

Abstract
Background and Purpose
The paradigm that GPCRs are able to prolong or initiate cellular signalling through intracellular receptors recently emerged. Melatonin binds to G protein‐coupled MT1 and MT2 receptors. In contrast to most other hormones targeting GPCRs, melatonin and its synthetic analogues are amphiphilic molecules easily penetrating into cells, but the existence of intracellular receptors is still unclear mainly due to a lack of appropriate tools.
Experimental Approach
We therefore designed and synthesized a series of hydrophilic melatonin receptor ligands coupled to the Cy3 cyanin fluorophore to reliably monitor its inability to penetrate cells. Two compounds, one lipophilic and one hydrophilic, were then functionally characterized in terms of their affinity for human and murine melatonin receptors expressed in HEK293 cells and their signalling efficacy.
Key Results
Among the different ligands, ICOA‐13 showed the desired properties as it was cell‐impermeant and bound to human and mouse MT1 and MT2 receptors. ICOA‐13 showed differential activities on melatonin receptors ranging from partial to full agonistic properties for the Gi/cAMP and ERK pathway and β‐arrestin 2 recruitment. Notably, ICOA‐13 enabled us to discriminate between Gi/cAMP signalling of the MT1 receptor initiated at the cell surface and neuronal mitochondria.
Conclusions and Implications
We report here the first cell‐impermeant melatonin receptor agonist, ICOA‐13, which allows us to discriminate between signalling events initiated at the cell surface and intracellular compartments. Detection of mitochondrial MT1 receptors may have an important impact on the development of novel melatonin receptor ligands relevant for neurodegenerative diseases, such as Huntington disease.
Abbreviations
- 2‐[125I]‐MLT
2‐[125I]‐iodomelatonin
- Fsk
forskolin
Introduction
Melatonin, N‐acetyl‐5‐methoxytryptamin, is derived from 5‐HT (serotonin) in a two‐step biosynthetic pathway that exists in most organisms. In mammals, melatonin is primarily produced by the pineal gland in a circadian manner with high levels during the night. Due to its lipophilic property, melatonin rapidly distributes all over the brain and in peripheral tissues (Reiter et al., 2014) playing an important role on the synchronization of physiological rhythms and on the regulation of various neuronal, retinal, immunological and metabolic functions (Dubocovich et al., 2010; Tosini et al., 2014). Most effects of melatonin are mediated by melatonin MT1 and MT2 receptors belonging to the large family of GPCRs. Both melatonin receptors are preferentially coupling to proteins of the Gi/o sub‐family (Jockers et al., 2016).
The lipophilic characteristic of melatonin raised since its discovery the intriguing possibility that this neurohormone might bind to molecular targets located not only at the cell surface but also in intracellular compartments. Several putative intracellular melatonin binding proteins have been reported such as calmodulin, the enzyme quinone reductase 2 (Jockers et al., 2008) and RORα/RZR nuclear receptors (Becker‐Andre et al., 1994). However, the interaction between melatonin and most of these targets remains highly controversial (Jetten, 2009). In addition, increasing evidence indicates that GPCRs are able to signal through heterotrimeric G proteins not only at the plasma membrane but also in intracellular compartments (Vilardaga et al., 2014; Irannejad et al., 2015). Recent evidence suggests that the MT1 receptor has mitochondrial functions in neurons that might be mediated by intracellular receptors (Zhang et al., 2013) and that are altered in Huntington disease (Wang et al., 2011). Moreover, significant levels of endogenous melatonin have been measured in brain mitochondria (Venegas et al., 2012), raising the possibility of an autocrine action. A significant amount of data evidencing melatonin‐dependent regulation of mitochondria function supports this scenario (reviewed by Tan et al., 2016). Reduced melatonin production is commonly observed in neurodegenerative diseases, including Huntington and Alzheimer's disease (Liu et al., 1999; Kalliolia et al., 2014), which launched the therapeutic use of melatonin and its analogues in several pathologies. However, only modest beneficial effects are usually reported, pointing to the need of a better understanding of the mechanism of action underlying the effects of melatonin. To better distinguish between cell surface and intracellular melatonin targets, we designed a series of melatonin derivatives coupled to the hydrophilic Cy3 cyanin fluorophore with the goal to generate a hydrophilic melatonin receptor ligand that would not penetrate into cells.
Methods
HEK293 cell culture
HEK293 cells were grown in complete DMEM medium, supplemented with 10% (v·v−1) FBS, 4.5 g·L−1 glucose, 100 U·mL−1 penicillin and 0.1 mg·mL−1 streptomycin and maintained at 37°C (95% O2, 5% CO2). Transient transfection of cells was performed with the JetPEI reagent according to the supplier's instructions (Polyplus‐transfection, New York, NY, USA).
Radioligand binding experiments
Membranes from HEK293 cells transiently expressing human or murine MT1 or MT2 receptors were prepared as previously described (Ayoub et al., 2004). 2‐[125I]‐iodomelatonin (2‐[125I]‐MLT) saturation binding experiments were performed in the range of 1–1000 pM, and specific binding was defined as binding displaced by 10 μM iodomelatonin. Competition curves were performed by simultaneous incubation of 2‐[125I]‐MLT (100 pM) and increasing concentrations of the respective ligands. Assays were carried out in duplicates for 120 min at 37°C, followed by rapid filtration through GF/F glass fibre filters (Whatman, Clifton, NJ, USA). Filter‐retained radioactivity was determined with a γ‐counter LB2111 (Berthold Technologies, Bad Wildbad, Germany). K i values were calculated from IC50 values using the Cheng–Prussof formula: K i = IC50/[1 + (L/K d)] where L represents the ligand concentration and K d the dissociation constant. K d values were 148 ± 8; 397 ± 30; 330 ± 128; and 95 ± 20 pM (n = 3) for hMT1, hMT2, mMT1 and mMT2 receptors respectively.
Fluorescence microscopy
Non‐transfected HEK293 cells (105 cells per well) were plated on 4‐well chamber‐slide plates (Nunc® Lab‐Tek®, Sigma‐Aldrich) and maintained at 37°C, 5% CO2 overnight. Cells were washed in PBS, fixed in 4% paraformaldehyde (15 min, −20°C), followed by treatment with TritonX‐100 (0.2%, 1 h) in order to permeabilize the cells, while another group of wells were kept intact. Cells were incubated in the presence of the fluorescent compounds ICOA‐9 or ICOA‐13 (100 nM, 4 h), washed with PBS and incubated with DAPI (1:2000 dilution, 5 min; Santa Cruz Biotechnology, Dallas, TX, USA) to stain the cell nuclei. The slides were mounted and analysed by fluorescence microscopy. Images were acquired under 63× oil objective using specific excitation/emission filters to image ICOA‐9 (ex/em 508/516 nm), or ICOA‐13 (ex/em 565/580 nm) and DAPI (ex/em 358/461 nm). Images were analysed using ImageJ software (National Institutes of Health, USA).
ERK1/2 activation assays
Total ERK and phospho‐ERK1/2 was detected by SDS‐PAGE/immunoblot analysis, as previously described (Cecon et al., 2015). Briefly, HEK293 cells transfected to express mMT1, mMT2, hMT1 or hMT2 receptors were starved (overnight) before addition of the ligands melatonin (100 nM), ICOA‐9 (1 μM) or ICOA‐13 (1 μM) for 5 min (37°C 5% CO2). Pertussis toxin treatment (10 ng·μL−1) was performed during the starvation. After the incubation, cells were washed in PBS, harvested in lysis buffer, and denaturated proteins were resolved in 12% SDS‐PAGE gels. Following the transfer of proteins to nitrocellulose membranes, the membranes were immunoblotted with antibodies against ERK (1:3000 dilution, sc‐154, Santa Cruz Biotechnology) and phospho‐ERK1/2 (1:1000 dilution, sc‐7383; Santa Cruz Biotechnology) and developed using appropriate secondary antibodies. Membranes were analysed using the Odyssey LI‐COR infrared fluorescent scanner (LI‐COR Biosciences, Lincoln, NE, USA).
For quantitative analysis of ERK activation, we used the P‐ERK AlphaScreen SureFire Assay kit (TGR BioSciences, Adelaide, Australia; and PerkinElmer, Waltham, MA, USA). Cells were stimulated as described above, collected in lysis buffer, and 4 μL of each sample was used for the assay, following the supplier's instructions. The plate was read using the Infinite M1000 Tecan microplate reader. No effect was observed in non‐transfected HEK293 cells up to 10 μM of the two fluorescent compounds (ICOA‐9, ICOA‐13). Data were fitted by non‐linear regression to determine Emax and EC50 values and normalized to the basal levels using GraphPad Prism software (La Jolla, CA, USA).
