

Abstract
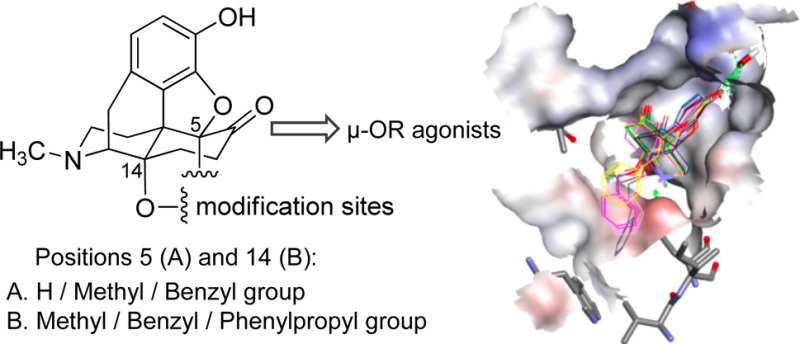
Among opioids, morphinans are of major importance as the most effective analgesic drugs acting primarily via μ-opioid receptor (μ-OR) activation. Our long-standing efforts in the field of opioid analgesics from the class of morphinans led to N-methylmorphinan-6-ones differently substituted at positions 5 and 14 as μ-OR agonists inducing potent analgesia and fewer undesirable effects. Herein we present the first thorough molecular modeling study and structure–activity relationship (SAR) explorations aided by docking and molecular dynamics (MD) simulations of 14-oxygenated N-methylmorphinan-6-ones to gain insights into their mode of binding to the μ-OR and interaction mechanisms. The structure of activated μ-OR provides an essential model for how ligand/μ-OR binding is encoded within small chemical differences in otherwise structurally similar morphinans. We reveal important molecular interactions that these μ-agonists share and distinguish them. The molecular docking outcomes indicate the crucial role of the relative orientation of the ligand in the μ-OR binding site, influencing the propensity of critical non-covalent interactions that are required to facilitate ligand/μ-OR interactions and receptor activation. The MD simulations point out minor differences in the tendency to form hydrogen bonds by the 4,5α-epoxy group, along with the tendency to affect the 3–7 lock switch. The emerged SARs reveal the subtle interplay between the substituents at positions 5 and 14 in the morphinan scaffold by enabling the identification of key structural elements that determine the distinct pharmacological profiles. This study provides a significant structural basis for understanding ligand binding and μ-OR activation by the 14-oxygenated N-methylmorphinan-6-ones, which should be useful for guiding drug design.
Keywords: μ-Opioid receptor, agonist, morphinan, ligand binding, molecular docking, structure−activity relationships
Introduction
Opioids, such as morphine (Figure 1), have a long history of clinical use as the most effective analgesic drugs for the alleviation of moderate to severe acute and chronic pain.1 Since the structure elucidation of morphine many years ago, its skeleton and its conversion to new analogues have been intensively explored.2,3 Consequently, the morphinan skeleton (Figure 1) has been the basis of numerous drug developments, and several molecules with distinctive pharmacology are available for patient use or employed as molecular probes in vitro and in vivo.2−4 The morphinan class of opioid analgesics includes naturally occurring alkaloids (e.g., morphine, codeine), semisynthetic derivatives (e.g., oxycodone, oxymorphone, buprenorphine), and synthetic analogues (e.g., levorphanol, butorphanol). They exert the analgesic action primarily via activation of the μ-opioid receptor (μ-OR).1,4 However, the desired analgesic effect is accompanied by undesirable side effects (e.g., respiratory depression, sedation, nausea, or constipation), and a considerable proneness to the development of tolerance and dependence is well-known, albeit most often associated with their long-term use.1,4,5 The μ-OR, as a member of the Family A (rhodopsin-like) G protein-coupled receptors (GPCRs) with a common seven transmembrane (7TM) helical architecture,6,7 has received constantly significant attention as a prominent drug discovery target toward pain treatment.
Figure 1.

Structures of morphine, the morphinan scaffold, and 14-oxygenated N-methylmorphinan-6-ones, along with sites targeted for derivatization (i.e., positions 5 and 14).
