

Abstract
Background
Hydrodistillation has been traditionally used to extract volatile fraction in traditional Chinese medicine. However, with the development of Soxhlet extraction (SE), microwave (MW), ultrasound (US), and cold maceration (CM), hydrodistillation (HD) is being replaced to meet some practical requirements. In this study, we investigated the effect of the five methods on the volatile fraction extract of Lonicera macranthoides.
Methods
Volatile fraction from the flower buds of Lonicera macranthoides was obtained by using different extraction methods, HD, SE, MW, US, and CM. The compositions of volatile fraction were analyzed by gas chromatography–mass spectrometric and further compared among extraction methods.
Results
Extracts obtained by the five methods reveal the qualitative and quantitative diversity in their compositions, especially for the low-content compositions. According to the results, SE shows the great value in the research where the high molecular-mass compound is of primary interest, and MW offers a way for the isolation of specific compound like octadecadienoic acid and hexadecanoic acid. HD, US, and CM have the advantage over SE and MW for the integrity of the constituents, whereas the phenomenon of compound degradation seems not so serious in solvent extraction methods such as US or CM as HD. Additionally, US and CM show superiority over time or material saving and diversity of the constituent.
Conclusion
HD is still the best choice for the pure volatile fraction without organic solvent pollution. However, when it comes to some specifically actual demands, it can be replaced by the four methods for the volatile fraction extraction process, especially for production of certain compound groups.
Keywords: Lonicera macranthoides, volatile fraction, extraction method, hydrodistillation, traditional Chinese medicine
1. Introduction
Lonicera macranthoides, one of the most important varieties in the Lonicera family, is widely cultivated and applied as a substitute for Lonicerae japonicae in China for its similar chemical compositions, pharmacological effects but relatively higher yield and lower cost.1, 2, 3 It has been reported that the effective components in L. macranthoides bud possess diverse biological activities, such as antibacterial,4 antipyretic,5 antioxidant,6 and hepatoprotective ability.7 It has been reported that volatile fractions together with chlorogenic acid are the main active ingredients in L. macranthoides.8 Other than the normal medical value, volatile fractions in L. macranthoides also attract intensive attention in fields such as cosmetics, shampoos, beverages, flower tea, and baked goods, all of which are owing to its properties of thirst-quenching, heat-clearing, and detoxifying, and render L. macranthoides highly appreciated in Chinese herbal medicine.
It is generally known that the budding flower of flower-employed Chinese medicine, rather than leaves or other parts of plants, is conventionally selected as the material for volatile fraction extraction and always ranks first in volatile fraction yields. Previous research also indicates that genetic and environment factors may influence the content and composition of volatile fraction in plants,9 as do the development stages.10 In addition, the volatile fraction also varies with extraction methods.11
With increasing energy consumption and the drive to improve efficiency, industries and research institutions are challenged to find ways which can simplify operation procedure, meet low cost requirements and achieve good quality. Apart from conventional techniques such as hydrodistillation (HD),12 Soxhlet extraction (SE),13 ultrasound (US),14 microwave (MW),15 and cold-maceration (CM),16, 17 some relatively new methods such as supercritical CO2 extraction18 and head space analysis19 have been employed for volatile fraction extraction research in laboratories. However, both supercritical CO2 extraction and head-space analysis need special equipment, thus making them too expensive for large-scale volatile fraction extraction when compared with traditional methods. For example, HD is widely treated as the conventional application of the volatile fraction in food and Chinese medicine20 and usually takes about 6–8 hours with distilled water for the whole extraction progress. The fraction obtained by this way is completed and pure without organic solvent pollution when compared with ultrasound and microwave. Likewise, SE progress is similar to HD but with organic extraction reagent and mainly used in the volatile fraction extraction of the material that is rich in fat, such as seed or spice.14, 21 Similar research for the volatile fraction extraction on flower materials has also been reported by Guan et al.22 Unlike HD and SE, both MW and US extractions offer a faster and simpler procedure and require less plant material.23 Thus, the two methods are widely applied for fast extraction at the cost of compounds integrity.
Additionally, CM always results in an odor similar to that in the original plant material without causing degradation of the thermo labile compounds present in the fraction due to the low extraction temperature similar to cold pressing,24 rather than heating in HD or SE, which makes heating a factor investigated for extraction of volatile fractions from aromatic flowers. However, the exact differences about the volatile fractions extracted via the five methods mentioned above have not been reported yet.
Over the years, procedures such as US and MW extraction have replaced some of the conventional processes such as HD and SE that have been used in industries and laboratories for decades. We wonder if the relatively new methods are exactly fit for the volatile fraction extraction in Chinese medicine, because it is the entirety of constituent rather than a single compound that cures diseases. In this paper, we present a comparative study of the content and composition of volatile fraction extracted by different methods from L. macranthoides in order to find the differences in terms of their quantity and quality.
2. Methods
2.1. Chemicals and plant material
Analytical grade anhydrous sodium sulfate and ethyl acetate were purchased from Kelong Chemical (Chengdu, China). The buds of L. macranthoides were authenticated by Professor Xingfu Chen in Sichuan Agricultural University (Sichuan, China) and collected in July 2012 from Suining in Sichuan province of China. The materials were dried and pulverized to a fine powder using a mechanical grinder.