Accumulative cAMP assay
The cAMP assay was performed as previously described (Kamal et al., 2015). Briefly, HEK293 cells expressing mMT1, mMT2, hMT1 or hMT2 receptors were dispensed into a 384‐well plate (4000 cells per well) and stimulated with 2 μM forskolin in the presence of increasing concentrations of melatonin, ICOA‐9 or ICOA‐13 for 30 min at room temperature in PBS buffer supplemented with 1 mM IBMX (Sigma‐Aldrich, St Quentin, France). Cells were then lysed, and cAMP levels were determined following the manufacturer's instruction (Cisbio Bioassays, Codolet, France). The plate was read using the Infinite F500 Tecan microplate reader. No effect was observed in non‐transfected HEK293 cells up to 10 μM of ICOA‐9 and 1 μM of ICOA‐13. Data were fitted by non‐linear regression to determine Emax and EC50 values and normalized to forskolin‐induced response (100%) and melatonin‐induced maximum inhibition (0%) using GraphPad Prism software.
Time‐course measurements of cAMP production
cAMP was assessed in live neuroblastoma N2a cells using a fluorescent resonance energy transfer (FRET)‐based assay. cAMP levels were measured either at the plasma membrane or at the outer mitochondrial membrane by transfecting the cells with FRET‐based biosensors (DiPilato et al., 2004; Feinstein et al., 2013). Cells plated on poly‐D‐lysine‐coated glass were mounted in Attofluor cell chambers and transferred on the Nikon Ti‐E confocal microscope. Cyan Fluorescent Protein (CFP) and Yellow Fluorescent Protein (YFP) were excited using a mercury lamp, and the emissions (filters: 480 ± 20 nm for CFP and 535 ± 15 nm for YFP) were simultaneously detected with a LUCAS EMCCD camera (Andor Technology). Fluorescence data were extracted from single cells using Nikon Element Software (Nikon Corporation, Minato‐ku, Tokio, Japan), as previously described (Ferrandon et al., 2009). Data were normalized to the forskolin‐induced response. Cells were continuously perfused with buffer or with the corresponding ligand for the indicated time.
ß‐arrestin2 recruitment assay
The PathHunter™ assay from the DiscoverX company was used to measure the recruitment of β‐arrestin2 to melatonin receptors (DiscoverX, Birmingham, United Kingdom). Briefly, HEK293 parental cells stably expressing a fusion protein of β‐arrestin2 and the larger N‐terminal deletion mutant of β‐gal were transiently transfected with hMT1 or hMT2 receptors fused at their C‐terminal part with the small enzyme fragment ProLink tag. Two days after transfection, the cells were treated with increasing concentrations of melatonin, ICOA‐9 or ICOA‐13 for 2–3 h at 37°C. β‐arrestin2 recruitment resulted in the complementation of the two enzyme fragments and the formation of an active β‐gal enzyme. Luminescence signals were determined after 60 min incubation at room temperature. No effect was observed in parental HEK293/ß‐arrestin2 cells in the absence of melatonin receptors up to 10 μM of ICOA‐9 and 1 μM of ICOA‐13. Data were fitted by non‐linear regression to determine Emax and EC50 values and normalized to basal levels (0%) and to maximum melatonin‐induced response (100%) using GraphPad Prism software.
Bias calculation
The dose–response curves were fitted to the operational model of agonism designed by Kenakin and Christopoulos (Kenakin and Christopoulos, 2013; Kenakin et al., 2012) to obtain log(τ/KA) transduction ratios that are a unique index of coupling efficiency (or efficacy) of the agonist. To eliminate the influence of the system and the observation biases, the activity of the compounds at a given signalling pathway was compared with that of melatonin which served as the reference compound, and subsequently, values are expressed as Δlog(τ/KA). The bias between two ligands is expressed as 10ΔΔlog(τ/KA) as described previously (van der Westhuizen et al., 2014). The significance of the ligand biases was determined by Student's two‐tailed unpaired t‐test. Because we did not reach a plateau for the two fluorescent compounds for ERK activation, we used the extrapolated data for EC50 and Emax as derived from GraphPad Prism using a non‐linear regression analysis to obtain an estimate of the bias factor at hMT1 receptors, following the same procedure as previously applied by the Christopoulos lab (Baltos et al., 2016). The estimated pEC50 values were −6.35 ± 0.52 (Emax = 112 ± 6) and −5.60 ± 0.32 (Emax = 128 ± 15) for ICOA‐9 and ICOA‐13 respectively.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as means ± SEM of n independent experiments, performed at least in duplicates to ensure the reliability of single values. EC50 and Emax values were obtained following non‐linear regression (curve fit) with four parameters of data from a minimum of eight different concentrations per experiment, repeated at least three times independently. All assays performed in this study were previously validated and demonstrated the robustness and variability of the procedure using this number of independent experiments for concentration–response curves. Statistical analysis was performed by Student's t‐test when comparing only two groups. Values of P < 0.05 were considered statistically significant. When possible, data were normalized to maximal and minimum responses using melatonin response as a reference in order to avoid unwanted sources of variations, as differences in the amplitude of melatonin effect between independent experiments. Data and statistical analysis were performed using GraphPad Prism software version 6.
Materials
Melatonin and iodomelatonin were purchased from Sigma‐Aldrich (St. Louis, MO, USA) and 2‐[125I]‐MLT from PerkinElmer (Waltham, MA, USA).
cDNA constructs
Expression vectors for Flag‐hMT1, hMT2, Flag‐mMT1 and 6Myc‐mMT2, hMT1‐GFP have been validated and described previously (Petit et al., 1999; Guillaume et al., 2008; Baba et al., 2013). Expression vectors for 3Myc‐MT2‐PK2 and Flag‐MT1‐PK2 were obtained from DiscoverX Corporation Ltd (Birmingham, UK).
Organic synthesis
All reagents were purchased from the commercial suppliers Sigma‐Aldrich (St. Louis, MO, USA) and Alfa Easer (Karlsruhe, Germany) and were used without further purification.
Tetrahydrofuran was purified with a dry station GT S100, dichloromethane was distillated over calcium hydridre, triethylamine and diisopropylethylamine and dimethylformamide were distillated over CaH2. The reactions were monitored by TLC analysis using silica gel (60 F254) plates. Compounds were visualized by UV irradiation and/or spraying with a solution of cerium molybdate, or phosphomolubdic acid, followed by heating at 200°C. Flash column chromatography was performed on silica gel 60 (230–400 mesh, 0.040–0.063 mm). Reversed‐phase column flash‐chromatographies were performed on octadecyl‐functionalized silica gel (mean pore size 60 Å) from Aldrich. Cy 3.29 was synthesized according to reported literature procedure (Mujumdar et al., 1993). 1H and 13C NMR spectra were recorded on a spectrometer at 250 MHz (13C, 62.9 MHz) or 400 MHz (13C, 100 MHz). The 1H and 13C NMR spectra for compounds 2, 5 and 7 (ICOA‐13) are provided in Supplementary Information Figures S7–S9. Chemical shifts are given in parts per million from tetramethylsilane as internal standard. The following abbreviations are used for the proton spectra multiplicities: s, singulet; d, doublet; t, triplet; q, quartet; qt, quintuplet; m, multiplet; br, broad. Coupling constants (J) are reported in Hertz (Hz). Signals were assigned as far as possible by means of two‐dimensional NMR spectroscopy: 1H‐1H‐COSY, 1H‐13C‐COSY (HSQC, heteronuclear single quantum coherence).
Ionspray methodology was used to record mass spectra. High Resolution Mass Spectrometry (HRMS) spectra were recorded on a Maxis Bruker 4G. The infrared spectra of compounds were recorded on a Thermo Scientific Nicolet iS10. Melting points [mp (°C)] were taken on open capillary tubes and are uncorrected, performed on a Electrothermal IA 9100.
(E)‐1‐(6‐(4‐(3‐(2‐acetamidoethyl)‐5‐methoxy‐1H‐pyrrolo[3,2‐b]pyridin‐2‐yl)benzylamino)‐6‐oxohexyl)‐2‐((E)‐3‐(1‐ethyl‐3,3‐dimethyl‐5‐sulfo‐3H‐indolium‐2‐yl)allylidene)‐3,3‐dimethylindoline‐5‐sulfonate (compound 2)
Under Argon, DIEA (25.0 μL, 0.14 mmol, 3.00 eq.) was added to a solution of Cy3.29 (30.0 mg, 47.0 μmol, 1.00 eq.) and TSTU (14.0 mg, 48.0 μmol, 1.01 eq.) in dry DMF (1 mL). The resulting solution was protected from the light and stirred 2 h at room temperature. A solution of the amine 1 (47.0 mg, 0.14 mmol, 3.00eq.) dissolved in dry DMF (1.5 mL) was then added to the mixture which was stirred overnight at room temperature. After removing the DMF under reduced pressure, the residue was purified on C18 reverse‐phase chromatography with a gradient (methanol/water from 25:75 to 4:6). The fractions containing the desired compound were lyophilized to provide the compound 2 as a pink solid (18.0 mg, 40% yield).
