

Abstract
Azoreductases are flavoenzymes that have been characterized in a range of prokaryotes and eukaryotes. Bacterial azoreductases are associated with the activation of two classes of drug, azo drugs for the treatment of inflammatory bowel disease and nitrofuran antibiotics. The mechanism of reduction of azo compounds is presented; it requires tautomerisation of the azo compound to a quinoneimine and provides a unifying mechanism for the reduction of azo and quinone substrates by azoreductases. The importance of further work in the characterization of azoreductases from enteric bacteria is highlighted to aid in the development of novel drugs for the treatment of colon related disorders. Human azoreductases are known to play a crucial role in the metabolism of a number of quinone‐containing cancer chemotherapeutic drugs. The mechanism of hydride transfer to quinones, which is shared not only between eukaryotic and prokaryotic azoreductases but also the wider family of NAD(P)H quinone oxidoreductases, is outlined. The importance of common single nucleotide polymorphisms (SNPs) in human azoreductases is described not only in cancer prognosis but also with regard to their effects on the efficacy of quinone drug‐based cancer chemotherapeutic regimens. This highlights the need to screen patients for azoreductase SNPs ahead of treatment with these regimens.
Linked Articles
This article is part of a themed section on Drug Metabolism and Antibiotic Resistance in Micro‐organisms. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.14/issuetoc
Abbreviations
- 5‐ASA
5‐aminosalicylate
- AcpD
acyl carrier protein phosphodiesterase
- ecAzoR
Escherichia coli azoreductase
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- hNQO1/2
human NAD(P)H quinone oxidoreductase 1 and 2
- IBD
inflammatory bowel disease
- NfsB
nitrofurazone sensitive protein B
- NRH
dihydronicotinamide riboside
- paAzoR
Pseudomonas aeruginosa azoreductase
- ppAzoR
Pseudomonas putida azoreductase
- WrbA
tryptophan repressor binding protein A
Table of Links
| LIGANDS | |
|---|---|
| 5‐fluorouracil | Idarubicin |
| Chloroquine | Imatinib |
| Doxorubicin | Streptonigrin |
| Etoposide | Tanespimycin |
| Famitinib | Troglitazone |
| Gemcitabine |
This Table lists key ligands in this article, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016).
Introduction to azoreductases
Azoreductases are a group of diverse enzymes found in many bacterial and eukaryotic organisms [Figure 1 (Ryan et al., 2014)]. The azoreductases discussed in this review are flavin‐dependent [typically flavin mononucleotide (FMN)] enzymes that are able to reductively cleave compounds containing an azo bond. Azo compounds are defined as those that contain an R1‐N = N‐R2 group, where R1 and R2 are typically aromatic groups. Azoreductases are primarily cytosolic enzymes; however, they have been shown to be secreted during exposure of bacteria to azo dyes (Morrison and John, 2015). The physiological role of these enzymes in the bacteria remains unclear; however, they are constitutively expressed during growth in vitro (Chen et al., 2005; Wang et al., 2007), which suggests a role in homeostasis. The majority of azoreductase research is focused on their use in bioremediation, where they can be used for the treatment of waste water contaminated with azo dyes from the textile and cosmetics industries (Singh et al., 2015). This review is unique in focusing on their role in the activation of several classes of drug.
Figure 1.
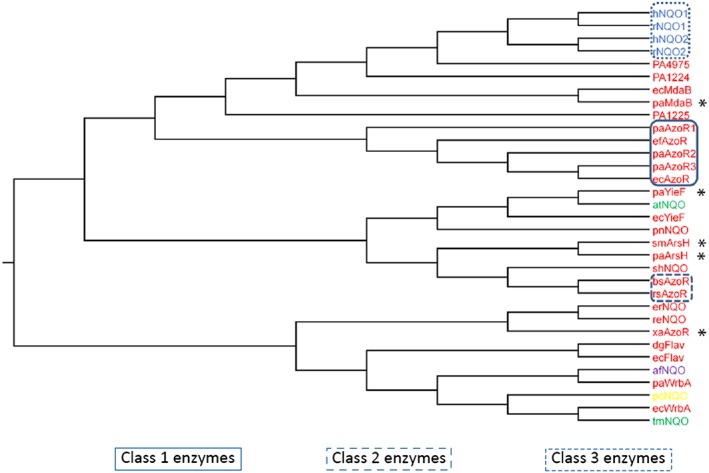
Phylogenetic tree showing relationships between azoreductases. Those enzymes in red text are from bacteria, those in blue are from mammals and those in green from plants. * Indicates a characterized azoreductase outside the three defined classes. paYieF, paArsH and paMdaB are azoreductases from P. aeruginosa. paWrbA, PA1224, PA1225, and PA4975 are NAD(P)H quinone oxidoreductases from P. aeruginosa. bsAzoR, efAzoR and rsAzoR are azoreductases from B. subtilis, E. faecalis and R. sphaeroides. rNQO1 and rNQO2 are rat azoreductases. xaAzoR is a flavin‐independent azoreductase from X. azovorans. ecMdaB, ecYieF and ecWrbA are NAD(P)H quinone oxidoreductases from E. coli. afNQO, pnNQO, tmNQO, pcNQO and atNQO are NAD(P)H quinone oxidoreductases from Archaeoglobus fulgidus, Paracoccus denitrificans, Triticum monococcum, Phanerochaete chrysosporium and Arabidopsis thaliana respectively. smArsH is an azoreductase from Sinorhizobium meliloti. dgFlav and ecFlav are flavodoxins from Desulfovibrio gigas and E. coli, respectively. shNQO, reNQO and erNQO are uncharacterized proteins from Staphylococcus haemolyticus, Ralstonia eutropha and Erwinia chrysanthemi. Adapted from Ryan et al. (2014).
To date, the majority of studies on azoreductases have been performed on enzymes from aerobic bacteria (Nakanishi et al., 2001; Crescente et al., 2016); in contrast, only a single azoreductase from a strict anaerobe has been characterized (Morrison et al., 2012). Several species of gut bacteria have been identified to have azoreductase activities (Wang et al., 2004); however, studies on individual azoreductases from enteric bacteria are very limited. Due to the anaerobic environment of the gut it is important to improve our understanding of the azoreductases, which predominate in these bacteria, in order to help design novel drugs.
Reduction of substrates by azoreductases requires the use of either NADH or NADPH as an electron donor in a bi‐bi ping pong mechanism (Nakanishi et al., 2001; Binter et al., 2009; Wang et al., 2010). Reduction is an obligate two electron process where a hydride is transferred from NAD(P)H to FMN and then on to the second substrate. The reduction of an azo substrate requires two NAD(P)H per azo substrate (Figure 2A). As well as azo compounds, the enzymes have been shown to reduce a range of other substrates including quinones (Ryan et al., 2010b; Gonçalves et al., 2013; Ryan et al., 2014) and nitroaromatics (Liu et al., 2007a; Ryan et al., 2011; Prosser et al., 2013). The physiological substrate of azoreductases remains unclear; however, the high specific activity of azoreductases when reducing quinones (Ryan et al., 2014) and increased survival of Escherichia coli overexpressing AzoR during treatment with menadione (Liu et al., 2008) suggest that detoxification of quinones is an important physiological function.
Figure 2.
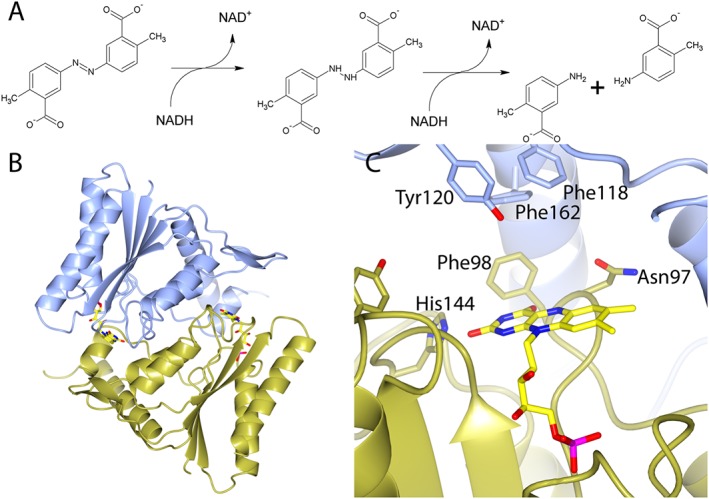
The structure of a typical bacterial azoreductase and its mechanism of azoreduction. (A) Mechanism for the reduction of an azo drug (olsalazine) by flavin‐dependent azoreductases. (B) Characteristic homodimeric flavodoxin fold of ecAzoR, a class 1 bacterial azoreductase. (C) Detailed view of the active site of ecAzoR. In (B) and (C), monomers are coloured blue and gold. The FMN with sticks and yellow carbon atoms. In (C), residues surrounding the active site are shown as sticks and labelled. (B) and (C) are based upon the structure of ecAzoR PDB 1V4B (Ito et al., 2006) and were generated in CCP4MG (McNicholas et al., 2011).
