity of farnesyl pyrophosphate synthase (FPPS) (Laezza et al., 2006). Moreover, i6A exhibited immunomodulatory properties as it selectively expanded and directly targeted human natural killer cells through modulation of FPPS (Ciaglia et al., 2013).
FPPS is a key enzyme involved in the mevalonate pathway and in the downstream prenylation of proteins. It catalyses the two‐step synthesis of the C15 isoprenoid, trans,trans‐farnesyl diphosphate (FPP), a crucial precursor for the synthesis of several classes of essential metabolites, such as sterols, ubiquinones and carotenoids. Therefore, FPPS plays a critical role in many vital cell reactions such as protein prenylation, synthesis of cell membrane constituents and signal transduction components (Sacchettini and Poulter, 1997; Szkopinska and Plochocka, 2005). FPPS is currently the main biochemical target of bisphosphonates for the treatment of bone‐related disorders, such as osteoporosis diseases, and metastatic bone‐related tumours. However, as it is involved in many cancer‐related pathways, and appears deregulated in many tumours (Thurnher et al., 2012), it has attracted the interest of pharmaceutical companies and has led to the identification of new, specific, anticancer compounds (Fournier et al., 2008; Jahnke et al., 2010). In our search for the molecular targets of i6A, using in silico inverse virtual screening (Chen and Zhi, 2001; Lauro et al., 2011; Wang et al., 2012), we recently gained additional evidence that FPPS is an important molecular target for i6A (Scrima et al., 2014). Saturation transfer difference (STD) NMR experiments and enzymic assays indicated that i6A bound to the active site of FPPS, exerting a moderate inhibition of its enzymic activity (vide infra) (Laezza et al., 2006; Ciaglia et al., 2013). Indeed, molecular docking calculations showed that i6A was able to occupy the FPPS active site, with a significant contribution from the isopentenyl portion of the molecule (Scrima et al., 2014).
Encouraged by these data, we have searched for a derivative exhibiting greater activity than i6A and have designed and synthesized i6A derivatives, characterized by the introduction of new substituents in the N6 position of adenosine (Scheme 1; see Supporting Information). These structural changes included the insertion of a phenyl ring characterized by increasing steric hindrance at N6 position of adenosine (FP11, FP13 and FP16), the insertion, in the same position, of a benzyl ring (CM223) and the elimination of a double bond (CM224) (Figure 1A; Ottria et al., 2010).
Figure 1.
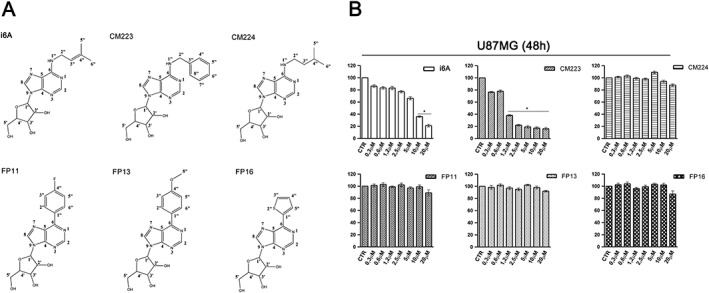
Effect of i6A and its analogues on cellular proliferation of human glioma cell line (U87MG). (A) i6A analogues: chemical structure of FP11, FP13, FP16, CM223, CM224 and of i6A for comparison. (B) U87MG cells were cultured for 48 h in the presence of the indicated concentrations (0–20 μM) of i6A, CM223, CM224, FP11, FP13 or FP16 before analysis of cell proliferation by BrdU incorporation assay. Results are expressed as means ± SD of five independent experiments performed in triplicate and reported as percentage of the untreated control (100%). *P < 0.05, significantly different from control; ANOVA with Tukey's test.
All the compounds were tested for their anti‐proliferative activity on U87MG cells and on primary cultures of glioma cells derived from patients. Glioblastoma (GBM) is the most common and highly malignant brain tumour, and recent work has highlighted the importance of the isoprenoid pathway, which is regulated by FPPS enzyme, for glioma development, progression and chemoresistance (Woo et al., 2010; Laezza et al., 2015). Very recently it has been shown that GBM cells rely on cholesterol uptake for survival (Villa et al., 2016) . This provided new insights into more effective therapeutic strategies for the treatment of GBM, suggesting the repositioning of old drugs or the synthesis of new compounds that modulate de novo cholesterol biosynthesis.
Here we have demonstrated that CM223, an analogue of i6A that includes a benzyl group at the N6 position of adenine, exhibited a marked cytostatic and cytotoxic activity, greater than the parent compound i6A. NMR‐based structural analysis and molecular docking calculations, showing a clear structural interaction of CM223 with FPPS, further delineated the molecular basis of the interaction between i6A analogues and FPPS protein and the key events of antiglioma action of these compounds. Thus, the anti‐glioma action of CM223 was mediated primarily by the FPPS‐dependent disruption of those lipid moieties which seem to be critical for the functionality and activity of the EGF receptor (EGFR) (Warren and Landgraf, 2006) and its downstream signalling pathway, for the growth and maintenance of glioma tumours (Furnari et al., 2015).
Methods
Cells
For experiments on normal cells, we used NHA cells (Clonetics™ Astrocyte Cell System), derived from normal fetal human brain tissue and cultured in the recommended medium Clonetics™ AGM™ BulletKit™ (Lonza; Basel, Switzerland). The human glioma cell lines U87MG (U87) were obtained from CLS Cell Lines Service GmbH (Eppelheim, Germany) or were kindly provided by Dr Daniela Parolaro (University of Insubria, Italy). The U87MG cells were cultured in EMEM (Lonza) supplemented with 10% heat‐inactivated FBS (Euroclone), 1% L‐glutamine, 1% antibiotic mixture, 1% sodium pyruvate, 1% non‐essential aminoacids (Euroclone). All cell cultures were maintained at 37°C in a humidified 5% CO2 atmosphere.
Clinical samples
All tissue samples were collected in accordance with the ethical standards of the Institutional Committee. The patients had been informed about the establishment of cellular models from their tumours and had given informed consent in written form. Small pieces of brain tissue containing tumour were collected at the time of craniotomy for tumour resection at the Neurosurgery Service of ‘G. Rummo’ Medical Hospital (Benevento, Italy) and the Neurosurgery Service of ‘San Giovanni di Dio Ruggi d'Aragona’ Medical Hospital (Salerno, Italy), divided into one portion immediately processed to generate primary tumour cell lines and another portion stored at −80°C for subsequent Western blot analysis. A second sample from each patient was also taken for clinical diagnosis performed by expert neuropathologists in accordance with the International Classification of CNS tumours drafted under the auspices of the World Health Organization (WHO). The tumours were diagnosed as astrocytoma (WHO grade I‐III; n = 3), glioma (WHO grade II, n = 2) or GBM multiforme (WHO grade IV; n = 18). There were no significant differences by gender, or age between the different groups (Ciaglia et al., 2015). The preparation of adherent primary cultures of brain tumour cells (designated as GBM) was conducted as reported elsewhere. (Ciaglia et al., 2015).
Cell cultures were maintained at 37°C in a humidified 5% CO2 atmosphere.
Determination of glioma cell proliferation
U87MG glioma cells or NHA cells (4 × 103 cells per well), were cultured for 24 h in 96‐well plates before addition of i6A or analogues at the indicated concentrations and cultured for additional 24, 48 and 72 h at 37°C. Cell proliferation was evaluated by measuring BrdU incorporation into DNA (BrdU colorimetric assay kit; Roche Applied Science, South San Francisco, CA, USA). Newly synthesized BrdU‐DNA was determined on an ELISA plate reader (ThermoScientific) at 450 nm. All experiments were performed in triplicate, and the relative cell growth was expressed as a percentage of the untreated control cells (set to 100%) to allow for unwanted source of variation.
Analysis of apoptosis
Quantitative assessment of apoptosis of U87MG glioma cell line or NHA was analysed by anti‐human Annexin V (BioLegend, San Diego, CA, USA) and PI staining and processed as previously described (Malfitano et al., 2013). Briefly, cells grown in 100 mm dishes for 24, 48 and 72 h in EMEM containing 2% FBS or in recommended medium AGM™ BulletKit™ were harvested with trypsin and washed in PBS. Following 24 h of incubation, immunofluorescence staining was performed. The cells were resuspended in Annexin V binding buffer (10 mM HEPES/NaOH, pH 7; 140 mM NaCl; and 2.5 mM CaCl2) and stained with Annexin V‐FITC for 20 min at room temperature and then with PI at room temperature, for an additional 15 min in the dark. The cells were acquired by flow cytometer within 1 h after staining. At least 10 000 events were collected, and the data were analysed by Cell‐Quest Pro software (Becton Dickinson, San Jose, CA). Data are expressed as logarithmic values of fluorescence intensity.