1H NMR (400 MHz, MeOD): δ 8.54 (t, J = 13.4 Hz, 1H), 7.99–7.94 (m, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.7 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.38 (dd, J = 15.4, 8.4 Hz, 4H), 6.67 (d, J = 8.7 Hz, 1H), 6.51 (dd, J = 13.4, 10.1 Hz, 2H), 4.41 (s, 2H), 4.18 (dd, J = 7.1, 3.8 Hz, 4H), 4.02 (s, 3H), 3.50 (t, J = 7.3 Hz, 2H), 3.09 (t, J = 7.3 Hz, 2H), 2.32 (t, J = 7.1 Hz, 2H), 1.98–1.66 (m, 16H), 1.57–1.46 (m, 2H), 1.39 (t, J = 6.6 Hz, 3H).
13C NMR (101 MHz, MeOD): δ 175.3, 175.1, 174.3, 171.8, 159.3, 151.4, 143.3, 142.8, 142.7, 142.6, 140.9, 140.8, 138.8, 131.4, 127.8, 127.7, 126.8, 125.4, 120.1, 120.1, 110.7, 110.5, 103.2, 103.1, 44.0, 42.4, 39.8, 39.2, 35.2, 26.8, 26.7, 25.9, 25.0, 23.3, 21.2, 17.3, 15.9, 11.2.
IR: νmax (cm−1) = 3273, 2930, 1645, 1553, 1433, 1415, 1173, 1109, 1064, 1019, 926, 686.
HRMS (ESI): m/z = 951.3779 [M + H]+ calculated for C50H59N6O9S2, found: m/z = 951.3764.
Melting point: 271°C.
(E)‐2‐((E)‐3‐(1‐(6‐((2‐((3‐(2‐acetamidoethyl)‐1H–pyrrolo[3,2‐b]pyridin‐5‐yl)oxy)ethyl)amino)‐6‐oxohexyl)‐3,3‐dimethyl‐5‐sulfo‐3H‐indol‐1‐ium‐2‐yl)allylidene)‐1‐ethyl‐3,3‐dimethylindoline‐5‐sulfonate (compound 5)
Under Argon, DIEA (25.0 μL, 0.14 mmol, 3.00 eq.) was added to a solution of Cy3.29 (30.0 mg, 47.0 μmol, 1.00 eq.) and TSTU (14.0 mg, 48.0 μmol, 1.01 eq.) in dry DMF (1 mL). The resulting solution was protected from the light and stirred for 2 h at room temperature. A solution of the amine 4 (37.0 mg, 0.14 mmol, 3.00 eq.) dissolved in dry DMF (1.5 mL) was then added to the mixture which was stirred overnight at room temperature. After removing of the DMF under reduced pressure, the residue was purified on C18 reverse‐phase chromatography with a gradient (methanol/water from 25:75 to 4:6). The fractions containing the desired compound were lyophilized to provide the compound 5 as a pink solid (22.0 mg, 53% yield).
1H NMR (400 MHz, D2O): δ 8.38 (t, J = 13.6 Hz, 1H, H19), 7.87 (s, 1H, H21), 7.83 (s, 1H, H21), 7.55 (d, J = 8.7 Hz, 1H, H7), 7.26 (d, J = 8.1 Hz, 1H), 7.21 (s, 1H), 7.17 (d, J = 8.5 Hz, 1H), 6.59 (d, J = 8.7 Hz, 1H, H6), 6.26 (d, J = 13.4 Hz, 1H, H20), 6.14 (d, J = 13.2 Hz, 1H, H18), 4.53–4.48 (m, 1H), 4.32 (t, J = 5.4 Hz, 1H, H11), 4.02 (d, J = 7.0 Hz, 2H, H17), 3.72 (d, J = 6.8 Hz, 2H, H24), 3.70–3.64 (m, 1H), 3.54 (t, J = 4 Hz, 2H, H12), 3.30 (t, J = 7.2 Hz, 2H, H8), 3.00–2.94 (m, 2H, H14), 2.75 (t, J = 7.2 Hz, 2H, H9), 2.27 (t, J = 6.4 Hz, 2H, H13), 1.86 (bs, 2H,), 1.65 (s, 6H, 2 x CH3), 1.60 (s, 6H, 2 x CH3), 1.31 (dd, J = 16.0, 8.6 Hz, 3H, H25), 1.26 (dd, J = 7.1, 6.0 Hz, 2H, H 15).
13C NMR (101 MHz, MeOD): δ 175.41, 175.10, 174.67, 171.68, 158.63, 151.43, 143.11, 142.70, 142.68, 142.64, 141.33, 140.88, 140.76, 126.84, 125.34, 125.27, 121.98, 120.07, 111.71, 110.69, 110.47, 104.10, 103.22, 103.06, 63.73, 43.89, 40.00, 39.21, 38.85, 35.25, 26.84, 26.67, 26.62, 25.65, 24.96, 23.84, 21.32, 11.22.
HRMS (ESI): m/z = 873.3320 [M‐H]− calculated for C44H53N6O9S2, found: m/z = 873.3312.
IR: νmax (cm−1) = 3292, 2923, 2853, 1651, 1556, 1434, 1415, 1175, 1109, 1065, 1020, 927, 687.
Melting point: 186°C.
E‐1‐(6‐(2‐(3‐(2‐acetamidoethyl)‐5‐methoxy‐1H‐pyrrolo[3,2‐b]pyridin‐1‐yl)ethylamino)‐6‐oxohexyl)‐2‐((E)‐3‐(1‐ethyl‐3,3‐dimethyl‐5‐sulfo‐3H‐indolium‐2‐yl)allylidene)‐3,3‐dimethylindoline‐5‐sulfonate (compound 7)
Dry DIEA (16 μL, 12 mg, 95 μmol, 3 eq.) was added to a solution of Cy3.29 (20 mg, 32 μmol, 1 eq.) and TSTU (10 mg, 32 μmol, 1.01 eq.) in dry DMF (1 mL). The resulting solution was protected from the light and stirred 2 h at room temperature under an atmosphere of argon. Thereafter, a solution of the amine 6 (26 mg, 95 μmol, 3 eq.) in dry DMF 1 mL was added, and the mixture was stirred overnight at room temperature. After removing the DMF, the residue was purified by reversed flash chromatography with 25 to 40% of MeOH in ultra‐pure water as eluent. The product‐containing fractions were lyophilized to provide the compound 7 as a pink solid (25 mg, 89% yield).
1H NMR (400 MHz, D2O): δ = 8.35 (t, J = 13.5, 1H), 7.85 (m, 4H), 7.67 (d, J = 8.9 Hz, 1H), 7.26 (m, 3H), 6.49 (d, J = 8.9 Hz, 1H), 6.24 (d, J = 13.4 Hz, 2H), 4.07 (t, J = 6.2 Hz, 2H), 3.90 (m, 4H), 3.73 (s, 3H), 3.41 (t, J = 6.0 Hz, 2H), 3.21 (m, 4H), 2.71 (t, J = 7.0 Hz, 2H), 2.05 (t, J = 6.9 Hz, 2H), 1.85 (s, 3H), 1.65 (t, J = X Hz, 2H), 1.41 (m, 14 H), 1.21 (t, J = 7.2 Hz, 3H).
13C NMR (101 MHz, D2O): δ 176.1, 175.6, 175.0, 173.4, 159.4, 151.5, 143.7, 143.2, 141.3, 141.3, 141.0, 139.9, 139.8, 129.3, 126.7, 126.0, 121.4, 119.8, 119.7, 111.3, 111.2, 111.1, 103.1, 102.9, 53.9, 49.2, 49.1, 48.7, 44.8, 43.8, 39.9, 39.4, 35.3, 27.0, 26.7, 26.3, 25.1, 24.5, 23.1, 21.9, 20.0, 11.5.
IR: νmax (cm−1) = 928, 1022, 1111, 1186, 1435, 1555, 1648, 2935, 3294.
HRMS (ESI): m/z = 443.1712 [M‐2H]2− calculated for C45H56N6O9S2, found: m/z = 443.1697.
Melting point: 281°C.