The structures of several bacterial azoreductases have been solved, and all share a characteristic homodimeric short‐flavodoxin fold (Figure 2B). The active sites of these enzymes are situated at the dimer interface and are formed by residues from both monomers (Figure 2C). One molecule of flavin is bound within each active site and is required for activity. The structure of E. coli azoreductase (ecAzoR) depicted in Figure 2 was the first to be solved (Ito et al., 2006) and is typical of the structures of those azoreductases that were subsequently solved [Figure 3 for examples (Wang et al., 2007; Liu et al., 2007b; Binter et al., 2009; Gonçalves et al., 2013; Yu et al., 2014)]. Azoreductases can be encoded by a diverse range of sequences [Figure 1 (Ryan et al., 2014)]; as a result, there are very few conserved residues in the protein and the ones that are present are thought to provide structural stability (Ryan et al., 2010a). The substrate binding pocket is typically lined by hydrophobic and aromatic residues (Figure 2C). Several of these aromatic residues are part of the β‐hairpin that forms the ‘lid’ of the active site, and these have been shown, via mutagenesis, to play an important role in determining the substrate specificity of the enzyme (Wang et al., 2010). The FMN is anchored by a series of sequence‐independent hydrogen bonds to a structural motif referred to as the FMN binding cradle (Ryan et al., 2010a), which is conserved in both bacterial and eukaryotic azoreductases.
Figure 3.
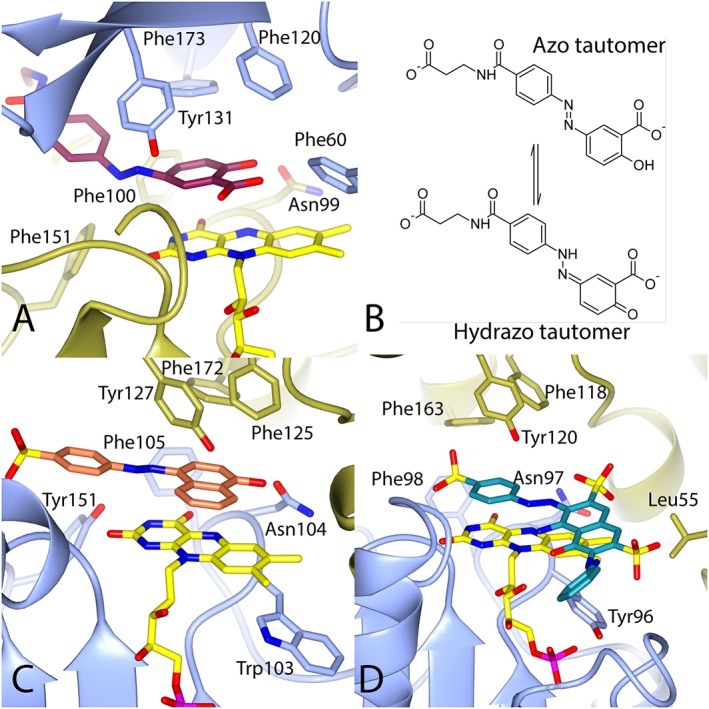
The binding of azo substrates to azoreductases. (A) The structure of balsalazide bound to paAzoR1. (B) The tautomeric forms of balsalazide that occur in solution. (C) The structure of Orange I bound to AzrC from Bacillus sp 29. (D) The structure of reactive black 5 bound to ppAzoR. The colouring is as in Figure 2. The structures are based upon PDBs (A) 3LT5 (Ryan et al., 2010a), (C) 3 W79 (Yu et al., 2014) and (D) 4C14 (Gonçalves et al., 2013).
The structures of several azoreductases with bound substrates have been solved, and these include azo compounds [Figure 3 (Wang et al., 2007; Wang et al., 2010; Ryan et al., 2010a; Gonçalves et al., 2013; Yu et al., 2014)], nitroaromatics (Ryan et al., 2011) and quinones (Gonçalves et al., 2013; Ryan et al., 2014). These substrates all lie sandwiched between the isoalloxazine rings of the flavin, with which they form π‐π stacking interactions, and the aromatic residues of the β‐hairpin (Figure 3). In general, the substrates of azoreductases do not make many specific hydrophilic interactions, which would explain the ability of the active site to accommodate a range of hydrophobic substrates. So far, no structure has been solved with a nicotinamide cofactor bound to an azoreductase. The structure of azoreductase bound to cibacron blue, a competitive NAD(P)H inhibitor, has been solved (Yu et al., 2014), but this does not provide details of the binding due to the structural differences between it and NAD(P)H.
Phylogeny of azoreductases
Defining which enzymes have azoreductase activity is a complex matter. In 2014, a phylogenetic tree was published that brought together all of the flavin‐dependent azoreductases characterized and a number of related NAD(P)H quinone oxidoreductases [adapted in Figure 1 (Ryan et al., 2014)]. Using this tree, three distinct classes of azoreductases can be defined. Class 1 is the most widespread and studied class of bacterial azoreductases. These enzymes have, since the sequencing of the first bacterial genomes, been misidentified as acyl carrier protein phosphodiesterases (AcpD). Although several of these enzymes have been tested, none have been shown to have AcpD activity (Nakanishi et al., 2001; Wang et al., 2007). In Figure 1, class 1 azoreductases include Pseudomonas aeruginosa azoreductases 1–3 [paAzoR1–3 (Ryan et al., 2010a; Wang et al., 2007)], Enterococcus faecalis azoreductase (Chen et al., 2004) and ecAzoR (Nakanishi et al., 2001). All members of this class share at least 30–40% sequence identity and are readily identifiable in most bacterial genomes. Class 2 bacterial azoreductases are much less common in bacterial genomes and share no significant sequence identity with class 1 enzymes. Class 2 enzymes do, however, share the same enzymatic activities and overall fold as the enzymes from class 1 (Binter et al., 2009). In Figure 1, class 2 are represented by azoreductases from Bacillus subtilis (Binter et al., 2009) and Rhodobacter sphaeroides (Bin et al., 2004). Class 3 enzymes are the mammalian azoreductases and include the human NAD(P)H quinone oxidoreductase 1 and 2 (hNQO1 and hNQO2) that will be discussed in subsequent sections.
The reason for inclusion of the NAD(P)H quinone oxidoreductases in Figure 1 was that a novel mechanism for azoreduction has been proposed [see below (Ryan et al., 2010a)], which suggested that the quinone and azo reduction shared the same mechanism in these enzymes (Ryan et al., 2010b). To support this, enzymes were identified in P. aeruginosa from four families [ArsH, modulator of drug activity B, tryptophan repressor binding protein A (WrbA) and YieF], which are NAD(P)H quinone oxidoreductases that have been characterized in other bacteria (Ryan et al., 2014). The enzymes identified in P. aeruginosa were cloned, expressed and characterized, and all, except WrbA (the most distantly related homologue), showed azoreductase activity (Crescente et al., 2016). This indicates that the azoreductase family is larger than originally thought, and more work is needed to characterize further members.