Accordingly, our long-standing interest and research efforts in the field of opioid analgesics from the class of morphinans led to innovative molecules with new substitution patterns and more favorable pharmacological properties, potent analgesia, and fewer undesirable effects.1,4,8,9 Modifications at position 14 of the morphinan skeleton were targeted and opened a new realm of prospects for drug discovery and development. Though naturally occurring opioids like morphine are unsubstituted at position 14 (Figure 1), strategies for functionalizing this site gave rise to pharmacologically attractive molecules. Moreover, position 5 in the morphinan scaffold is recognized to represent a feasible site for tuning functional activities by influencing interactions with opioid receptors in this class of ligands.8,9
Recent advances in structural biology integrating new methodological approaches together with the development of more and more powerful computational systems enabled the elucidation of X-ray crystal structures of many GPCRs in different conformations.10−12 GPCRs constitute the largest integral membrane protein family in the human genome and the most frequently targeted receptor class for therapeutic interventions.10−12 The emerged structural results are generally considered to be highly significant when a novel three-dimensional (3D) structure of a rhodopsin-like GPCR is reported, as they allow an atomic-level investigation of the structural features that promote ligand binding and selectivity and ultimately give insights into the mechanism(s) of receptor activation.13−15 Such 3D structures encompass distinctive conformations (i.e., the apo form and forms with ligands bound to the binding site of the GPCR as either agonists or antagonists),10,13,14 providing profound insight and nowadays allowing increased accuracy regarding computational modeling in GPCR drug design.10,11 The X-ray crystal structures of protein–ligand complexes reveal the location of the binding site, which was shown to be predominately located within the transmembrane region in the case of rhodopsin-like GPCRs, while also a certain variability was noticed.10,14 Deciphering the modifications that occur during activation revealed a subtle alteration in the binding site among members of this prominent GPCR family, a contraction in most cases, along with a gross and more uniform change in the conformation in the region forming the cytosolic surface.10,11,15 Furthermore, with increasing knowledge of the structural biology of the GPCRs, the essential role of distinct structural motifs, called molecular switches, in receptor activation was proposed.14,16 Distinct modifications in the relative orientations of the amino acid residues constituting these molecular switches are considered to be pivotal, as these changes are reflected to be accompanied by substantial alterations in receptor conformation and ultimately in its function. Specifically, ligand binding to the GPCR results in molecular switches disrupting stabilized intramolecular interactions, with notable examples including the tyrosine toggle switch, the ionic lock mimicking hydrogen, and the 3–7 lock.14,16,17 As the current understanding is mainly based on information from experimentally determined X-ray structures, 3D structures of GPCRs in distinct conformations are awaited with considerable interest, as they may open up new perspectives in structure-based drug design,12−14,17 including the thorough characterization of the role of molecular switches, which typically are composed of a few predominately conserved amino acid residues within the family of rhodopsin-like GPCRs.14,16
In 2012, the first X-ray crystal structure of the murine μ-OR was published (PDB ID 4DKL), in which the μ-OR is in complex with the irreversible morphinan antagonist β-funaltrexamine.6 Whereas this initial structure represented the receptor in an inactive state, current research takes advantage of the 3D structure of the μ-OR in the active conformation, reported in 2015, where the receptor is cocrystallized with the morphinan agonist BU72 (PDB ID 5C1M).7 Structure-based discovery campaigns have used the high-resolution μ-OR structures to computationally dock large libraries of molecules, seeking ligands with new chemotypes as well as to elucidate the mechanism(s) by which known ligands (i.e., peptides and small molecules) bind to the μ-OR and activate the receptor.18−21 In this study, we have addressed the active μ-OR structure for structure-based docking of μ-OR agonists from the class of N-methylmorphinan-6-ones. Encouraged by the interesting outcomes on the in vitro and in vivo pharmacology of N-methylmorphinan-6-ones differently substituted at positions 5 and 14 designed by our group,8,9 we report for the first time on an explorative structure–activity relationship (SAR) study aiming to gain mechanistic insights via molecular docking and molecular dynamics (MD) simulations of ligand/μ-OR interactions of these potent μ-OR agonists.
Results and Discussion
Structural Description and Pharmacology of 14-Oxygenated N-Methylmorphinan-6-ones
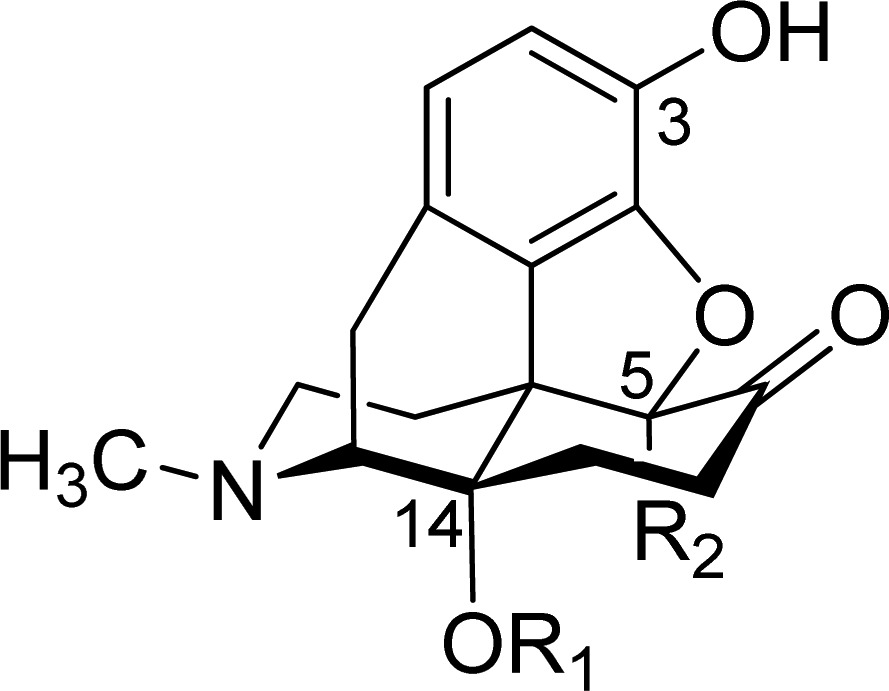
An outline of the substitution patterns, pharmacology, and key SARs toward identification of new μ-OR agonists from the class of N-methylmorphinan-6-ones as effective analgesics with reduced adverse effects is presented herein. The targeted 14-oxygenated N-methylmorphinan-6-ones in this study are listed in Table 1. In examining the specific functional groups at positions 14 and 5, we observed several important trends. We found that substitution of the hydroxyl group at position 14 of the clinically used μ-OR agonist oxymorphone (OM, 1) with a methoxy group, leading to 14-O-methyloxymorphone (14-OMO, 2a),22 not only increases the μ-OR affinity by ca. 9-fold but also results in ca. 40-fold improved antinociceptive potency (Table 1). However, compound 2a induces the typical opioid-like side effects.22−24 14-O-Ethyloxymorphone (14-OEO, 3a) (Table 1) displayed similar μ-OR affinity and selectivity compared to its 14-methoxy analogue 2a but exhibited reduced antinociceptive potency, although the latter was still ca. 300-fold higher than that of morphine.25 Replacement of the 14-methoxy group in 2a with a benzyloxy group resulted in 14-O-benzyloxymorphone (14-OBO, 4a),23 which retained the high affinity for the μ-OR, whereas its antinociceptive potency was comparable to that of 2a and ca. 50-fold and 700-fold greater compared with 1 (OM) and morphine, respectively (Table 1). The 14-benzyloxy substituted derivative 4a is a μ-OR agonist eliciting limited inhibition of gastrointestinal motility in mice at analgesic doses. It exhibited 2.5-fold less constipation than morphine, and it was ca. 7-fold less potent than 2a in this respect.23
Table 1. Structures, Binding Affinities and Selectivities for the μ-OR, and Antinociceptive Potencies of Oxymorphone (1) and Investigated 14-Oxygenated N-Methylmorphinan-6-ones (2–6)a.