Photophysical measurements
UV–visible spectra were obtained on a Varian Cary 50 scan spectrophotometer by using a rectangular quartz micro cell (Hellma, 104‐QS, light path: 10 mm, 1.0 mL). Fluorescence spectroscopic studies (emission/excitation spectra) were performed with a Jasco FP‐8100 spectrophotometer with a fluorescence quartz ultra‐micro cell (Hellma, 105.250‐QS, light path: 10 × 2 mm, 50 μL). Emission spectra were recorded under the same conditions after excitation at the corresponding wavelength (see Supporting Information Figure S3 and Table S1, excitation and emission filters: auto, excitation and emission slit = 5 nm) in DMSO. Relative quantum yields were measured in DMSO at 25°C by a relative method using a suitable standard (see Supporting Information Table S1). The following equation was used to determine the relative fluorescence quantum yield:
where A is the absorbance (in the range 0.01–0.1 A.U.), F is the area under the emission curve, n is the refractive index of the solvents (at 25°C) used in measurements, and the subscripts s and x represent standard and unknown respectively.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Since all synthetic melatonin receptor ligands, including fluorescent ligands, are all lipophilic (Wu et al., 2007; Thireau et al., 2014; Zlotos et al., 2014; de la Fuente Revenga et al., 2015), we set out to generate a new series of hydrophilic fluorescent melatonin receptor ligands by introducing the Cy3 cyanin fluorophore known to be hydrophilic (Licha et al., 2000; Tatikolov and Costa, 2002). Melatonin derivatives bearing the Cy3 fluorophore at positions N‐1, C‐2 or C‐5 were synthesized to determine the fluorophore position most compatible with biological function. Compound 1 was coupled at the C‐2 position of the indole ring with an activated acid of Cy3. Compound 2 was isolated in 40% yield (Figure S1). In order to synthesize C‐5 tagged and N‐1 tagged cyanine ligands, amines 4 and 6 were reacted with activated Cy3 to provide probes 5 and 7 in 53 and 89% yield respectively (Figure 1A, Supporting Information Figure S2). To generate a fluorescent lipophilic melatonin receptor probe, compound 1 was coupled with an activated acid of the BODIPY‐FL® dye. This reference probe 3 was isolated as previously described (Figure 1A) (Viault et al., 2016).
Figure 1.
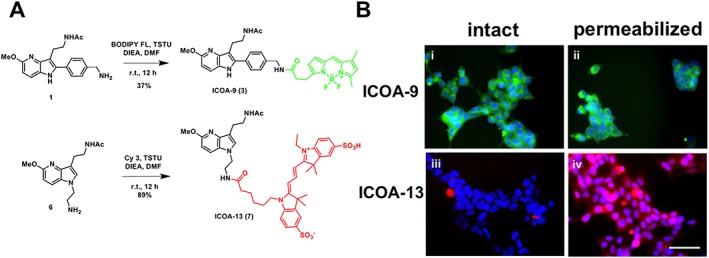
Chemical synthesis pathway and permeability properties of fluorescent melatonin receptor ligands. (A) Chemical synthesis of ICOA‐9 (3) and ICOA‐13 (7). (B) Representative fluorescence microscopy images of intact (i, iii) and permeabilized (ii, iv) non‐transfected HEK293 cells incubated with 100 nM ICOA‐9 (i, ii) (green) or ICOA‐13 (iii, iv) (red). Cell nuclei are stained with DAPI (blue). Scale bar = 50 μm.
The spectroscopic characterization showed that the Cy3‐tagged compounds 2, 5 and 7 have a very good brightness at low energy excitation and emission wavelengths in accordance with the fluorescent properties of the cyanine 3 family (Supporting Information Figure S3 and Table S1). Compound 3 with the BODIPY fluorophore showed also excellent fluorescent properties.
The affinity of compounds 2, 3, 5 and 7 for human MT1 and MT2 receptors was then determined in competition binding experiments with 2‐[125I]iodomelatonin (2‐[125I]‐MLT) in crude membranes prepared from HEK293 cells transiently expressing either receptor. Preliminary experiment showed that C‐2 and C‐5 tagged Cy3 ligands 2 and 5 were unable to compete 2‐[125I]‐MLT binding at both receptors at concentrations up to 1 μM. However, ligand 7 (henceforward named ICOA‐13) had micromolar affinity for both receptors (Table 1, Supporting Information Figure S4). Ligand 3 (henceforward named ICOA‐9) containing the BODIPY‐FL fluorophore displays sub‐micromolar affinities for both melatonin receptors (Table 1, Supporting Information Figure S4A, B). Similar affinities were observed for mouse MT1 and MT2 receptors (supporting Information Figure S4C, D).
Table 1.
Functional characteristics of ICOA‐9 and ICOA‐13 in HEK293 cells expressing the melatonin receptors indicated
| Receptor | Ligand | 2‐[125I]‐MLT compet. pKi | ERK act. pEC50 (Emax) | cAMP inh. pEC50 (Emax) | ß‐ARR recrut. pEC50 (Emax) |
|---|---|---|---|---|---|
| hMT1 | Melatonin | −8.61 ± 0.19 | −8.46 ± 0.41 (100 ± 9) | −9.37 ± 0.22 (100 ± 5) | −9.67 ± 0.17 (100 ± 5) |
| ICOA‐9 (3) | −5.76 ± 0.16 | > −6 (n.d.) | −9.18 ± 0.47 (49 ± 8) | −7.60 ± 0.48 (95 ± 7) | |
| ICOA‐13 (7) | −4.97 ± 0.28 | > −6 (n.d.) | −7.76 ± 0.69 (37 ± 8) | −7.13 ± 0.05 (57 ± 5) | |
| hMT2 | Melatonin | −9.3 ± 0.20 | −9.16 ± 0.57 (100 ± 8) | −9.49 ± 0.11 (100 ± 5) | −9.53 ± 0.16 (100 ± 6) |
| ICOA‐9 (3) | −7.41 ± 0.05 | −6.65 ± 0.21 (156 ± 23) | n.d. (n .d.) | −7.91 ± 0.72 (79 ± 8) | |
| ICOA‐13 (7) | −5.48 ± 0.50 | −6.58 ± 0.38 (133 ± 14) | n.d. (n.d.) | −7.76 ± 0.49 (49 ± 8) | |
| mMT1 | Melatonin | −9.42 ± 0.28 | −9.22 ± 0.86 (100 ± 14) | −9.65 ± 0.45 (100 ± 12) | n.d. |
| ICOA‐9 (3) | −6.88 ± 0.29 | −6.90 ± 0.12 (146 ± 13) | −9.88 ± 0.34 (81 ± 12) | n.d. | |
| ICOA‐13 (7) | −5.81 ± 0.33 | −7.12 ± 0.10 (164 ± 15) | −9.60 ± 0.94 (92 ± 12) | n.d. | |
| mMT2 | Melatonin | −9.52 ± 0.18 | −8.90 ± 0.47 (100 ± 9) | −10.19 ± 0.22 (100 ± 9) | n.d. |
| ICOA‐9 (3) | −7.39 ± 0.36 | −6.69 ± 0.24 (105 ± 13) | −9.60 ± 0.94 (94 ± 12) | n.d. | |
| ICOA‐13 (7) | −6.24 ± 0.29 | −7.24 ± 0.30 (84 ± 14) | −7.47 ± 0.65 (62 ± 8) | n.d. |
Concentration–response curves were analysed by non‐linear regression. Binding affinity was measured with 2‐[125I]‐MLT and is expressed as mean pKi ± SEM (M). Agonist potency is expressed as pEC50 ± SEM (M). While the maximal efficacy, Emax, is expressed as a percentage of the maximal effect observed with melatonin (=100%). Data are mean of at least three independent experiments, each of them performed using at least eight different ligand concentrations. n.d., not determined.
In accordance with the reported hydrophilic property of Cy3 (Licha et al., 2000; Tatikolov and Costa, 2002), ICOA‐13 containing the Cy3 fluorophore was much more hydrophilic than ICOA‐9 as it did not penetrate into intact cells unless permeabilized (Figure 1B). In contrast, ICOA‐9 was readily penetrating into intact and permeabilized cells (Figure 1B). Therefore, ICOA‐13 showed the desired properties in terms of melatonin receptor binding and absence of cell penetration. Unfortunately, the use of ICOA‐13 as a fluorescent probe to label melatonin receptors for fluorescence microscopy imaging was not successful, probably due to its low binding affinity, as no specific staining was observed in melatonin receptor‐transfected cells.