Azo drugs
The use of azo compounds as drugs stretches back to the first commercially available antibiotic, prontosil (Supporting Information Fig. 1), a sulphonamide prodrug, which was identified in the 1930s (Colebrook et al., 1936), and was later used to identify the first azoreductase (Fouts et al., 1957). Prontosil was among the first drugs to be used to illustrate the importance of the mammalian gut microflora in drug metabolism (Gingell et al., 1971). In modern clinics, azo drugs, such as olsalazine, are used to treat inflammatory bowel disease (IBD) and ulcerative colitis (Lautenschlager et al., 2014). Azo drugs for the treatment of IBD are pro‐drugs that, upon reduction, release the non‐steroidal anti‐inflammatory 5‐aminosalicylate [5‐ASA (Makins and Cowan, 2001)]. In these prodrugs, 5‐ASA (Supporting Information Fig. 1) is covalently linked via an azo bond to an inert carrier to prevent rapid adsorption from the digestive tract (Haagen Nielsen and Bondesen, 1983). These drugs rely upon cleavage of the azo bond by azoreductases secreted by the gut microflora in order to release 5‐ASA (Peppercorn and Goldman, 1972).
The use of azo‐linked compounds for specific drug delivery to the gut remains an area of great interest. As well as anti‐inflammatory compounds, azo chemistry has been used to target a range of other drugs and they include antibiotics (Kennedy et al., 2011; Deka et al., 2015) and anticancer drugs (Sharma et al., 2013; Plyduang et al., 2014). Development continues on new azo‐bonded carriers for drugs (Ruiz et al., 2011b; Kim et al., 2016) as well as linking pairs of drugs together (Ruiz et al., 2011a). Studies are making use of the azo linkage in new systems such as using azo polymers as a coating material, which is degraded to release the drug (Saphier and Karton, 2010). Other studies are investigating the use azo‐linked nanoparticles to release drugs into the colon (Naeem et al., 2014), or azo containing hydrogels to release olsalazine upon reduction (Li et al., 2010).
The mechanism of azoreduction
The basic mechanism for azoreduction by all FMN‐dependent azoreductases is described in Figure 2A. In this mechanism, N5 of FMN accepts a hydride during oxidation of NAD(P)H and donates it upon reduction of the substrate (Figure 4). The structures of three azoreductases bound to azo substrates have been solved (Figure 3), and in each, the azo bond is not in an optimal position for hydride transfer (distance from N5 of FMN varies 4.8–6.3 Å, transfer distance should be ~3.5 Å). There is unlikely to be a significant shift in the position of the substrate as a result of FMN reduction as a comparison of ecAzoR in the oxidized and reduced states showed little conformational change to the residues surrounding the active site (Ito et al., 2008). This makes direct transfer of the hydride to the substrate azo bond unlikely and leaves the question of what is the site of electron transfer.
Figure 4.
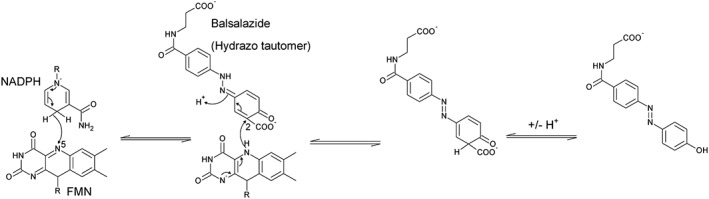
Proposed mechanism for the reduction of balsalazide by paAzoR1. For simplicity, only the isoalloxazine ring of FMN and the nicotinamide group of NADPH are shown.
The answer was provided when the structure of balsalazide bound to paAzoR1 was solved [Figure 3A (Ryan et al., 2010a)]. The clue was that, although balsalazide should be planar due to its conjugated system, the electron density clearly indicated a 50° bend in the molecule at the azo bond. In order to account for this anomaly, an alternative explanation was put forward in which the azo compound was in fact in the hydrazo tautomer (Figure 3B). Tautomerisation introduces an sp3 hybridized nitrogen into the azo bond, which would account for the bend in the electron density. Tautomerisation also means the salicylate ring forms a quinoneimine structure that is in a more optimal position to be reduced and would account for the ability of these enzymes to reduce both quinones and azo compounds (Ryan et al., 2014). As a result, a novel mechanism of azoreduction was proposed based upon the reduction of the quinoneimine‐containing tautomer (Figure 4). With this mechanism, the hydride is transferred to the carbon at position 2 of the quinonimine ring of balsalazide as the covalently bonded carboxylate would make the carbon δ+.
Subsequently, two structures of azoreductases complexed with their azo substrates (Figure 3C and D) have been published (Gonçalves et al., 2013; Yu et al., 2014), which show the azo compounds in a more planar conformation. Both azo drugs should be able to tautomerise to form a hydrazo tautomer. In both structures, a carbon atom from the quinoneimine ring formed via tautomerisation of the azo substrate is in a similar position to carbon 2 of balsalazide and at an optimal distance for hydride transfer (3.4 Å and 3.7 Å), consistent with the mechanism in Figure 4.
Nitrofuran and other nitroaromatic drug activation by azoreductases
The number of drugs that incorporate a nitroaromatic group is relatively small due to their toxicity, which stems from their ability to generate ROS via redox cycling via single electron reduction. Four electron reduction of the nitro group generates a reactive hydroxylamine, which can covalently modify either proteins or DNA (Kovacic and Somanathan, 2014). Among the most heavily studied nitroaromtic azoreductase substrates are the nitrofuran antibiotics. The most commonly studied nitrofurans are nitrofurazone, which is a topical antibiotic for treating burns (Ungureanu, 2014), and nitrofurantoin (Supporting Information Fig. 1), which is used for treating urinary tract infections (Garau, 2008). Although typically used as antibiotics, nitrofurans are also able to kill trypanosomatids (Patterson and Wyllie, 2014).
All nitrofurans must be activated via the reduction of their nitro group to a reactive hydroxylamine (Whiteway et al., 1998). In bacteria, this can be achieved either via dedicated nitroreductases such as nitrofurazone sensitivity protein B (NfsB) in E. coli (Race et al., 2005) or azoreductases (Ryan et al., 2011). The structure of paAzoR1 was solved in complex with nitrofurazone [Figure 5A (Ryan et al., 2011)]. The nitro group of nitrofurazone is positioned over the N5 of FMN to allow optimal hydride transfer (3.6 Å, Figure 5B and C). The nitro group is within the hydrogen bonding range (3 Å) of the side chain of Asn99, which would stabilize the reduced form, and also a water molecule, which could donate a proton required for reduction.
Figure 5.
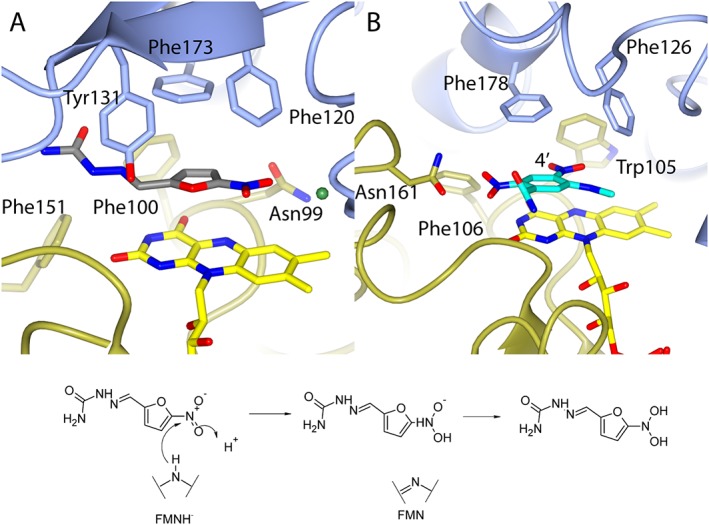
Reduction of nitro compounds by azoreductases. (A) The structure of nitrofurazone bound to paAzoR1; (B) the structure of CB1954 bound to hNQO2. (C) Proposed mechanism of nitrofurazone reduction in paAzoR1. In (A) and (B), protein colour coding is as in Figure 2. For ease of interpretation, only one of the two possible binding orientations for nitrofurazone is shown in (A) with grey carbon atoms, while water is shown as a green ball. In (B), CB1954 is shown with turquoise carbon atoms and the 4′ nitrate is labelled. These images are based upon PDB files (A) 3R6W (Ryan et al., 2011) and (B) 1XI2 (Fu et al., 2005).