Western blot analysis
For analysis of protein levels from cells they were grown in p60 tissue culture plates at a density of 2×104 cells cm−2 for 24 h. Tumour cells were then incubated with vehicle, i6A, CM223 or zoledronic acid in the presence or absence of EGF. After incubation, cells were washed with PBS, harvested and lysed in ice‐cold RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 0.5% Triton X‐100, 0.5% deoxycholic acid, 10 mg•mL−1 leupeptin, 2 mM phenylmethylsulfonyl fluoride, and 10 mg•mL−1 aprotinin). Tumour pieces were disrupted for protein extraction by gentle homogenization (Potter‐Elvehjem Pestle) in cold RIPA buffer. After removal of cell debris by centrifugation (14 500 g for 20 min at 4°C), the proteins were estimated. About 30 μg of proteins was loaded on 10, 12 or 15% SDS–polyacrylamide gels under reducing conditions and then transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat dry milk (Bio‐Rad, Richmond, CA, USA) in Tris‐buffered saline containing 0.1% Tween‐20 (TBST) for 1 h at RT and incubated with the specific antibody. Immunodetection of specific proteins was carried out with horseradish peroxidase‐conjugated donkey anti‐mouse or anti‐rabbit IgG (Biorad, Hercules, CA), using the enhanced chemiluminescence system (Amersham Pharmacia Biotech, Piscataway, NJ) according to the manufacturer's instructions and then exposed to X‐ray film (Amersham Biosciences). Immunoreactive bands were quantified with Quantity One 1D analysis software (Bio‐Rad). To ascertain equal protein loading in Western blots of cell lysates, membranes were probed with an antibody raised to α‐tubulin (Sigma‐Aldrich, St. Louis, MO, USA) or β‐actin (Abcam, Cambridge, UK).
siRNA‐mediated knockdown
FPPS siRNA transfection was carried out according to the manufacturer's instructions. First, siRNA FPPS (Ambion) and negative control siRNA (Silencer Negative Control, Ambion) were dissolved in Opti‐MEM serum‐free media. Both FPPS and scrambled siRNA were delivered into the U87 cell cultures plated 18 h before transfection (approximately 80% confluency) via Lipofectamine 3000. The final concentration of FPPS and scrambled siRNA in culture was 100 nM. The cells were incubated with the transfection reagents for 48 h. Then, cell media was replaced with serum‐free media to induce starvation and treated with 10 μM CM223 for 8 h. After that EGF was added (final concentration 50 ng•mL−1) for 10 min. The cells were then harvested for analysis of protein knockdown by Western blotting and tested for cell signalling experiments.
FPPS gene expression
The plasmid p11, transformed into BL21(DE3)‐pLysS cells, was purchased from SGC‐Oxford and contained the T7/Lac promoter and ampicillin resistance. FPPS was expressed in Escherichia coli as a fusion protein (67–419 residues) with an N‐terminal poly‐histidine tail and a mutation (threonine with serine) on residue 266, molecular weight 43 kDa. For expression in E. coli, bacterial clones were grown in 1 L of LB (Luria‐Bertani) medium containing 50 μg•mL−1 ampicillin. Cell growth was monitored spectrophotometrically by measuring OD600 nm periodically. When growth was performed to an OD600 of 0.7 at 37°C, isopropyl‐D‐thiogalactoside (IPTG; 1mM) was added. After 6 h of cell growth, cells were pelleted by centrifugation and re‐suspended in lysis buffer [50 mL of 5% glycerol, 5 mM imidazole, 500 mM NaCl, 50 mM PBS (pH 7.5)] and sonicated. Protein was purified with His‐Trap HP column at 1 mL•min−1 using an AKTA purifier system, the soluble extract was applied to a nickel‐chelated agarose affinity column that had been equilibrated with the same buffer. The protein was eluted from the column with elution buffer [5% glycerol, 250 mM imidazole, 500 mM NaCl, 50 mM PBS (pH 7.5)]. Affinity chromatography on a nickel‐chelated agarose column permitted a simple one‐step protein purification. Then FPPS was transferred into Vivaspin 20 concentrator, cut‐off 3 kDa, to exchange the buffer for NMR studies.
NMR sample preparation
All chemicals were purchased from Sigma‐Aldrich. i6A was purchased from Iris Biotech GMBH and zoledronic acid from Sigma‐Aldrich.The i6A analogues were synthesized at University of Pisa and University of Milano . STD‐NMR and WaterLOGSY experiments were recorded at 25°C on Bruker AV600 MHz spectrometer at a 1H resonance frequency of 600 MHz equipped with a 5 mm triple resonance 1H(13C/15N), z‐axis pulsed‐field gradient probe head. For characterization purposes, i6A samples consisted of a 5 mM solution in 25 mM d‐Tris, pH 7.4, 0.5 mM MgCl2 and 25 mM NaCl with 1% dimethyl sulfoxide‐d6 as a co‐solvent, and the spectra were referenced to residual solvent. 1H − 1D spectra were acquired at a resolution of 16 k complex points in the time domain with 32 accumulations each (sw = 6000 Hz, d1 = 3 s). The FPPS protein at 8 μM concentration was titrated with i6A‐analogues to have protein/ligands molar ratios 1:10, 1:20, 1:30, 1:50, 1:70 and 1:100. For each addition of ligands, STD build‐up experiment was carried out using different saturation times (0.50, 1.00, 1.50, 2.00, 3.00, 4.00 and 5.00 s and different relaxation delay 1.50, 2.00, 2.50, 3.00, 4.00, 5.00 and 6.00 s). For each experiment in the frequency list (FQ2LIST), the on‐resonance and off‐resonance pulse were 320 and 50 000 Hz respectively. Briefly, two free induction decay (FID) data sets were collected in an interleaved manner to minimize temporal fluctuations with the protein irradiation frequency set on‐resonance (−0.5 p.p.m.) and off‐resonance (40 p.p.m.) respectively (sw = 6000 Hz, 16 steady state scans, 2048 transients, 4 k complex points, d1 = 3 s). Protein saturation was obtained using a train of individual 50 ms long, frequency selective Gaussian radio frequency pulses separated by an inter pulse delay of 1 ms. The FID acquired with off‐resonance irradiation generated the reference spectrum (Ioff), whereas the difference FID (off‐resonance on‐resonance) yielded the STD spectrum (ISTD = Ioff − Ion). Spectra were processed with an exponential apodization function (1Hz line broadening) and zero‐filling to 8 k complex points before Fourier transformation and baseline correction with a third order Bernstein polynomial fit. The STD measurements were done in duplicate, and all data were processed and analysed using TopSpin software (Bruker v 1.3).
FPPS colourimetric assay
The colourimetric assays were performed in 96‐well plates, flat bottom. 200 ng of fresh pure FPPS was assayed in a final volume of 100 μL buffer (50 mM Tris pH 7.5, 2 mM MgCl2, 1 mM DTT, 5 μg•mL−1 BSA) with or without pre‐incubation with inhibitors (i6A 1‐10 mM, CM223 0.1–5.0 mM, zoledronic acid 1–2 μM as positive control) for 30 min. at 37°C. The reaction was initiated by the addition of the substrates dimethylallyl pyrophosphate (50 μM) and isopentenyl pyrophosphate (50 μM) and proceeded for 1 h at 37°C. 10 μL of 2.5% ammonium molybdate reagent (in 5 N H2SO4) were added and incubated for 10 min to allow the formation of the pyrophosphate(PPi)‐molybdate complex, finally the complex was reduced by 10 μL of 0.5 M 2 ‐mercaptoethanol and 5 μL of Eikonogen reagent (0.25 g of sodium sulfite and 14.7 g of meta‐bisulfite were dissolved in 100 mL water). Plates were incubated with gentle mixing on a plate shaker for 20 min. The absorbance was measured at 580 nm employing a microplate reader. The control experiments were carried out with incubation mixture in the absence of substrate or FPPS for background deduction. To set the conditions, a standard curve using Na2P2O7 as the source of PPi (Figure S6) was constructed.
Docking studies
AutoDock version 4.2 (Morris et al., 2009) and AutoDock Vina version 1.0 (Trott and Olson, 2010) in combination with the LGA were used for all docking calculations. The starting conformations for docking studies of CM223 and FP13 ligands were built with Maestro (version 9.6). (Schrödinger, 2009). 3D starting model of each compounds for the subsequent docking calculations, were preliminary optimized by conjugate gradient, (0.05 Å convergence threshold).
3D FPPS protein model was obtained from Protein Data Bank database (PDB code: 1ZW5) (Kavanagh et al., 2006). Water molecules were removed, and the obtained file was then processed with Autodock Tools 1.5.6, merging non‐polar hydrogens and adding Gasteiger charges.
For both docked ligands, all bonds were considered rotatable. For an exhaustive exploration of conformational space, six docking calculations consisting of 250 runs were performed, yielding 1500 structures. An initial population of 450 randomly placed individuals, a maximum number of 5 × 106 energy evaluations, and a maximum number of 4 × 106 generations were taken into account. The mutation and crossover rates used were 0.02 and 0.80 respectively. The local search probability was 0.26. For all docking calculations, a grid box size of 53 65 57 points (spacing between the grids points of 0.375 Å) was used, centred on the centre of the target binding site.