We next determined the functional properties of ICOA‐9 and ICOA‐13 in comparison to melatonin in HEK293 cells expressing human or murine MT1 and MT2 receptors. We first determined time‐courses of ERK1/2 phosphorylation by melatonin, ICOA‐9 and ICOA‐13. A peak of phosphorylation was observed at 5 min for all the compounds at a saturating ligand concentration (Figure 2A). A quantitative assessment of ERK activation at 5 min was performed with the P‐ERK AlphaScreen SureFire Assay that monitors P‐ERK levels directly in cell lysates. ERK activation by melatonin ranged between 1.5‐ to 2.5‐fold on human and mouse melatonin receptors, respectively, compared with the basal. ICOA‐9 and ICOA‐13 exhibited potencies in the sub‐micromolar range except for hMT1, for which micromolar potencies were observed (Figure 2, Table 1). The response of all ligands was abolished in the presence of pertussis toxin, indicating that activation of the ERK pathway is Gi/o protein‐dependent (Supporting Information Figure S5).
Figure 2.
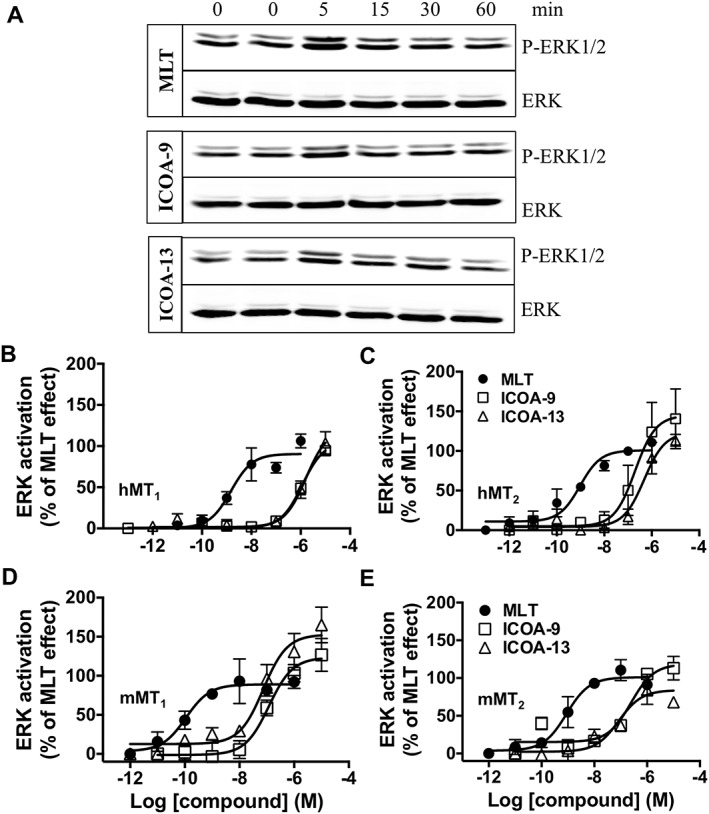
ERK1/2 activation triggered by melatonin, ICOA‐9 and ICOA‐13. Detection of phospho‐ERK1/2 (P‐ERK1/2) in HEK293 cells expressing hMT1 (A, B), hMT2 (C), mMT1 (D) or mMT2 (E) receptors incubated for 0 to 60 min with melatonin (100 nM), ICOA‐9 (1 μM), ICOA‐13 (1 μM) (Western blot; A) or at 5 min at different concentrations of the indicated ligands (Alpha‐Screen assay; B, C, D, E). Data are expressed as mean ± SEM from three to four independent experiments [(B) melatonin n = 4, ICOA‐9 and ICOA‐13 n = 3; (C) all groups n = 3; (D, E) melatonin n = 4, ICOA‐9 n = 3, ICOA‐13 n = 4]. Receptor expression levels were 155 ± 62, 52 ± 22, 136 ± 63 and 112 ± 22 fmol·mg−1 of protein for hMT1, hMT2, mMT1 and mMT2 receptors respectively.
We next measured the capacity of ICOA‐9 and ICOA‐13 to inhibit forskolin‐stimulated cAMP production. Melatonin inhibited forskolin‐stimulated cAMP production at all four receptors with similar EC50 values in the sub‐nanomolar range (Figure 3, Table 1). ICOA‐9 behaved as a partial agonist (Emax = 49–90%), and potencies varied between receptors (pEC50: between 10−8 M and 10−10 M). ICOA‐13 is a full agonist for this pathway on mMT1 receptors with potency in the sub‐nanomolar range, and partial agonist for hMT1 and mMT2 (pEC50 = −7.76 ± 0.69 and −7.47 ± 0.65 respectively). For hMT2, EC50 and Emax could not be determined as the colour of ICOA‐9 and ICOA‐13 interfered with the assay at concentrations higher than 1 μM respectively.
Figure 3.
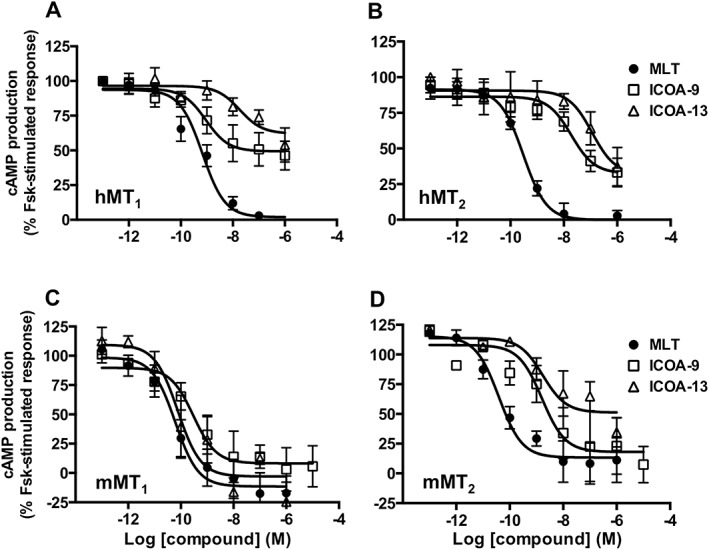
Inhibition of cAMP production by melatonin, ICOA‐9 and ICOA‐13. Inhibitory effect of melatonin, ICOA‐9 and ICOA‐13 on forskolin (Fsk)‐stimulated cAMP production in HEK293 cells expressing hMT1 (A), hMT2 (B), mMT1 (C) or mMT2 (D) receptors. Data are expressed as mean ± SEM from 5 to 10 independent experiments [(A) melatonin n = 10; ICOA‐9 and ICOA‐13 n = 6; (B) melatonin n = 7, ICOA‐9 and ICOA‐13 n = 6; (C) all groups n = 6; (D) all groups n = 5]. Data are presented as percentage of forskolin‐stimulated response and are normalized to the maximal and minimal melatonin effect. The amplitude of cAMP inhibition varied between 25 and 50%, depending on the receptor.
To explore another major signalling event of GPCRs, namely, the recruitment of β‐arrestins, we monitored β‐arrestin 2 recruitment to hMT1 and hMT2 receptors in response to melatonin, ICOA‐9 and ICOA‐13 (Figure 4A, B, Table 1). We used the PathHunter assay (DiscoverX) that is based on the complementation between the PK2 tag (fused to the C‐terminus of receptors) and the β‐arrestin 2‐β‐gal‐enzyme acceptor fusion protein. Addition of the PK2 tag did not modify the capacity of hMT1 and hMT2 to inhibit forskolin‐induced cAMP production. ICOA‐9 behaved as a full agonist on hMT1 and partial agonist on hMT2 receptors. ICOA‐13 is a partial agonist for both receptors with potencies in the sub‐micromolar range as ICOA‐9.
Figure 4.
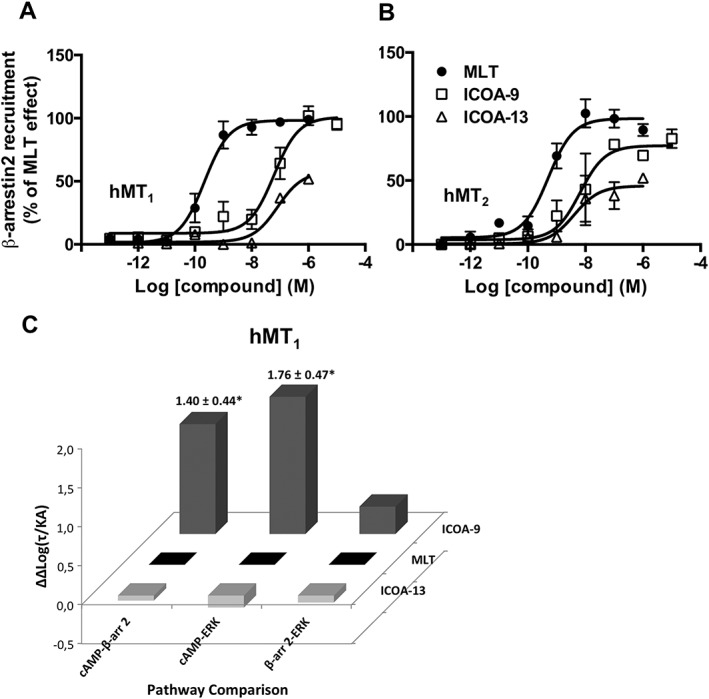
Recruitment of ß‐arrestin2 by melatonin, ICOA‐9 and ICOA‐13 and signalling bias of ICOA‐9. Recruitment of ß‐arrestin2 to hMT1 (A) or hMT2 (B) receptors induced by melatonin, ICOA‐9 and ICOA‐13 in HEK293 cells. Data are expressed as mean ± SEM from three to four independent experiments [(A) melatonin and ICOA‐9 n = 4; ICOA‐13 n = 3; (B) melatonin n = 4, ICOA‐9 and ICOA‐13 n = 3] and are presented as percentage of melatonin effect. (C) Signalling bias factor of ICOA‐9 and ICOA‐13 for hMT1 receptors presented as ∆∆Log(t/KA) ratios, calculated from each individual experiment on hMT1 receptor signalling shown in Figures 2B, 3A and 4A. *P < 0.05.
