

Abstract
Background and Purpose
With the emergence of extensively drug‐resistant tuberculosis, there is a need for new anti‐tubercular drugs that work through novel mechanisms of action. The meta cleavage product hydrolase, HsaD, has been demonstrated to be critical for the survival of Mycobacterium tuberculosis in macrophages and is encoded in an operon involved in cholesterol catabolism, which is identical in M. tuberculosis and M. bovis BCG.
Experimental Approach
We generated a mutant strain of M. bovis BCG with a deletion of hsaD and tested its growth on cholesterol. Using a fragment based approach, over 1000 compounds were screened by a combination of differential scanning fluorimetry, NMR spectroscopy and enzymatic assay with pure recombinant HsaD to identify potential inhibitors. We used enzymological and structural studies to investigate derivatives of the inhibitors identified and to test their effects on growth of M. bovis BCG and M. tuberculosis.
Key Results
The hsaD deleted strain was unable to grow on cholesterol as sole carbon source but did grow on glucose. Of seven chemically distinct ‘hits’ from the library, two chemical classes of fragments were found to bind in the vicinity of the active site of HsaD by X‐ray crystallography. The compounds also inhibited growth of M. tuberculosis on cholesterol. The most potent inhibitor of HsaD was also found to be the best inhibitor of mycobacterial growth on cholesterol‐supplemented minimal medium.
Conclusions and Implications
We propose that HsaD is a novel therapeutic target, which should be fully exploited in order to design and discover new anti‐tubercular drugs.
Linked Articles
This article is part of a themed section on Drug Metabolism and Antibiotic Resistance in Micro‐organisms. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.14/issuetoc
Abbreviations
- 4,9‐DHSA
4,5–9,10‐diseco‐3‐hydroxy‐5,9,17‐trioxoandrosta‐1(10), 2‐diene‐4‐oic acid
- ADC
albumin‐dextrose‐catalase
- BCG
Bacillus Calmette–Guérin
- DSF
differential scanning fluorimetry
- FBDD
fragment‐based drug discovery
- FCS
fetal calf serum
- HOPDA
2‐hydroxy‐6‐oxo‐6‐phenylhexa‐2,4‐dienoic acid
- LB
Luria‐Bertani
- MB
Middlebrook
- MCP
meta‐cleavage product
- MDR‐TB
multidrug‐resistant tuberculosis
- MIC
minimum inhibitory concentration
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAT
arylamine N‐acetyltransferase
- SPOTi
spot culture growth inhibition assay
- TB
tuberculosis
Introduction
Tuberculosis (TB) remains a global health emergency with an estimated 9 million new cases and 1.5 million deaths every year (WHO, 2015). Importantly, 3.7% of new cases and 20% of previously treated patients are infected with multidrug‐resistant TB (MDR‐TB). Moreover, up to 9% of these MDR‐TB cases are resistant to nearly all known treatments and considered as extensively drug resistant TB. Current treatment modalities for drug‐sensitive TB cases require continuous dosing for at least 6 months of chemotherapy leading to poor patient compliance, which further increases the rate of drug resistance (WHO, 2015). There is a clear need for new anti‐tubercular drugs, particularly those that shorten the treatment period. However, the long‐term and indefinite intracellular survival of Mycobacterium tuberculosis in different physiological states has made the development of novel therapeutics extremely challenging.
Cholesterol has been identified as important in the infection process of M. tuberculosis (Peyron et al., 2000; Ouellet et al., 2011; Crowe et al., 2015; Lovewell et al., 2016; Fineran et al., 2016) and is used as a carbon source during survival within macrophages (Van der Geize et al., 2007; Pandey and Sassetti, 2008; Rohde et al., 2012). In previous work, we demonstrated that an operon consisting of hsaA (Rv3567), hsaB (Rv3570), hsaC (Rv3568), hsaD (Rv3569) (Van der Geize et al., 2007) is involved in cholesterol metabolism. This operon was first identified in M. tuberculosis and also includes the gene encoding arylamine N‐acetyltransferase (nat, Rv3566) (Payton et al., 2001; Anderton et al., 2006) and then further characterized in other mycobacterial species (Evangelopoulos et al., 2014). It is currently believed that the NAT protein may metabolize the alkyl chain generated by cholesterol catabolism (Lack et al., 2009), whilst the other proteins are involved in the degradation of the sterol nucleus of cholesterol (Van der Geize et al., 2007; Griffin et al., 2011; 2012). Studies using transposon mutagenesis have shown that HsaC and HsaD are required for the survival of M. tuberculosis within macrophages (Rengarajan et al., 2005). The expression of hsaC gene was later demonstrated to be essential both in vitro and in vivo for infection of M. tuberculosis in guinea pigs (Yam et al., 2009). More recent studies have also implicated a role for the operon and HsaD in mycobacterial growth linked to cholesterol catabolism (Griffin et al., 2011). HsaD has also been shown to be overexpressed in hypervirulent strains of M. bovis Bacillus Calmette–Guérin (BCG) during mouse infection (Blanco et al., 2009) and compounds with antitubercular activity have been identified as potential HsaD inhibitors via virtual screening (Rebollo‐Lopez et al., 2015).
HsaD belongs to the superfamily of αβ‐hydrolases that consists of the meta‐cleavage product (MCP) hydrolase subfamily (Lack et al., 2008). HsaD catalyses the hydrolytic cleavage of carbon–carbon bonds through a serine protease‐like catalytic triad (Lack et al., 2008; 2010) of the MCP 4,5‐9,10‐diseco‐3‐hydroxy‐5,9,17‐trioxoandrosta‐1(10),2‐diene‐4‐oic acid (4,9‐DHSA) in the cholesterol metabolism pathway (Van der Geize et al., 2007). In a previous study, we demonstrated that mechanism‐based serine protease inhibitors can inhibit HsaD (Ryan et al., 2014) suggesting that an acyl enzyme intermediate may be formed during the reaction process in line with studies on the mechanism of other C─C bond hydrolases (Ruzzini et al., 2012; 2013). As the HsaC protein (Yam et al., 2009) is unstable in oxygen, HsaD is a promising pharmacological target essential for M. tuberculosis infection. It is highly soluble as a stable recombinant protein (Lack et al., 2009), and there are multiple crystal structures available (Lack et al., 2008; 2010). Moreover, HsaD has a very large active site cavity that makes it a good target for fragment‐based drug discovery (FBDD) (Silvestre et al., 2013). FBDD combines NMR, differential scanning fluorimetry (DSF) and crystallography to use weakly binding small molecules (<300 Da) as building blocks to develop potent small molecular inhibitors. It is firmly established that fragment screening is a powerful approach to obtain useful start points for inhibitor discovery. Once the biological rationale for a given target is established, then fragment based approaches are warranted as one of the most effective methods to identify tractable starting points for drug design (Ciulli and Abell, 2007; Ciulli, 2013). This approach has shown tremendous success in modern drug discovery (Hudson et al., 2012; Hung et al., 2016) and has been particularly recommended for developing agents against TB (Marchetti et al., 2016; Mendes and Blundell, 2016).
In this study, we investigate HsaD as a novel pharmacological target for TB through generation of a specific gene deletion mutant and describe the identification of compounds as inhibitors of HsaD by a FBDD approach.
Methods
Bacterial growth conditions
M. tuberculosis, M. bovis BCG and M. smegmatis mc2155 liquid cultures were grown in Middlebrook (MB) 7H9 broth containing 10% (v·v−1) albumin‐dextrose‐catalase (ADC), 0.2% (v·v−1) glycerol and 0.05% (v·v−1) Tween‐80. Mycobacterial strains were also grown on MB7H10 agar containing 10% (v·v−1) oleic ADC and 0.5% (v·v−1) glycerol. M. tuberculosis cultures were grown in 10 mL broth in a 30 mL vials as standing cultures, M. bovis BCG in 100 mL broth in a roller bottle rolling cultures at 2 r.p.m. and M. smegmatis in 10 mL in a 50 mL centrifuge tubes rotating at 180 r.p.m. all in a 37°C incubator, unless specified otherwise. ΔhsaD M. bovis BCG was further supplemented with 50 μg·mL−1 hygromycin.
Growth curve comparison of the wild‐type and ΔhsaD M. bovis BCG were done in minimal medium containing 0.05% (v·v−1) tyloxapol and 0.05% (v·v−1) ethanol with either (i) no additional carbon source, (ii) 100 μg·mL−1 glycerol or, (iii) 100 μg·mL−1 cholesterol. An equivalent number of bacteria (6 × 108) were added to start the cultures and the bacterial growth rate was measured by monitoring the OD600 daily for 16 days.
Escherichia coli JM109 cells were grown in Luria‐Bertani (LB) broth 10 mL of a centrifuge tube rotating at 180 r.p.m. a 37°C incubator. LB agar was used for solid growth of E. coli strains at 37°C, unless specified otherwise.