Mammalian azoreductases
Like bacteria, eukarya including mammals have azoreductases. In humans the azoreductases are referred to as hNQO1 and hNQO2 (Wu et al., 1997). Although they share limited sequence identity (<10%) to either class 1 or 2 azoreductases, they are able to reduce many of the same classes of substrates via the same bi‐bi ping pong mechanism (Wu et al., 1997). Similarly to bacterial azoreductases, hNQO1 and hNQO2 have flavodoxin‐like folds and form homodimers in solution [Figure 6A (Faig et al., 2000)]. Both hNQO1 and hNQO2 use flavin adenine dinucleotide (FAD) as a cofactor rather than FMN (Figure 6B). hNQO1 is an important phase II drug metabolizing enzyme that is expressed in many tissues throughout the body (Siegel and Ross, 2000). hNQO1 is overexpressed in many cancers including lung (Li et al., 2015), breast (Yang et al., 2014) and pancreatic tumours (Lewis et al., 2005). hNQO1 is known to control the degradation of a range of proteins by the proteasome including the tumour suppressors p53 (Asher et al., 2001) and p73 (Asher et al., 2005), which is likely to contribute to its role in tumourigenesis. There is also an association between the common [allelic frequency 0.22 in Caucasians and 0.45 in Asian populations (Kelsey et al., 1997)] C609T NQO1 single nucleotide polymorphism (SNP) (P187S mutant) and cancer (Fagerholm et al., 2008; Lajin and Alachkar, 2013). The P187S mutation results in a reduced affinity of hNQO1 for FAD and thus reduced enzymic activity and an increased susceptibility to proteolysis (Lienhart et al., 2014).
Figure 6.
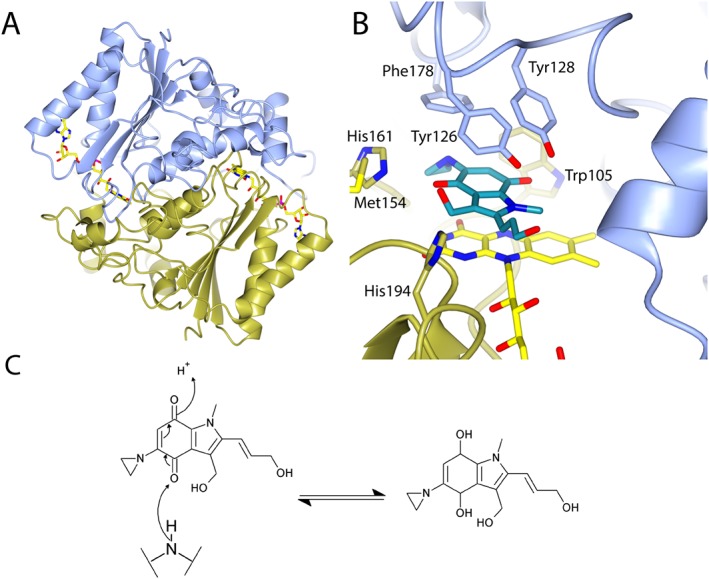
Binding of the chemotherapeutic EO9 to hNQO1. (A) Overall structure of human hNQO1. (B) Binding of EO9 to hNQO1. (C) Mechanism of hydride transfer during the reduction of EO9. Protein colour coding is as in Figure 2 and in (B) EO9 has turquoise carbon atoms. (A) and (B) are based upon PDB file 1GG5 (Faig et al., 2001).
The role of hNQO2 in cells is less clear than for hNQO1. hNQO2 expression is less widespread than hNQO1 and is mainly found in muscle and kidney (Jaiswal, 1994). hNQO2 is unusual in that it utilizes neither NADH nor NADPH, but instead uses dihydronicotinamide riboside [NRH (Wu et al., 1997)]. Like hNQO1, hNQO2 is involved with the regulation of proteasomal degradation of some proteins such as cyclin D1 (Hsieh et al., 2012). hNQO2 has a common SNP (C659T or L47F) that does not affect enzymic activity but makes hNQO2 more susceptible to proteolysis (Megarity et al., 2014). hNQO2 SNPs have been linked to prostate cancer (Mandal et al., 2012), colorectal cancer (Chen et al., 2016) and prognosis in breast cancer (Hubackova et al., 2012). hNQO2 has been associated with the activation of chemotherapeutics (Celli et al., 2006; Jamieson et al., 2011), and hNQO2 SNPs are associated with the cardiotoxicity of the anthraquinone idarubicin (Megias et al., 2015). Both imatinib [K i ~40 nM (Bantscheff et al., 2007)] and chloroquine [K i = 0.6 μM (Kwiek et al., 2004)] are known inhibitors of NQO2, and hNQO2 is likely to be a secondary target for both drugs in the cell.
Mammalian azoreductases and chemotherapeutic drugs
Azoreductases in bacteria have been shown to have 10‐ to 100‐fold greater activity against quinones than against azo substrates (Ryan et al., 2014; Crescente et al., 2016). Quinones are highly cytotoxic due to their ability to undergo redox cycling via one or two electron reduction but can also cause alkylation of cellular protein and DNA (Bolton et al., 2000). As a result, quinones are not used as antibiotics; however, several quinones are either in use as cancer chemotherapeutics, for example anthracyclines (Hortobagyi, 1997), or in clinical trials, for example β‐lapachone [currently in phase I/II trials for a range of cancers (Li et al., 2014)]. Quinones such as atovaquone are also used for the treatment of malaria (Looareesuwan et al., 1996). Metabolites of several drugs, including etoposide (Smith et al., 2014), famitinib (Xie et al., 2013) and troglitazone (Yamamoto et al., 2002) have been identified as having either quinone or related quinoneimine functional groups that have been linked to their hepatotoxicity. As a result, it is important to understand the role of human azoreductases in the cytotoxicity of quinone drugs.
The cytotoxicity of a range of quinone‐based cancer chemotherapeutics are altered by hNQO1 activity (Table 1). Many of these drugs are cytotoxic as they undergo redox cycling via reduction by NQO1 to their quinol form before oxidation back to the quinone and release of ROS (Docampo et al., 1979). In contrast, the reduction of tanespimycin to its quinol makes it a more potent inhibitor of its target Hsp90 (Guo et al., 2005). Although hNQO1 is primarily linked with the toxicity of quinone‐based chemotherapeutics, it has been linked to some nucleoside analogues (Table 1). The reason for the change in resistance to nucleoside analogues is also thought be linked to their ability to generate ROS (Aresvik et al., 2010) and the antioxidant role played by hNQO1 (Bauer et al., 2012). As both overexpression of NQO1 and SNPs which inactivate NQO1 are commonly found in tumours we suggest that, treatment of cancer with chemotherapeutics activated by hNQO1 is best undertaken after genotyping the patient. Alternatively fluorogenic substrates are under development that could circumvent the need for genotyping, significantly speeding up the process (Silvers et al., 2013; Best et al., 2016).
Table 1.
hNQO1 and its association with cancer chemotherapy toxicity
| Drug type | Druga | Role of NQO1 | Tumour type | Citation |
|---|---|---|---|---|
| Quinone | β‐lapachone | Activation | Breast | (Glorieux et al., 2016) |
| Lung | (Bey et al., 2007) | |||
| Skin | (Li et al., 2014) | |||
| Liver | (Li et al., 2014; Park et al., 2014) | |||
| Pancreatic | (Ough et al., 2005) | |||
| Doxorubicin | Protection | Bile duct | (Zeekpudsa et al., 2014) | |
| Bladder | (Matsui et al., 2010) | |||
| Epirubicin | Activation | Breast | (Fagerholm et al., 2008) | |
| Streptonigrin | Activation | Colon | (Beall et al., 1996) | |
| Breast | (Dehn et al., 2003) | |||
| Pancreatic | (Lewis et al., 2005) | |||
| Tanespimycin | Activation | Brain | (Gaspar et al., 2009) | |
| Colon & Ovarian | (Kelland et al., 1999) | |||
| Oesophageal | (Hadley and Hendricks, 2014) | |||
| Pancreatic | (Siegel et al., 2011) | |||
| Nucleoside analogues | Gemcitabine | Protection | Bile duct | (Buranrat et al., 2010; Zeekpudsa et al., 2014) |
| 5‐fluorouracil | Protection | Bile duct | (Zeekpudsa et al., 2014) | |
| Gastric | (Peng et al., 2016) | |||
| Liver | (Sutton et al., 2015) |
The structures of all drugs are shown in Figure 1.