| in vitro μ-OR bindingc |
|||||
|---|---|---|---|---|---|
| ligand | R1, R2b | μ-OR affinity (Ki, nM) | μ-OR selectivity vs δ-OR | μ-OR selectivity vs κ-OR | antinociceptive potencyd |
| OM (1) | H, H | 0.97 | 83 | 63 | 13,e 18,f 10g |
| 14-OMO (2a) | Me, H | 0.10 | 48 | 102 | 810,e 300,f 126,g |
| 14-MM (2b) | Me, Me | 0.15 | 89 | 168 | 99,e 82,f 94g |
| 14-OEO (3a) | Et, H | 0.15 | 60 | 91 | 316e |
| 14-EM (3b) | Et, Me | 0.46 | 26 | 94 | 46e |
| 14-OBO (4a) | Bz, H | 0.12 | 18 | 10 | 697g |
| 14-BM (4b) | Bz, Me | 0.18 | 20 | 14 | 103g |
| PPOM (5) | PhPr, Me | 0.20 | 0.7 | 2 | 2500,e 24000,f 8500g |
| BOMO (6) | Me, Bz | 0.31 | 42 | 73 | 53,f 50g |
Bz, benzyl; Et, ethyl; Me, methyl; PhPr, phenylpropyl.
Determined by in vitro radioligand binding assays with rat brain membranes.
Relative to morphine, determined in mice after s.c. administration using the indicated tests.
Acetic acid-induced writhing test.
Tail-flick test.
Hot-plate test.
Of particular attention is another oxymorphone analogue, 14-methoxymetopon (14-MM, 2b) (Table 1). Further chemical derivatization in the class of N-methylmorphinan-6-ones using 2a as the lead targeted position 5 by introducing a 5-methyl group, giving rise to the new μ-OR agonist 2b.26 14-MM maintained the high affinity for the μ-OR in the subnanomolar range shown by its 5-unsubstituted analogue 2a as well as μ-OR-mediated agonism, while showing increased μ-OR selectivity (Table 1). Pharmacologically, it is highly efficacious as a μ-opioid analgesic in various pain models in animals.23,24,27−30 Moreover, 2b was generally described to cause less pronounced opioid adverse actions in terms of respiratory depression,28 hypotension,28 bradycardia,28 constipation,29 physical dependence,27 addiction potential,27 and tolerance27,31 in comparison with conventional μ-opioid analgesics such as morphine. Similar observations on the induction of minimal physical dependence and less development of tolerance to analgesia compared with morphine were also made for its 14-ethoxy-substituted analogue 14-ethoxymetopon (14-EM, 3b).27
In view of the interesting in vitro and in vivo functional profile of 2b, our work in the field of opioid morphinans was directed toward the design of 14-MM analogues. Chemical work targeted 14-arylalkyloxy-substituted derivatives of 2b, resulting in 14-benzyloxymetopon (14-BM, 4b)23 and the 14-phenylpropoxy-substituted analogue (PPOM, 5)32 (Table 1). These derivatives bind with very high affinity to the μ-OR, comparable to that of 2b. The μ-OR selectivity was considerably decreased for the 14-benzyloxy derivative 4b, and a complete loss of μ-OR selectivity was observed for 5 (Table 1). The two 14-arylalkyloxy substituted morphinans 4b and 5 showed very high antinociceptive activity in mice.23,32 Remarkable was the observation that PPOM is an extremely potent agonist in vivo, with not only considerably improved analgesic potency compared with 2b (up to 400-fold) and morphine (up to 24000-fold) (Table 1) but also greater efficacy even than etorphine (up to 25-fold),32 a μ-OR agonist used in veterinary medicine for anesthesia.
The switch from a methyl group (2b) to a benzyl group at position 5 resulted in analogue 6 (BOMO)24 (Table 1). This exchange left the affinity for the μ-OR largely unchanged (Ki = 0.15 nM for 2b vs 0.31 nM for 6) and produced only a modest decrease (2-fold) in analgesic potency (Table 1). Behavioral studies showed that contrary to morphine, 14-OMO (2a), and 14-MM (2b), no significant alterations in motor activity were induced by 6 at analgesic doses.24 These observations indicated that replacing the 5-methyl group in 2b with a benzyl group led to a potent antinociceptive agent with reduced propensity to cause unwanted motor impairment.