Generation of the ΔhsaD gene deletion
Deletion of the hsaD gene was performed using specialized transduction as previously described (Bardarov et al., 2002). Briefly, the upstream and downstream flanking DNA regions of the hsaD gene were amplified by PCR using the following pair of oligonucleotide primers for the upstream flanking DNA region 5′‐TTTTTTTTGCATAAATTGCAGGCACCGTAGGCCAT‐3′ and 5′‐TTTTTTTTGCATTTCTTGCAGTGACGTCCATTCAACA‐3′ as the forward and reverse primers respectively with a BstAPI restriction site (underlined). The primers for the downstream flanking region were 5′‐TTTTTTTTCACAGAGTGCTTGACCGAGGCAATTGGAGAC‐3′ and 5′‐TTTTTTTTCACCTTGTGCACCTGTTGGGCGGGC‐3′ as the forward right and reverse primers respectively with a DraIII restriction site (underlined). The upstream and downstream products were subsequently digested with DraIII and BstAPI, respectively, and ligated with PfIMI‐digested p0004s vector arms. The knockout construct was linearised following digestion with PacI and cloned into phAE159. Both pAES and phAE159 were subsequently digested with PacI, gel purified and ligated with T4 DNA ligase. The ligation mixture was then packaged into λ phage heads by in vitro packaging, transduced into E. coli and plated on LB agar supplemented with hygromycin. Following validation the phasmids were then electroporated into M. smegmatis and grown at a permissive temperature (30°C) to generate mycobacteriophages. The resulting high‐titre mycobacteriophages were then used to transduce the recipient mycobacteria at 37°C (non‐permissive temperature). The correct identity of loss‐of‐function mutations was confirmed by PCR amplifications with primers against the internal hsaD gene (forward: 5′ AAGTCGGCTCCGGC 3′ reverse: 5′ TGGCCGTCGACCAGC 3′) and the region flanking the hsaD deletion (forward: 5′ GATGCTCATCTGCCACC 3′ reverse: 5′ ATGACAGCTACCGAGGAAT 3′).
Intracellular survival of ΔhsaD M. bovis BCG in macrophages
The RAW264.7 mouse macrophage cells were grown as a monolayer in RPMI‐1640 media (GIBCO, Paisley, UK), supplemented with 1% (w·v−1) L‐glutamine and 10% (v·v−1) fetal calf serum (FCS) at 37°C with 5% CO2. For the intracellular survival assays, RAW264.7 cells were plated approximately 24 h prior to infection on 6‐well plates in the presence of 2 mL RPMI (supplemented with 10% v·v−1 FCS) as described previously (Bhakta et al., 2004; Westwood et al., 2010). Cells were subsequently washed with fresh media, infected with M. bovis BCG at a multiplicity of infection of 20 (i.e. for every 1 RAW264.7 cell, 20 cells of M. bovis BCG were added) and incubated for 2 h. Cells were then washed twice with PBS and re‐suspended in fresh medium. The medium was later removed, and the cells were lysed with 1 mL of sterile distilled water for 10 min at 0, 24, 48, 72 and 120 h. The number of intracellular bacteria was determined by serially diluting the lysate, plating it on MB7H10 agar plates and incubating at 37°C for 3–4 weeks.
Effect of inhibitors against M. tuberculosis grown in cholesterol
The minimum inhibitory concentrations (MIC) of selected inhibitors were determined using the spot culture growth inhibition assay (SPOTi). This method has been compared favourably with other methods of MIC determination (Evangelopoulos and Bhakta, 2010). Briefly, mycobacteria were plated in 24 well plates on minimal agar based media containing: asparagine (0.5 g·L−1), KH2PO4 (1.0 g·L−1), Na2HPO4 (2.5 g·L−1), ferric ammonium citrate (50 mg·L−1), MgSO4,7H2O (0.5 g·L−1), CaCl2 (0.5 mg·L−1), ZnSO4 (0.1 mg·L−1), agar (1.5% w·v−1) and either glycerol (0.1% v·v−1) or cholesterol (0.01% w.v−1). Fragments dissolved in DMSO (0.0125, 0.025, 0.05, 0.1 and 0.2 μg·mL−1) or DMSO alone were mixed with 2 mL of the minimal media with cholesterol per well. The final DMSO concentration in the wells containing fragments was 0.2%. In addition a well with 0.2% DMSO only as well as isoniazid at various concentrations (0.001, 0.01, 0.1, 1 and 10 μg·mL−1) were used as positive controls. The inhibitors and isoniazid were prepared by one experimentalist and supplied as a numbered array such that growth experiments were carried out blinded. The plates were then inoculated with 2 μL (approximately 103 viable cells) of a mid‐log phase culture of either M. bovis BCG or M. tuberculosis that was added in the centre of each well and allowed to soak before the plates were sealed with para‐film, wrapped with aluminium foil and incubated at 37°C for 2 weeks until a spot of colonies were developed in the control wells. The MIC was determined as the lowest concentration in wells where mycobacterial growth was not evident. This was repeated in triplicate in three independent experiments and the same MIC values were obtained in all experiments.
Effect of inhibitors against E. coli
An overnight culture of E. coli JM109 cells grown as stated before was diluted in LB media to a final OD600 of 0.1 and acted as a working culture. Then 10 μL of this working culture was used to inoculate a 96 well plate containing 90 μL of LB media with a range of inhibitor concentrations up to 100 μg·mL−1. The maximum DMSO concentration in the well was 1.7% (v·v−1). The plates were then transferred into the Tecan M200 Infinite Pro spectrophotometer where they grew at 37°C as standing cultures with absorbance measurements at 600 nm taken every hour for 4 h.
Cytotoxicity against mammalian cells
Cytotoxicity was tested using CCL‐228 colorectal cancer cells (American Tissue Culture Collection Manassas, VA). Cells were cultured in DMEM (GIBCO, UK), supplemented with 10% (v·v−1) FBS (Sigma‐Aldrich, Poole, UK), 10 000 U·mL−1 penicillin, 10 mg·mL−1 streptomycin in 0.9% NaCl (Sigma‐Aldrich), and maintained at 37°C in a humidified atmosphere with 5% CO2. Cells were seeded in 96‐well plates (20 000 cells per well) and treated for 48 h after which they were incubated with 100 μL of 50 μg·mL−1 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) solution in phenol red free DMEM (GIBCO) for 3 h in the dark at 37°C. The MTT was removed and the crystals solubilised with 100 μL DMSO for 15 min at room temperature with gentle shaking. The intensity was measured colorimetrically at 570 nm using the M200 Infinite Pro plate reader (Tecan) and percent growth was calculated. These studies were carried out blind by one experimenter using numbered samples provided by AR with the numbers randomised in five separate experiments.
Fragment‐based drug discovery
DSF was performed on a total of 1258 fragments as described previously (Niesen et al., 2007), using 0.4–1 μM HsaD in the presence of 2.5× SYPRO orange (Invitrogen) and 2.5 mM of compound (0.5 μL from a 0.1 M stock) at a final volume of 100 μL per well (final DMSO concentration of 2.5%) in 50 mM HEPES 300 mM NaCl pH 7.5. Fluorescence was monitored using an Agilent Mx3005p qPCR instrument (Santa Clara, USA). The fluorescence excitation and emission wavelengths were 483 and 533 nm. The output data was plotted as derivative of the fluorescence versus temperature, and the maximum of such derivative over temperature was determined as the melting temperature (TM). The primary DSF screen was carried out three times in triplicate, and the repeat screen was also carried out three times in triplicate making a total of six independent determinations in triplicate for each compound identified as a hit. Hits were identified by the shift in TM compared to HsaD with vehicle according to previously described methodologies (Ciulli, 2013).
NMR fragment screening
Confirmed hits were validated by ligand‐observed NMR spectroscopy (Dalvit et al., 2001; Ciulli et al., 2008; Ferguson et al., 2013). All NMR experiments were carried out at 278 K on a Bruker Avance 700 MHz equipped with a 5 mm triple TXI cryoprobe with z gradients. Relaxation‐edited NMR experiments incorporated a CPMG spin‐lock time of 200 ms before the acquisition period (Carr and Purcell, 1954). Saturation Transfer Difference NMR experiments employed a 40 ms selective Gaussian 180° shaped pulse at a frequency alternating between ‘on resonance’ (0.5 ppm) and ‘off resonance’ (~80 ppm) after every scan (Mayer and Meyer, 1999). Water suppression was achieved by using a W5 Watergate gradient spin‐echo pulse sequence (Piotto et al., 1992). WaterLOGSY experiments (Dalvit et al., 2000; 2001) employed a 20 ms selective Gaussian 180° shaped pulse at the water frequency and an NOE mixing time of 1 s. Water suppression was achieved using a double‐gradient echo excitation sculpting sequence with gradients. The resulting spectra were analysed using Topspin 2.0 software (Bruker). All samples were made up to a total volume of 200 μL in 3 mm capillaries, and contained 1.2 mM fragment, 50 mM Tris–HCl buffer at pH 7.5, 10% v·v−1 D2O, and 2.4% v·v−1 DMSO‐d 6. (Trimethylsilyl)‐propionic acid‐d 4 was present at 20 μM concentration in all samples for calibration purposes. A protein only control sample was run as buffer control. Each fragment was run as three samples (A–C). (A) Fragment only; (B) fragment + protein (10 μM HsaD); (C) fragment + HsaD + displacer compound PMSF (100 μM).
Enzymatic assays
Following the initial identification of fragments from the fragment‐based drug design library, inhibition assays were carried out as previously described (Ryan et al., 2014) in the presence of 1 mM with compounds 1–7. Subsequently, compounds that were capable of inhibiting the cleavage of HOPDA by HsaD were further selected for IC50 determination using a range of concentrations (1, 0.5, 0.375, 0.25, 0.125, 0.05 and 0 or 3.5, 3, 2.5, 2, 1.5, 1, 0.5 and 0 mM). Briefly pure HsaD (16 μg·mL−1 in 100 mM phosphate buffer pH 7.5, 20 mM NaCl) was added to the reaction mixture in the presence of 100 μM HOPDA (final concentration of 1% ethanol v·v−1) and inhibitor (final DMSO concentration of 5% v·v−1) in a final volume of 100 μL. The enzymatic activity was measured by monitoring the absorbance at 450 nm at 21°C using a M200 Infinite Pro plate reader (Tecan), and the IC50 was determined using GraphPad Prism. The ε450 of HOPDA was measured as 13 200 M−1 cm−1. N values refer to the number of independent experiments.