The mechanism of quinone reduction by hNQO1 and other azoreductases
In order to better understand the mechanism of quinone reduction by flavoproteins, the structures of a number of quinones bound to bacterial (Gonçalves et al., 2013; Ryan et al., 2014) and mammalian azoreductases (Faig et al., 2000) have been solved, as well as, more recently, the structure of E. coli WrbA (Degtjarik et al., 2016). There are also structures of several quinones under development as chemotherapeutics complexed with hNQO1 (Faig et al., 2001; Pidugu et al., 2016). EO9 or apaziquone is a good example of an NQO1 substrate (Walton et al., 1991); it has entered phase 3 clinical trials for the treatment of bladder cancer (Phillips et al., 2013). The structure of EO9 bound to hNQO1 is typical of other quinones bound to bacterial and mammalian azoreductases. In the structure of EO9 bound to hNQO1, the quinone oxygen is 3.6 Å from the N5 of FMN in an ideal location for electron transfer (Figure 6A). A quinone oxygen is similarly positioned in structures of paAzoR1 (3.7 Å (Ryan et al., 2014)) and ppAzoR (3.6 Å (Gonçalves et al., 2013)) bound to anthraquinone‐2‐sulphonate. Transfer of the hydride to the carbonyl oxygen of the quinone as the mechanism of quinone reduction (Figure 6C) is supported by recent quantum mechanical calculations, which were performed on a high resolution structure of WrbA bound to benzoquinone (Degtjarik et al., 2016).
Mammalian azoreductases and nitroaromatic drugs
As discussed above, nitroaromatics are not commonly used in treatment due to their cytotoxic side effects; however, like quinones there are ongoing efforts to use them for the treatment of intractable diseases. A good example of this is the prodrug CB1954 (Supporting Information Fig. 1). CB1954 has undergone clinical trials for the treatment of prostate cancer with virally encoded E. coli NfsB (Patel et al., 2009). The use of viraly encoded E. coli NfsB in combination with CB1954 is an example of gene‐directed enzyme prodrug therapy, as CB1954 toxicity is reliant upon its activation by E. coli NfsB expressed by tumour cells (Williams et al., 2015). CB1954 has also undergone clinical trials in a range of tumours with NRH (Middleton et al., 2010). As with nitrofurazone, one or other of the nitro groups must be reduced to a hydroxylamine in order to activate the compound. hNQO2 selectively reduces the 4′ nitro of CB1954 [Figure 5B (Fu et al., 2005)]. Bacterial azoreductases also reduce the CB1954 (Liu et al., 2007a; Prosser et al., 2013), but at both nitro groups equally. The reason for the nitro group selectivity in hNQO2 is believed to be Asn161, which orients the molecule within the active site [Figure 5B (AbuKhader et al., 2005)]. The 4′ nitro group of CB1954 is positioned in close contact with the N5 of FMN (3.5 Å) suggesting the mechanism of reduction is as described for nitrofurazone (Figure 5C).
There is a lot of research targeting difficult to treat infections using nitroaromatic drugs. In neglected tropical diseases such as malaria (Cakmak et al., 2011) and sleeping sickness (Torreele et al., 2010), nitroaromatic compounds are selectively activated by parasite‐specific nitroreductases. BTZ043 is the first of a novel class of nitroaromatic drugs (Lechartier et al., 2012) that are showing promise for the treatment of extensively drug resistant Mycobacterium tuberculosis (Pasca et al., 2010) and is now entering phase I clinical trials. As a result, it is important to improve our understanding of nitroreduction by the human enzymes to avoid side effects resulting from host‐specific interactions with these novel classes of drugs.
Conclusions
Azoreductases are a diverse and adaptable protein family that play an important role in the metabolism of drugs by the gut microflora. In the area of bacterial azoreductases, one of the key challenges remains the characterization of azoreductases encoded by bacteria, found in the natural gut microflora, in order to improve the design of novel azo prodrugs for the treatment of not only IBD but also other diseases of the colon.
In recent years, the importance of the human azoreductases in the area of cancer chemotherapy is coming to the fore. As increasing numbers of chemotherapy drugs rely on hNQO1 and hNQO2 for toxicity, the need to genotype patients for the presence of inactivating mutations must now be considered not only for treatment with quinone‐based drugs but also nucleoside analogues.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Figure S1 Structures of compounds mentioned in the review. Numbered atoms mentioned in the text are labelled.
Fig. 1 Supplementary info item
Ryan, A. (2017) Azoreductases in drug metabolism. British Journal of Pharmacology, 174: 2161–2173. doi: 10.1111/bph.13571.
References
- AbuKhader M, Heap J, De Matteis C, Kellam B, Doughty SW, Minton N et al. (2005). Binding of the anticancer prodrug CB1954 to the activating enzyme NQO2 revealed by the crystal structure of their complex. J Med Chem 48: 7714–7719. [DOI] [PubMed] [Google Scholar]
- Aresvik DM, Pettersen RD, Abrahamsen TG, Wright MS (2010). 5‐fluorouracil‐induced death of Jurkat T‐cells ‐ A role for caspases and MCL‐1. Anticancer Res 30: 3879–3887. [PubMed] [Google Scholar]
- Asher G, Lotem J, Cohen B, Sachs L, Shaul Y (2001). Regulation of p53 stability and p53‐dependent apoptosis by NADH quinone oxidoreductase 1. Proc Natl Acad Sci U S A 98: 1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Tsvetkov P, Kahana C, Shaul Y (2005). A mechanism of ubiquitin‐independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev 19: 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S et al. (2007). Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 25: 1035–1044. [DOI] [PubMed] [Google Scholar]
- Bauer KM, Lambert PA, Hummon AB (2012). Comparative label‐free LC–MS/MS analysis of colorectal adenocarcinoma and metastatic cells treated with 5‐fluorouracil. Proteomics 12: 1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall HD, Liu Y, Siegel D, Bolton EM, Gibson NW, Ross D (1996). Role of NAD(P)H:quinone oxidoreductase (DT‐diaphorase) in cytotoxicity and induction of DNA damage by streptonigrin. Biochem Pharmacol 51: 645–652. [DOI] [PubMed] [Google Scholar]
- Best QA, Johnson AE, Prasai B, Rouillere A, McCarley RL (2016). Environmentally robust rhodamine reporters for probe‐based cellular detection of the cancer‐linked oxidoreductase hNQO1. ACS Chem Biol 11: 231–240. [DOI] [PubMed] [Google Scholar]
- Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L et al. (2007). An NQO1‐ and PARP‐1‐mediated cell death pathway induced in non‐small‐cell lung cancer cells by β‐lapachone. Proc Natl Acad Sci U S A 104: 11832–11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin Y, Jiti Z, Jing W, Cuihong D, Hongman H, Zhiyong S et al. (2004). Expression and characteristics of the gene encoding azoreductase from Rhodobacter sphaeroides AS1.1737. FEMS Microbiol Lett 236: 129–136. [DOI] [PubMed] [Google Scholar]
- Binter A, Staunig N, Jelesarov I, Lohner K, Palfey BA, Deller S et al. (2009). A single intersubunit salt bridge affects oligomerization and catalytic activity in a bacterial quinone reductase. FEBS J 276: 5263–5274. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ (2000). Role of quinones in toxicology. Chem Res Toxicol 13: 135–160. [DOI] [PubMed] [Google Scholar]