2.2. Extraction procedures
The extraction of volatile fraction from L. macranthoides was performed using five different methods, and each test was carried out in triplicate.
2.2.1. HD
A 20 g sample of L. macranthoides was subjected to hydrodistillation, according to the China Pharmacopeia,21 and extracted with 200 mL of distilled water for 6 hours (until no more volatile fraction was obtained). The volatile fraction was collected, dried with anhydrous sodium sulfate and stored at 4 °C until used.
2.2.2. SE
The samples (20 g) were weighed and transferred into the Soxhlet apparatus (SOX500, Haineng, China, with 6 individual extractors), then extracted with 200 mL ethyl acetate for about 6 hours at 60 °C.14 After extraction, the mixture was combined, concentrated, and stored at 4 °C until analysis.
2.2.3. US extraction
According to Pingret et al,25 an ultrasonic cleaner with temperature control (SB5200DT; Ningbo Xinzhi Biotechnology Co., Ltd., Ningbo, China) was used for ultrasonic extraction. For each experiment, 6 g of crushed material with 30 mL ethyl acetate were allowed to stand overnight and then placed into the reactor. Then the US was twice applied for 30 minutes operating at a frequency of 25 kHz and 45 °C. The two extracts were filtered with speed quantitative filter and mixed together as the final extract, which was then concentrated and stored at 4 °C until analysis.
2.2.4. MW extraction
The MW extraction experiment was performed in a MM-2270MG MW extractor (Haier, Qingdao, China), with the time, temperature, and power controlled.16 In a MW procedure performed at atmospheric pressure, 6 g of L. macranthoides with 30 mL ethyl acetate, which was let stand overnight first, was heated twice at 200 W for 10 minutes. To facilitate rigorous comparison, each final extract was concentrated and stored in the same way as in US extraction.
2.2.5. CM
By reference to Liang et al16 and Du et al,17 6 g of L. macranthoides, with 30 mL ethyl acetate, were placed in a conical flask sealed with a stopper and wraps. The material was soaked in 30 mL ethyl acetate twice for 24 hours each time. The extracts were combined as the final extract for each material. To facilitate rigorous comparison, each final extract was concentrated and stored in the same way as for US extraction.
2.3. Gas chromatography–mass spectrometric identification
Gas chromatography–mass spectrometric analyses were performed on a QP2010 system (Shimadzu, Kyoto, Japan) by electronic ionization at 70 eV, employing a chromatographic column (DB-5MS, 30 mm × 0.25 mm, 0.25 μm). Each extract (1 μL) was injected using the splitless injection method. The column temperature was increased from 60 °C to 200 °C at the rate of 6 °C/min (held at 200 °C for 6 minutes), then raised to 220 °C by 5 °C/min, next to 250 °C by 4 °C/min, and finally held at 250 °C for 5 minutes. The injection port was set at 250 °C. Helium gas was used as the carrier at a flow rate of 1 mL/min.
Relative percentage data were obtained from electronic integration of peak areas without the use of a correction factor. The software adopted to handle mass spectra and chromatograms was GC-MS Solutions. Retention indices were determined using retention times of normal alkanes that had been injected after the oil under the same chromatographic conditions mentioned above. The compounds were identified by comparison of their mass spectra with the NIST05.LIB database or with published mass spectra.
3. Results
3.1. Composition of volatile fraction
The results indicate that both similarities and differences are present in the compositions of the volatile fraction collected via the five methods (see Table 1). Table 1 lists the constituents (content > 0.5%) of volatile fractions extracted by different methods. The relative content of the volatile fraction is determined by area normalization. The compounds identified account for 99.98%, 99.96%, 99.96%, 97.58%, and 91.62% in HD, SE, MW, US, and CM, respectively. Therefore, the total proportion of all the other compounds, every single compound of which accounts for < 0.5% and has not been exhibited in our result, was 0.02%, 0.04%, 0.04%, 2.42%, and 8.38%, respectively.
Table 1.