The resulting data, differing by less than 3.5 Å in positional RMSD, were clustered together and represented by the result with the most favourable free energy of binding. All 3D models were depicted using Python and VMD (Humphrey et al., 1996).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analysis was performed in all the experiments shown, using the GraphPad prism 6.0software for Windows (GraphPad software). For each type of assay or phenotypic analysis, data obtained from multiple experiments are calculated as mean ± SD and analysed for statistical significance using the two‐tailed Student t‐test , for independent groups, or ANOVA followed by Tukey's post hoc test for multiple comparisons, when F achieved was P < 0.05, and there was no significant variance inhomogeneity. Minimal statistical significance was set at P < 0.05. Some results were normalized to avoid unwanted sources of variation. To ensure reproducibility of the results and minimize bias, the use of authenticated U87MG and NHA at the same passage and wherever possible, the same manufactures' kit and reagents were used for selected experiments. For the control of bias, all data generated during study have been recorded directly, promptly, accurately and legibly by a dedicated individual entering the data.
Materials
Erlotinib, i6A, IPTG, methyl‐β‐cyclodextrin (MβCD), simvastatin and zoledronic acid monohydrate were purchased from Sigma–Aldrich. Human EGF was purchased from Peprotech (London, UK) and was diluted in a buffer containing a stabilizer (5% trehalose) and added to cell cultures at the indicated concentrations. Lipofectamine 3000 was from Invitrogen (San Diego, CA, USA);. Opti‐MEM media were from Gibco (#51985; Grand Island, NY, USA). For Western blot analysis, the following antibodies were used: rabbit polyclonal anti‐human β‐actin, rabbit polyclonal anti‐human ras and rabbit polyclonal anti‐human FPPS were purchased from Abcam, mouse monoclonal anti‐human α‐Tubulin from Sigma‐Aldrich Inc.; rabbit polyclonal anti‐human cleaved caspase‐3, rabbit polyclonal anti‐human Rap1A, rabbit polyclonal anti‐human caspase‐3, rabbit monoclonal anti human cleaved caspase‐9, rabbit monoclonal anti‐human caspase‐9, rabbit monoclonal anti‐human caspase‐7, rabbit monoclonal anti‐human cleaved caspase‐7, rabbit monoclonal anti‐human cleaved caspase‐8, rabbit monoclonal anti‐human Bim, rabbit monoclonal anti‐human Bcl‐2, rabbit monoclonal anti‐human Bcl‐xL, rabbit polyclonal anti‐human phospho‐STAT3 (p‐STAT3; Tyr705), rabbit monoclonal anti‐human STAT3, rabbit monoclonal anti‐human cleaved PARP (Asp214), rabbit monoclonal anti‐human PARP, rabbit monoclonal anti‐human phospho‐p44/42 MAPK (p‐Erk1/2; Thr202/Tyr204), rabbit monoclonal anti‐human p44/42 MAPK (Erk1/2), rabbit monoclonal anti‐human phospho‐Akt (p‐Akt; Ser473), rabbit monoclonal anti‐human Akt, rabbit monoclonal anti‐human phospho‐EGF receptor (p‐EGFR; Tyr1068) and rabbit monoclonal anti‐human phospho‐EGF receptor were purchased from Cell Signaling Technology (Danvers, MA). Secondary HRP‐linked goat anti‐mouse or goat anti‐rabbit IgG, were also purchased from Cell Signaling Technology.
Results
Chemistry
CM223 and CM224 were prepared and purified as described previously (Ottria et al., 2010). FP11, FP13 and FP16 were prepared modifying the synthetic route previously reported (Scheme S1; see Supporting Information) (Hocek et al., 2000). Purity of all compounds (≥ 99%) was verified by HPLC, NMR and mass spectrometry measurements. Structures of these compounds are presented in Figure 1. Compounds FP11, FP13 and FP16 were obtained by the synthetic route described in the Scheme S1 (see Supporting Information).
Biological assays
Dose–response studies of the effects of i6A and its analogues on DNA synthesis in the U87 cell line
The nucleoside i6A and its analogues CM223, CM224, FP11, FP13 and FP16 were tested as inhibitors of the growth of U87MG cells, a cell line derived from human gliomas. The U87 cells were incubated for 48 h, with the indicated compounds over a concentration range (0.3–20 μM) chosen because i6A had shown inhibitory activity in this range. As shown in Figure 1B, i6A showed a concentration‐dependent inhibition of proliferation, compared with untreated cells, using the BrdU incorporation assay, with a significant effect at a concentration of about 7.5 μM. Interestingly the compound CM223 elicited a more pronounced inhibition than i6A, as a significant response was already achieved at a concentration of 1.2 μM. Notably, the other compounds tested, CM224, FP11, FP13 and FP16, showed no comparable anti‐proliferative activity on U87 cells.
Analysis of the cytostatic and cytotoxic effects of the i6A derivative CM223 on U87 glioma cells and normal primary astrocytes
The growth inhibitory effects of CM223 were further investigated at different time points by proliferation and clonogenic assay, using i6A as a reference compound. Figure 2A shows the inhibitory effect s of i6A or CM223 (0.3–20 μM), sampled at 24, 48 and 72 h, on U87MG cells. Although inhibition by either compound increased over time, the inhibitory effects of CM22 were always greater than those of i6A, even after 72 h treatment. Interestingly, CM223 also was more effective than i6A on the formation of clones (Figure 2B). In these assays, the number and dimension of clones was decreased with the lowest concentrations of CM223 (1 and 2.5 μM).
Figure 2.
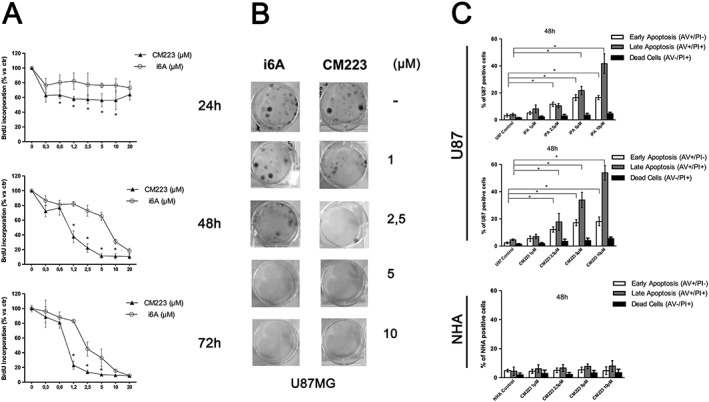
Comparison of the effect of i6A and CM223 on proliferation of human glioma cell line (U87MG) and NHA cells. (A) U87MG cells were cultured for 24, 48 and 72 h in the presence of the indicated concentrations (0–20 μM) of i6A or CM223 before analysis of cell proliferation by BrdU incorporation assay. Results are expressed as means ± SD of five independent experiments performed in triplicate and reported as percentage of the untreated control (100%). *P < 0.05, significantly different from control; ANOVA with Tukey's test. (B) Representative images of the clonogenic assay on glioma cell line U87MG, 10 days after treatment with i6A and CM223 at the indicated concentrations (0–10 μM). The experiment was conducted twice, in triplicate. (C) Induction of apoptosis measured by Annexin V and PI double staining through flow cytometry in i6A or CM223‐treated U87MG cells (upper panels) and in CM223‐treated NHA (lower panel) after 48 h. Histograms indicate total percentage of early (Annexin V‐positive cells/ PI‐negative cells) and late apoptotic events (Annexin V/ PI‐double positive cells) as well as necrotic cells (Annexin V‐negative cells/PI‐positive cells). Results are representative of five independent experiments performed in duplicate and expressed as mean ± SD. *P < 0.05, significantly different from control; ANOVA with Tukey's test.
To assess whether the inhibition of cell proliferation by i6A and, especially, by CM223 was also associated with the induction of apoptosis, we performed a cytofluorimetric cell death analysis, using Annexin‐V and propidium iodide (PI) staining. i6A and CM223 exhibited a dose‐dependent induction of apoptosis when U87cells were treated for 48 h (Figure 2C, upper panel). Interestingly, even in this experiment, the percentage of cells in early and late apoptosis after CM223 treatment was significantly higher than those after i6A treatment overall, suggesting a better cytostatic and cytotoxic potential for this i6A derivative. Importantly, CM223 elicited no significant reduction of viability in normal human astrocyte (NHA) cells, compared with U87 cells, under the same conditions (Figure 2C, lower panel).
Structural interactions between FPPS and CM223; studies with NMR spectroscopy
We have shown that i6A suppresses growth of KiMol xenograft tumours by inhibiting FPPS expression and activity (Laezza et al., 2006)and that the immunomodulatory activity of i6A is due to modulation of FPPS activity (Ciaglia et al., 2013). Preliminary NMR data and colorimetric enzymic assays showed that i6A interacts with the binding site of FPPS, inhibiting its activity.
We therefore analysed by STD‐NMR experiments (Mayer and Meyer, 1999; Mayer and Meyer, 2001) the interactions of the i6A analogues with FPPS protein. (Figure 3A). STD is a powerful NMR technique that enables the determination of K D values for protein‐ligand interactions. The NMR sample of FPPS (8.0 μM) was titrated with the i6A analogues to have STD build‐up at protein‐ligand molar ratios of 1:10, 1:20, 1:30, 1:50, 1:70 and 1:100. For each titration point, STD experiments were carried out using different saturation times (0.50, 1.00, 1.50, 2.00, 3.00, 4.00 and 5.00 s).(Angulo and Nieto, 2011) Standard 1H monodimensional and 2D correlated spectroscopy NMR experiments allowed the 1H chemical shift assignment of the proton signals from the i6A‐analogues (Figure S2–S6; see Supporting Information).