Model for Binding of 14-Oxygenated N-Methylmorphinan-6-ones to the μ-OR: Molecular Docking, Molecular Dynamics, and SAR Exploration
In order to rationalize our above-described empirical SAR observations in the series of 14-oxygenated N-methylmorphinan-6-ones 1–6 (Table 1), we explored the ligand/μ-OR interactions and dynamics of the μ-OR upon ligand binding. We conducted molecular modeling studies by means of docking and MD simulations, with the latter being nowadays an important methodology when studying GPCR versatility associated with functioning and ligand recognition.12,33 In this study, following MD simulations, the snapshots collected during the last 10% of a 1 ns trajectory were analyzed in order to elucidate which functional groups in the targeted morphinans 1–6 form intermolecular hydrogen bonds and to characterize the propensity of these moieties to be involved in polar interactions. The influence on an important molecular switch was also examined, specifically, the 3–7 lock, a link between TM3 and TM7 characterized as a hydrogen-bonding interaction formed between D1473.32 and Y3267.43 in the μ-OR.14,34,35 Opening of the 3–7 lock was earlier proposed by the group of Khorana to be the first switch activated in rhodopsin, and possibly it is one of the first switches that can be activated upon ligand binding in some other rhodopsin-like GPCRs.35 In the case of the μ-OR, this switch was predicted to be considerably affected upon binding of an agonist, resulting in breaking of the hydrogen bond.34
Our computational approach was initiated by docking of oxymorphone (1) into the binding pocket of the active structure of the murine μ-OR. In line with in vitro experimental data on the high μ-OR affinity (Ki = 0.97 nM),23 the results of the docking study revealed that 1 binds to the μ-OR in a highly favorable manner with all of the predicted interaction patterns (Table 2). The polar group at position 3 of 1 is involved in a hydrogen-bonding network formed with a few water molecules that are present in the binding pocket and mediate interactions from the ligand to the receptor via H2976.52. When we overlaid the docking solution for 1 with the experimentally determined pose of BU72,7 a very good overlap was noticed for the 3-phenol group, the ligand moiety proposed to be involved in a hydrogen-bonding network (Figure 2). This water-mediated hydrogen-bonding interaction with H2976.52 is seemingly common to ligands in the morphinan class.7 Our docking study also revealed that the hydroxyl group at position 14 in 1 forms a hydrogen bond with D1473.32, while as well the involvement in a charge-enhanced hydrogen bond was predicted, which again is formed to D1473.32. In this case, the polar interaction is made by the basic nitrogen. Additionally, the aromatic ring is embedded by several hydrophobic residues (M1513.36, V2365.42, I2966.51, and V3006.55), that are responsible for hydrophobic interactions (Table 2 and Figures 3A and 4A). Interestingly, during the MD simulations, we observed a characteristic for 1 that was not found among the other morphinan ligands, as additional polar contacts were formed by the 3-phenol group with considerable tendency (Table 3). Furthermore, these results indicated that the 3–7 lock switch is not broken in the presence of 1, which is comparable to the other investigated molecules 2–6 in this class of morphinans.
Table 2. Ligand/μ-OR Interaction Pharmacophores Inferred from Molecular Docking Solutions of Oxymorphone (1) and the Investigated 14-Oxygenated N-Methylmorphinan-6-ones (2–6).
| hydrophobic
interactions |
hydrogen bonds |
|||
|---|---|---|---|---|
| ligand | inferred from the phenol | inferred from the introduced group | charge-enhanced hydrogen bond | interactions mediated by water molecules |
| OM (1) | M151, V236, I296, V300 | NDa | D147 | H297 |
| 14-OMO (2a) | M151, V236, I296, V300 | ND | D147 | H297 |
| 14-MM (2b) | M151, V236, I296, V300 | ND | D147 | H297 |
| 14-OEO (3a) | M151, V236, I296, V300 | ND | D147 | H297 |
| 14-EM (3b) | M151, V236, I296, V300 | I322 | D147 | H297 |
| 14-OBO (4a) | M151, V236, I296, V300 | I144 | D147 | H297 |
| 14-BM (4b) | M151, V236, I296, V300 | I144 | D147 | H297 |
| PPOM (5) | M151, V236, I296, V300 | W133, V143, I144 | D147 | H297 |
| BOMO (6) | M151, V236, I296, V300 | I322 | D147 | H297 |
ND denotes not deduced.
Figure 2.

Docking pose of oxymorphone (1) (cyan) compared with that of the agonist BU72 (magenta) bound to the active μ-OR crystal structure. Both ligands are oriented in the binding pocket in a way that the phenolic hydroxyl groups overlay very well, and interact via water molecules with H2976.52. Amino acid residues involved in intermolecular hydrogen bonds, i.e., D1473.32 and H2976.52, are shown. Hydrogen bonds are depicted as green dashed lines, and the binding pocket surface is color-coded according to interpolated charge (blue/red = positive/negative).
Figure 3.

Binding modes presented for the investigated 14-oxygenated N-methylmorphinan-6-ones: (A) 1 (OM), (B) 2a (14-OMO; X = H) and 2b (14-MM; X = Me), (C) 3a (14-OEO; X = H; 14-O-Et group shown with dashed lines) and 3b (14-EM; X = Me), (D) 4a (14-OBO; X = H) and 4b (14-BM; X = Me), (E) 5 (PPOM), and (F) 6 (BOMO). The binding pocket amino acid residues predicted to be involved in critical non-covalent interactions are also shown. Me, methyl; Et, ethyl. Amino acid residues are labeled using one-letter amino acid codes.
Figure 4.

Docking of the investigated 14-oxygenated N-methylmorphinan-6-ones to the active crystal structure of the μ-OR. Shown are the binding poses of (A) 1 (OM, c), (B) 2a (14-OMO, c) and 2b (14-MM, b), (C) 3a (14-OEO, c) and 3b (14-EM, b), (D) 4a (14-OBO, c) and 4b (14-BM, b), (E) 5 (PPOM, c), and (F) 6 (BOMO, c), where c/b denotes cyan/blue. Hydrogen bonds are depicted as green dashed lines, and the binding pocket surface is color-coded according to the interpolated charge (blue/red = positive/negative).