Volatile compounds (accounting for > 0.5%) of Lonicera macranthoides analyzed by gas chromatography–mass spectrometry
| No. | Identified compounds | Molecular formula | RMM* | Relative content (%) |
||||
|---|---|---|---|---|---|---|---|---|
| HD | SE | MW | US | CM | ||||
| 1 | Dimethyl phthalate | C10H10O4 | 194 | 14.07 | 13.32 | 17.00 | 19.28 | 7.65 |
| 2 | Dibutyl phthalate | C16H22O4 | 278 | 23.10 | 16.10 | 18.40 | 17.86 | 7.09 |
| 3 | Hexadecanoic acid | C16H32O2 | 256 | 4.56 | 17.86 | 25.72 | 20.66 | 12.25 |
| 4 | Octadecanoic acid | C18H36O2 | 284 | 1.39 | 3.47 | 2.85 | 2.83 | 18.22 |
| 5 | Tetradecanoic acid | C14H28O2 | 228 | 11.90 | 0.61 | 0.66 | 0.89 | 0.73 |
| 6 | 9,12-Octadecadienoic acid | C18H32O2 | 280 | 3.98 | —† | 27.48 | 12.98 | 12.04 |
| 7 | 9,12,15-Octadecatrienoic acid, methyl ester | C19H32O2 | 292 | 1.95 | — | 7.19 | 4.13 | 8.00 |
| 8 | 1,2,3-Propanetriol, monoacetate | C5H10O4 | 134 | — | 1.19 | — | 8.42 | 2.86 |
| 9 | 9,12-Octadecadien-1-ol | C18H34O | 266 | — | 24.57 | — | — | 22.41 |
| 10 | 10-Nonadecanol | C19H40O | 284 | — | 4.28 | — | — | 3.64 |
| 11 | Heneicosane | C21H44 | 296 | — | 3.24 | — | — | 1.50 |
| 12 | Hexadecanoic acid, butyl ester | C20H40O2 | 312 | 1.32 | — | 0.66 | 1.77 | — |
| 13 | 3,7,11,15-Tetramethyl-2-hexadecen-1-ol | C20H40O | 296 | — | 0.84 | — | 0.79 | — |
| 14 | Phenylethyl alcohol | C8H10O | 122 | 0.91 | — | — | 0.82 | — |
| 15 | 1-Hexadecanol | C16H34O | 242 | 1.56 | 1.79 | — | — | — |
| 16 | Heptadecane,2,6,10,15-tetramethyl- | C21H44 | 296 | 1.12 | 0.72 | — | — | — |
| 17 | 3-Hydroxy-2,2,6-trimethyl-6-vinyltetrahydropyran | C10H18O2 | 170 | 3.65 | — | — | 1.60 | — |
| 18 | 2,6-Octadien-1-ol, 3,7-dimethyl- | C10H18O | 154 | 4.20 | — | — | — | — |
| 19 | Benzoic acid, 4-formyl-, methyl ester | C9H8O3 | 164 | 0.79 | — | — | — | — |
| 20 | Undecanoic acid | C11H22O2 | 186 | 1.19 | — | — | — | — |
| 21 | Hexadecanoic acid, methyl ester | C17H34O2 | 270 | 1.52 | — | — | — | — |
| 22 | Hexadecanoic acid, ethyl ester | C18H36O2 | 284 | 12.70 | — | — | — | — |
| 23 | 12,15-Octadecadienoic acid, methyl ester | C19H34O2 | 294 | 1.49 | — | — | — | — |
| 24 | 9,12,15-Octadecatrienoic acid, methyl ester | C14H26 | 194 | 1.88 | — | — | — | — |
| 25 | 9,12,15-Octadecatrien-1-ol | C18H32O | 264 | 1.44 | — | — | — | — |
| 26 | Nonadecane | C19H40 | 268 | 3.12 | — | — | — | — |
| 27 | Hexadecane | C16H34 | 226 | 2.14 | — | — | — | — |
| 28 | 1-Heptacosanol | C27H56O | 396 | — | 7.35 | — | — | — |
| 29 | Pentatriacontane | C35H72 | 492 | — | 4.62 | — | — | — |
| 30 | Pentanoic acid, ethyl ester | C7H14O2 | 130 | — | — | — | 1.20 | — |
| 31 | Hexanoic acid | C6H12O2 | 116 | — | — | — | 1.30 | — |
| 32 | 1,2-Benzenedicarboxylic acid, bis(2-methylpropyl) ester | C16H22O4 | 278 | — | — | — | 0.90 | — |
| 33 | 2-Nonadecanone | C19H38O | 282 | — | — | — | 1.61 | — |
| 34 | Tetracosane | C24H50 | 338 | — | — | — | 1.88 | — |
| 35 | Octadecanoic acid, butyl ester | C22H44O2 | 340 | — | — | — | 1.08 | — |
| 36 | Acetic acid, octadecyl ester | C20H40O2 | 312 | — | — | — | — | 0.99 |
| 37 | Citronellyl isobutyrate | C14H26O2 | 226 | — | — | — | — | 1.59 |
| 38 | Eicosanoic acid | C20H40O2 | 312 | — | — | — | — | 1.03 |
| Total | 99.98 | 99.96 | 99.96 | 97.58 | 91.62 | |||
Relative molecular mass.
Less than 0.5% or undetectable.
HD, hydrodistillation; SE, Soxhlet extraction; US, ultrasound extraction; MW, microwave extraction; CM, cold-maceration.
As to the type of compounds, HD extract appears to rank first with 22 different compounds, followed by SE, US, and CM with 14–16 compounds; however, only eight compounds are present in the MW, which means that MW may lead to reduction in compound complexity. Therefore we can conclude that MW may make it easier for the isolation of some specific compounds such as hexadecanoic acid and octadecadienoic acid. Moreover, compounds with high relative molecular mass (RMM) in SE, such as 1-heptacosanol (RMM = 396) and pentatriacontane (RMM = 492), accounted for 7.35% and 4.62%, respectively. The other methods appear to focus on extracting lower molecular-mass compounds, the RMM of which vary from 116 to 340.