Figure 3.
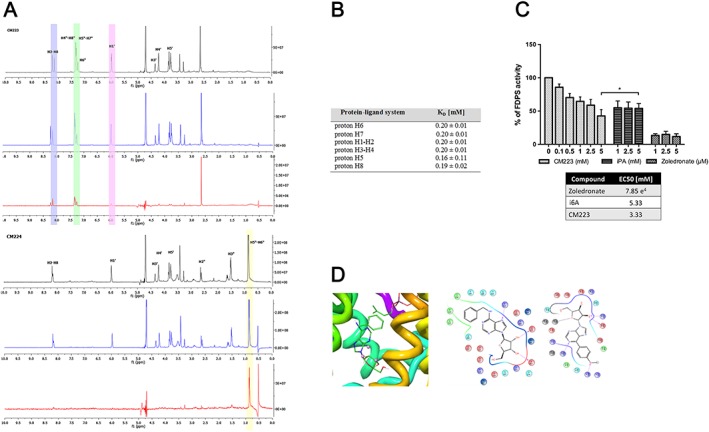
Evaluation of the biological significance of the FPPS‐CM223 interaction in U87MG glioma cells. (A) STD‐NMR spectra of FPPS‐CM223 (left) and FPPS‐CM224 (right), in black the off‐resonance spectra, in blue the on‐resonance spectra and in red the STD spectra. (B) K D of the FPPS‐CM223 complex. (C) FPPS activity after the pretreatment with CM223 (from 0.1 to 5 mM, 30 min 37°C) and i6A (from 1 to 5 mM, 30 min 37°C). The potent FPPS inhibitor, zoledronic acid (1–2 μM, 30 min 37°C) was used as positive control. The inhibitory activity of CM223 with respect to i6A was evaluated. Results are expressed as mean ± SD of n = 5 experiments. *P < 0.05, significantly different from i6A; ANOVA with Tukey's test. (D) Left: Molecular docking prediction of CM223 ligand in the FPPS binding site (CM223 coloured by atom type: C green, O red, H white, N blue, Mg2+ pink). FPPS backbone was represented as ribbon. Centre and Right: 2D interactions panel showing the particular interaction of CM223 and FPPS residues (centre) and 2D interactions panel showing peculiar interaction of FP13 and FPPS residues (right) (Red: charged negative, Violet: charged positive, Cyano: polar, Green: Hydrophobic, Blue: Mg2+).
Analysis of STD‐NMR experiments for the compounds CM223 (benzyl at N6) and CM224 (isopentyl at N6) indicated a marked interaction with the enzyme, with an important participation of the adenosine moiety (in particular protons H2‐H8, H4”‐H8”, H5”‐H7”, H6” and H1’). These effects reached the significant value of 30% for CM223 and 20% for CM224 (Figure 3A) The compounds of the FP series, containing an aromatic ring directly linked to the N6 of adenosine, showed less marked interactions (Supporting Information Figure S1). To confirm that the i6A analogues bound to the active site of FPPS and not to an allosteric binding site (Jahnke et al., 2010), the FPPS‐ligand complexes were titrated with increasing concentrations of the known allosteric modulator. 3‐carboxymethyl‐5,7‐dichloro‐1H‐indole‐2‐carboxylic acid (IC50 = 6.0 μM) (Jahnke et al., 2010) or zoledronic acid, high‐affinity FPPS inhibitor (data not shown) (Scrima et al., 2014). The quantitative evaluation of STD data confirmed that the compound CM223 interacted with the FPPS binding site with a consistent contribution from the N6 substituent.
We calculated the K D for the complex FPPS‐CM223 interaction based on the quantitative measurement of STD effects (Figure 3B). We collected experiments at different protein‐ligand ratios and saturation time conditions (see above), according to the methodology developed by Angulo and Nieto (2011). Using this procedure, the calculation of protein‐ligand affinities is independent of contingent experimental factors, such as STD saturation time, ligand residence time in the complex, and the intensity of the signal (Angulo and Nieto, 2011). The mean K D value calculated for the single protons of CM223 (Figure 3B) was 0.19 mM. This value, which is fivefold lower than the K D value for the i6A‐FPPS interaction (Scrima et al., 2014), indicates a stronger binding to the FPPS target for CM223 and suggests that the benzyl substituent at N6 may lead to more potent FPPS inhibitors.
We then carried out molecular docking calculations (AutoDock 4.2 software) (Morris et al., 2009), using the interprotonic distances calculated on the basis of STD NMR experiments, as restraints. Compounds CM223 and FP13, respectively representing the strongest and weakest binding to FPPS, were subjected to molecular docking simulations against the 3D FPPS protein model (PDB code: 1ZW5), selected in inverse virtual screening procedure in our previous work (Kavanagh et al., 2006; Scrima et al., 2014). For an exhaustive exploration of conformational space, iterative docking calculations consisting of 250 runs were executed, yielding 1500 structures. The resulting binding poses were clustered together according to positional root‐mean‐square deviation (RMSD < 3.5 Å). Docking analysis revealed a favourable accommodation of the i6A analogues in the binding site of FPPS through a large set of both hydrophobic and polar interactions (Figure 3D).
The ribose sugar moiety establishes a network of H‐bonds with Asp118, Asp257 and Lys271., whereas the adenine core of the molecule is oriented between the three Mg2+ ions co‐crystallized with FPPS, making polar contacts with Lys214, Asp257 and H‐bonding with Gln254. In CM223, the benzyl group, as observed with the isopentenyl moiety of i6A, is placed in the deeper region of the binding site and engages in π‐stacking interactions with Phe113, and Tyr218 of the protein (Scrima et al., 2014) The increased binding of CM223 (K D 0.19 mM) as compared to i6A (K D 1.0 mM) may be due to these interactions that, with CM223, are stabilized by the presence of the aromatic benzyl ring, instead of the isopentenyl chain. Concerning FP13, the difference in the binding mode as compared to i6A and CM223 is due to the phenyl ring that represents a low flexibility point for this molecule. STD NMR data, on the contrary show that the weak binding of FP13 to the FPPS catalytic pocket is based on interactions of the ribose sugar moiety.
Colorimetric assay of FPPS activity
To test whether the interaction of CM223 with FPPS, observed in NMR data, might result in an enhancement of enzyme inhibition compared to i6A, we performed a colorimetric assay. (Gao et al., 2010). The histograms in Figure 3C show FPPS activity after pretreatment with CM223 (0.1 ‐ 5 mM) or i6A (1.0 –0.5 mM), for 30 min at 37°C. The enzymic activity of FPPS with or without the high affinity FPPS inhibitor, zoledronic acid (1–2 μM, 30 min 37°C) provided the negative and positive controls. As expected, zoledronic acid was much more effective than either i6A or CM223, but the data indicated inhibition of FPPS activity by i6A and, to a greater extent, by CM223, with IC50 values of 5.33 and 3.33 mM respectively. This would suggest that the inhibition of FPPS activity by CM223 is really more effective that by its isoprenoid analogue, i6A, even if we cannot rule out effects on other enzymes of the pathway, beyond FPPS.
CM223 acts on U87 glioma cells via FPPS modulation and disruption of lipid dependent‐EGF receptor signalling pathway
In order to confirm that the anti‐proliferative and pro‐ apoptotic effects of CM223 on U87 glioma cells were associated with its capacity to interfere with the mevalonate pathway through FPPS modulation, we tested the effects of CM223 on post‐translational modification of small GTP‐binding proteins, by analysing the prenylation status of Ras‐proximate‐1 or Ras‐related protein 1 (Rap1A). Rap1A is a protein prenylated exclusively by geranyl geranyl transferase and its unprenylated form is recognized by a specific unprenylated anti‐Rap1A antibody. We incubated U87MG cells with i6A, CM223 or zoledronic acid, for 24 h. In control, EGF‐treated, U87 cells, Rap1A was in the processed prenylated forms, so it was not detected in our Western blot analysis. As expected, by acting through FPPS modulation, treatment with zoledronic acid or i6A and, to a greater extent, with CM223 inhibited the processing of Rap1A, increasing levels of the unprenylated Rap1A form (Figure 4A). Interestingly, transfection of U87MG cells with FPPS siRNA increased the sensitivity of these cells to CM223. Indeed, while FPPS siRNA transfection alone did not modify the levels of unprenylated Rap1A, probably due to a residual activity of the enzyme after its knock‐down that provided sufficient synthesis of isoprenoid (or other lipid) moiety (Wang et al., 2011), after FPPS siRNA‐CM223 co‐treatment, the levels of unprenylated Rap1A were more evident (Figure 4B), suggesting a more complete inhibition of FPPS activity achieved by the combination. At the same time, a parallel accumulation of Ras in its unprenylated form, seen as an ‘up‐smearing’ on immunoblots, has been also highlighted. Overall, these data point to FPPS as a possible target of action of CM223.