Solution of the structures of HsaD with fragments 2, 27 and 32
Crystals of apo‐HsaD were grown as described previously (Lack et al., 2008). All fragments were dissolved directly into the cryoprotectant containing 30% (w·v−1) PEG‐3000, 0.1 M N‐cyclohexyl‐2‐aminoethanesulfonic acid pH 9.5, 25% (v·v−1) glycerol, at concentrations of between 15 and 50 mM. Crystals were transferred into the cryoprotectant containing one of the fragments and allowed to soak overnight at 21°C before flash freezing for analysis.
Data collection for the structures of HsaD bound to 2, 27 and 32 were all performed at the Diamond Light source (Oxon, UK) using beamline I24 in the case of the structure with 2 and using beamline I03 for the others. In all cases, initial processing was performed via Xia2 (Winter, 2010) using XDS (Kabsch, 2010) to index and either Scala (2) (Evans, 2006) or Aimless (Collaborative Computational Project, 1994; Winn et al., 2011) (27 and 32), to merge and scale the data. The structure was solved by molecular replacement using PHASER (McCoy et al., 2007) using the structure of apo‐HsaD (PDB code, 2vf2) (Lack et al., 2008) stripped of all ligands as a search model. The atomic models were rebuilt in COOT (Emsley et al., 2010) and refined using REFMAC (Murshudov et al., 1997) and PHENIX (Adams et al., 2010). Model restraints for 2, 27 and 32 were generated in eLBOW (Moriarty et al., 2009).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Samples were randomised in all 96 well experiments (Westwood et al., 2011), and sample identity was not known to the investigator. Data are shown as the mean ± SD, and IC50 values in enzymic inhibition assays were determined via non‐linear fit analysis in Graphpad Prism.
Software tools to fit raw data obtained from DSF for data visualization and analysis were used based on Microsoft Excel and were obtained from the Structural Genomics Consortium Oxford (ftp://ftp.sgc.ox.ac.uk/pub/biophysics), see also (Niesen et al., 2007). The entire fluorescence raw data from the thermal melt curve was accurately fitted to a Maxwell–Boltzman distribution in Graphpad Prism.
Materials
All reagents were purchased from Sigma Aldrich (Poole, UK) unless otherwise stated. 3,5‐dichlorobenzenesulfonamide, 3‐chloro‐4‐hydroxybenzoate, 4‐toluenesulfonamide and 2‐oxovaleric acid were obtained from TCI Europe. 3,5‐diflurobenzenesulfonamide, 3,5‐dichlorobenzamide and 3‐bromobenzenesulfonamide were obtained from Apollo Scientific, and ethyl‐3,5–dichloro‐4‐hydroxybenzoate was obtained from Maybridge. The fragment library used in this study was composed of 1258 compounds from commercial libraries supplied by Maybridge (Thermo Fisher Scientific: http://www.maybridge.com/), as previously described (Ciulli, 2013; Van Molle et al., 2012). All library compounds were dissolved and stored in d 6‐DMSO.
All inhibitors were dissolved in DMSO. The DMSO final concentration in the assays was 5% (v·v−1) for the enzymatic assays and between 0.02% and 1.7% (v·v−1) for the growth experiments. The synthetic substrate, 2‐hydroxy‐6‐oxo‐6‐phenylhexa‐2,4‐dienoic acid (HOPDA) was synthesized by Almac Sciences (Craigavon, Northern Ireland) as previously described and dissolved in ethanol (1% v·v−1) (Lack et al., 2009). The fragment library was prepared as previously described (Van Molle et al., 2012; Ciulli, 2013).
Nomenclature of targets and ligands
Key ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016).
Results
Growth
Deletion of hsaD in M. bovis BCG (ΔhsaD) was generated using a mycobacteriophage‐based knockout method (Bardarov et al., 2002). All of the hygromycin resistant colonies tested were the correct mutant. Our results are in accordance with previous studies, which have reported that approximately 95% of antibiotic‐resistant transductants were the correct genotype (Bardarov et al., 2002).
When grown in MB7H9 broth (Figure 1A) or in minimal medium (Figure 1B) the deletion of hsaD had no apparent effect on bacterial growth when compared with M. bovis BCG. However, when the minimal medium was supplemented with cholesterol (Figure 1C), a significant difference between the two strains was observed. Whilst the growth of the wild type strain was very similar to that in minimal medium alone, the growth of ΔhsaD M. bovis BCG was significantly inhibited in the presence of cholesterol as compared to M. bovis BCG (Figures 1B,C). The growth of ΔhsaD M. bovis BCG in the presence of cholesterol was suppressed compared with growth in minimal medium alone.
Figure 1.
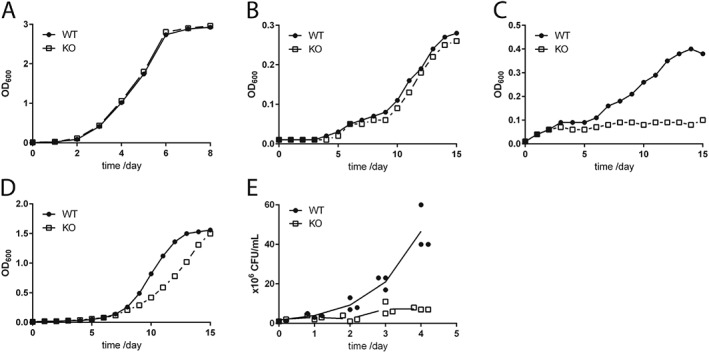
Effect of deletion of hsaD gene on M. bovis BCG. Growth rates of ΔhsaD and wildtype M. bovis BCG in MB7H9 (A) on minimal medium containing 0.05% (v·v−1) tyloxapol, 0.05% (v·v−1) ethanol with no additional carbon source (B) or supplemented with, 100 μg·mL−1 cholesterol (C) or 100 μg·mL−1 glycerol (D). The symbols show the average values of three independent experiments, and the symbols encompass the range of values obtained. The intracellular survival of ΔhsaD M. bovis BCG relative to the wild‐type in RAW264.7 cells (E) was determined as described in the Methods section. The results in (E) are the results of three independent experiments in which the mean values are plotted in a staggered plot. All symbols represent the same sampling time points (days 0, 1, 2, 3 and 4) but are shown as a cluster for clarity.
Growth in minimal medium with glycerol as the sole carbon source is supported in both the ΔhsaD strain and M. bovis BCG. The gene deleted strain showed a slight lag in rate of growth although it caught up by day 16, reaching the same optical density (OD600 = 1.5) (Figure 1D).
The intracellular survival of the wildtype and ΔhsaD strains of M. bovis BCG within RAW246.7 shows that, similar to ΔhsaC and Δnat strains, there was a reduction in intracellular growth of the ΔhsaD strain as compared to M. bovis BCG (Figure 1E).
We also deleted hsaD from M. smegmatis and when it was grown on cholesterol as carbon source the culture turned pink (Supporting Information Figure S1).
Identification of chemical fragments as inhibitors of HsaD
A commercial library of rule‐of‐three compliant 1258 fragments (Silvestre et al., 2013) was screened by DSF. The melting temperature of pure recombinant HsaD was monitored in the presence of the fragments. The melting temperature of HsaD in previous studies (Lack et al., 2009) had been determined by circular dichroism and loss of enzymatic activity of the protein itself to be between 69 and 72°C. The high melting temperature was confirmed by DSF monitored by SYPRO Orange to be 72.7 (average mean value from quadruplicates across 17 plates) ±0.21°C (average SD from quadruplicates across 17 plates) in the presence of solvent and no added fragment. Fragments were identified as potential binders to purified HsaD when a shift was monitored in the melting temperature of greater than two standard deviations from the mean melting temperature of pure HsaD alone (Table 1). Hit compounds (18) from the primary screen were re‐tested in duplicates in the thermal shift assay, from which eight were confirmed. The validated fragments were also tested as inhibitors of HsaD at a concentration of 1 mM of the compound (Figure 2) and were in addition tested by saturation transfer NMR for displacement by PMSF, an already identified inhibitor (Ryan et al., 2014) (Table 1 and Figure 2A).
Table 1.
Fragments from a 1258 member library producing a significant shift in HsaD TM and where saturation NMR demonstrated displacement of the ligand by PMSF
| Compound | TM shiftsa | NMR | PMSF displacement | Structure |
|---|---|---|---|---|
| 1 | 1.78; 2.64; 2.64 | Hit | Displaced |
(1).
|
| 2 | 0.77; 0.38; 0.38 | Hit | Displaced |
(2).
|
| 3 | 0.55; 0.76; 0.76 | Hit | Displaced |
(3).
|
| 4 | 0.55; ‐2.26; ‐2.26 | Hit | Displaced |
(4).
|
| 5 | 0.92; 1.13; 1.13 | Hit | Not displaced (PMSF bound) |
(5).
|
| 6 | 0.70; 1.13; 1.13 | Hit | Not displaced (PMSF not bound) |
(6).
|
| 7 | 0.82; 0.38; 0.76 | Hit | Displaced |
(7).
|
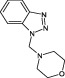

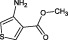
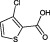
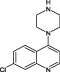
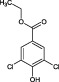

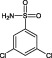
The compounds are numbered sequentially: 4‐((1H–benzo[d][1,2,3] triazol‐1‐yl)methyl)morpholine compound 1; 3,5,‐dichloro‐benzenesulfonamide compound 2; methyl 4‐aminothiophene‐3‐carboxylate compound 3; 3‐chlorothiophene‐2‐carboxylic acid, compound 4; 7‐chloro‐4‐(piperazin‐1‐yl)quinolone, compound 5; ethyl 3,5‐dichloro‐4‐hydroxybenzoate, compound 6; 4‐(piperidin‐1‐ylsulfonyl)aniline, compound 7.