As shown in Table 1, hexadecanoic acid, reported as dominating first (21.52%) in the HD extract,26 was present in all the volatile fractions extracted by the methods we used. However, the total percentage of volatile fractions varied with methods and was 4.56%, 17.86%, 25.72%, 20.66%, and 12.25%, respectively. Meanwhile, we also found 17.1% of the derivatives from hexadecanoic acid in HD. Although the five extracts contained the same components, such as dimethyl phthalate, dibutyl phthalate, hexadecanoic acid, octadecanoic acid, and tetradecanoic acid, their proportions varied more or less according to the extraction technique used, as did the most abundant component isolated by different methods. The most abundant compounds in HD, SE, US, MW, and CM were dibutyl phthalate (23.10%), octadecadien-1-ol (24.57%), octadecadienoic acid (27.48%), hexadecanoic acid (20.66%), and octadecadien-1-ol (22.41%), respectively. The relative content of octadecadienoic acid is very different among the five methods and is 3.98% (HD), not detectable or < 0.5% (SE), 27.48% (MW), 12.98% (US), and 12.04% (CM). However, we can also notice the presence of octadecatrien-1-ol in HD (1.44%), SE (24.57%), and CM (22.41%). This difference is possibly due to the degradation through the extracting procedure or may vary with method.
As shown in Table 2, the volatile fraction of L. macranthoides can be divided into aliphatic, aromatic, and terpene, which is consistent with the result of HD extract by Wang et al.26, 27 The functional groups of the constituents from SE and MW are similar to each other but different from the other three methods, especially for the proportion of terpene group, which is < 0.5% or not detectable in our samples. The terpene groups of volatile fractions from L. japonica has been reported as only sharing 0.526% of the total volatile fraction in the result of Vereshchagin et al,28 meaning that the terpenes occupy a small proportion in the volatile fraction. Furthermore, most of the terpene group occupied < 0.5% of the total yield in the result of Wang et al26 and Tong et al.29
Table 2.
Percentages of functional groups of the constituents of essential oils from Lonicera macranthoides analyzed by gas chromatography–mass spectrometry
| Compound |
Relative content (%) |
|||||
|---|---|---|---|---|---|---|
| Category | Group | HD | SE | MW | US | CM |
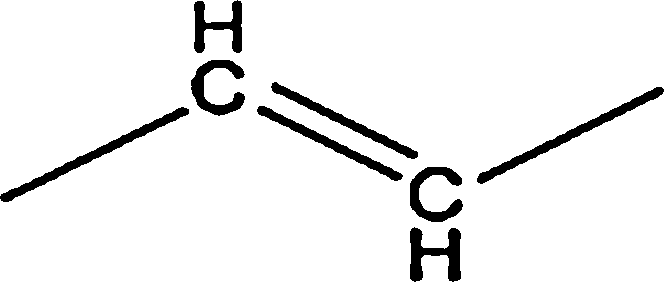
| Aliphatic | 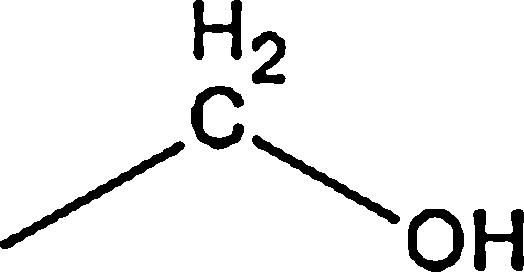 |
3.00 | 38.83 | — * | 0.79 | 26.05 |
 |