Figure 4.
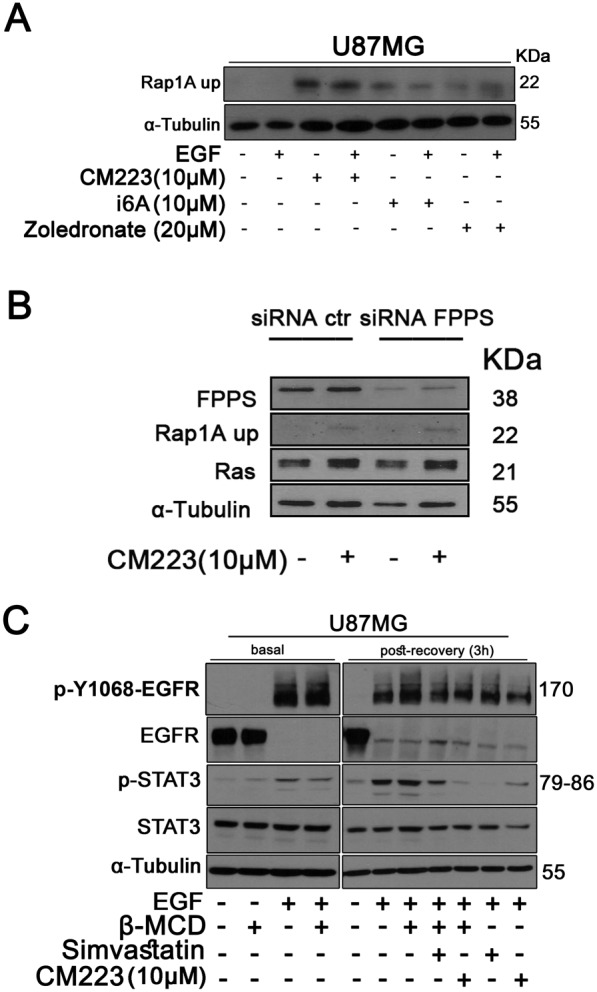
Analysis of CM223‐induced block of isoprenylation and its effects on EGFR status. (A) Western blot analysis of Rap1A‐up on whole cell extracts from cell line U87MG cultured for 24 h in the presence of CM223 (10 μM), i6A(10 μM) or zoledronate (20 μM), in the presence or absence of EGF (50 ng·mL−1); α‐tubulin was used as control of protein loading. Panel shows a representative Western blot of three different experiments performed with similar results. (B) Western blot analysis of Rap1A‐up and Ras in siCTR‐ and siFPPS transfected U87MG cells cultured alone or in the presence of CM223 (10 μM) for 8 h; α‐tubulin was used as control of protein loading. Panel shows a representative Western blot of three different experiments performed with similar results. (C) Western blot analysis for p‐Y1068 EGFR, total EGFR, p‐STAT3 and total STAT3 on whole cell extracts from U87MG cells stimulated for 10 min with EGF (50 ng·mL−1). U87MG cells were serum starved for 18 h and then incubated with and without methyl‐β‐cyclodextrin (β‐MCD; 10 mM) for 15 min at 37°C. After that, in basal condition (left panel) U87MG cells were stimulated for 10 min with EGF (50 ng·mL−1) or alternatively (right panel) the cells were left to recover signalling in serum free control medium in presence or absence of CM223 and/or simvastatin (20 μM) for 3 h and then stimulated with EGF for 10 min. α‐tubulin was used as control of protein loading. The panel shows a representative Western blot of two different experiments performed with similar results.
As the lipid environment of plasma membranes and lipid–proteins interactions have been proposed as being molecular platforms important for oncogenic signalling processes (Warren and Landgraf, 2006) , we then tested the effects of CM223 on the activity of receptors embedded in the membrane. One of these is the EGF receptor (EGFR, also referred to as ERBB1 or HER1) a well‐known oncoprotein belonging to the HER superfamily of receptor tyrosine kinases, along with ERBB2, ERBB3 and ERBB4. This receptor constitutes the most common genetic alteration associated with malignant glioma where over‐activation and/or overexpression of EGFR leads to the deregulation of pleiotropic cellular processes, including cell differentiation, metabolism, proliferation, and survival (Furnari et al., 2015). As expected, stimulation of U87 cells with EGF induced an early activation of EGFR (10 min) shown by increased phosphorylation on Tyr1068 of this receptor (Figure 4C). As a consequence, the STAT3 signalling pathway was activated. In this experimental setting, the importance of the lipid environment to the function of the EGFR was demonstrated by treatment of the cells with the membrane impermeable, small cyclic oligosaccharide methyl‐β‐cyclodextrin (MβCD), which binds cholesterol and thus disorganizes lipid rafts in the plasma membrane. In our system, MβCD altered EGFR signalling and decreased the phosphorylation of STAT3, in response to EGF (Figure 4C, left panel). The recovery from cholesterol depletion and inhibition by MβCD was complete by 3 h after removal of MβCD, even in the absence of added serum, mainly due to lipid synthesis in the cells. Simvastatin, by inhibiting cholesterol synthesis, delayed the recovery of EGF‐induced STAT3 signalling, as did CM223 (Figure 4C, right panel). These data strengthen the hypothesis that the i6A analogue CM223 significantly interferes with the mevalonate metabolic pathway.
CM223 selectively inhibits the oncogenic Akt and STAT3 signalling and induces apoptosis, activating the intrinsic caspase cascade in cultured U87 glioma cells
It is well characterized that the PKB/Akt, STAT and Ras‐MAPK (ERK) pathways, which regulate cellular growth, proliferation and survival, are downstream components of EGFR signalling (Hackel et al., 1999). In order to investigate if the effects of CM223 on EGFR signalling significantly changed this downstream pathway, we treated U87 cells for 24 h with increasing concentrations of the nucleoside, prior to assaying proteins by Western blot analysis. We found that, at all doses tested, CM223 significantly suppressed Akt and STAT3 activation in cultured U87 glioma cells, reflected by the reduction of the phosphorylation of AKT (Ser473) and of STAT3 (Tyr705) (Figure 5A).
Figure 5.
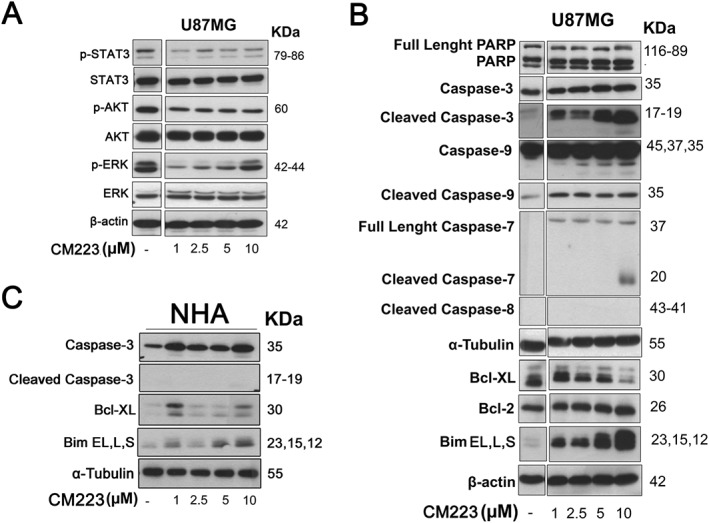
CM223‐mediated modulation of oncogenic signalling pathways and proteins of the apoptotic process in U87 and NHA. (A) Western blot analysis for p‐STAT3, total STAT3, p‐AKT, total Akt, p‐ERK and total ERK on whole cell extracts from U87MG cells, cultured for 24 h in the presence of the indicated concentrations (0–20 μM) of CM223; β‐actin was used as control of protein loading. Panel shows a representative Western blot of 3 different experiments performed with similar results. (B) Western blot analysis for PARP, caspase‐3, cleaved caspase‐3, caspase‐9, cleaved caspase‐9, full length and cleaved caspase‐7, cleaved caspase‐8, Bcl‐XL, Bcl‐2 and Bim, on whole cell extracts from cell line U87MG cultured for 24 h in the presence of the indicated concentrations (0–20 μM) of CM223. β‐actin and α‐tubulin were used as control of protein loading. Panel shows a representative Western blot of three different experiments performed with similar results. (C) Western blot analysis for caspase‐3, cleaved caspase‐3, Bcl‐XL and Bim on whole cell extracts from cell line NHA cultured for 24 h in the presence of the indicated concentrations (0–20 μM) of CM223; α‐tubulin was used as control of protein loading. Panel shows a representative Western blot of three different experiments performed with similar results.