The TM shifts in °C are shown for each compound tested with a positive result for DSF, performed as described in the Methods section at 2.5 mM final fragment concentration per well. Structures are drawn with ChemDraw Ultra 12.0 (CambridgeSoft). Hits from the primary DSF screen were re‐tested twice to confirm results of the initial screen and finally validated for protein binding using NMR spectroscopy (as shown in Figure 2A) (after Ciulli and Abell, 2007; Mayer and Meyer, 1999).
Figure 2.
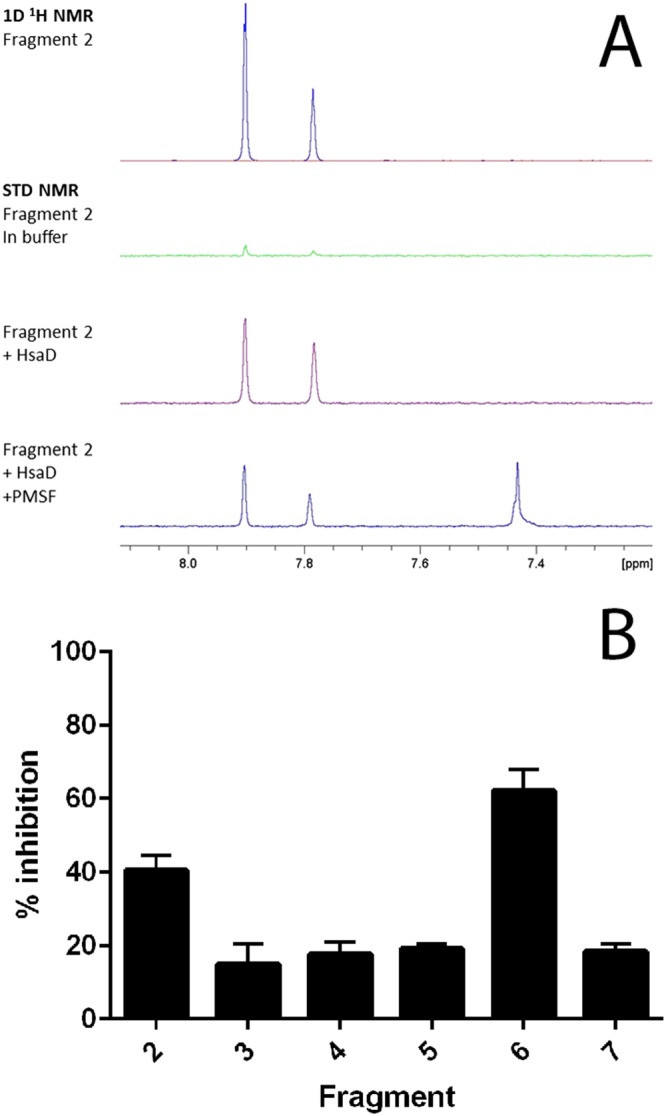
(A) NMR evidence of fragment binding. Water‐suppressed 1D 1H and STN NMR screening spectra for a representative fragment hit (fragment 2, at 1.2 mM) in the presence and absence of HsaD (10 μM) plus PMSF (100 μM). Only the fragment resonances in the aromatic region of the spectra are shown. The strong positive fragment signals in the presence of HsaD (purple spectrum) indicate binding, and this interaction is reduced by the addition of PMSF. (B) Inhibition of HsaD by DSF hits. Fragments identified by DSF as binding to HsaD were assayed for inhibition as described in Methods at 1 mM. The mean of six independent experiments (each carried out in triplicate) ± SD is shown. Fragments are numbered as shown in Table 1; 100% activity was defined as the rate of enzymatic HOPDA hydrolysis by 16 μg·mL−1 HsaD with vehicle (no compound), which was 0.08 μmol·min−1·(mg protein)−1.
From this combination of methods, a series of seven distinct fragments were identified as binders. Fragment 1 was not pursued further due to its insolubility at 1 mM. In addition to the ‘hits’ from the screen, a selection of 20 compounds from the library, which showed no evidence of binding by the DSF criteria, were also tested at 1 mM as inhibitors of HsaD, and none were found to inhibit the enzyme (data not shown). From the initial inhibition studies at 1 mM, two compounds looked promising – compound 2 and compound 6, which produced 38% and 60% inhibition respectively (Figure 2). Fragment 2 is a sulfonamide, and fragment 6 is a hydroxybenzoate.
When tested at a range of concentrations, compound 2 (3,5‐dichlorobenzenesulfonamide) is an inhibitor of HsaD with an IC50 of 0.41 mM (Table 2) and fragment 6 (ethyl 3,5‐dichloro‐4‐hydroxybenzoate), despite it not being displaced by PMSF (Table 1), is also an inhibitor of HsaD with an IC50 of 0.52 mM (Table 2).
Table 2.
Effects of (A) fragment 2 and its derivatives and (B) fragment 6 and a derivative on HsaD enzymic activity and growth of mycobacteria on cholesterol as carbon source
| ID | Compound | Structure | IC50 (mM) | MIC (μg·mL−1) growth on cholesterol | |
|---|---|---|---|---|---|
| M. bovis BCG | M. tuberculosis | ||||
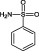
| 2 | 3,5,‐dichloro‐benzenesulfonamide |
(8).
|
0.41 ± 0.02 | <12.5 | 25 |
| 8 | Benzenesulfonamide |
(9).
|
NI | NI | NI |
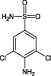
| 24 | 3,5‐dichlorosulfanilamide |
(10).
|
0.16 ± 0.01 | 25 | 200 |
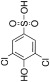
| 27 | 3,5‐dichloro‐4‐hydroxybenzenesulphonic acid |
(11).
|
0.27 ± 0.01 | >200 | >200 |
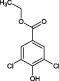
| 6 | ethyl 3,5‐dichloro‐4‐hydroxybenzoate |
(12).
|
0.52 ± 0.02 | <12.5 | <12.5 |
| 32 | 3,5‐dichloro‐4‐hydroxybenzoic acid |
(13).
|
0.54 ± 0.01 | <12.5 | 200 |
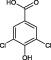
NI indicates no inhibition observed at 5 mM for inhibition of enzyme activity and no inhibition of growth at 200 μg·mL−1 as described in the Methods section. IC50 values shown as average ± SD (n = 6). See Supporting Information Table S1 for full list.
The fragments that were identified were distinct from previously identified substrates (Lack et al., 2010) and also distinct from putative inhibitors identified from a virtual screen (Rebollo‐Lopez et al., 2015) but, interestingly, bear some resemblance to the central moiety of an inhibitor identified as targeting the HsaAB protein complex of the same operon (VanderVen et al., 2015).
A small series of compounds related to each of the two classes of hit fragments 2 and 6 was tested for inhibition of HsaD (Supporting Information Table S1).
Derivatives of fragment 2 as inhibitors of HsaD
We investigated the small series of fragments modified around Compound 2, which has two chlorine atoms at the meta position. We investigated the effects of altering the position and number of chlorines in the molecule as well as substituting different halide groups. Removal of one chlorine from position 3 (Compound 9) was detrimental to the inhibitory potency and the IC50 was increased more than 10‐fold to 5 mM (Supporting Information Table S1). Removal of both the chlorine atoms as in compound 8 (Table 2 and Supporting Information Table S1), destroyed inhibition of HsaD completely. Substitution of the chlorines by fluorine (fragment 10) also resulted in loss of inhibition of HsaD (Supporting Information Table S1). Having a single chlorine in the para position (Fragment 13) did not possess any inhibitory effects on HsaD (Supporting Information Table S1).
Altering the position of chlorine from meta to the ortho position (fragment 11) likewise resulted in a decreased inhibitory effect (IC50 1.89 mM) compared with fragment 2 and changing the chlorines to trifluoromethyl compounds (fragment 12) showed poorer inhibition as compared to the initial compound with less than 50% inhibition being reached. (Supporting Information Table 1). Mono‐substituted benzenesulfonamides with a bromine in position 3 (fragment 14), like the mono‐chlorinated fragment, produced a similar IC50 of 2.69 mM.
The library of fragments used for these studies also incorporated sulfonamide fragments with the addition of nitro‐, amino‐, methyl‐, methoxy‐, hydroxy‐ and carboxy‐ groups on various positions of the benzene ring. None of the modifications including these side groups were capable of inhibiting HsaD (fragments 15–23) with the exception of fragment 20 which, with an IC50 of 92.6 mM, was an extremely poor inhibitor.
A derivative of fragment 2, 3,5‐dichlorosulfanilamide in which an amide group was incorporated in position 4 of the benzene ring, was able to inhibit HsaD with an IC50 of 0.16 mM (fragment 24) (Table 2 and Supporting Information Table S1). Addition of a dimethyl group on the nitrogen (Fragment 25), however, reduced the inhibitory effect on HsaD with an IC50 of 3.68 mM. Replacement of the chlorines with bromines (Fragment 26) reduced the inhibitory potency compared with the parent compound to IC50 2.10 mM.
The important features for inhibition of HsaD by derivatives of 2 appear to be a para hydroxyl group on the aromatic ring and two chlorines as meta substituents and the inhibitory potency (IC50 of 0.27 mM) of fragment 27 supports this conclusion. Fragment 27 also has a sulfonate group at position 1 (Table 2).
Derivatives of compound 6 as inhibitors of HsaD
A small group of hydroxybenzoate analogues were tested for comparison with compound 6 as inhibitors of HsaD.
Like compound 2, compound 6 has meta (3,5) substituted chlorine atoms and the position and number of chlorine substituents on the aromatic ring was altered as well as the presence of the 4‐hydroxyl group.