23.02 | 21.94 | 56.73 | 38.66 | 44.27 | |
 |
9.25 | 25.41 | 34.67 | 17.90 | 42.45 | |
 |
— | — | — | 1.61 | — | |
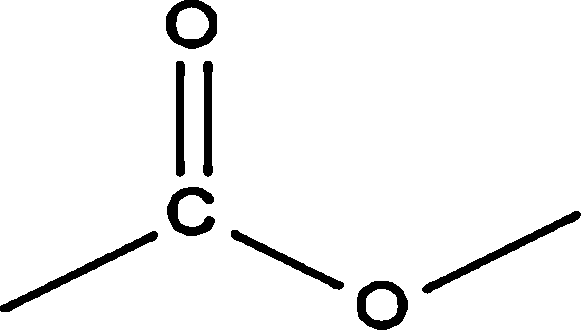 |
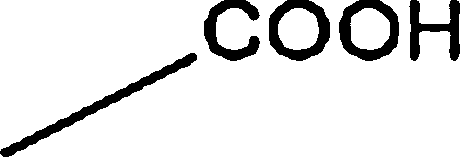
17.49 | 1.19 | 7.19 | 16.60 | 8.99 | |
 |
6.39 | 8.58 | — | 1.88 | 1.50 | |
| Total | 59.15 | 95.95 | 98.59 | 77.44 | 123.26 | |
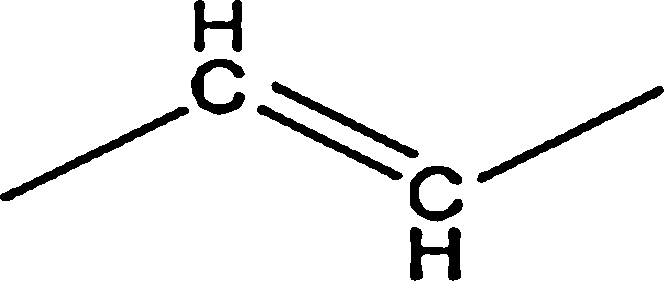
| Aromatic |  |
0.92 | — | — | 0.82 | — |
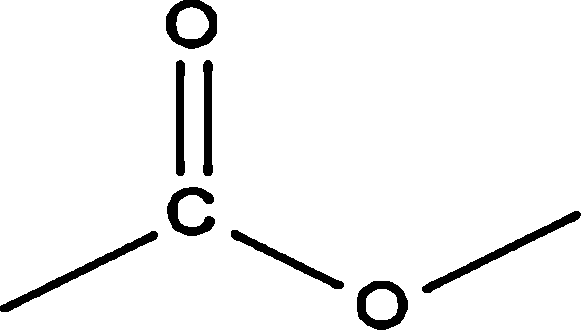 |
39.45 | 29.46 | 35.42 | 38.04 | 14.74 | |
 |
1.49 | — | — | — | — | |
| Total | 41.86 | 29.46 | 35.42 | 38.86 | 14.74 | |
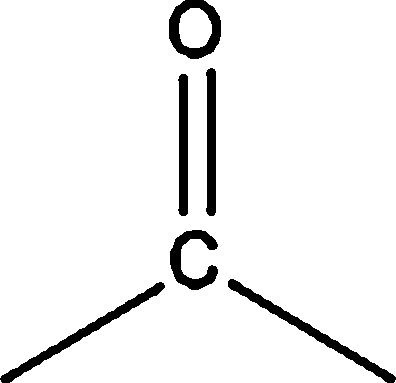
| Terpene | 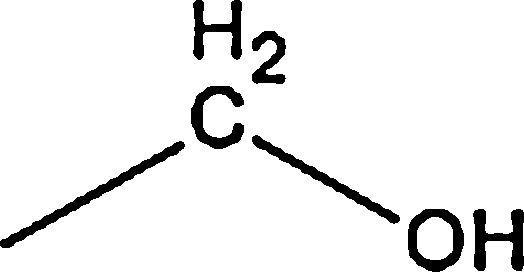 |
7.85 | — | — | 1.60 | — |
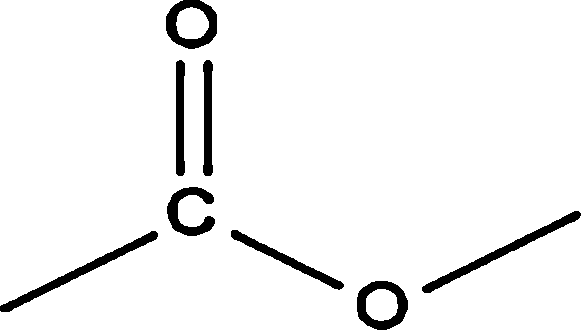 |
— | — | — | — | 1.59 | |
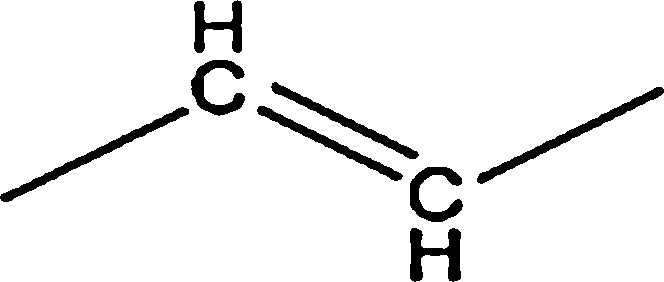 |
7.85 | — | — | 1.60 | — | |
| Total | 15.70 | — | — | 3.20 | 1.59 | |
Trace or undetectable.
HD, hydrodistillation distillation; SE, Soxhlet extraction; US, ultrasound extraction; MW, microwave extraction; CM, cold-maceration.
3.2. Analysis of the extraction conditions
According to Table 3, only 6 g of the sample are needed for US, MW, and CM, which indicates that they may be more appropriate where the amount of material is limited, when compared with the other two methods. Only the traditional HD employs water rather than organic solvent for the whole extraction procedure. When HD and SE, both of which were performed with the same sample amount and under the same extraction time condition are compared, the higher extraction temperature at 100 °C with water as extraction solvent in HD may have accelerated the terpene groups extracting procedure. Likewise, 3.20% and 1.59% of terpene groups were also obtained in US and CM, respectively, which indicates that ultrasound or longer extraction time even at 4 °C with 24 hours by CM may have a similar impact on the terpene group extracting procedure, and 10 minutes in MW is not long or good enough for the terpene group extraction in the volatile fraction.
Table 3.
Extraction conditions of different methods
| Condition | Sample amount (g) | Extraction time (min) | Extraction temperature (°C) | Extraction solvent (mL) |
|
|---|---|---|---|---|---|
| Water | Ethyl acetate | ||||
| HD | 20 | 360 | 100 | 200 | — |
| SE | 20 | 360 | 60 | — | 200 |
| US | 6 | 30 | 45 | — | 30 |
| MW | 6 | 10 | 45 | — | 30 |
| CM | 6 | 1,440 | 4 | — | 30 |
HD, hydrodistillation distillation; SE, Soxhlet extraction; US, ultrasound extraction; MW, microwave extraction; CM, cold-maceration.