- Buranrat B, Prawan A, Kukongviriyapan U, Kongpetch S, Kukongviriyapan V (2010). Dicoumarol enhances gemcitabine‐induced cytotoxicity in high NQO1‐expressing cholangiocarcinoma cells. World J Gastroenterol 16: 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakmak R, Durdagi S, Ekinci D, Senturk M, Topal G (2011). Design, synthesis and biological evaluation of novel nitroaromatic compounds as potent glutathione reductase inhibitors. Bioorg Med Chem Lett 21: 5398–5402. [DOI] [PubMed] [Google Scholar]
- Celli CM, Tran N, Knox R, Jaiswal AK (2006). NRH:quinone oxidoreductase 2 (NQO2) catalyzes metabolic activation of quinones and anti‐tumor drugs. Biochem Pharmacol 72: 366–376. [DOI] [PubMed] [Google Scholar]
- Chen D, Sun Q, Cheng X, Zhang L, Song W, Zhou D et al. (2016). Genome‐wide analysis of long noncoding RNA (lncRNA) expression in colorectal cancer tissues from patients with liver metastasis. Canc Med . doi:10.1002/cam1004.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Hopper SL, Cerniglia CE (2005). Biochemical and molecular characterization of an azoreductase from Staphylococcus aureus, a tetrameric NADPH‐dependent flavoprotein. Microbiology 151: 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wang RF, Cerniglia CE (2004). Molecular cloning, overexpression, purification, and characterization of an aerobic FMN‐dependent azoreductase from Enterococcus faecalis . Protein Expr Purif 34: 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colebrook L, Buttle GAH, O'Meara RAQ (1936). The mode of action of p‐aminobenzenesulphonamide and prontosil in haemolytuc Streptococcal infections. Lancet 2: 1323–1326. [Google Scholar]
- Crescente V, Holland Sinead M, Kashyap S, Polycarpou E, Sim E, Ryan A (2016). Identification of novel members of the bacterial azoreductase family in Pseudomonas aeruginosa . Biochem J 473: 549–558. [DOI] [PubMed] [Google Scholar]
- Degtjarik O, Brynda J, Ettrichova O, Kuty M, Sinha D, Kuta Smatanova I et al. (2016). Quantum calculations indicate effective electron transfer between FMN and benzoquinone in a new crystal structure of E. coli WrbA. J Phys Chem B . doi:10.1021/acs.jpcb.1025b11958. [DOI] [PubMed] [Google Scholar]
- Dehn DL, Siegel D, Swann E, Moody CJ, Ross D (2003). Biochemical, cytotoxic, and genotoxic effects of ES936, a mechanism‐based inhibitor of NAD(P)H:quinone oxidoreductase 1, in cellular systems. Mol Pharmacol 64: 714–720. [DOI] [PubMed] [Google Scholar]
- Deka SR, Yadav S, Mahato M, Sharma AK (2015). Azobenzene‐aminoglycoside: Self‐assembled smart amphiphilic nanostructures for drug delivery. Colloids Surf B Biointerfaces 135: 150–157. [DOI] [PubMed] [Google Scholar]
- Docampo R, Cruz FS, Boveris A, Muniz RP, Esquivel DM (1979). β‐Lapachone enhancement of lipid peroxidation and superoxide anion and hydrogen peroxide formation by sarcoma 180 ascites tumor cells. Biochem Pharmacol 28: 723–728. [DOI] [PubMed] [Google Scholar]
- Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjakoski K et al. (2008). NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet 40: 844–853. [DOI] [PubMed] [Google Scholar]
- Faig M, Bianchet MA, Talalay P, Chen S, Winski S, Ross D et al. (2000). Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: species comparison and structural changes with substrate binding and release. Proc Natl Acad Sci U S A 97: 3177–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faig M, Bianchet MA, Winski S, Hargreaves R, Moody CJ, Hudnott AR et al. (2001). Structure‐based development of anticancer drugs: complexes of NAD(P)H:quinone oxidoreductase 1 with chemotherapeutic quinones. Structure 9: 659–667. [DOI] [PubMed] [Google Scholar]
- Fouts JR, Kamm JJ, Brodie BB (1957). Enzymatic reduction of prontosil and other azo dyes. J Pharmacol Exp Ther 120: 291–300. [PubMed] [Google Scholar]
- Fu Y, Buryanovskyy L, Zhang Z (2005). Crystal structure of quinone reductase 2 in complex with cancer prodrug CB1954. Biochem Biophys Res Commun 336: 332–338. [DOI] [PubMed] [Google Scholar]
- Garau J (2008). Other antimicrobials of interest in the era of extended‐spectrum β‐lactamases: fosfomycin, nitrofurantoin and tigecycline. Clin Microbiol Infect 14: 198–202. [DOI] [PubMed] [Google Scholar]
- Gaspar N, Sharp SY, Pacey S, Jones C, Walton M, Vassal G et al. (2009). Acquired resistance to 17‐allylamino‐17‐demethoxygeldanamycin (17‐AAG, tanespimycin) in glioblastoma cells. Cancer Res 69: 1966–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell R, Bridges JW, Williams RT (1971). The role of the gut flora in the metabolism of prontosil and neoprontosil in the rat. Xenobiotica 1: 143–156. [DOI] [PubMed] [Google Scholar]
- Glorieux C, Sandoval JM, Dejeans N, Ameye G, Poirel HA, Verrax J et al. (2016). Overexpression of NAD(P)H:quinone oxidoreductase 1 (NQO1) and genomic gain of the NQO1 locus modulates breast cancer cell sensitivity to quinones. Life Sci 145: 57–65. [DOI] [PubMed] [Google Scholar]
- Gonçalves AMD, Mendes S, de Sanctis D, Martins LO, Bento I (2013). The crystal structure of Pseudomonas putida AzoR: the active site revisited. FEBS J 280: 6643–6657. [DOI] [PubMed] [Google Scholar]
- Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D (2005). Formation of 17‐allylamino‐demethoxygeldanamycin (17‐AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17‐AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res 65: 10006–10015. [DOI] [PubMed] [Google Scholar]
- Haagen Nielsen O, Bondesen S (1983). Kinetics of 5‐aminosalicylic acid after jejunal instillation in man. Br J Clin Pharmacol 16: 738–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley KE, Hendricks DT (2014). Use of NQO1 status as a selective biomarker for oesophageal squamous cell carcinomas with greater sensitivity to 17‐AAG. BMC Cancer 14. doi:10.1186/1471-2407-1114-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi GN (1997). Anthracyclines in the treatment of cancer. An overview. Drugs 54 (Suppl 4): 1–7. [DOI] [PubMed] [Google Scholar]
- Hsieh TC, Yang CJ, Lin CY, Lee YS, Wu JM (2012). Control of stability of cyclin D1 by quinone reductase 2 in CWR22Rv1 prostate cancer cells. Carcinogenesis 33: 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubackova M, Vaclavikova R, Ehrlichova M, Mrhalova M, Kodet R, Kubackova K et al. (2012). Association of superoxide dismutases and NAD(P)H quinone oxidoreductases with prognosis of patients with breast carcinomas. Int J Cancer 130: 338–348. [DOI] [PubMed] [Google Scholar]
- Ito K, Nakanishi M, Lee WC, Sasaki H, Zenno S, Saigo K et al. (2006). Three‐dimensional structure of AzoR from Escherichia coli. An oxidereductase conserved in microorganisms. J Biol Chem 281: 20567–20576. [DOI] [PubMed] [Google Scholar]
- Ito K, Nakanishi M, Lee WC, Zhi Y, Sasaki H, Zenno S et al. (2008). Expansion of substrate specificity and catalytic mechanism of azoreductase by X‐ray crystallography and site‐directed mutagenesis. J Biol Chem 283: 13889–13896. [DOI] [PubMed] [Google Scholar]
- Jaiswal AK (1994). Human NAD(P)H:quinone oxidoreductase 2. Gene structure, activity, and tissue‐specific expression. J Biol Chem 269: 14502–14508. [PubMed] [Google Scholar]
- Jamieson D, Cresti N, Bray J, Sludden J, Griffin MJ, Hawsawi NM et al. (2011). Two minor NQO1 and NQO2 alleles predict poor response of breast cancer patients to adjuvant doxorubicin and cyclophosphamide therapy. Pharmacogenet Genomics 21: 808–819. [DOI] [PubMed] [Google Scholar]
- Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P (1999). DT‐Diaphorase expression and tumor cell sensitivity to 17‐allylamino, 17‐demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst 91: 1940–1949. [DOI] [PubMed] [Google Scholar]