Given the fact that the action of the cell‐impermeable ICOA‐13 is spatially restricted as compared with the lipophilic melatonin, we determined possible signalling bias of ICOA‐9 and ICOA‐13, by fitting dose–response curves of functional assays to the operational model of agonism designed by Kenakin and Christopoulos (Kenakin and Christopoulos, 2013) to obtain log(τ/KA) transduction ratios, which is a unique index of coupling efficiency (or efficacy) of an agonist. To eliminate the influence of the system and the observation biases, the activity of the compounds at a given signalling pathway was compared with that of melatonin, which served as the reference compound, and subsequently, values were expressed as Δlog(τ/KA). Ligand bias for two pathways, relative to melatonin, was expressed as 10ΔΔlog(τ/KA) as previously described (van der Westhuizen et al., 2014) (Supporting Information Table S2). No bias was observed for ICOA‐9 and ICOA‐13 for most receptors, with the exception of hMT1 for which ICOA‐9 showed significant bias (25‐ and 57‐fold) on the cAMP over β‐arrestin and ERK pathways, respectively, when using extrapolated EC50 and Emax values for ERK activation as previously reported (Baltos et al., 2016) (see Methods section) (Figure 4C, Supporting Information Table S2). Taken together, this analysis shows that the cell‐permeable ICOA‐9 shows bias at hMT1 and the cell‐impermeable ICOA‐13 does not display any detectable signalling bias compared with the cell‐permeable melatonin.
Recently, MT1 receptors have been described to be located in brain mitochondria (Wang et al., 2011). Here, we confirmed the localization of MT1 receptors in mitochondria by its extensive colocalization with the mitochondria targeted red fluorescent protein (mito‐RFP) (Figure 5A). We further measured the capacity of melatonin and ICOA‐13 to inhibit forskolin‐stimulated cAMP production. To this end, we used FRET‐based cAMP sensors localized either in the plasma membrane (PM‐ICUE) (DiPilato et al., 2004) or in the outer mitochondrial membrane (OMM‐ICUE) (Figure 5B, C). Stimulation of MT1 receptors by melatonin and ICOA‐13 resulted in a comparable (40–50%) reduction of the forskolin‐stimulated signal of the PM cAMP sensor (Figure 5B, C). In contrast, signals detected by the OMM cAMP sensor in the presence of melatonin and ICOA‐13 were very different. Melatonin reduced the signal by 80% and ICOA‐13 only by 25%. ICOA‐9, similarly to melatonin, decreased the signal of the OMM cAMP sensor by 80% (Supporting Information Figure S6). These data show that the cell‐permeable melatonin readily decreases cAMP concentrations at the OMM, whereas cell‐impermeable ICOA‐13 has only a limited capacity to do so. These data validate the usefulness of the ICOA‐13 compound to discriminate between plasma membrane and intracellular receptors and are consistent with the idea of mitochondrial MT1 receptors.
Figure 5.
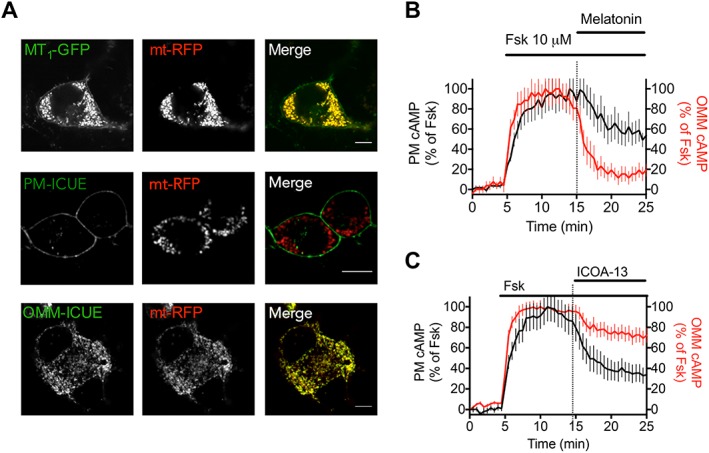
Mitochondrial MT1 receptor signalling and localization. (A) Confocal imaging of N2a cells expressing MT1‐GFP or the FRET‐based cAMP sensors that localized either at the plasma membrane (PM‐ICUE) or the outer mitochondrial membrane (OMM‐ICUE) (green) together with the mitochondrial marker mt‐RFP (red). Scale bar, 10 μm. (B, C) Averaged time courses of forskolin‐mediated (10 μM) cAMP production in N2a cells expressing MT1 receptors and the FRET‐based cAMP biosensors localized either at the plasma membrane (PM) or the outer mitochondrial membrane (OMM) and challenged by melatonin (100 nM) (B) and ICOA‐13 (1 uM) (C). Data are expressed as the mean value ± SD, from n = 19 (MT, PM), n = 34 (MT, OMM), n = 24 (ICOA13, PM) and n = 41 (ICOA13, OMM).
Discussion
We describe here the synthesis and functional characterization of the first melatonin receptor cell‐impermeant agonist that activates receptors exclusively at the cell surface. Despite the generation of numerous melatonin receptor ligands with various chemical scaffolds, all of them are lipophilic and cell permeant (Zlotos et al., 2014). Introduction of a Cy3 cyanin fluorophore allowed us to significantly increase the hydrophilicity of the compound and to introduce a fluorescent label. Due to the small size of melatonin, introduction of fluorophores is technically challenging and often results in loss of binding affinity, as observed in our study for compounds 2 and 5. ICOA‐13 and ICOA‐9 showed micromolar and sub‐micromolar affinities, respectively, for both melatonin receptors, which were compatible with functional studies on ERK phosphorylation, cAMP inhibition and ß‐arrestin2 recruitment. For fluorescence microscopy studies, ICOA‐13 was inappropriate, as no specific cell surface labelling was observed in cells expressing melatonin receptors.
Emerging evidence shows that GPCR signalling can be prolonged and/or initiated in intracellular compartments such as endosomes following receptor endocytosis (Vilardaga et al., 2014; Tsvetanova et al., 2015). Other studies reported the presence of GPCRs in neuronal mitochondria as shown in the case of the type‐1 cannabinoid receptor (Benard et al., 2012; Ma et al., 2015). The lipophilic nature of most cannabinoids implies that receptor–ligand interactions might occur not only at plasma membranes but also inside cells. Mitochondrial localization has been also suggested for P2Y‐like, angiotensin II and 5‐HT4 receptors (Belous et al., 2004; Abadir et al., 2011; Wang et al., 2016). In addition, several components of the cAMP pathway such as Gα and Gβ subunits of heterotrimeric G proteins and PKA, among others, have been identified in mitochondria by proteomic approaches (Chen et al., 2013).
These examples indicate a new emerging paradigm of intracellular GPCR signalling in mitochondria. Melatonin receptors and its cell‐penetrating ligand (melatonin) are excellent candidates for such a scenario. Indeed, melatonin has well‐established protective effects on mitochondrial energy metabolism and on limiting oxidative stress (Pacini and Borziani, 2016), but the underlying molecular mechanisms are unknown. A recent study suggests that the MT1 receptor is located in brain mitochondria (Wang et al., 2011). Our data on transfected N2a cells are consistent with this finding. More rapid progress on the subcellular localization of melatonin signalling is hampered by the low expression levels of these receptors and poor quality of available tools like anti‐receptor antibodies and selective ligands. The development of the ICOA‐13 agonist represents a major progress in this respect. The comparison of this cell‐impermeant fluorescent ligand ICOA‐13 and its permeant homologue ICOA‐9 showed no difference in affinity but more in efficacy and potency between human and mouse melatonin receptors. Indeed, on the cAMP pathway, ICOA‐9 is more potent but partial on hMT1 compared with mMT1 receptors. In contrast, ICOA‐13 has a better potency and is a full agonist on mMT1 receptor versus hMT1 receptor.