4. Discussion
In terms of the variety of compounds accounting for > 0.5%, the major components obtained by the five methods turn out to be similar to each other, albeit in different amounts, such as dimethyl phthalate, dibutyl phthalate, and hexadecanoic acid. It is widely accepted that dibutyl phthalate or diisobutyl phthalate and hexadecanoic acid are the principal constituents in volatile fractions of L. japonica and L. macranthoides extracted by HD and supercritical fluid extraction, whereas their relative proportions vary with methods.27, 30, 31 The same result was described in the L. japonica volatile fraction obtained at different harvest times (from June to August),32 different flower stages (buds, white flower, and yellow flower),33 and different origins (Shandong, Henan, and Jiangsu provinces in China).34
Some of the organic acids and esters, which have special smells, are relevant to the flavor in flowers and fruits. Ethyl acetate, accounting for 1.13%,35 was reported as one of the major constituents in the volatile fractions of kumquats. Citronellyl formate and citronellyl acetate were considered as the characteristic scent related compounds of the fractions from peels according to flavor dilution factors, relative flavor activity, and GC-sniffing. Similar results occurred in the research of Nguyen et al.36 Dibutyl phthalate itself is a kind of colorless, transparent and oily liquid with special fragrant smell. Thus, we suspect dibutyl phthalate and hexadecanoic acid as the likely scent-related compounds in the volatile fractions from L. macranthoides.
The constituents in CM are most complicated and there are fewer differences in the variety of volatile fractions by US, MW, and HD. This phenomenon may be related to the heating directly or indirectly during the extracting process, causing the loss of trace compounds to different degree by different methods. The reason for the loss of some compounds in the other methods compared with CM is probably not that these compounds are not extracted but rather that the reduction in extraction time and heating. Therefore we can conclude that CM enjoys the advantage in extracting the low-percentage rather than high-percentage compounds. Some new compounds may be more easily found during the CM and US extraction procedure than that of the other three methods.
Indeed, for an extract to be classified as volatile fraction, heat and water may be used in its extraction from traditional Chinese medicine. HD is a classical technology and appears to be a good extraction method for volatile fractions from aromatic plants in China. Nevertheless, some compounds such as linalyl acetate in the volatile fraction extracted via HD are well known to be thermally sensitive and vulnerable to chemical changes. Linalyl acetate has been reported by Périno-Issartier et al23 to be likely to degrade into linalool via HD and the degradation reaction would appear to be more limited with microwave extraction, probably because the lavandin was not in direct contact with water and extracted with a lower temperature. In our study, we observe that the volatile fraction collected by HD consists of five kinds of hexadecanoic acid and their derivatives, namely, hexadecanoic acid (4.56%), hexadecanoic acid, methyl ester (1.52%), hexadecanoic acid, ethyl ester (12.70%), 1-hexadecanol (1.56%), and hexadecanoic acid, butyl ester (1.32%), whereas all the four derivatives are not detectable in CM. Only 1.79% of 1-hexadecanol has been collected in SE, and 0.66% and 1.77% of hexadecanoic acid, butyl ester in MW and US, respectively. Besides, alcohol and acids can easily generate into esters, and this reaction is reversible, according to Hu et al.37 Therefore, we can conclude that water directly mixed with L. macranthoides can be used as an initiator of the reaction causing the degradation of hexadecanoic acid, and this situation seems to be not so serious in the other four solvent extraction methods applied in this study.
In the light of previous study, octadecadienoic acid is an essential fatty acid with great health benefits in humans and its derivatives possess antihypertensive activities and the function of accelerating the blood microcirculation and depressing blood-fat, which make it an excellent cure for hypertension, hyperlipidemia, angina, and obesity. These data suggest that volatile fraction extracts under the proper conditions from L. macranthoides may possess similar activities as octadecadienoic acid and its derivatives, and higher activities may occur in the volatile fractions collected via organic solvent, rather than the direct contact with water as with HD. Likewise, the study of Huang et al38 indicated that heating can destroy the anti-inflammation effective components in the comparison of heated and unheated volatile fractions of L. japonica. It also exhibited good antipyretic, anti-inflammatory and analgesic activities according to Ni et al.39 Besides, L. macranthoides used to be regarded as excellent substitute for L. japonica in China in consideration of its similar pharmacology activities, high production, and lower cost. Thus, we speculate that the volatile fractions of L. macranthoides may possess similar activities as that of L. japonica. With further study and the accumulation of base data, more attention will be paid to the bioactivities of volatile fractions in L. macranthoides.
In this study, the volatile fractions of L. macranthoides have been extracted using five different methods and a comparison of the different techniques has been carried out. The results indicate that the different methods may be considered as the optimum process according to some specific request and the classical HD method is the best way to purify volatile fraction without pollution, which can sometimes be replaced by the volatile fraction extraction process. For example, SE shows great value in research where the high molecular-mass compounds are of primary interest, and MW offers a method for the isolation of specific compound such as octadecadienoic acid and hexadecanoic acid. In addition, HD, US, and CM have the advantage over SE and MW for the integrity of the constituents, whereas the phenomenon of compound degradation seems not so serious in solvent extraction such as US or CM as in HD. Moreover, US and CM show superiority over time or material saving and diversity of constituents.