Ethyl 3,5‐dichloro‐4‐hydroxybenzoate (fragment 6) inhibited HsaD (IC50 0.52 mM). When the hydroxyl group was removed, there was no inhibition of HsaD (Fragment 28), and when either one or both of the chlorine atoms were removed, no inhibition was then observed. Fragment 29 with only one chlorine at the 3 position was not inhibitory, and the same was true when both chlorines were missing (Compound 30).
An analogue in which both the chlorines are present and the 4 hydroxyl is present but there is a change in the ester substituent on the 1 position to a carboxylate group (3,5‐dichloro‐4‐hydroxybenzoic acid ‐ fragment 32) has the same IC50 value of 0.54 mM as the parent compound (Table 2B and Supporting Information Table S1). If the hydroxyl group in this charged compound is substituted by an amino group (fragment 33), the IC50 remains essentially the same (0.55 mM). Interestingly, altering the ethyl substituent on the carboxylate group to a methyl group (compound 31) was inhibitory but with a slightly higher IC50 (0.71 mM).
To further elucidate the importance of the chlorines on positions 3 and 5, these were rearranged into positions 1 and 3 with the addition of a nitro group in position 6 (fragment 35). The resulting compound was less inhibitory, with an IC50 of 2.15 mM. Removal of the chlorines and only the presence of the carboxylic acid and amine group in positions 1 and 4 of the benzene ring respectively (fragment 36) and the amine in position 3 (fragment 37), completely reversed the inhibitory effect (Supporting Information Table S1).
Structural studies of HsaD with inhibitors
In order to identify the mode of binding of the inhibitors that were identified, structural studies were carried out using three different inhibitors: Compound 2 (IC50, 0.52 mM) and an analogue (compound 27, IC50, 0.27 mM) were chosen as representatives of the sulfonamide series, and compound 32 (IC50, 0.54 mM) was chosen as a representative of the hydroxybenzoate class. The latter compound was chosen in preference to the parent compound because it is more soluble. Crystals of HsaD were soaked with the inhibitory fragments.
The crystal structure of HsaD with compound 2 was obtained to a resolution of 2.1 Å. The crystal structure with 3,5‐dichloro‐4‐hydroxybenzenesulphonic acid (fragment 27) was obtained at a resolution of 2.68 Å, and the structure of HsaD was also obtained where the ligand bound was a derivative of hydroxybenzoic acid (3,5‐dichloro‐4‐hydroxybenzoic acid, fragment 32), at a resolution of 2.27 Å. Processing and refinement statistics for all three structures are shown in Table 3.
Table 3.
Processing and refinement statistics for X‐ray crystallographic structures
| Structure | HsaD – 2 | HsaD – 27 | HsaD – 32 |
|---|---|---|---|
| PDB code | 5JZB | 5JZ9 | 5JZS |
| Space group | I 212121 | I 212121 | I 212121 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| a, b, c (Å) | 82.03, 82.23, 194.25 | 81.73, 82.32, 193.58 | 81.92, 82.27, 195.13 |
| Processing statistics | |||
| Resolution range (Å) | 50.9–2.1 | 75.75–2.68 | 97.57–2.27 |
| Unique reflections a | 38 700 | 18 699 (2445) | 30 015 (2024) |
| R merge | 0.134 (0.469) | 0.231 (0.895) | 0.109 (0.453) |
| <I/σ(I)>a | 8 (2.6) | 4.4 (1.5) | 7.7 (1.4) |
| Completeness %a | 100 (100) | 99.6 (99.0) | 99.3 (57.4) |
| Multiplicity a | 6.3 (5.3) | 4.4 (4.5) | 4.9 (3.1) |
| Refinement statistics | |||
| R work % | 23.51 | 24.05 | 23.48 |
| R free % | 20.55 | 21.27 | 20.25 |
| RMS bond angle (°) | 1.28 | 0.89 | 1.20 |
| RMS bond length (Å) | 0.005 | 0.004 | 0.009 |
| Ramachandran statisticsb | |||
| Preferred region % | 97.5 | 98.2 | 98.6 |
| Allowed region % | 2.3 | 1.8 | 1.4 |
| Outliers % | 0.2 | 0 | 0 |
Numbers in parentheses are for the highest resolution shell.
Ramachandran statistics were calculated using MolProbity (Chen et al., 2010).
The crystal structures of each of these ligand bound structures can be compared with the structure of HsaD alone (Figure 3A). With the exception of a 55° rotation of the side chain of Trp270, which is only observed in the structure of HsaD with compound 2, there are no major conformational changes compared with structure of apo‐HsaD (overall RMSD 0.2 – Figure 3). The binding of substrate molecules to HsaD revealed the presence of two different conformations, an open conformation for the apo‐enzyme and a closed conformation when the substrate was bound. However, there was no change in the juxtaposition of the active site residues in the main catalytic pocket. The same is true on the binding of the inhibitors which have been investigated here (Figure 3A).
Figure 3.
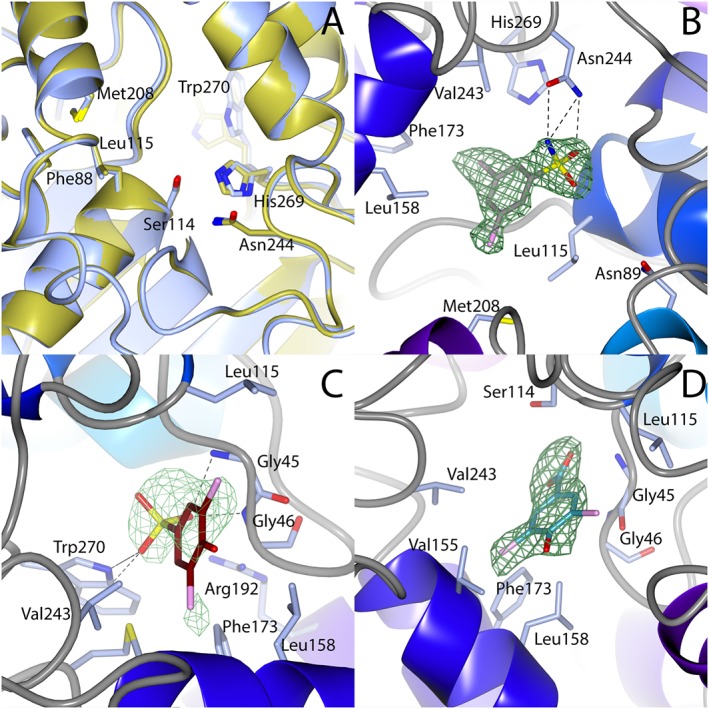
Binding of inhibitors to HsaD. (A) The active site residues of apo HsaD (Lack et al., 2008) are overlaid onto the active site residues of HsaD bound to fragment 27 (fragment not shown). (B) Fragment 2, (C) fragment 27 and (D) fragment 32 each bound to HsaD. In (A), HsaD structure with compound 2 has blue carbon atoms; apo‐HsaD has gold carbon atoms. In (B)–(D), green mesh is unbiased positive difference density contoured at 3σ; the protein is shown with blue carbon atoms, 2 is shown with grey carbon atoms, 27 with brown carbon atoms and 32 with cyan carbon atoms. Chlorines are coloured pink and hydrogen bonds are black dashed lines. Interacting residues are labelled. All figures were generated in CCP4MG (McNicholas et al., 2011).
Two molecules of fragments 2 and 27 were observed bound to HsaD in their respective structures: one molecule was bound in each of the active sites of the protomers in the dimer in the asymmetric unit. In each of these cases, the orientation of the ligand was superimposable with its orientation in each of the two active sites in the dimer.
Compound 32 was found in only one of the active sites of the dimer in the asymmetric unit, with the A chain active site being unoccupied. This reflects the data obtained with the substrate bound structures (Lack et al., 2010). The inhibitor binding sites each overlap with the binding site of the substrate from previous structures (Figure 4D) and are close to the active site Ser (Figure 3B–D). These structural studies confirm the specificity of the screening process in identifying inhibitors of HsaD.
Figure 4.
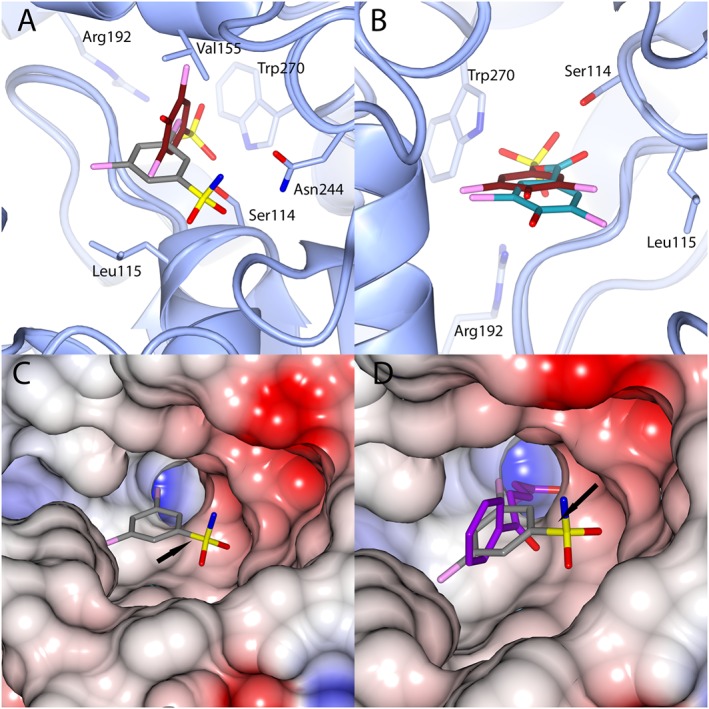
Comparison of the orientation of binding of fragments to the HsaD active site. (A) Shows the overlaid binding orientations of compounds 27 and 32. (B) Shows the overlaid binding orientations of compounds 2 and 27. (C) Shows the electrostatic potential surface map of HsaD in the pocket around compound 2. (D) An overlay of the structure of HsaD bound to 2 with the structure of HsaD bound to HOPDA (PDB code 2wug) (Lack et al., 2010). Colour coding of carbon atoms is as in Figure 3. HOPDA is shown with purple carbon atoms. CCP4MG was used to perform secondary structure alignments for as well as generating the electrostatic potential maps for (C) and (D). In frames (A) and (B), the active site serine residue 114 is shown in stick format. In frames (C) and (D), the serine residue 114 position is indicated by an arrow.