We also quantified the degree of signalling bias of each of the compounds using the operational model of agonism to determine the potential impact of the spatial restriction of ICOA‐13 to the plasma membrane to signalling bias as compared with the cell‐permeable ICOA‐9 and melatonin. In our study, the quantification of biased agonism was calculated at a single time point for each signalling pathway which corresponds to the maximum activation of that pathway, or when the signal reaches a steady state. Different experimental conditions such as different kinetics contexts can influence the bias factor, as reported recently for a series of agonists for the dopamine D2 receptor (Klein Herenbrink et al., 2016). Despite these obvious limitations, we were able to conclude that the spatial restriction of ICOA‐13 did not introduce any detectable bias compared with ICOA‐9 and melatonin since all three ligands were tested in parallel using the same time points for each signalling pathway. Significant bias was observed for ICOA‐9 on hMT1 receptors with preferences for the Gi/cAMP pathway over the ß‐arrestin2 and ERK pathways.
Melatonin has pleiotropic effects ranging from antioxidant and neuroprotective effects to the regulation of glucose homeostasis, circadian rhythms, sleep and retinal physiology (Dubocovich et al., 2010; Tosini et al., 2014). How melatonin exerts these effects remains in many cases elusive. Dysregulation of the melatonin system is associated with autoimmune disorders such as multiple sclerosis and neurodegenerative diseases such as Parkinson, Alzheimer and also Huntington disease for which mitochondrial MT1 receptors have been suggested to play an important role (Wang et al., 2011). Cell‐impermeant ligands like ICOA‐13 will be precious tools to determine the contribution of mitochondrial versus plasma membrane receptors and the signalling pathways involved. This information will be essential for the design of new drugs and therapeutic strategies for melatonin‐associated diseases including a controlled enhancement of melatonin's antioxidant‐related effects.
Author contributions
E.C. and F.G. performed functional assays. R.G. and R.J. performed radioligand binding assays. G.V. performed synthesis and characterization of compounds 2, 3, 5 and 7. F.J.A. performed experiments in N2a cells. A.K. performed analysis of biased signalling. G.G. elaborated the synthesis pathway. P.D. and R.M.F. participated in the study design. J.P.V. supervised studies in N2a cells. F.S. designed the fluorescent melatonin ligands and supervised the chemical synthesis. E.C. and R.J. wrote the article. F.G., E.C., A.L., F.S. and J.P.V. participated in the reviewing and editing of the article. R.J. elaborated and supervised the project.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Synthesis of probe 2 “C2 tagging”.
Figure S2 Synthesis of probes 5 “C5 tagging”.
Figure S3 Absorption (Abs) and emission (Em) spectra of compounds 2, 3, 5 and 7 recorded in DMSO at 25°C.
Figure S4 Competition of 2‐[125I]‐MLT binding by melatonin (MLT), ICOA‐9 and ICOA‐13. Crude membranes of HEK293 cells expressing hMT1 (A), hMT2 (B), mMT1 (C) or mMT2 (D) receptors were incubated with 0.1 nM 2‐[125I]‐MLT and increasing concentrations of the indicated compounds. Data are expressed as mean ± S.E.M. from 3–8 independent experiments (A. melatonin n = 4, ICOA‐9 n = 4, ICOA‐13 n = 3; B. all groups n = 4; C. melatonin n = 8, ICOA‐9 n = 7, ICOA‐13 n = 6; D. melatonin n = 8, ICOA‐9 and ICOA‐13 n = 7). Data are represented as percentage of maximal binding in the absence of compounds and normalized to melatonin maximum effect.
Figure S5 ERK1/2 activation in the absence or presence of PTX. Detection of phospho‐ERK1/2 in HEK293 cells expressing hMT1 (A), hMT2 (B), mMT1 (C) or mMT2 (D) receptors incubated for 5 min with MLT (100 nM), ICOA‐9 (1 μM), ICOA‐13 (1 μM) in the absence or presence of PTX (10 ng/μL, overnight). Data are expressed as mean ± S.E.M. from 5 independent experiments. *P < 0.05 compared to respective control by Student t‐test, one‐tail analysis.
Figure S6 Effect of ICOA‐9 on cAMP production in neuroblastoma cells. Averaged time courses of forskolin‐mediated (10 μM) cAMP production in N2a cells expressing MT1 and the FRET‐based cAMP biosensors localized at the outer mitochondrial membrane (OMM) and challenged by ICOA‐9 (1 μM). Data expressed as the mean value ± s.d. from n = 48.
Figure S7 1H and 13C NMR spectra of compound 2.
Figure S8 1H and 13C NMR spectra of compound 5.
Figure S9 1H and 13C NMR spectra of compound 7 (ICOA‐13).
Table S1 Photophysical and physicochemical properties of compounds 2, 3, 5 and 7.
Table S2 ΔΔlog(τ/KA) ratios and bias factors for ICOA‐9 and ICOA‐13 at the hMT1, hMT2, mMT1 and mMT2.
Acknowledgements
This work was supported by the Agence Nationale de la Recherche (ANR 2011‐BSV1‐012‐01 “MLT2D” and ANR‐2011‐META “MELA‐BETES”, ANR‐12‐RPIB‐0016 “MED‐HET‐REC‐2”), the Fondation pour la Recherche Médicale (Equipe FRM DEQ20130326503, to R.J.), Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS) and the Who am I? laboratory of excellence No.ANR‐11‐LABX‐0071 funded by the French Government through its “Investments for the Future” programme operated by The French National Research Agency (ANR) under grant No.ANR‐11‐IDEX‐0005‐01 (to R.J.); La Région Centre (APR2009‐LOIREMEL to G.G. and APR2012‐LIFERMEL to F.S.), Labex SynOrg (ANR‐11‐LABX‐0029 to F.S.), the National Institutes of Health (NIH) under Award numbers R01 DK087688 (J.P.V.) and R01 DK102495 (J.P.V.) and the Cotswold Foundation Fellowship Award (F.J.A.).
Gbahou, F. , Cecon, E. , Viault, G. , Gerbier, R. , Jean‐Alphonse, F. , Karamitri, A. , Guillaumet, G. , Delagrange, P. , Friedlander, R. M. , Vilardaga, J.‐P. , Suzenet, F. , and Jockers, R. (2017) Design and validation of the first cell‐impermeant melatonin receptor agonist. British Journal of Pharmacology, 174: 2409–2421. doi: 10.1111/bph.13856.