In conclusion, US, HD, and CM can be applied in the volatile fraction extraction of L. macranthoides for the benefit of time or material saving, integrity, and variety of constituents, respectively. However, HD is still the best choice for pure volatile fraction without organic solvent pollution.
Conflicts of interest
All authors declare no conflicts of interest.
Acknowledgments
This study was funded by a grant from Sichuan Provincial Crop breeding research project Application in the 12th 5-Year Period (No. 2011NZ0098–12–01).
References
- 1.Hou M., Tang Q., Zhang M.H., Li Y., Xu X.Y. Comparative studies on pharmacodynamics of Lonicera macranthoides and Lonicera japonica. Chin Trad Herbal Drugs. 2013;44:309–314. [Google Scholar]
- 2.Liu J.H. Comparative studies on the quality and safety of Lonicerae Flos and Lonicerae japonicae Flos. Beijing University of Chinese Medicine (Master's thesis) 2014 [Google Scholar]
- 3.Zhang X.N. Comparative studies on the chemical components and pharmacological effects of Lonicera macranthoides Hand-Mazz. and Lonicera japonica Thunb. Southwest University (Master's thesis); 2014 [Google Scholar]
- 4.Lei Z.J., Zhou R.B., He Y.S., Zeng R. Comparison of antibacterial action between large flower-like Honeysuckle Flower and the certified Honeysuckble. Guiding J TCM. 2005;11:8–9. [Google Scholar]
- 5.Lei Z.J., Zhou R.B., Zeng R., He Y.S. Comparative experiments on antipyretic effect between Lonicera macranthoides Hands Mazz. and the certified Flos Lonicera. J TCM Univ Hunan. 2005;25:14–15. [Google Scholar]
- 6.Zhang W.M., Wei J., Hu Z., Zhong G. Study on extraction and purification of chlorogenic acid from Flos Lonicerae flowers and its antioxidation properties. Food Sci. 2008;29:109–112. [Google Scholar]
- 7.Shi J.Z., Chen X.F., Wan L. Hepatoprotective effect of several constituents of Lonicera fulvotomentosa Hsu et S. C. Cheng, and L. macranthoides Hand. -Mazz. on CCl4 and D-galactosamine induced liver injuries in mice and rats. Zhongguo Zhong Yao Za Zhi. 1999;24:363–364. [In Chinese] [PubMed] [Google Scholar]
- 8.Liu W.J., Chen Y., Ma X., Feng X., Liang J.Y. Progress in the research on chemical constituents of Lonicera macranthoides Hand.-Maza. Chinese Wild Plant Resources. 2013;32:6–10. [Google Scholar]
- 9.Bernath J. Production ecology of secondary plant products. In: Craker L.E., Simon J.E., editors. Herbs, spices and medicinal plants: recent advances in botany, horticulture, and pharmacology. Oryx Press; Phoenix: 1986. pp. 185–234. [Google Scholar]
- 10.Burbott A.J., Loomis W.D. Effects of light and temperature on the monoterpenes of peppermint. Plant Physiol. 1967;42:20–28. doi: 10.1104/pp.42.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guenther E. The production of essential oils. In: Guenther E., editor. The essential oils. Krieger Publishing Co.; Malabar: 1972. p. 87. [Google Scholar]
- 12.Bounatirou S., Smiti S., Miguel M.G., Faleiro L., Rejeb M.N., Neffati M. Chemical composition, antioxidant and antibacterial activities of the essential oils isolated from Tunisian Thymus capitatus Hoff. et Link. Food Chem. 2007;105:146–155. [Google Scholar]
- 13.Singh G., Marimuthu P., De Heluani C.S., Catalan C.A. Antioxidant and biocidal activities of Carum nigrum (seed) essential oil, oleoresin, and their selected components. J Agric Food Chem. 2006;54:174–181. doi: 10.1021/jf0518610. [DOI] [PubMed] [Google Scholar]
- 14.Pingret D., Fabiano-Tixier A.S., Chemat F. An improved ultrasound Clevenger for extraction of essential oils. Food Anal Methods. 2014;7:9–12. [Google Scholar]
- 15.Costa S.S., Gariepy Y.R., Sandra C.S. Microwave extraction of mint essential oil—Temperature calibration for the oven. J Food Engin. 2014;126:1–6. [Google Scholar]
- 16.Liang C.Y., Fu H., Li W.L., Xia B., Wu J.L. Comparison of different extraction methods of volatile oil from Mentha haplocalyx Briq., Lishizhen Med Mater Med Res. 2007;18:2085–2086. [Google Scholar]
- 17.Du Z.X., Wu H.E., Li F.Y., Guo M., Wu P.C., Gong S.J. Chemical constituents of volatile oil from Porella setigera (steph.) Hatt., Lishizhen Med Mater Med Res. 2010;21:336–338. [Google Scholar]
- 18.Vagi E., Simandi B., Suhajda A., Héthelyic É. Essential oil composition and antimicrobial activity of Origanum majorana L. extracts obtained with ethyl alcohol and supercritical carbon dioxide. Food Res Int. 2005;38:51–57. [Google Scholar]