Binding of 2 and its analogue 27 to HsaD
The structure of HsaD bound to fragment 2 allowed the unambiguous determination of the fragment's binding orientation. Fragment 2 binds within the active site with its sulfonamide group making short‐range hydrogen bonds with the side chain of Asn244 (Figure 3B). It also forms a hydrogen bond with the backbone amide of Gly140 and a water‐bridged interaction with the side chain of Asn89. The benzene ring forms hydrophobic contacts with Gly46 and the side chains of Leu115, Leu158 and Val243. One chlorine forms contacts with the side chains of Met208 and Phe220, while the other contacts the side chains of Phe173 and His269.
The number of hydrophobic contacts made by the chlorines explains why removal of one or both has a detrimental effect on fragment binding (Fragments 8 and 9) and also why replacement of the chlorines with polar functional groups is energetically unfavourable (fragments 16, 18 and 20). The importance of the interactions of chlorines at positions 3 and 5 also explains the inability of 17, 21 and 22 to inhibit HsaD. The hydrophobic contacts with Met208 and Phe220 are relatively long range (4.3 and 4.8 Å respectively), which indicates why the bromine of the inhibitory fragment 14 could be accommodated.
The short‐range hydrogen bond formed by the sulfonamide nitrogen explains the detrimental effect observed by double methylation of this group in fragment 25. Accommodation of the sulphanilamide of 24 in the current orientation would not be possible due to the distance between the fragment's ring and Gly46 being too short to allow accommodation of an amine group in between (3.7 Å).
Fragment 27 binds with its acidic group at the entrance of the polar sub‐pocket (Figure 3C) that binds the dienoate moieties of HOPDA and 4,9‐DHSA (Lack et al., 2010). The sulfonate group of 27 forms hydrogen bonds with the side chain of Trp270 as well as the backbone amides of Gly45 and Gly46 (Figure 3C). The sulfonate also forms a salt bridge interaction with the side chain of Arg192 (4.5 Å). One of the chlorines of 27 fits into a hydrophobic pocket formed by the side chains of Leu158, Phe173, Met177 and Val243 (all distances <4 Å). The other chlorine interacts with Gly45 and the side chain of Leu115.
The acidic compound 27, binds in a different orientation to the uncharged sulfonamide 2 (Figure 4B). The charged sulfonate group of 27 may drive its binding to the polar pocket of the active site where it is able to form the salt bridge with Arg192 that could not be formed with the neutral sulfonamide of 2. This clear distinction in the binding modes of fragments that have charged versus simply polar groups provides useful information for modifying the fragments to improve affinity.
Binding of fragment 32 to HsaD
Fragment 32 binds in an almost coplanar conformation to 27 (Figure 3D). However, its binding position is shifted by around 1.8 Å outward from the polar subsite (Figure 4B). As a result, the carboxyl group of 32 is not inserted as far into the pocket as the sulfonate of 27. The carboxyl group forms hydrogen bonds with the side chain of Ser114 and the backbone amides of Gly45 and Gly46 (Figure 3D). This also means the fragment is unlikely to form a salt bridge with Arg192 (separation >6 Å). The altered binding conformation also removes the interaction of the chlorine with Gly45, leaving only the contact with Leu115. The other chlorine still contacts Leu158, Phe173 and Val243 but does not contact Met177; rather, it interacts with Val155.
The extensive hydrophobic contacts made by both chlorines of 32 explains their importance in the binding of the fragment. In view of the proposed importance for the 4‐hydroxy group of 32 in the inhibition of HsaD, it was unexpected to find the hydroxyl group makes no interactions with either the side chain or backbone of HsaD.
The binding of 32 (Figure 3D), like the other acidic inhibitor investigated (27 Figure 3C), shows a distinct binding orientation from the uncharged sulfonamide (fragment 2) Figure 3B.
From these comparisons of binding sites, using fragment 2 as a starting point (Figure 4), there are two possible areas of expansion. The first is into the polar pocket that is accessed by the acidic groups of 27 and 32 (Figure 4C). The second area of expansion would be into the strongly negatively pocket in close proximity to the sulfonamide group of 2. Expansion into this polar pocket would mimic that of HOPDA (Figure 4D), which extends deep into the pocket and makes a shorter range salt bridge with Arg192 (3.1 Å).
Effects of inhibitors on growth of M. bovis BCG and M. tuberculosis H37Rv
In order to evaluate the specific anti‐mycobacterial activity of the compounds, we tested them for inhibition of growth of E. coli . There was no inhibition observed at up to 100 μg·mL−1 (Supporting Information Figure S2). We also tested for any cytotoxic effect against RAW264.7 murine macrophage cells and again found no evidence of eukaryotic cell toxicity at concentrations up to 100 μg·mL−1 (data not shown).
None of the HsaD inhibitors had any effect on the growth of M. bovis BCG or M. tuberculosis when cultured in enriched MB7H9 broth. This observation is consistent with the effect seen in the gene deleted mutant strains. In order to test the proof of principle that inhibitors of HsaD would have an effect on the growth of M. bovis BCG and M. tuberculosis on cholesterol, we tested the fragments at concentrations ranging from 12.5 to 200 μg·mL−1 in minimal growth media supplemented with cholesterol, using the SPOTi assay. Fragment 6, which is one of the most potent inhibitors of HsaD, was also the most effective at inhibiting growth of both M. bovis BCG and M. tuberculosis in the presence of cholesterol, with growth only at 12.5 μg·mL−1. In contrast, fragment 8, which showed no inhibition of HsaD, was also ineffective at concentrations up to 200 μg·mL−1 at inhibiting the growth of either M. bovis BCG or M. tuberculosis on cholesterol.
Fragment 2 was capable of inhibiting the growth of M. bovis BCG cells with an MIC of <12.5 μg·mL−1, whereas fragment 24, inhibited the growth at 25 μg·mL−1. In contrast, fragment 27 did not appear to have an effect, with growth evident even at the highest concentration (200 μg·mL−1). As with fragment 2, both fragments 6 and 32, completely halted growth, with an MIC below 12.5 μg·mL−1. Isoniazid, gave an MIC of 0.1 mg·mL on bacteria grown on cholesterol.
Similarly, the effect on M. tuberculosis growth following treatment with the fragments was also investigated during growth on cholesterol. Treatment with fragments 2 and 6 gave an MIC of 50 and 25 μg·mL−1 respectively, whilst fragments 24 and 32, 200 μg·mL−1. No inhibition was observed with fragments 27 and 8 (growth present at 200 μg·mL−1) therefore, the MIC could not be calculated. Isoniazid was again found to have an MIC of 0.1 mg·mL−1. Fragment 27 is negatively charged and is ineffective in the inhibition of growth (see discussion below).
Discussion and conclusions
We have investigated the role of hsaD in mycobacteria and determined whether the gene product is a suitable target for developing anti‐tuberculars using a fragment based approach to identifying inhibitors of the HsaD enzyme. We have confirmed effects on growth of deleting the hsaD gene and used structural studies to demonstrate the binding of inhibitors identified by this method. We have also demonstrated that inhibitors of the enzyme inhibit the growth of M. tuberculosis on cholesterol.
Role of HsaD in growth
There is growing evidence from whole genome studies of the importance of cholesterol for intracellular survival of M. tuberculosis and recurring evidence for the importance of the operon encoding HsaD as being essential for intracellular survival of M. tuberculosis and M. bovis BCG. We show here that this is the case in studies where we have created a targeted deletion of hsaD from M. bovis BCG. We demonstrate that the growth of ΔhsaD M. bovis BCG on miminmal medium plus cholesterol is inhibited (Figure 1C).
In addition to the effects on the growth pattern of the ΔhsaD M. bovis BCG strain on cholesterol, we also generated a ΔhsaD strain of M. smegmatis and we observed a pink colour when the latter strain was grown on minimal medium plus cholesterol in liquid culture (Supporting Information Figure S1). The same observation has been made when a ΔhsaC M. tuberculosis H37Rv strain is grown in minimal medium with cholesterol (Yam et al., 2009). Therefore, similar to the ‘pink’ metabolite of the ΔhsaC M. tuberculosis, we propose that deletion of hsaD results in inhibition of growth of M. bovis BCG on cholesterol as a result of accumulation of an inhibitory metabolite.
In addition to effects on growth on cholesterol, we have also noted a lag in growth on glycerol in minimal medium of the ΔhsaD strain (Figure 1D). This mirrors previous findings on deletion of hsaD and other essential genes involved in cholesterol catabolism from M. tuberculosis (Griffin et al., 2011). Additionally, it has been observed that hsaD expression is significantly increased in M. smegmatis when grown on cholesterol (Uhía et al., 2012), and genome wide studies with M. smegmatis grown on cholesterol have also pointed to an increase in HsaD expression on growth on cholesterol (Li et al., 2016).
We have also investigated growth in macrophages. Whole genome studies have previously identified the role of HsaD in the survival of M. tuberculosis within macrophages (Sassetti et al., 2003). It has also been demonstrated that other genes in the operon (hsaC and nat) are essential for survival of M. bovis BCG in macrophage (Bhakta et al., 2004; Anderton et al., 2006).