As for the MAPK/ERK pathway, its inhibition, in terms of reduction of phosphorylation of activation loop residues Thr202/Tyr204, was achieved only at the lower concentrations of CM223 (1 ‐ 5 μM). At the highest concentration of CM223 (10 μM), there was a sustained ERK phosphorylation. In this context, Wei et al., (2011) have suggested that ERK activation can also play an active role in mediating apoptosis, functioning upstream of caspase activation to initiate the apoptotic signal. In T98G cells, activation of ERK was observed in response to cisplatin and UV light and was shown to strongly promote apoptosis in these GBM cell line (Hamdi et al., 2008). To test this hypothesis and to corroborate the pro‐apoptotic effect of CM223, we examined the expression of caspases and other proteins essential to apoptosis induction. Treatment of U87 cells for 24 h with CM223 strongly induced the expression of cleaved caspase‐9, cleaved caspase‐7 and ‐3, as well as cleaved PARP, in a concentration‐dependent manner (Figure 5B), without affecting caspase 8 activity, suggesting that CM223 stimulated the intrinsic apoptotic programme. This suggestion was supported by the decreased expression of the anti‐apoptotic protein Bcl‐XL, the canonical transcriptional target of Akt, and the marked increase in the levels of the three major Bim isoforms of pro‐apoptotic BIM (Bim EL, L, S). In keeping with these observations, phosphorylation of ERK was down‐regulated by CM223 treatment which lead to a concomitant induction of the levels of the pro‐apoptotic Bim and to a clear increase of caspase 3 cleavage (Figure 5B). Interestingly, in control, normal, NHA cells, we were hardly able to detect Bim and caspase 3 cleavage, compared with levels in the U87 cells, under the same conditions (Figure 5C). These results are compatible with the inability of CM223 to affect NHA cell viability (Figure 2C), implying this compound would exert its actions selectively on tumour cells, with minimal cytotoxic effects on normal cells or tissues.
Analysis of sensitivity of primary cultures of glioma cells derived from patients, to CM223 treatment
To test its efficacy in a more physiopathological context ex vivo, we tested the sensitivity of two representative glioma cell lines (GBM37 and GBM50) derived from patients, to CM223 treatment. As shown, CM223 (10 μM) inhibited the phosphorylation of STAT3 in all glioma cells, with or without stimulation by exogenous EGF (50 ng·mL−1). The latter condition, as expected, results in the physiological degradation of the receptor in order to protect the cell from overstimulation (Figures 4C and 6A) (Sigismund et al., 2013). Interestingly, the degree of inhibition was more pronounced in primary GBM cells, compared with the U87 glioma cell line. Accordingly in GBM cells (Figure 6B), CM223 achieved an IC50 value lower (1.2 μM in GBM37; 0.45 μM in GBM50 cells) than that in U87 cells (1.9 μM). Even though CM223 and the EGFR tyrosine kinase inhibitor erlotinib both inhibit EGFR signalling, they do so by different mechanisms. We therefore tested the effects of combinations of these compounds in U87 and GBM cells. For erlotinib, the fixed concentration of 0.6 μM was chosen. At this concentration (0.6 μM), CM223 alone was more effective than erlotinib alone in reducing proliferation of glioma cells (Figure 6B) but the combination was more effective than either compound alone at the lower concentrations of CM223.
Figure 6.
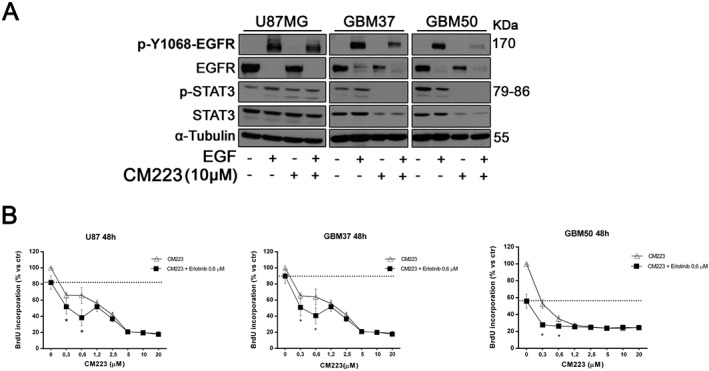
CM223 inhibits EGF/EGFR signalling in primary cultures of glioma cells derived from patients. (A) Western blot analysis for p‐Y1068‐EGFR, EGFR, p‐STAT3 and total STAT3 in whole cell extracts from two patient‐derived, primary cell lines (GBM37 and GBM50) serum starved and cultured for 18 h with CM223 (10 μM) and then stimulated or not with EGF (50 ng·mL−1) for 10 min. α‐tubulin was used as control of protein loading. Panels show representative Western blots of two different experiments performed with similar results. (B) U87MG cells and two patient‐derived, primary cell lines (GBM37 and GBM50) were treated for 48 h with erlotinib (0.6 μM) in the presence or absence of the indicated concentrations (0–20 μM) of CM223 before analysis of cell proliferation by BrdU incorporation assay. Results, reported as percentage, are expressed as mean ± SD of six independent experiments performed in triplicate. *P < 0.05, significantly different from untreated control; ANOVA with Tukey's test.
Discussion
We have demonstrated that the modified adenosine compound i6A exerts cytotoxic, antiangiogenic and proapoptotic activity in different cell lines (Laezza et al., 2006; Spinola et al., 2007; Laezza et al., 2009; Laezza et al., 2010; Pisanti et al., 2014; Ciaglia et al., 2017). Earlier reports by our group highlighted the involvement of FPPS in i6A action in different physiopathological settings (Laezza et al., 2006; Ciaglia et al., 2013). More recently, we documented for the first time the antitumor activity of i6A on human glioma cells (Ciaglia et al., 2017). In our work, i6A clearly displayed antiglioma action through AMP‐activated protein kinase (AMPK)‐dependent internalization and degradation of EGFR but we could not exclude the possibility that i6A might also elicit other effects. Indeed, i6A reduces the growth of xenograft tumours induced by KiMol by inhibiting FPPS, a key enzyme of the isoprenoid biosynthetic pathway and protein prenylation (Laezza et al., 2006), which are crucial events for the function and activity of EGFR and its downstream signalling. Moreover, using an inverse virtual screening approach and NMR spectroscopy experiments, we have shown that i6A binds to the active site of FPPS and produces a moderate enzyme inhibition. All these results and, in particular, the study of i6A binding to the FPPS active site, led to the design of new analogues of i6A with different substituents at the N6 position of the purine ring. As we had demonstrated an important contribution from the isopentenyl moiety to the binding of i6A with the FPPS protein, we designed and synthesized the analogues shown in Figure 1A, including a benzyl ring (CM223) and an isopentyl moiety (CM224) or a diversely substituted phenyl ring (compounds FP11, FP13 and FP14), (Ottria et al., 2010), at the N6 position.
In order to improve targeted drug therapy for GBM, which is the most common and malignant CNS tumour, these analogues of i6A were assessed for their inhibition of cell growth in the U87MG human glioma cell line and in primary glioma cells, derived from patients. In these assays, only CM223 exhibited a marked cytostatic effect significantly higher than that of i6A, whereas the other compounds showed no relevant activity. According to STD‐NMR experiments and enzymic colorimetric assays, the cytostatic activity of CM223, like that of i6A, was related to the modulation of FPPS. Interaction of CM223 with the active site of FPPS had a K D fivefold lower, and an EC50 twofold lower, than those for i6A. Interestingly, compounds CM224 and FP13 showing no significant cytostatic effect, exhibited no significant STD effects, confirming the relationship between the antiproliferative activity and FPPS binding. Interpretation of these data in terms of SAR, corroborated by molecular modelling calculation, indicated that the increased binding of CM223 (K D 0.19 mM) compared to i6A (K D 1.0 mM) was due to the presence of the aromatic benzyl ring that made more stable π‐stacking interactions with Phe113 and Tyr218 of the FPPS protein. For the analogue FP13, the difference in its binding from that of i6A and CM223, is due to the phenyl ring that provides a low flexibility point in this molecule.
A more detailed investigation of the antiproliferative action of CM223 has disclosed a marked anti‐clonogenic effect, associated with higher pro‐apoptotic action, compared with i6A. Exploration of the cellular pathway through which CM223 elicited these effects showed the suppression of Akt and STAT3 activation, two proteins whose signalling is critical for glioma cell proliferation and survival. The effect of CM223 on the MAPK/ERK pathway was concentration‐dependent, as it appeared inhibitory at lower concentrations (1‐to 5 μM), but stimulatory at 10 μM. According to earlier reports (Hamdi et al., 2008; Wei et al., 2011), these results may reflect the fact that ERK activation can also induce apoptosis, upstream of caspase activation. CM223 exerted several molecular actions, coherent with the execution of the intrinsic apoptotic cell program, such as induction of cleaved caspase‐9, cleaved caspase‐7 and ‐3, and induction of PARP cleavage, without effect on capase 8. Moreover, CM223 increased the levels of the proapoptotic three major Bim isoforms (Bim EL, L, S) and decreased the expression of antiapoptotic Bcl‐XL.