Table 3. Results from MD Trajectories Inspecting the Propensity To Form Intermolecular Hydrogen Bonds of Oxymorphone (1) and the Investigated 14-Oxygenated N-Methylmorphinan-6-ones (2–6).
| basic nitrogen |
4,5α-epoxy group | 3-phenol group |
|||||
|---|---|---|---|---|---|---|---|
| ligand | D147 | Y326 | Y148 | H54 | Y148 | K233 | K303 |
| OM (1)a | 82% | 4% | 0% | 82% | 0% | 0% | 60% |
| 14-OMO (2a) | 78% | 40% | 20% | 0% | 0% | 0% | 0% |
| 14-MM (2b) | 82% | 32% | 6% | 0% | 0% | 0% | 0% |
| 14-OEO (3a) | 76% | 18% | 2% | 0% | 0% | 0% | 0% |
| 14-EM (3b) | 86% | 14% | 0% | 0% | 0% | 0% | 0% |
| 14-OBO (4a) | 46% | 18% | 8% | 0% | 0% | 0% | 0% |
| 14-BM (4b) | 80% | 8% | 0% | 0% | 0% | 0% | 0% |
| PPOM (5) | 56% | 26% | 10% | 0% | 6% | 2% | 0% |
| BOMO (6) | 16% | 74% | 34% | 0% | 38% | 4% | 0% |
Truncated, as intermolecular hydrogen bonds formed by the 14-hydroxyl group are not included (as this moiety is unique to OM and not present in the other molecules of the investigated morphinan class).
Next, to attain insights into the ligand/μ-OR interaction patterns of targeted 14-O-alkyl- and 14-O-arylalkyl-substituted oxymorphone derivatives 2–6 designed and synthesized in our laboratory (Table 1), we directly docked them into the empty binding pocket of the active receptor. A summary of the identified ligand/μ-OR interactions is shown in Table 2. Whereas derivatives 2–6 showed similar binding modes, we also observed fairly substantial predicted interaction pattern differences related to specific structural features. Our docking strategy involved SAR examinations of the influence of the substituents at positions 5 and 14 on ligand binding.
Analogous to the parent compound oxymorphone (1), all of the active ligands 2–6 at the μ-OR form a charged interaction with D1473.32 and a hydrogen bond to H2976.52 via a water network (Table 2). We also observed that the presence of small 14-alkoxy groups, such as methoxy and ethoxy (2a and 3a, respectively), did not substantially alter the ligand/receptor interaction outline compared to 1, as the polar contacts of these ligands were comparable to those of 1 except for the hydrogen bond formed by the 14-hydroxyl group of 1 to D1473.32. This is a unique characteristic, as in the other molecules this polar group was modified during derivatization. Intriguingly, in the case of the 14-benzyloxy-substituted derivative 4a (14-OBO), which displayed potent analgesia while being less constipative in mice,23 the intermolecular hydrogen bonds were predicted to be maintained on the basis of a comparison of the proposed binding mode of this analogue to the docking solution for compound 2a (14-OMO). In addition, a further hydrophobic interaction to I1443.29 could be formed by 4a (Table 2). Thus, the importance of the nature of the substituent at position 14 (i.e., alkyl vs arylalkyl) for the binding to the receptor was evident not only from the SAR assessment within the series of compounds 2a–4a but also for 2b–4b. Morphinan 5 carrying a bulky group (i.e., phenylpropoxy) at position 14 was recognized to present additional ligand/μ-OR interactions, as listed in Table 2.
The relevance of the substitution pattern at position 5 to the binding mode of the investigated morphinans was examined with interesting SAR outcomes. In our previous in vitro activity studies, we found that replacement of the hydrogen (2a–4a) with a methyl group at position 5 (2b–4b) has a minor influence on the binding affinity to the μ-OR. An improved selectivity for the μ-OR over the δ-OR and κ-OR was shown by 2b compared with 2a. In the case of the 14-ethoxy analogues 3a and 3b, the μ-OR versus δ-OR selectivity was decreased for 5-methyl-substituted 3b, while the μ-OR versus κ-OR selectivity remained unchanged. From a comparison of 14-benzyloxy-substituted 4a and 4b, it was apparent that a methyl group at position 5 does not considerably alter the μ-OR selectivity over the other receptor subtypes (Table 1). Additionally, the exchange of H for Me at position 5 (2a vs 2b) results in a marked improvement in the side-effect profile.27−29,31 Thus, we noticed that the function of the receptor following ligand/μ-OR interaction and activation is affected by analogues of the two corresponding series 2a–4a and 2b–4b in a different manner, as ligands from the latter series are somewhat less effective concerning analgesic potency (Table 1). This may be a consequence of the different orientation relative to the receptor due to the additional group at position 5. In turn, this could result in orientations of certain ligand groups relative to the receptor that are as not optimal for the formation of non-covalent interactions between the ligand and the receptor. Specifically, the proposed binding modes of 2a (14-OMO) and 2b (14-MM) differed in their relative orientation, although the observed differences were minor, when we accounted for the non-covalent interactions (Table 2 and Figure 3B). The observed difference in the analgesic potency is considerable in the case of these two analogues, which differ only in the substituent at position 5 (Table 1). Thus, we recognized that the variation in the relative orientation appears to have an important influence on the analgesic potency (Figure 4B). Then when we evaluated the MD simulation outcomes, a subtle difference between these potent N-methylmorphinan-6-ones was observed, indicating that 2a forms a hydrogen bond between the 4,5α-epoxy group and Y1483.33, which was inferred in ca. 20% of the snapshots. Contrarily, the propensity to form a polar contact to this amino acid residue was comparatively marginal for 2b (Table 3).