- Kelsey KT, Ross D, Traver RD, Christiani DC, Zuo ZF, Spitz MR et al. (1997). Ethnic variation in the prevalence of a common NAD(P)H quinone oxidoreductase polymorphism and its implications for anti‐cancer chemotherapy. Br J Cancer 76: 852–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DA, Vembu N, Fronczek FR, Devocelle M (2011). Synthesis of mutual azo prodrugs of anti‐inflammatory agents and peptides facilitated by α‐aminoisobutyric acid. J Org Chem 76: 9641–9647. [DOI] [PubMed] [Google Scholar]
- Kim W, Nam J, Lee S, Jeong S, Jung Y (2016). 5‐aminosalicylic acid azo‐linked to procainamide acts as an anticolitic mutual prodrug via additive inhibition of NFκB. Mol Pharm 13: 2126–2135. [DOI] [PubMed] [Google Scholar]
- Kovacic P, Somanathan R (2014). Nitroaromatic compounds: environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism. J Appl Toxicol 34: 810–824. [DOI] [PubMed] [Google Scholar]
- Kwiek JJ, Haystead TA, Rudolph J (2004). Kinetic mechanism of quinone oxidoreductase 2 and its inhibition by the antimalarial quinolines. Biochemistry 43: 4538–4547. [DOI] [PubMed] [Google Scholar]
- Lajin B, Alachkar A (2013). The NQO1 polymorphism C609T (Pro187Ser) and cancer susceptibility: a comprehensive meta‐analysis. Br J Cancer 109: 1325–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschlager C, Schmidt C, Fischer D, Stallmach A (2014). Drug delivery strategies in the therapy of inflammatory bowel disease. Adv Drug Deliv Rev 71: 58–76. [DOI] [PubMed] [Google Scholar]
- Lechartier B, Hartkoorn RC, Cole ST (2012). In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob Agents Chemother 56: 5790–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AM, Ough M, Hinkhouse MM, Tsao MS, Oberley LW, Cullen JJ (2005). Targeting NAD(P)H : quinone oxidoreductase (NQO1) in pancreatic cancer. Mol Carcinog 43: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Ke Y, Misra HP, Trush MA, Li YR, Zhu H et al. (2014). Mechanistic studies of cancer cell mitochondria‐ and NQO1‐mediated redox activation of β‐lapachone, a potentially novel anticancer agent. Toxicol Appl Pharmacol 281: 285–293. [DOI] [PubMed] [Google Scholar]
- Li X, Li J, Gao Y, Kuang Y, Shi J, Xu B (2010). Molecular nanofibers of olsalazine form supramolecular hydrogels for reductive release of an anti‐inflammatory agent. J Am Chem Soc 132: 17707–17709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Jin T, Men J, Lin Z, Qi P et al. (2015). NQO1 protein expression predicts poor prognosis of non‐small cell lung cancers. BMC Cancer 15. doi:10.1186/s12885-015-1227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhart WD, Gudipati V, Uhl MK, Binter A, Pulido SA, Saf R et al. (2014). Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase. FEBS J 281: 4691–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhou J, Jin R, Zhou M, Wang J, Lu H et al. (2008). Enhancing survival of Escherichia coli by expression of azoreductase AZR possessing quinone reductase activity. Appl Microbiol Biotechnol 80: 409–416. [DOI] [PubMed] [Google Scholar]
- Liu G, Zhou J, Lv H, Xiang X, Wang J, Zhou M et al. (2007a). Azoreductase from Rhodobacter sphaeroides AS1.1737 is a flavodoxin that also functions as nitroreductase and flavin mononucleotide reductase. Appl Microbiol Biotechnol 76: 1271–1279. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Chen H, Shaw N, Hopper SL, Chen L, Chen S et al. (2007b). Crystal structure of an aerobic FMN‐dependent azoreductase (AzoA) from Enterococcus faecalis . Arch Biochem Biophys 463: 68–77. [DOI] [PubMed] [Google Scholar]
- Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ (1996). Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54: 62–66. [DOI] [PubMed] [Google Scholar]
- Makins RJ, Cowan RE (2001). 5‐amino‐salicylate in the management of inflammatory bowel disease. Colorectal Dis 3: 218–222. [DOI] [PubMed] [Google Scholar]
- Mandal RK, Nissar K, Mittal RD (2012). Genetic variants in metabolizing genes NQO1, NQO2, MTHFR and risk of prostate cancer: a study from North India. Mol Biol Rep 39: 11145–11152. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Watanabe J, Ding S, Nishizawa K, Kajita Y, Ichioka K et al. (2010). Dicoumarol enhances doxorubicin‐induced cytotoxicity in p53 wild‐type urothelial cancer cells through p38 activation. BJU Int 105: 558–564. [DOI] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, Noble MEM (2011). Presenting your structures: the CCP4mg molecular‐graphics software. Acta Crystallogr D67: 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megarity CF, Gill JRE, Caraher MC, Stratford IJ, Nolan KA, Timson DJ (2014). The two common polymorphic forms of human NRH‐quinone oxidoreductase 2 (NQO2) have different biochemical properties. FEBS Lett 588: 1666–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megias JE, Montesinos P, Herrero MJ, Moscardo F, Boso V, Martinez‐Cuadron D et al. (2015). Influence of single nucleotide polymorphisms in anthracycline metabolism pathway in standard induction of acute myeloid leukemia. Blood 126: 4845. [Google Scholar]
- Middleton MR, Knox R, Cattell E, Oppermann U, Midgley R, Ali R et al. (2010). Quinone oxidoreductase‐2‐mediated prodrug cancer therapy. Sci Transl Med 2. 40ra50 [DOI] [PubMed] [Google Scholar]
- Morrison JM, John GH (2015). Non‐classical azoreductase secretion in Clostridium perfringens in response to sulfonated azo dye exposure. Anaerobe 34: 34–43. [DOI] [PubMed] [Google Scholar]
- Morrison JM, Wright CM, John GH (2012). Identification, isolation and characterization of a novel azoreductase from Clostridium perfringens . Anaerobe 18: 229–234. [DOI] [PubMed] [Google Scholar]
- Naeem M, Kim W, Cao J, Jung Y, Yoo JW (2014). Enzyme/pH dual sensitive polymeric nanoparticles for targeted drug delivery to the inflamed colon. Colloids Surf B Biointerfaces 123: 271–278. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Yatome C, Ishida N, Kitade Y (2001). Putative ACP phosphodiesterase gene (acpD) encodes an azoreductase. J Biol Chem 276: 46394–46399. [DOI] [PubMed] [Google Scholar]
- Ough M, Lewis A, Bey EA, Gao JM, Ritchie JM, Bornmann W et al. (2005). Efficacy of β‐lapachone in pancreatic cancer treatment ‐ Exploiting the novel, therapeutic target NQO1. Cancer Biol Ther 4: 95–102. [DOI] [PubMed] [Google Scholar]
- Park EJ, Min KJ, Lee TJ, Yoo YH, Kim YS, Kwon TK (2014). β‐Lapachone induces programmed necrosis through the RIP1‐PARP‐AIF‐dependent pathway in human hepatocellular carcinoma SK‐Hep1 cells. Cell Death Dis 5: e1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca MR, Degiacomi G, Ribeiro AL, Zara F, De Mori P, Heym B et al. (2010). Clinical isolates of Mycobacterium tuberculosis in four european hospitals are uniformly susceptible to benzothiazinones. Antimicrob Agents Chemother 54: 1616–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, Young JG, Mautner V, Ashdown D, Bonney S, Pineda RG et al. (2009). A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954. Mol Ther 17: 1292–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S, Wyllie S (2014). Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol 30: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng LZ, Duan YX, Zhang YB, Zhao DD, Wen YH, Yao JB et al. (2016). Expression of Nrf2 and NQO1 in human gastric cancer and their clinical significance. Int J Clin Exp Pathol 9: 1635–1643. [Google Scholar]
- Peppercorn MA, Goldman P (1972). The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181: 555–562. [PubMed] [Google Scholar]