Similar to ΔhsaC and Δnat strains, there was a reduction in intracellular growth of the ΔhsaD strain as compared to the M. bovis BCG (Figure 1E). This is consistent with the whole genome studies and studies identifying the role of other members of this operon in the metabolism of cholesterol, which has been identified as controlled as the Kst‐Regulon (Kendall et al., 2007; Kendall et al., 2007; Uhía et al., 2012).
Identification of chemical fragments as inhibitors of HsaD
The studies described previously add to the growing weight of evidence that the operon in which hsaD is encoded along with the nat gene in the same gene cluster (Anderton et al., 2006) are essential for intracellular survival of mycobacteria and also that the gene product are required for cholesterol metabolism in M. bovis BCG and M. tuberculosis (Van der Geize et al., 2007; Yam et al., 2009; Kendall et al., 2010). Given this importance, we speculated that the hsaD gene is a potential target for anti‐tubercular agents. Supporting drug design, the crystal structure of HsaD has been established to better than 2 Å resolution with the substrate HOPDA bound and shows that the active site cleft is over 2100 Å3, which is a good prospect for a fragment based approach to identifying inhibitors (Lack et al., 2008). HsaD is also a stable soluble protein that crystallizes readily (Ryan et al., 2014). Moreover, chemical inhibitors of HsaAB were found to dramatically decrease the intracellular proliferation of M. tuberculosis in macrophages (VanderVen et al., 2015).
FBDD relies on having crystallographic data. The structure of HsaD has been determined alone (Lack et al., 2008) to 2.6 Å and the structure of an inactive S114A mutant of HsaD has been determined at resolutions of better than 2 Å with one of several different substrates bound (Lack et al., 2010). It was necessary to use the inactive version of HsaD in order for the substrate/protein complex to be stable enough for crystallization. In each of the crystal structures, the asymmetric unit consists of two protomers of identical polypeptide chains with one protomer being assigned as the A chain and the other as the B chain. These protomers are identical and each folds to form an α/β hydrolase fold, which contains an active site including the catalytic triad (Ser114, His269 and Asp241). (Lack et al., 2008; 2010).
We used a medium throughput platform for screening a well‐defined fragment library (Ciulli and Abell, 2007; Ciulli, 2013), and following subsequent enzyme inhibition studies, we identified two fragments namely fragment 2 (3,5‐dichlorobenzenesulfonamide) and fragment 6 (ethyl 3,5‐dichloro‐4‐hydroxybenzoate) (Table 2), which each have IC50 values of less than 1 mM. We investigated these compounds and a series of chemical derivatives by both enzymological methods and by determining their crystallographic structures to identify the binding mode. In addition, we determined their effects on growth of mycobacteria and E. coli for comparison, and this is discussed below.
From the series of compounds that we tested, we were able to identify key features of fragment 2 and fragment 6, which were important for inhibition. The important features for inhibition of HsaD by derivatives of 2 appear to be the introduction of a para hydroxyl group on the aromatic ring and the presence of two chlorines as meta substituents, and the inhibitory potency (IC50 of 0.27 mM) of fragment 27 supports this conclusion. The potency of derivatives of fragment 6 support the conclusion that the presence of chlorine substituents on the ring at the meta positions and the presence of a hydroxyl at the para position are important features for inhibition of HsaD.
We obtained the crystal structures of HsaD with compound 2 and a derivative, compound 27, bound. The structure of HsaD with compound 6 could not be determined, but we obtained a high resolution structure of HsaD with compound 32 bound. Compound 32 is a derivative of our hit fragment 6.
Compound 27, a sulfonate, and compound 32, a carboxylate, bind to HsaD in a very similar orientation (Figure 4A) which is distinct from, but overlapping with, the binding site of Compound 2, an uncharged but polar sulfonamide (Figure 4B). Specifically, the negatively charged moieties bind in the section of the active site pocket that was identified as the polar sub pocket, which accommodates the negatively charged section of each of the negatively charged substrates, including its physiological substrate from cholesterol degradation (Lack et al., 2010).
Whilst each of these inhibitory compounds is bound in the active site, the adjacent nature of these binding sites provides an excellent starting point for growth into new areas of the active site pocket which is a key feature of fragment based drug development (Marchetti et al., 2016).
We have also tested these compounds identified from the screen and their derivatives as inhibitors of growth of E. coli, M. bovis BCG and M. tuberculosis. None of the compounds inhibited growth of E. coli at 100 μg·mL−1 (Supporting Information Figure S2). In addition, the compounds were found to be non‐cytotoxic. Inhibition of growth of both M. bovis and M. tuberculosis on cholesterol was observed, and the potency of inhibition of enzymic activity broadly mirrored the effects on growth. This was the case apart from the potent and negatively charged derivative of compound 2, namely compound 27. Its ineffectiveness in the whole‐cell inhibition may possibly be due to a lack of permeation through the lipid‐rich mycobacterial cell wall.
We conclude that HsaD is a valid target for antitubercular drug discovery. In view of the growing threat of multi drug resistant TB, no viable target that has characteristics like HsaD should be ignored.
Author contributions
E.P., D.E., N.L., S.B., A.R., R.B., A.H., S.K., A.C., C.S., E.L. and O.E. carried out the experiments. E.P., D.E., N.L., S.B., A.R., E.S., R.B., A.C., C.S., E.L. and O.E. analysed the data. E.P., D.E., N.L., S.B., A.R., A.A. and A.C. co‐wrote the manuscript. A.R., W.J. and T.M. co‐ordinated the research. E.S. co‐ordinated the research as PI and wrote the manuscript. A.A. advised on the experiments. R.B., A.H., W.J., S.K., T.M. and C.S. contributed to the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Cultures of Mycobacterium smegmatis grown on minimal medium containing cholesterol as carbon source are coloured when HsaD is deleted. Wildtype (WT) – on the left and ΔHsaD on the right Mycobacterium smegmatis were grown in 7H9 (containing 50 μg/mL hygromycin for knockout) to an OD = 1, washed twice with sterile PBS and then used to inoculate (1/100 dilution) a200mL culture of minimal medium containing 0.05% (v:v) EtOH, 0.05% (v:v) tyloxapol and 50 μg/ml cholesterol. And grown at 37 °C for 36 h.
Figure S2 Effect of fragments on the growth of E. coli JM109 in LB medium (A) fragment 2; (B) fragment 24; (C) fragment 27; (D) fragment 32; and (E) fragment 6. Liquid cultures in LB were diluted to an OD 600 of 0.1 and fragments were added Briefly, an overnight culture was sub‐cultured and was diluted to an OD600 of 0.1. in LB containing 12.5 μg/mL, 25 μg/mL, 50 μg/mL, or 100 μg/mL of fragments. Absorbance readings at 600 nm represent means of triplicate determinations in one experiment and the symbols encompass the spread of values obtained. The experiments were repeated 3 times each confirming the results shown.
Table S1 Supplementary Inhibition of HsaD enzymic activity by a sublibrary of compounds based on fragments 2 and 6 from the initial screen (Table 1). The values for IC50 were determined from the inhibition of HsaD enzymic activity by the fragments as indicated in Methods. The values shown are averages +/− standard deviation of six independent determinations (N = 6). No inhibition indicates that the compounds were not inhibitory at either 5 mM or at the highest soluble concentration which could be obtained. The * indicates that inhibition was observed but that it did not reach 50%.