Looking for a possible molecular mechanism, we consider that the anti‐glioma action of CM223 was related to inhibition of the mevalonate pathway and, at least in part, to blockade of FPPS. The observation of increased levels of unprenylated form of the small GTPase Rap1A in response to CM223 (Figure 4B), and the more complete inhibition of FPPS activity achieved by the FPPS siRNA‐CM223 combination (Figure 4C), along NMR interaction data, would suggest a direct effect on the FPPS enzymic activity. However, there was a discrepancy between the potency of the compound in NMR and FPPS colorimetric assays, where the direct interactions between CM223 and FPPS was explored, and the potency exhibited in a more complex cellular model. Here, the higher efficiency of CM223 in inhibiting the cell growth of tumour cells through cholesterol depletion (Figure 4C) suggests that CM223 does really interfere with the mevalonate metabolic pathway. However, the FPPS might not be the only target of CM223 and a bystander influence of other key enzymes, and intermediate metabolites of the mevalonate and isoprenoid pathway, modulated by CM223, cannot be ruled out. On the other hand, the greater effectiveness of the compound in cultured cells might be due to its conversion in a more active metabolite inside the cells, as previously documented for i6A, which needs to be phosphorylated to 5′‐i6A‐monophosphate by adenosine kinase, in order to exert its inhibitory effects on HUVEC (Pisanti et al., 2014).
Several reports show the correlation between inhibition of FPPS and cytostatic and/or pro‐apoptotic effects. Of note, overactive prenylated Rap1A enhanced the proliferative capacity of certain cell types contributing to tumourigenesis (Frische and Zwartkruis, 2010; Jeyaraj et al., 2011). Moreover, accumulation of unprenylated Rap1A due to the inhibition of farnesyl‐ or geranyl geranyl transferase in vitro induced apoptosis in human plasma cells, suggesting that geranylgeranylated proteins, like Rap1A, play a role in signalling pathways that prevent cell death (Gordon et al., 2002). Here, we showed that the depletion of lipid moieties by CM223‐mediated FPPS inhibition was critical for the functionality and activity of EGFR (Warren and Landgraf, 2006) and that depletion potently impaired the main intracellular signalling pathways responsible for the maintenance of the neoplastic phenotype. Prenylation is indeed required for the proper function and localization of several downstream critical mediators of the EGFR signalling cascade, including the Ras, Rho and Rab families, which require farnesyl and geranylgeranyl post‐translational modifications for their activity (Gibbs et al., 1994). Moreover, it is well known that the isoprenoid compound dolichol is involved in the N‐linked glycosylation of EGFR, which facilitates its proper conformation and efficient ligand binding and activation of EGFR (Carlberg et al., 1996; Ceresa and Bahr, 2006). Accordingly, we found that the CM223‐treated cells were slower to properly re‐establish the endogenous mevalonate and isoprenoid levels and recover EGFR signalling after washout of MβCD (Figure 4C). Consistent with these mechanisms of action, we observed that CM223 was highly effective at blocking tumour cell growth of EGFR‐activated primary GBM cells in which the EGF‐induced signalling sensitizes them to the effects of CM223. Last, by indirectly targeting EGFR signalling, CM223 improved the response to the EGFR tyrosine kinase inhibitor erlotinib, when used together (Figure 6B).
In view of the results collected in our experiments and considering the published evidence, we consider that the antiproliferative and proapoptotic effects of CM223 in glioma cells may be in part ascribable to the disruption of EGFR signalling, as a consequence of the modulation of the mevalonate metabolic pathway of and, in particular, of FPPS, a key branch point enzyme. Indeed, we demonstrated by STD‐NMR experiments that CM223 binds and inhibits FPPS activity. Interactions of CM223 with the FPPS active site had a K D fivefold lower and an EC50 twofold lower, than those of i6A. Interestingly compounds CM224 and FP13 showing no significant cytostatic effect, failed to bind FPPS enzyme in STD‐NMR experiments, confirming the relationship between the antiproliferative activity and the FPPS binding. Interpretation of these data in a SAR perspective, corroborated by molecular modelling calculation, indicates that the difference in the binding of CM223, compared to the compounds of FP series is due to the phenyl ring that confers rigidity to this part of the molecule.
In conclusion, our data confirm that i6A and, to a greater extent, its benzyl analogue CM223 exhibit a promising antitumor activity in GBM cells. The modifications we made to the i6A scaffold leads to molecules that can be considered lead compounds in the search for antiglioma agents. Indeed, the in vitro efficacy of CM223, its lack of cytotoxicity in healthy brain cells, its additional activity in patient‐derived primary GBM cells that more accurately reflect the physiopathological context, make CM223 an interesting compound for the development of novel chemotherapeutic agents in glioma and other EGFR‐driven cancers.
All the biological pathways controlled by i6A (Laezza et al., 2006; Ciaglia et al., 2013) and CM223 are related to the inhibition of FPPS activity. Therefore, the development of i6A analogues characterized by increased antitumor activity may take advantage of the FPPS‐CM223 structural interaction data.
Author contributions
E.C. wrote the paper, designed and conducted biological assays and analysed and interpreted data; M.G. conducted FPPS gene expression procedure, NMR studies and analysed and interpreted data; M.A. performed biological research, analysed data and reviewed critically the paper; M.S. performed Molecular Docking studies; C.L. performed research, R.R., S.P. and P.G. performed siRNA transfection procedure and established primary derived cells. M.R., C.M. and P.C. conducted the synthesis of new i6A derivatives and gave substantial contribution to analysis and interpretation of data; A.M.D. and M.B. supervised the project in its entirety, wrote and reviewed critically the paper and provided financial support. All authors approved the final version to be published.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 STD‐NMR spectra of FPPS/FP11, FP13 and FP16, respectively, in black the offresonance spectra, in blue the on‐resonance spectra and in red the STD spectra.
Figure S2 1H NMR spectrum (600 MHz T= 298K) of FP11 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S3 1H NMR spectrum (600 MHz T= 298K) of FP13 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S4 1H NMR spectrum (600 MHz T= 298K) of FP16 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S5 1H NMR spectrum (600 MHz T= 298K) of CM223 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S6 1H NMR spectrum (600 MHz T= 298K) of II (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Scheme 1 Synthesis of 6‐substituted purine derivatives FP11, FP13 and FP16.
Acknowledgements
This study was supported by Associazione Italiana Ricerca sul Cancro (AIRC; IG 18999 and IG13312 to M. Bifulco,). E. Ciaglia was supported by a fellowship from Fondazione Umberto Veronesi (FUV 2017, cod.1072). The authors thank Dr Francesco Montella and Dr Ylenia Senatore (University of Salerno) for their continual support.
Ciaglia, E. , Grimaldi, M. , Abate, M. , Scrima, M. , Rodriquez, M. , Laezza, C. , Ranieri, R. , Pisanti, S. , Ciuffreda, P. , Manera, C. , Gazzerro, P. , D'Ursi, A. M. , and Bifulco, M. (2017) The isoprenoid derivative N6‐benzyladenosine CM223 exerts antitumor effects in glioma patient‐derived primary cells through the mevalonate pathway. British Journal of Pharmacology, 174: 2287–2301. doi: 10.1111/bph.13824.
Contributor Information
Anna Maria D'Ursi, Email: dursi@unisa.it.