References
- Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A et al. (2011). Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A 108: 14849–14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub MA, Levoye A, Delagrange P, Jockers R (2004). Preferential formation of MT1/MT2 melatonin receptor heterodimers with distinct ligand interaction properties compared with MT2 homodimers. Mol Pharmacol 66: 312–321. [DOI] [PubMed] [Google Scholar]
- Baba K, Benleulmi‐Chaachoua A, Journe AS, Kamal M, Guillaume JL, Dussaud S et al. (2013). Heteromeric MT1/MT2 melatonin receptors modulate photoreceptor function. Sci Signal 6: ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltos JA, Paoletta S, Nguyen AT, Gregory KJ, Tosh DK, Christopoulos A et al. (2016). Structure‐activity analysis of biased agonism at the human adenosine A3 receptor. Mol Pharmacol 90: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker‐Andre M, Wiesenberg I, Schaeren WN, Andre E, Missbach M, Saurat JH et al. (1994). Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J Biol Chem 269: 28531–28534. [PubMed] [Google Scholar]
- Belous A, Wakata A, Knox CD, Nicoud IB, Pierce J, Anderson CD et al. (2004). Mitochondrial P2Y‐like receptors link cytosolic adenosine nucleotides to mitochondrial calcium uptake. J Cell Biochem 92: 1062–1073. [DOI] [PubMed] [Google Scholar]
- Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria‐Gomez E et al. (2012). Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat Neurosci 15: 558–564. [DOI] [PubMed] [Google Scholar]
- Cecon E, Chen M, Marcola M, Fernandes PA, Jockers R, Markus RP (2015). Amyloid beta peptide directly impairs pineal gland melatonin synthesis and melatonin receptor signaling through the ERK pathway. FASEB J 29: 2566–2582. [DOI] [PubMed] [Google Scholar]
- Chen X, Cui Z, Wei S, Hou J, Xie Z, Peng X et al. (2013). Chronic high glucose induced INS‐1beta cell mitochondrial dysfunction: a comparative mitochondrial proteome with SILAC. Proteomics 13: 3030–3039. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente Revenga M, Herrera‐Arozamena C, Fernandez‐Saez N, Barco G, Garcia‐Orue I, Sugden D et al. (2015). New coumarin‐based fluorescent melatonin ligands. Design, synthesis and pharmacological characterization. Eur J Med Chem 103: 370–373. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J (2004). Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A 101: 16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubocovich ML, Delagrange P, Krause DN, Sugden D, Cardinali DP, Olcese J (2010). International union of basic and clinical pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein‐coupled melatonin receptors. Pharmacol Rev 62: 343–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Yui N, Webber MJ, Wehbi VL, Stevenson HP, King JD Jr et al. (2013). Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem 288: 27849–27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT et al. (2009). Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5: 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaume JL, Daulat AM, Maurice P, Levoye A, Migaud M, Brydon L et al. (2008). The PDZ protein mupp1 promotes Gi coupling and signaling of the Mt1 melatonin receptor. J Biol Chem 283: 16762–16771. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tsvetanova NG, Lobingier BT, von Zastrow M (2015). Effects of endocytosis on receptor‐mediated signaling. Curr Opin Cell Biol 35: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten AM (2009). Retinoid‐related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal 7: e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G et al. (2016). Update on melatonin receptors. IUPHAR Review. Br J Pharmacol 173: 2702–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockers R, Maurice P, Boutin JA, Delagrange P (2008). Melatonin receptors, heterodimerization, signal transduction and binding sites: what's new? Br J Pharmacol 154: 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliolia E, Silajdzic E, Nambron R, Hill NR, Doshi A, Frost C et al. (2014). Plasma melatonin is reduced in Huntington's disease. Mov Disord 29: 1511–1515. [DOI] [PubMed] [Google Scholar]
- Kamal M, Gbahou F, Guillaume JL, Daulat AM, Benleulmi‐Chaachoua A, Luka M et al. (2015). Convergence of melatonin and serotonin (5‐HT) signaling at MT2/5‐HT2C receptor heteromers. J Biol Chem 290: 11537–11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz‐Medina V, Christopoulos A, Novick S (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem Nerosci 3: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J et al. (2016). The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7: 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licha K, Riefke B, Ntziachristos V, Becker A, Chance B, Semmler W (2000). Hydrophilic cyanine dyes as contrast agents for near‐infrared tumor imaging: synthesis, photophysical properties and spectroscopic in vivo characterization. Photochem Photobiol 72: 392–398. [DOI] [PubMed] [Google Scholar]
- Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF (1999). Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer's disease, and apolipoprotein E‐epsilon4/4 genotype. J Clin Endocrinol Metab 84: 323–327. [DOI] [PubMed] [Google Scholar]
- Ma L, Jia J, Niu W, Jiang T, Zhai Q, Yang L et al. (2015). Mitochondrial CB1 receptor is involved in ACEA‐induced protective effects on neurons and mitochondrial functions. Sci Rep 5: 12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS (1993). Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug Chem 4: 105–111. [DOI] [PubMed] [Google Scholar]
- Pacini N, Borziani F (2016). Oncostatic‐cytoprotective effect of melatonin and other bioactive molecules: a common target in mitochondrial respiration. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit L, Lacroix I, deCoppet P, Strosberg AD, Jockers R (1999). Differential signaling of human Mel1a and Mel1b melatonin receptors through the cyclic guanosine 3 '‐5'‐monophosphate pathway. Biochem Pharmacol 58: 633–639. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Tan DX, Kim SJ, Cruz MH (2014). Delivery of pineal melatonin to the brain and SCN: role of canaliculi, cerebrospinal fluid, tanycytes and Virchow‐Robin perivascular spaces. Brain Struct Funct 219: 1873–1887. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan DX, Manchester LC, Qin L, Reiter RJ (2016). Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatikolov AS, Costa SM (2002). Photophysics and photochemistry of hydrophilic cyanine dyes in normal and reverse micelles. Photochem Photobiol Sci 1: 211–218. [DOI] [PubMed] [Google Scholar]
- Thireau J, Marteaux J, Delagrange P, Lefoulon F, Dufourny L, Guillaumet G et al. (2014). Original design of fluorescent ligands by fusing BODIPY and melatonin neurohormone. ACS Med Chem Lett 5: 158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosini G, Owino S, Guillaume JL, Jockers R (2014). Understanding melatonin receptor pharmacology: latest insights from mouse models, and their relevance to human disease. Bioessays 36: 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanova NG, Irannejad R, von Zastrow M (2015). G protein‐coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem 290: 6689–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M (2014). Quantification of ligand bias for clinically relevant beta2‐adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85: 492–509. [DOI] [PubMed] [Google Scholar]
- Venegas C, Garcia JA, Escames G, Ortiz F, Lopez A, Doerrier C et al. (2012). Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res 52: 217–227. [DOI] [PubMed] [Google Scholar]
- Viault G, Poupart S, Mourlevat‐Le Magrex S, Lagaraine C, Devavry S et al. (2016). Design, synthesis and biological evaluation of fluorescent ligands for MT1 and/or MT2 melatonin receptors. RSC Adv 6: 62508–62521. [Google Scholar]
- Vilardaga JP, Jean‐Alphonse FG, Gardella TJ (2014). Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10: 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang H, Xu H, Guo D, Shi H, Li Y et al. (2016). 5‐HTR3 and 5‐HTR4 located on the mitochondrial membrane and functionally regulated mitochondrial functions. Sci Rep 6: 37336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J et al. (2011). The melatonin MT1 receptor axis modulates mutant Huntingtin‐mediated toxicity. J Neurosci 31: 14496–14507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PW, Cheng YM, Hsieh WT, Wang YH, Wei CY, Chou PT (2007). 7‐azamelatonin: efficient synthetic routes, excited‐state double proton transfer properties and biomedical implications. ChemMedChem 2: 1071–1075. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cook A, Kim J, Baranov SV, Jiang J, Smith K et al. (2013). Melatonin inhibits the caspase‐1/cytochrome c/caspase‐3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 55: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotos DP, Jockers R, Cecon E, Rivara S, Witt‐Enderby PA (2014). MT1 and MT2 melatonin receptors: ligands, models, oligomers, and therapeutic potential. J Med Chem 57: 3161–3185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Synthesis of probe 2 “C2 tagging”.
Figure S2 Synthesis of probes 5 “C5 tagging”.
Figure S3 Absorption (Abs) and emission (Em) spectra of compounds 2, 3, 5 and 7 recorded in DMSO at 25°C.
Figure S4 Competition of 2‐[125I]‐MLT binding by melatonin (MLT), ICOA‐9 and ICOA‐13. Crude membranes of HEK293 cells expressing hMT1 (A), hMT2 (B), mMT1 (C) or mMT2 (D) receptors were incubated with 0.1 nM 2‐[125I]‐MLT and increasing concentrations of the indicated compounds. Data are expressed as mean ± S.E.M. from 3–8 independent experiments (A. melatonin n = 4, ICOA‐9 n = 4, ICOA‐13 n = 3; B. all groups n = 4; C. melatonin n = 8, ICOA‐9 n = 7, ICOA‐13 n = 6; D. melatonin n = 8, ICOA‐9 and ICOA‐13 n = 7). Data are represented as percentage of maximal binding in the absence of compounds and normalized to melatonin maximum effect.
Figure S5 ERK1/2 activation in the absence or presence of PTX. Detection of phospho‐ERK1/2 in HEK293 cells expressing hMT1 (A), hMT2 (B), mMT1 (C) or mMT2 (D) receptors incubated for 5 min with MLT (100 nM), ICOA‐9 (1 μM), ICOA‐13 (1 μM) in the absence or presence of PTX (10 ng/μL, overnight). Data are expressed as mean ± S.E.M. from 5 independent experiments. *P < 0.05 compared to respective control by Student t‐test, one‐tail analysis.
Figure S6 Effect of ICOA‐9 on cAMP production in neuroblastoma cells. Averaged time courses of forskolin‐mediated (10 μM) cAMP production in N2a cells expressing MT1 and the FRET‐based cAMP biosensors localized at the outer mitochondrial membrane (OMM) and challenged by ICOA‐9 (1 μM). Data expressed as the mean value ± s.d. from n = 48.
Figure S7 1H and 13C NMR spectra of compound 2.
Figure S8 1H and 13C NMR spectra of compound 5.
Figure S9 1H and 13C NMR spectra of compound 7 (ICOA‐13).
Table S1 Photophysical and physicochemical properties of compounds 2, 3, 5 and 7.
Table S2 ΔΔlog(τ/KA) ratios and bias factors for ICOA‐9 and ICOA‐13 at the hMT1, hMT2, mMT1 and mMT2.