Ryan, A. , Polycarpou, E. , Lack, N. A. , Evangelopoulos, D. , Sieg, C. , Halman, A. , Bhakta, S. , Eleftheriadou, O. , McHugh, T. D. , Keany, S. , Lowe, E. D. , Ballet, R. , Abuhammad, A. , Jacobs, W. R. Jr , Ciulli, A. , and Sim, E. (2017) Investigation of the mycobacterial enzyme HsaD as a potential novel target for anti‐tubercular agents using a fragment‐based drug design approach. British Journal of Pharmacology, 174: 2209–2224. doi: 10.1111/bph.13810.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N et al. (2010). PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderton MC, Bhakta S, Besra GS, Jeavons P, Eltis LD, Sim E (2006). Characterization of the putative operon containing arylamine N‐acetyltransferase (nat) in Mycobacterium bovis BCG. Mol Microbiol 59: 181–192. [DOI] [PubMed] [Google Scholar]
- Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J et al. (2002). Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M bovis BCG and M smegmatis . Microbiology 148: 3007–3017. [DOI] [PubMed] [Google Scholar]
- Bhakta S, Besra GS, Upton AM, Parish T, Sholto‐Douglas‐Vernon C, Gibson KJ et al. (2004). Arylamine N‐acetyltransferase is required for synthesis of mycolic acids and complex lipids in Mycobacterium bovis BCG and represents a novel drug target. J Exp Med 199: 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FC, Nunez‐Garcia J, García‐Pelayo C, Soria M, Bianco MV, Zumárraga M et al. (2009). Differential transcriptome profiles of attenuated and hypervirulent strains of Mycobacterium bovis . Microbes Infect 11: 956–963. [DOI] [PubMed] [Google Scholar]
- Carr H, Purcell E (1954). Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev 94: 630. [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ et al. (2010). MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D 66: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulli A (2013). Biophysical screening for the discovery of small‐molecule ligands. Methods Mol Biol 1008: 357–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulli A, Abell C (2007). Fragment‐based approaches to enzyme inhibition. Curr Opin Biotechnol 18: 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulli A, Scott DE, Ando M, Reyes F, Saldanha SA, Tuck KL et al. (2008). Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. Chembiochem 9: 2606–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Crowe AM, Stogios PJ, Casabon I, Evdokimova E, Savchenko A, Eltis LD (2015). Structural and functional characterization of a ketosteroid transcriptional regulator of Mycobacterium tuberculosis . J Biol Chem 290: 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B (2001). WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J Biomol NMR 21: 349–359. [DOI] [PubMed] [Google Scholar]
- Dalvit C, Pevarello P, Tatò M, Veronesi M, Vulpetti A, Sundström M (2000). Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J Biomol NMR 18: 65–68. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010). Features and development of Coot. Acta Crystallogr D 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelopoulos D, Bhakta S (2010). Rapid methods for testing inhibitors of mycobacterial growth In: Antibiotic Resistance Protocols: Second Edition, Springer, New York: pp. 193–201. [DOI] [PubMed] [Google Scholar]
- Evangelopoulos D, Gupta A, Lack N, Maitra A, ten Bokum AMC, Kendall SL et al. (2014). Characterisation of a putative AraC transcriptional regulator from Mycobacterium smegmatis . Tuberculosis 94: 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P (2006). Scaling and assessment of data quality. Acta Crystallogr D 62: 72–82. [DOI] [PubMed] [Google Scholar]
- Ferguson FM, Fedorov O, Chaikuad A, Philpott M, Muniz JR, Felletar I et al. (2013). Targeting low‐druggability bromodomains: fragment based screening and inhibitor design against the BAZ2B bromodomain. J Med Chem 56: 10183–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran P, Lloyd-Evans E, Lack NA, Platt N, Davis LC, Morgan AJ et al (2016). Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann‐Pick type C disease cellular pathway. Wellcome Open Research 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM (2011). High‐resolution phenotypic profiling defines genes essential for Mycobacterial growth and cholesterol catabolism. PLoS Pathog 7: e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR et al. (2012). Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol 19: 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson SA, McLean KJ, Surade S, Yang YQ, Leys D, Ciulli A et al. (2012). Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew Chem Int Ed 51: 9311–9316. [DOI] [PubMed] [Google Scholar]
- Hung AW, Silvestre HL, Wen S, George GP, Boland J, Blundell TL et al. (2016). Optimization of Inhibitors of Mycobacterium tuberculosis pantothenate synthetase based on group efficiency analysis. ChemMedChem 11: 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr Section D 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SL, Burgess P, Balhana R, Withers M, ten Bokum A, Lott JS et al. (2010). Cholesterol utilization in mycobacteria is controlled by two TetR‐type transcriptional regulators: kstR and kstR2. Microbiology 156: 1362–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B et al. (2007). A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis . Mol Microbiol 65: 684–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack N, Lowe ED, Liu J, Eltis LD, Noble ME, Sim E et al. (2008). Structure of HsaD, a steroid‐degrading hydrolase, from Mycobacterium tuberculosis . Acta Crystallogr F 64: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack NA, Kawamura A, Fullam E, Laurieri N, Beard S, Russell AJ et al. (2009). Temperature stability of proteins essential for the intracellular survival of Mycobacterium tuberculosis . Biochem J 418: 369–378. [DOI] [PubMed] [Google Scholar]
- Lack NA, Yam KC, Lowe ED, Horsman GP, Owen RL, Sim E et al. (2010). Characterization of a carbon‐carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem 285: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Ge F, Tan Y, Zhang G, Li W (2016). Genome‐wide transcriptome profiling of Mycobacterium smegmatis MC2155 cultivated in minimal media supplemented with cholesterol, androstenedione or glycerol. Int J Mol Sci 17: 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovewell RR, Sassetti CM, VanderVen BC (2016). Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr Opin Microbiol 29: 30–36. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Chan DS, Coyne AG, Abell C (2016). Fragment‐based approaches to TB drugs. Parasitology. https://doi.org/10.1017/S0031182016001876. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B (1999). Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed Engl 38: 1784–1788. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007). Phaser crystallographic software. J Appl Cryst 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson K, Noble M (2011). Presenting your structures: the CCP4mg molecular‐graphics software. Acta Crystallogr D 67: 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes V, Blundell TL (2016). Targeting tuberculosis using structure‐guided fragment‐based drug design. Drug Discov Today. https://doi.org/10.1016/j.drudis.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Moriarty NW, Grosse‐Kunstleve RW, Adams PD (2009). electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D 65: 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997). Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2: 2212–2221. [DOI] [PubMed] [Google Scholar]
- Ouellet H, Johnston JB, de Montellano PR (2011). Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis . Trends Microbiol 19: 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM (2008). Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A 105: 4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton M, Gifford C, Schartau P, Hagemeier C, Mushtaq A, Lucas S et al. (2001). Evidence towards the role of arylamine N‐acetyltransferase in Mycobacterium smegmatis and development of a specific antiserum against the homologous enzyme of Mycobacterium tuberculosis . Microbiology 147: 3295–3302. [DOI] [PubMed] [Google Scholar]
- Peyron P, Bordier C, Elsa‐Noah N, Maridonneau‐Parini I (2000). Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol‐anchored proteins. J Immunol 165: 5186–5191. [DOI] [PubMed] [Google Scholar]
- Piotto M, Saudek V, Sklenár V (1992). Gradient‐tailored excitation for single‐quantum NMR spectroscopy of aqueous solutions. J Biomol NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Rebollo‐Lopez MJ, Lelièvre J, Alvarez‐Gomez D, Castro‐Pichel J, Martínez‐Jiménez F, Papadatos G et al. (2015). Release of 50 new, druglike compounds and their computational target predictions for open source anti‐tubercular drug discovery. PLoS One 10: e0142293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Bloom BR, Rubin EJ (2005). Genome‐wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102: 8327–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde KH, Veiga DFT, Caldwell S, Bala´ G, Russell DG (2012). Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Path 8: e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzini AC, Bhowmik S, Ghosh S, Yam KC, Bolin JT, Eltis LD (2013). A substrate‐assisted mechanism of nucleophile activation in a Ser–His–Asp containing C–C bond hydrolase. Biochemistry 52: 7428–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzini AC, Ghosh S, Horsman GP, Foster LJ, Bolin JT, Eltis LD (2012). Identification of an acyl‐enzyme intermediate in a meta‐cleavage product hydrolase reveals the versatility of the catalytic triad. J Am Chem Soc 134: 4615–4624. [DOI] [PubMed] [Google Scholar]
- Ryan A, Keany S, Eleftheriadou O, Ballet R, Cheng H‐Y, Sim E (2014). Mechanism‐based inhibition of HsaD: a CC bond hydrolase essential for survival of Mycobacterium tuberculosis in macrophage. FEMS Microbiol Lett 350: 42–47. [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48: 77–84. [DOI] [PubMed] [Google Scholar]
- Silvestre HL, Blundell TL, Abell C, Ciulli A (2013). Integrated biophysical approach to fragment screening and validation for fragment‐based lead discovery. Proc Natl Acad Sci 110: 12984–12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhía I, Galán B, Kendall SL, Stoker NG, García JL (2012). Cholesterol metabolism in Mycobacterium smegmatis . Environ Microbiol Rep 4: 168–182. [DOI] [PubMed] [Google Scholar]
- Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC et al. (2007). A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104: 1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Molle I, Thomann A, Buckley DL, So EC, Lang S, Crews CM et al. (2012). Dissecting fragment‐based lead discovery at the von Hippel‐Lindau protein: hypoxia inducible factor 1α protein‐protein interface. Chem Biol 19: 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB et al. (2015). Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog 11: e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood I, Bhakta S, Russell AJ, Fullam E, Anderton MC, Kawamura A et al. (2010). Identification of arylamine N‐acetyltransferase inhibitors as an approach towards novel anti‐tuberculars. Protein Cell 1: 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood IM, Kawamura A, Russell AJ, Sandy J, Davies SG, Sim E (2011). Novel small Molecule inhibitors of arylamine N‐acetyltransferases: Drug discovery by high throughput screening. Comb Chem High Throughput Screen 14: 117–124. [DOI] [PubMed] [Google Scholar]
- WHO (2015). Global tuberculosis Report 2015. Available at http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf.
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D 67: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G (2010). xia2: an expert system for macromolecular crystallography data reduction. J App Crystallogr 43: 186–190. [Google Scholar]
- Yam KC, D'Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V et al. (2009). Studies of a ring‐cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis . PLoS Pathog 5: e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cultures of Mycobacterium smegmatis grown on minimal medium containing cholesterol as carbon source are coloured when HsaD is deleted. Wildtype (WT) – on the left and ΔHsaD on the right Mycobacterium smegmatis were grown in 7H9 (containing 50 μg/mL hygromycin for knockout) to an OD = 1, washed twice with sterile PBS and then used to inoculate (1/100 dilution) a200mL culture of minimal medium containing 0.05% (v:v) EtOH, 0.05% (v:v) tyloxapol and 50 μg/ml cholesterol. And grown at 37 °C for 36 h.
Figure S2 Effect of fragments on the growth of E. coli JM109 in LB medium (A) fragment 2; (B) fragment 24; (C) fragment 27; (D) fragment 32; and (E) fragment 6. Liquid cultures in LB were diluted to an OD 600 of 0.1 and fragments were added Briefly, an overnight culture was sub‐cultured and was diluted to an OD600 of 0.1. in LB containing 12.5 μg/mL, 25 μg/mL, 50 μg/mL, or 100 μg/mL of fragments. Absorbance readings at 600 nm represent means of triplicate determinations in one experiment and the symbols encompass the spread of values obtained. The experiments were repeated 3 times each confirming the results shown.
Table S1 Supplementary Inhibition of HsaD enzymic activity by a sublibrary of compounds based on fragments 2 and 6 from the initial screen (Table 1). The values for IC50 were determined from the inhibition of HsaD enzymic activity by the fragments as indicated in Methods. The values shown are averages +/− standard deviation of six independent determinations (N = 6). No inhibition indicates that the compounds were not inhibitory at either 5 mM or at the highest soluble concentration which could be obtained. The * indicates that inhibition was observed but that it did not reach 50%.