- Phillips RM, Hendriks HR, Peters GJ, Grp E‐PMM (2013). EO9 (Apaziquone): from the clinic to the laboratory and back again. Br J Pharmacol 168: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidugu LSM, Mbimba JCE, Ahmad M, Pozharski E, Sausville EA, Emadi A et al. (2016). A direct interaction between NQO1 and a chemotherapeutic dimeric naphthoquinone. BMC Struct Biol 16. doi:10.1186/s12900-12016-10052-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plyduang T, Lomlim L, Yuenyongsawad S, Wiwattanapatapee R (2014). Carboxymethylcellulose‐tetrahydrocurcumin conjugates for colon‐specific delivery of a novel anti‐cancer agent, 4‐amino tetrahydrocurcumin. Eur J Pharm Biopharm 88: 351–360. [DOI] [PubMed] [Google Scholar]
- Prosser GA, Copp JN, Mowday AM, Guise CP, Syddall SP, Williams EM et al. (2013). Creation and screening of a multi‐family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR‐104 A. Biochem Pharmacol 85: 1091–1103. [DOI] [PubMed] [Google Scholar]
- Race PR, Lovering AL, Green RM, Ossor A, White SA, Searle PF et al. (2005). Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J Biol Chem 280: 13256–13264. [DOI] [PubMed] [Google Scholar]
- Ruiz JF, Kedziora K, Keogh B, Maguire J, Reilly M, Windle H et al. (2011a). A double prodrug system for colon targeting of benzenesulfonamide COX‐2 inhibitors. Bioorg Med Chem Lett 21: 6636–6640. [DOI] [PubMed] [Google Scholar]
- Ruiz JFM, Kedziora K, Windle H, Kelleher DP, Gilmer JF (2011b). Investigation into drug release from colon‐specific azoreductase‐activated steroid prodrugs using in‐vitro models. J Pharm Pharmacol 63: 806–816. [DOI] [PubMed] [Google Scholar]
- Ryan A, Laurieri N, Westwood I, Wang CJ, Lowe E, Sim E (2010a). A novel mechanism for azoreduction. J Mol Biol 400: 24–37. [DOI] [PubMed] [Google Scholar]
- Ryan A, Wang CJ, Laurieri N, Westwood I, Sim E (2010b). Reaction mechanism of azoreductases suggests convergent evolution with quinone oxidoreductases. Protein Cell 1: 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, Kaplan E, Laurieri N, Lowe E, Sim E (2011). Activation of nitrofurazone by azoreductases: multiple activities in one enzyme. Sci Rep 1. doi:10.1038/srep00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, Kaplan E, Nebel J‐C, Polycarpou E, Crescente V, Lowe E et al. (2014). Identification of NAD(P)H quinone oxidoreductase activity in azoreductases from P. aeruginosa: azoreductases and NAD(P)H quinone oxidoreductases belong to the same FMN‐dependent superfamily of enzymes. PLoS One 9: e98551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphier S, Karton Y (2010). Novel salicylazo polymers for colon drug delivery: dissolving polymers by means of bacterial degradation. J Pharm Sci 99: 804–815. [DOI] [PubMed] [Google Scholar]
- Sharma R, Rawal RK, Gaba T, Singla N, Malhotra M, Matharoo S et al. (2013). Design, synthesis and ex vivo evaluation of colon‐specific azo based prodrugs of anticancer agents. Bioorg Med Chem Lett 23: 5332–5338. [DOI] [PubMed] [Google Scholar]
- Siegel D, Ross D (2000). Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic Biol Med 29: 246–253. [DOI] [PubMed] [Google Scholar]
- Siegel D, Shieh B, Yan C, Kepa JK, Ross D (2011). Role for NAD(P)H:quinone Oxidoreductase 1 and manganese‐dependent Superoxide Dismutase in 17‐(Allylamino)‐17‐demethoxygeldanamycin‐induced heat shock protein 90 inhibition in pancreatic cancer cells. J Pharmacol Exp Ther 336: 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers WC, Prasai B, Burk DH, Brown ML, McCarley RL (2013). Profluorogenic reductase substrate for rapid, selective, and sensitive visualization and detection of human cancer cells that overexpress NQO1. J Am Chem Soc 135: 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RL, Singh PK, Singh RP (2015). Enzymatic decolorization and degradation of azo dyes – A review. Int Biodeter Biodegr 104: 21–31. [Google Scholar]
- Smith NA, Byl JAW, Mercer SL, Deweese JE, Osheroff N (2014). Etoposide quinone is a covalent poison of human topoisomerase IIβ. Biochemistry 53: 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton P, Evans J, Jones R, Malik H, Vimalachandran D, Palmer D et al. (2015). Proteomic analysis to identify biomarkers in the primary tumour that predict response to neoadjuvant chemotherapy in liver metastases. Lancet 385: 95–95. [DOI] [PubMed] [Google Scholar]
- Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazue G et al. (2010). Fexinidazole‐‐a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis 4: e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu M (2014). Concepts in local treatment of extensive paediatric burns. J Med Life 7: 183–191. [PMC free article] [PubMed] [Google Scholar]
- Walton MI, Smith PJ, Workman P (1991). The role of NAD(P)H: quinone reductase (EC 1.6.99.2, DT‐diaphorase) in the reductive bioactivation of the novel indoloquinone antitumor agent EO9. Cancer Commun 3: 199–206. [DOI] [PubMed] [Google Scholar]
- Wang CJ, Hagemeier C, Rahman N, Lowe E, Noble M, Coughtrie M et al. (2007). Molecular cloning, characterisation and ligand‐bound structure of an azoreductase from Pseudomonas aeruginosa . J Mol Biol 373: 1213–1228. [DOI] [PubMed] [Google Scholar]
- Wang CJ, Laurieri N, Abuhammad A, Lowe E, Westwood I, Ryan A et al. (2010). Role of Tyrosine 131 in the active site of paAzoR1, an azoreductase with specificity for the inflammatory bowel disease pro‐drug balsalazide. Acta Crystallogr F66: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Chen HH, Paine DD, Cerniglia CE (2004). Microarray method to monitor 40 intestinal bacterial species in the study of azo dye reduction. Biosens Bioelectron 20: 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway J, Koziarz P, Veall J, Sandhu N, Kumar P, Hoecher B et al. (1998). Oxygen‐insensitive nitroreductases: Analysis of the roles ofnfsA and nfsB in development of resistance to 5‐Nitrofuran derivatives in Escherichia coli . J Bacteriol 180: 5529–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EM, Little RF, Mowday AM, Rich MH, Chan‐Hyams JV, Copp JN et al. (2015). Nitroreductase gene‐directed enzyme prodrug therapy: insights and advances toward clinical utility. Biochem J 471: 131–153. [DOI] [PubMed] [Google Scholar]
- Wu K, Knox R, Sun XZ, Joseph P, Jaiswal AK, Zhang D et al. (1997). Catalytic properties of NAD(P)H:Quinone Oxidoreductase‐2 (NQO2), a dihydronicotinamide riboside dependent oxidoreductase. Arch Biochem Biophys 347: 221–228. [DOI] [PubMed] [Google Scholar]
- Xie C, Zhou JL, Guo ZT, Diao XX, Gao ZW, Zhong DF et al. (2013). Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients. Brit J Pharm 168: 1687–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Yamazaki H, Ikeda T, Watanabe T, Iwabuchi H, Nakajima M et al. (2002). Formation of a novel quinone epoxide metabolite of troglitazone with cytotoxic to HepG2 cells. Drug Metab Dispos 30: 155–160. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang Y, Wu Q, Cui X, Lin Z, Liu S et al. (2014). Clinical implications of high NQO1 expression in breast cancers. J Exp Clin Cancer Res 33. doi:10.1186/1756-9966-33-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Ogata D, Gai Z, Taguchi S, Tanaka I, Ooi T et al. (2014). Structures of AzrA and of AzrC complexed with substrate or inhibitor: insight into substrate specificity and catalytic mechanism. Acta Crystallogr D70: 553–564. [DOI] [PubMed] [Google Scholar]
- Zeekpudsa P, Kukongviriyapan V, Senggunprai L, Sripa B, Prawan A (2014). Suppression of NAD(P)H‐quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J Exp Clin Cancer Res 33. doi:10.1186/1756-9966-33-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Structures of compounds mentioned in the review. Numbered atoms mentioned in the text are labelled.
Fig. 1 Supplementary info item