When comparing docking of 3a (14-OEO) and the corresponding analogue with a 5-methyl group, 3b (14-EM), we noticed that in 3a the 14-O-ethyl group was not oriented in a way that a hydrophobic interaction could be formed, in contrast to 3b, which interacted with I3227.39 (Table 2 and Figures 3C and 4C). This result is in agreement with the ca. 3-fold affinity difference between the in vitro binding affinities and analgesic potencies of 3a and 3b (Table 1). Examination of 4a (14-OBO) and 4b (14-BM) revealed that the 14-O-benzyl group was in both cases oriented in such a manner that this moiety was involved in a hydrophobic interaction with I1443.29 (Table 2 and Figure 3D). Again, the difference in the relative orientation seems to have a substantial influence (Figure 4D), accounting for the reported analgesic potency difference between 4a and 4b (Table 1). Interestingly, following the analysis with MD simulations, alterations in the non-covalent interactions also became evident, as we observed that 4a forms a hydrogen bond to D1473.32 with comparatively low susceptibility, whereas 4b was predicted to bind to the μ-OR by forming a tight interaction with this residue. Furthermore, rather similar to the situation identified for the 14-ethoxy analogues (3a and 3b), we observed that a polar contact was formed to Y1483.33 in the case of 4a whereas not by 4b (Table 3).
Hence, the 5-methyl group introduced during targeted derivatization has a considerable influence on the ligand/μ-OR interactions (e.g., it results in enhanced hydrophobic ligand/receptor interactions and reduced analgesic potency), as the orientation of the oxymorphone derivatives relative to the receptor is influenced by this modification, which we consider as crucial. This may influence the orientation of further groups (i.e., at position 14) that are predicted to interact favorably with the μ-OR in some cases but not in others (Table 2). In addition, our results suggest that the susceptibility to affect the 3–7 lock switch upon agonist binding to the receptor may serve as a possible explanation for the reduced antinociceptive potency, as the tendency to affect this molecular switch exhibited by the 5-unsubstituted analogues (2a–4a) with high antinociceptive potency was comparatively low (50–76%), whereas the corresponding 5-methyl-substituted analogues (2b–4b) with decreased antinociceptive potency affected this molecular switch with comparatively high tendency (80–86%) (Figure 5 and Table 1). The characteristics of 5 and 6 are outlined below, including distinctive characteristics concerning their probability to affect the 3–7 lock switch. Another structural characteristic that appears to be critical and may contribute to the difference in the analgesic potency includes the hydrogen bond to Y1483.33. In case of the 14-methoxy analogues, we observed that this hydrogen bond was made to Y1483.33 by both analogues, the 5-unsubstituted 2a and the 5-methyl-substituted 2b (Table 3). Figure 6 shows a snapshot from the end of the MD trajectory in which the probability of intermolecular hydrogen bonding was evaluated for compound 2b (14-MM). As the difference in the analgesic potency is comparatively moderate (Table 1), this aspect seemingly influences the analgesic activity, albeit by evening out the difference in this case. In case of the 14-ethoxy and 14-benzyloxy analogues, our results indicated that the situation is different. However, a polar interaction could be observed with the 5-H derivatives (3a and 4a) but not the 5-methyl-substituted ones (3b and 4b) (Table 3), apparently contributing to the difference in the analgesic potency, which is fairly marked in case of these analogues (Table 1). Notably, compound 2b (14-MM), considered as a highly promising μ-OR agonist, exhibits improved tolerability, albeit with reduced analgesic potency. This decline in the antinociceptive potency shown by the 5-methyl-substituted analogues (2b–4b) may be a consequence of the higher tendency to affect the 3–7 lock switch (Figure 5). Interestingly, further light was shed on these aspects following the analyses of MD trajectories, as the emerged results indicated that the tendency to form a hydrogen bond between the 4,5α-epoxy group and Y1483.33 was not diminished in case of 2b (14-MM) but was in the case of the other 5-methyl substituted analogues, compounds 3b and 4b (Table 3). In turn, this may positively influence the ratio of analgesic potency to side effects, as we assume that this characteristic is highly relevant, evening out the difference regarding the analgesic potency in this case. These two characteristics together may account for the improved tolerability that was noticed for 2b.
Figure 5.
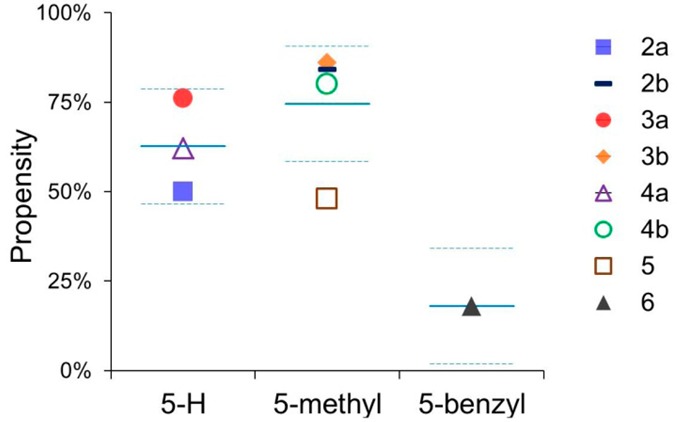
Tendencies to affect the 3–7 lock switch for derivatives with a 5-H (2a–4a), 5-methyl (2b–4b, 5), or 5-benzyl group (6). The mean (±1 standard deviation) is delineated with blue solid (dashed) lines.
Figure 6.