Maurizio Bifulco, Email: maubiful@unisa.it.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo J, Nieto PM (2011). STD‐NMR: application to transient interactions between biomolecules‐a quantitative approach. Eur Biophys J 40: 1357–1369. [DOI] [PubMed] [Google Scholar]
- Bifulco M, Malfitano AM, Proto MC, Santoro A, Caruso MG, Laezza C (2008). Biological and pharmacological roles of N‐6‐Isopentenyladenosine: an emerging anticancer drug. Anticancer Agents Med Chem 8: 200–204. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Dricu A, Blegen H, Wang M, Hjertman M, Zickert P et al. (1996). Mevalonic acid is limiting for N‐linked glycosylation and translocation of the insulin‐like growth factor‐1 receptor to the cell surface. Evidence for a new link between 3‐hydroxy‐3‐methylglutaryl‐coenzyme a reductase and cell growth. J Biol Chem 271: 17453–17462. [DOI] [PubMed] [Google Scholar]
- Castiglioni S, Casati S, Ottria R, Ciuffreda P, Maier JAM (2013). N6‐isopentenyladenosine and its analogue N6‐benzyladenosine induce cell cycle arrest and apoptosis in bladder carcinoma T24 cells. Anticancer Agents Med Chem 13: 672–678. [DOI] [PubMed] [Google Scholar]
- Ceresa BP, Bahr SJ (2006). rab7 activity affects epidermal growth factor: epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J Biol Chem 281: 1099–1106. [DOI] [PubMed] [Google Scholar]
- Chen YZ, Zhi DG (2001). Ligand‐protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins Struct Funct Genet 43: 217–226. [DOI] [PubMed] [Google Scholar]
- Ciaglia E, Abate M, Laezza C, Pisanti S, Vitale M, Seneca V et al. (2017). Antiglioma effects of N6‐isopentenyladenosine, an endogenous isoprenoid end product, through the Downregulation of Epidermal Growth Factor Receptor. Int J Cancer 140: 959–972. [DOI] [PubMed] [Google Scholar]
- Ciaglia E, Pisanti S, Picardi P, Laezza C, Malfitano AM, D'Alessandro A et al. (2013). N6‐isopentenyladenosine, an endogenous isoprenoid end product, directly affects cytotoxic and regulatory functions of human NK cells through FDPS modulation. J Leukoc Biol 94: 1207–1219. [DOI] [PubMed] [Google Scholar]
- Ciaglia E, Torelli G, Pisanti S, Picardi P, D'Alessandro A, Laezza C et al. (2015). Cannabinoid receptor CB1 regulates STAT3 activity and its expression dictates the responsiveness to SR141716 treatment in human glioma patients' cells. Oncotarget 6: 15464–15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier PGJ, Dauhine F, Lundy MW, Rogers MJ, Ebetino FH, Clezardin P (2008). Lowering bone mineral affinity of bisphosphonates as a therapeutic strategy to optimize skeletal tumor growth inhibition in vivo. Cancer Res 68: 8945–8953. [DOI] [PubMed] [Google Scholar]
- Frische EW, Zwartkruis FJT (2010). Rap1, a mercenary among the Ras‐like GTPases. Dev Biol 340: 1–9. [DOI] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS (2015). Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 15: 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JB, Chu XS, Qiu YG, Wu L, Qiao YQ, Wu JS et al. (2010). Discovery of potent inhibitor for farnesyl pyrophosphate synthase in the mevalonate pathway. Chem Commun 46: 5340–5342. [DOI] [PubMed] [Google Scholar]
- Gibbs JB, Oliff A, Kohl NE (1994). Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 77: 175–178. [DOI] [PubMed] [Google Scholar]
- Gordon S, Helfrich MH, Sati HIA, Greaves M, Ralston SH, Culligan DJ et al. (2002). Pamidronate causes apoptosis of plasma cells in vivo in patients with multiple myeloma. Br J Haematol 119: 475–483. [DOI] [PubMed] [Google Scholar]
- Hackel PO, Zwick E, Prenzel N, Ullrich A (1999). Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr Opin Cell Biol 11: 184–189. [DOI] [PubMed] [Google Scholar]
- Hamdi M, Popeijus HE, Carlotti F, Janssen JM, van der Burgt C, Cornelissen‐Steijger P et al. (2008). ATF3 and Fra1 have opposite functions in JNK‐and ERK‐dependent DNA damage responses. DNA Repair 7: 487–496. [DOI] [PubMed] [Google Scholar]
- Hocek M, Holy A, Votruba I, Dvorakova H (2000). Synthesis and cytostatic activity of substituted 6‐phenylpurine bases and nucleosides: application of the Suzuki‐Miyaura cross‐coupling reactions of 6‐chloropurine derivatives with phenylboronic acids. J Med Chem 43: 1817–1825. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pelle X, Geiser M et al. (2010). Allosteric non‐bisphosphonate FPPS inhibitors identified by fragment‐based discovery. Nat Chem Biol 6: 660–666. [DOI] [PubMed] [Google Scholar]
- Jeyaraj SC, Unger NT, Chotani MA (2011). Rap1 GTPases: an emerging role in the cardiovasculature. Life Sci 88: 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH et al. (2006). The molecular mechanism of nitrogen‐containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A 103: 7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten H (1984). On the biological significance of modified nucleosides in tRNA. Prog Nucleic Acid Res Mol Biol 31: 59–114. [DOI] [PubMed] [Google Scholar]
- Laezza C, Caruso MG, Gentile T, Notarnicola M, Malfitano AM, Di Matola T et al. (2009). N6‐isopentenyladenosine inhibits cell proliferation and induces apoptosis in a human colon cancer cell line DLD1. Int J Cancer 124: 1322–1329. [DOI] [PubMed] [Google Scholar]
- Laezza C, Caruso MG, Gentile T, Notarnicola M, Malfitano AM, Di Matola T et al. (2014). N6‐isopentenyladenosine inhibits cell proliferation and induces apoptosis in a human colon cancer cell line DLD1 (vol 124, pg 1322, 2009). Int J Cancer 135: E11–E11. [DOI] [PubMed] [Google Scholar]
- Laezza C, D'Alessandro A, Di Croce L, Picardi P, Ciaglia E, Pisanti S et al. (2015). p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis 6: e1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laezza C, Malfitano AM, Di Matola T, Ricchi P, Bifulco M (2010). Involvement of Akt/NF‐kappaB pathway in N6‐isopentenyladenosine‐induced apoptosis in human breast cancer cells. Mol Carcinog 49: 892–901. [DOI] [PubMed] [Google Scholar]
- Laezza C, Notarnicola M, Caruso MG, Messa C, Macchia M, Bertini S et al. (2006). N6‐isopentenyladenosine arrests tumor cell proliferation by inhibiting farnesyl diphosphate synthase and protein prenylation. FASEB J 20: 412–418. [DOI] [PubMed] [Google Scholar]
- Laten HM, Zahareasdoktor S (1985). Presence and source of free isopentenyladenosine in yeasts. Proc Natl Acad Sci U S A 82: 1113–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro G, Romano A, Riccio R, Bifulco G (2011). Inverse virtual screening of antitumor targets: pilot study on a small database of natural bioactive compounds. J Nat Prod 74: 1401–1407. [DOI] [PubMed] [Google Scholar]
- Malfitano AM, Laezza C, Saccomanni G, Tuccinardi T, Manera C, Martinelli A et al. (2013). Immune‐modulation and properties of absorption and blood brain barrier permeability of 1,8‐naphthyridine derivatives. J Neuroimmune Pharmacol 8: 1077–1086. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B (1999). Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed 38: 1784–1788. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B (2001). Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc 123: 6108–6117. [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottria R, Casati S, Baldoli E, Maier JA, Ciuffreda P (2010). N(6)‐alkyladenosines: synthesis and evaluation of in vitro anticancer activity. Bioorg Med Chem 18: 8396–8402. [DOI] [PubMed] [Google Scholar]
- Pisanti S, Picardi P, Ciaglia E, Margarucci L, Ronca R, Giacomini A et al. (2014). Antiangiogenic effects of N6‐isopentenyladenosine, an endogenous isoprenoid end product, mediated by AMPK activation. FASEB J 28: 1132–1144. [DOI] [PubMed] [Google Scholar]
- Sacchettini JC, Poulter CD (1997). Biochemistry – Creating isoprenoid diversity. Science 277: 1788–1789. [DOI] [PubMed] [Google Scholar]
- Schrödinger M (2009). LLC New York NY.
- Scrima M, Lauro G, Grimaldi M, Di Marino S, Tosco A, Picardi P et al. (2014). Structural evidence of N6‐isopentenyladenosine as a new ligand of farnesyl pyrophosphate synthase. J Med Chem 57: 7798–7803. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Algisi V, Nappo G, Conte A, Pascolutti R, Cuomo A et al. (2013). Threshold‐controlled ubiquitination of the EGFR directs receptor fate. EMBO J 32: 2140–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinola M, Colombo F, Falvella FS, Dragani TA (2007). N6‐isopentenyladenosine: a potential therapeutic agent for a variety of epithelial cancers. Int J Cancer 120: 2744–2748. [DOI] [PubMed] [Google Scholar]
- Szkopinska A, Plochocka D (2005). Farnesyl diphosphate synthase; regulation of product specificity. Acta Biochim Pol 52: 45–55. [PubMed] [Google Scholar]
- Thurnher M, Nussbaumer O, Gruenbacher G (2012). Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin Cancer Res 18: 3524–3531. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL et al. (2016). An LXR‐cholesterol axis creates a metabolic co‐dependency for brain cancers. Cancer Cell 30: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J‐C, Chu P‐Y, Chen C‐M, Lin J‐H (2012). idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide‐and‐conquer docking approach. Nucleic Acids Res 40: W393–W399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Panasiuk A, Grainger DW (2011). Small interfering RNA knocks down the molecular target of alendronate, farnesyl pyrophosphate synthase, in osteoclast and osteoblast cultures. Mol Pharm 8: 1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CM, Landgraf R (2006). Signaling through ERBB receptors: multiple layers of diversity and control. Cell Signal 18: 923–933. [DOI] [PubMed] [Google Scholar]
- Wei F, Yan J, Tang D (2011). Extracellular signal‐regulated kinases modulate DNA damage response ‐ a contributing factor to using MEK inhibitors in cancer therapy. Curr Med Chem 18: 5476–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo IS, Eun SY, Kim HJ, Kang ES, Kim HJ, Lee JH et al. (2010). Farnesyl diphosphate synthase attenuates paclitaxel‐induced apoptotic cell death in human glioblastoma U87MG cells. Neurosci Lett 474: 115–120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 STD‐NMR spectra of FPPS/FP11, FP13 and FP16, respectively, in black the offresonance spectra, in blue the on‐resonance spectra and in red the STD spectra.
Figure S2 1H NMR spectrum (600 MHz T= 298K) of FP11 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S3 1H NMR spectrum (600 MHz T= 298K) of FP13 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S4 1H NMR spectrum (600 MHz T= 298K) of FP16 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S5 1H NMR spectrum (600 MHz T= 298K) of CM223 (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Figure S6 1H NMR spectrum (600 MHz T= 298K) of II (5 mM) in 25 mM d‐Tris (pH 7.4), 0.5 mM MgCl2 and 25 mM NaCl.
Scheme 1 Synthesis of 6‐substituted purine derivatives FP11, FP13 and FP16.