- 19.Barboni T., Venturini N., Paolini J., Desjobert J.M., Chiaramonti N., Costa J. Characterisation of volatiles and polyphenols for quality assessment of alcoholic beverages prepared from Corsican Myrtus communis berries. Food Chem. 2010;122:1304–1312. [Google Scholar]
- 20.Chinese Pharmacopoeia Commission . People's Medical Publishing House; Beijing: 2010. Phamacopoeia of the People's Republic of China; p. 1311. [Google Scholar]
- 21.He D.M., Liu H.X., Huang C.S., Yang X.Y., Chen Q. Analysis of volatile oil of Illicium verum hook seed by gas chromatography-mass spectrometry. Chin Condiment. 2010;35:85–87. [Google Scholar]
- 22.Guan W.Q., Li S.F., Yan R.X., Tang S.K., Quan C. Comparison of essential oils of clove buds extracted with supercritical carbon dioxide and other three traditional extraction methods. Food Chem. 2007;101:1558–1564. [Google Scholar]
- 23.Périno-Issartier S., Ginies C., Cravotto G., Chemat F. A comparison of essential oils obtained from lavandin via different extraction processes: ultrasound, microwave, turbohydrodistillation, steam and hydrodistillation. J Chromatogr A. 2013;1305:41–47. doi: 10.1016/j.chroma.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Chen H.C., Peng L.W., Shen M.J., Lin L.Y., Chiang H.M., Wu C.T. Effects of hot water treatment on the essential oils of calamondin. J Food Drug Anal. 2013;12:363–368. [Google Scholar]
- 25.Pingret D., Fabiano-Tixier A.S., Chemat F. Degradation during application of ultrasound in food processing: a review. Food Control. 2013;31:593–606. [Google Scholar]
- 26.Wang T.Z., Li Y.M., Wang Z.X. A study about volatile oil in the buds of Lonicera mancranthoides. Chin Tradit Herbal Drugs. 2000;31:657–658. [Google Scholar]
- 27.Wang Z.Z., Bi Y.A., Shang Q. The GC-MS analysis of volatile constituents from Lonicera japonica and Lonicera macranthoides. Chin Tradit Herbal Drugs. 2008;39:672–674. [Google Scholar]
- 28.Vereshchagin P.V., Bezzubov A.A., Rodopulo A.K. Examination of the composition of essential oils of the edible honeysuckle (Lonicera edulis) Priklad Biokhim Mikrobiol. 1983;19:423–428. [Google Scholar]
- 29.Tong Q.Z., Zhou R.B., Luo Y.L., He Y.S., Qu W.H. Analysis of volatile oils of Lonicera macranthoides in Hunan Province by GC-MS. Chin Tradit Pat Med. 2005;27:52–55. [Google Scholar]
- 30.Tang L.J., Zhou R.B., Liu X.R. GC-MS analysis of volatile oil extracted from Lonicera macranthoides Hand-Mazz. with supercritical CO2 fluid extraction and stream distillation. J TCM Univ Hunan. 2010;30:109–113. [Google Scholar]
- 31.Su X.P., Gong D.C., Zhang Y.X. Study on the chemical constituents of essential oil from Lonicerae by supercritical CO2 extraction. Lishizhen Med Mater Med Res. 2007;18:2643–2644. [Google Scholar]
- 32.Yang M.L., Zhao Y.G. A comparative study of composition of volatile oil of Flos Lonicerae of Yuanzhou in Ningxia in different months. J Ningxia Univ. 2007;28:140–146. [Google Scholar]
- 33.Wang J.M., Ji Z.Q., Kang W.Y. The volatile oil from different parts of Lonicera japonica Thunb. Fine Chem. 2008;25:1075–1078. [Google Scholar]
- 34.Xing J.B., Li P., Zhang Z.Y. The GC-MS analysis of volatile oil in Lonicera japonica from different producing area. Chin Tradit Herbal Drugs. 2002;33:784–785. [Google Scholar]
- 35.Choi H.S. Characteristic odor components of kumquat (Fortunella japonica Swingle) peel oil. J Agric Food Chem. 2005;53:1642–1647. doi: 10.1021/jf040324x. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen T.L.P., Nguyen T.M.T., Chieko N., Masayoshi S. Characterisation of the odour volatiles in Citrus aurantifolia Persa lime oil from Vietnam. Dvpt Food Sci. 2006;43:193–196. [Google Scholar]
- 37.Hu J.P., Chu W., Qiu F.L. Study on esterification of aceticacid and butanol catalyzed by solidacid. Fine Chem. 2000;17:269–273. [Google Scholar]
- 38.Huang J.J., Song B.W., Zhu X.Y., Fu R.L. Analysis of components of volatile oils of Lonicera and heated VOL and comparison of their anti-inflammatory activities. J Zhejiang Univ Tech. 2009;37:126–130. [Google Scholar]
- 39.Ni L.J., Wu K.F., Shi W.Z., Zhang L.G. The pharmacodynamics study on volatile constituents of anti-pyretic and anti-inflammatory Chinese herbs. Chin Tradit Pat Med. 2008;29:1217–1221. [Google Scholar]