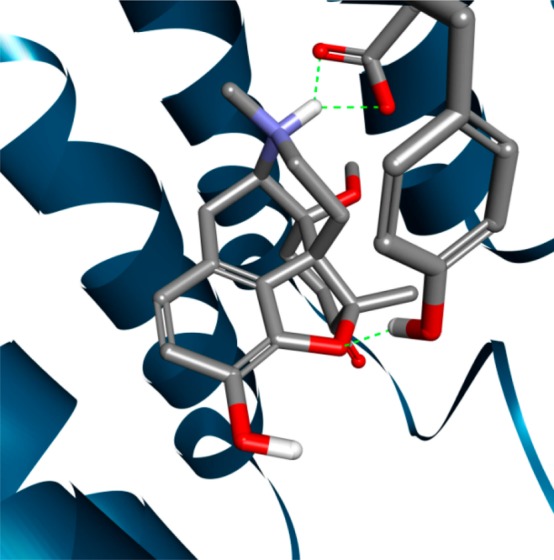
Compound 2b (14-MM) is depicted by showing a snapshot from the end of the MD trajectory, which was calculated to scrutinize the propensity to form intermolecular hydrogen bonds between the agonist and the μ-OR. Amino acid residues involved in polar interactions are illustrated (i.e., D1473.32 and Y1483.33) along with intermolecular hydrogen bonds (green dashed lines).
The 14-phenylpropoxy-5-methyl-substituted derivative 5 (PPOM) was reported by our group to have an extremely high antinociceptive potency,32 even greater than that of etorphine, a highly active opioid analgesic used in veterinary medicine to immobilize large animals. However, the selectivity profile was altered in an unfavorable manner, as the μ-OR selectivity was significantly reduced compared with those of the other 5-methyl-substituted analogues 2b–4b because of the introduction of the 14-phenylpropoxy group (Table 1). The in vitro and in vivo activity profiles of 5 as a high-affinity μ-OR agonist and one of the most effective opioids regarding its analgesic potency is consistent with the docking pose (Figure 4E). We detected that a further hydrophobic region was targeted, embedding the phenyl group of 5 in this region formed by hydrophobic or aromatic residues (i.e., W133, V1433.28, and I1443.29) (Table 2 and Figure 3E). In addition to the strong analgesic activity previously demonstrated for 5,32 we evaluated its propensity to interact with the conserved D1473.32 and detected that the trend to form a charge-enhanced hydrogen bond to this amino acid residue was only moderate (Table 3). Notably, we found that the probability of 5 to affect the 3–7 lock switch was comparatively low, comparable to that of 2a (14-OMO), representing an exception among the 5-methyl-substituted analogues (Figure 5). We thereby noticed that this finding is in agreement with our hypothesis, as both morphinans show very high (2a) or even remarkable analgesic potency (5) (Table 1).
In the SAR exploration on how the nature of the substituent at position 5 affects the ligand/μ-OR interaction profile, we compared 5-H (2a), 5-methyl (2b), and 5-benzyl (6) derivatives. Although they all exhibit similar μ-OR binding affinities (Table 1) and polar interactions with D1473.32 and with H2976.52 via a water network, other important ligand–receptor contacts are different or unexpected for 6 (BOMO) (Table 2 and Figures 3F and 4F), as especially evidenced by the analysis of MD simulations. We noticed that this potent μ-agonist interacts with D1473.32, albeit with reduced proneness, while the polar contact to the nearby Y3267.43 was formed rather frequently, which is a unique characteristic of 6. Furthermore, 6 shows a notable tendency to form a hydrogen bond to Y1483.33, by either the 3-phenol group, the 4,5α-epoxy group, or both during 60–70% of the MD trajectory (Table 3). Together with a comparatively low propensity to affect the 3–7 lock switch (Figure 5), these results indicated that 6 adopts a unique binding pose in the μ-OR distinct from those of the other investigated N-methylmorphinan-6-ones. However, we observed that the basic nitrogen reorients in an unexpected manner, moving toward Y3267.43 and away from the conserved D1473.32, the counterpart in the charge-enhanced hydrogen bond, which is (more or less) common among the other investigated opioid ligands (Table 3) and which basically is sought to consider the molecule promising, as polar core moieties should be complementary to the environment.
Conclusions
The newly available high-resolution structure of the μ-OR in the active state made it possible to analyze the SAR of targeted 14-oxygenated N-methylmorphinan-6-ones (1–6) by enabling the identification of key elements in the chemical structure that determine their pharmacological profiles. We have presented the first thorough molecular modeling study aided by docking and MD simulations of μ-OR agonists 1–6. This SAR analysis represents a basic approach that sheds light on the subtle interplay between substituents at positions 5 and 14 in the morphinan scaffold. The results from molecular docking indicate the crucial role of the relative orientation of the ligand in the μ-OR binding site, thus influencing the propensity of critical non-covalent interactions that are required to facilitate ligand/μ-OR interactions and receptor activation. MD simulations were utilized in order to rationalize the relevance of functional groups attached during optimization efforts as well as their interrelated dependence. We inspected molecular characteristics that could provide a clarification of the differences in ligand binding and analgesic potency. Our observations pointed toward the differences in the tendency to affect the 3–7 lock switch by the investigated morphinans, therefore providing grounds for an explanatory interaction mechanism to the μ-OR. Among the analogues unsubstituted at position 5, compounds 2a–4a, a relatively low tendency was observed, whereas a higher tendency was marked between the corresponding 5-methyl-substituted derivatives, compounds 2b-4b, for which reduced analgesic potency was noticed. In turn, the variation was comparatively small in the case of compound 2b (14-MM) regarding its analgesic potency. This may be explained by a second critical characteristic, as the propensity of a hydrogen bond formed by the 4,5α-epoxy group was minor, though not diminished in this case. Upon the introduction of a 14-phenylpropoxy group, the resulting μ-OR agonist, PPOM (5), showed remarkable analgesic potency. Docking of 5 to the μ-OR revealed that the bulky group at position 14 was favorably accommodated in a hydrophobic region of the binding site, hence explaining its remarkable antinociceptive activity. Explorations of 1–6 also allowed us to define an additional SAR at the key position 5 in the morphinan scaffold. The 5-benzyl-substituted analogue 6 adopted a unique binding mode in the μ-OR, distinct from the other investigated 14-oxygenated N-methylmorphinan-6-ones. The structure of the activated μ-OR provides a valuable model for how ligand/μ-OR binding is encoded within small chemical differences in otherwise structurally similar morphinan ligands. Altogether, our current analysis of the SAR that emerged from molecular modeling investigations on morphinans 1–6 confirms the SAR derived from pharmacological assessments, thereby providing a molecular understanding for their μ-OR activities, and opens up opportunities for structure-based design and discovery of new analgesics with improved pharmacological profiles and enhanced therapeutic efficacies.
Methods
Hardware and Software Specifications
All of the molecular modeling studies were conducted on a workstation running the Windows 7 operating system and equipped with an Intel Xeon E5-1607 v3 CPU, 32 GB of RAM, and high-end NVIDIA graphic devices (Quadro K620 and Quadro K5000). For molecular docking, preparation of ligands and μ-OR protein was conducted using Discovery Studio (version 3.0),36 and the docking was run by employing GOLD version 5.2.37,38 This was followed by optimization of the docking poses conducted with the Discovery Studio. The docking solutions that were retained were evaluated with LigandScout (version 3.1).39 MOPAC201640 was employed to perform calculations with a semiempirical quantum-mechanical method. For the calculation of MD simulations, we utilized the appropriate protocols within Discovery Studio.36
Ligand Preparation
The protonation state of the investigated morphinans was manually adjusted, assigning the basic nitrogen a positive charge, which was followed by the generation of conformational models. Hereto, the more exhaustive “BEST” method with a maximum of 10 conformers per molecule was employed,41 along with a variant of the generalized Born model (generalized Born with molecular volume, GBMV42) in order to account for solvent effects.
Molecular Docking
The X-ray crystal structure of the murine μ-OR in the active conformation (PDB ID 5C1M)7 was downloaded from the Protein Data Bank (http://rcsb.org/pdb), and the protein was prepared by employing the protocol “prepare protein”, followed by the adaptation of two issues, as the protonation state of H54 was assigned as neutral, accounting for chemical intuition, while the acidic group of D1473.32 (superscripts indicate Ballesteros and Weinstein numbering43) was adjusted to the ionized form.44,45 As non-covalent interactions to the conserved D1473.32 have a pivotal role,44,45 this amino acid residue was assigned as a constraint directing the ligand placement. The previous observations of Huang et al.7 suggested that a few water molecules are of relevance, as a hydrogen-bonding network from the receptor to the cocrystallized ligand BU72 was assumed to be mediated by those water molecules. Therefore, during the docking procedure the docking protocol was adapted to account for these water molecules, which surround the phenol moiety of BU72 (HOH505, HOH526, and HOH538), by assigning to them a versatile state (“toggle and spin”). Afterward, the prepared opioid ligands were docked into the binding pocket of the μ-OR, defined by protein residues surrounding the cocrystallized ligand BU72 within a radius of 6 Å, with up to 10 conformers per ligand and 15 runs per molecule. In addition, enhanced ligand flexibility was employed during the docking runs (“flip pyramidal N”). This was then followed by retaining the three docking solutions per ligand and conformer that were prioritized by the docking score, obtained using the ChemPLP function,46 for which reasonable docking and scoring performance was suggested;47,48 the consistency of the results was pointed out utilizing the program GOLD.49
Postprocessing and Evaluation
Docking solutions were next submitted to pose optimization using Discovery Studio. We employed the module CDOCKER50 in “full potential mode” along with the CHARMM force field.51 Furthermore, a distinct variant for the estimation of partial atomic charges was employed in the case of the investigated opioid ligands (i.e., by assigning charges from the CFF force field52), while in the case of the valuable and substantial μ-OR structure the semiempirical quantum-mechanical method PM753 was utilized. Finally, the 3D pharmacophores taken into account for the evaluation were inferred using LigandScout.39
Validation
BU72, the morphinan μ-OR ligand of the protein cocrystal active structure,7 was extracted, and following the preparation of this molecule in a comparable manner as for the other molecules (as atoms/groups were adjusted manually, which are ionized at physiological pH, and which was followed by the generation of a conformational model), BU72 was then docked into the binding pocket of the μ-OR, followed by a comparison of the docking solutions to the experimentally determined pose. Following redocking experiments, the results were evaluated with the calculation of RMSD values for the highest-ranked pose and the best pose along with the average RMSD of all of the retrieved poses. Thereby, reasonable performance of the docking protocol was suggested (RMSD of the highest-ranked and best pose = 0.870 Å; average RMSD = 0.917 Å), while further improvement was obtained by submitting the docking poses to pose optimization (RMSD of the highest-ranked and best poses = 0.546 and 0.515 Å, respectively; average RMSD = 0.582 Å).
Molecular Dynamics (MD) Simulations
The investigated morphinans and μ-OR complexes resulting from molecular docking and the pose optimization were submitted to MD simulations. Through the application of MD simulations, the insight gained through the molecular docking was extended, as MD is considered to serve as a more profound basis for molecular analysis of ligand recognition.54,55 We analyzed in more depth the non-covalent interactions formed between the targeted ligands and the active μ-OR, which was seen as the case when the propensity of intermolecular hydrogen bonding was taken into account. The snapshots collected during the last 10% of a 1 ns trajectory were evaluated following the MD simulations. The retained snapshots were inspected to elucidate which groups of morphinan ligands form intermolecular hydrogen bonds and to characterize the propensities of these ligand moieties to be involved in polar contacts. Calculations were performed with the CHARMM force field51 along with the SHAKE algorithm.56 The protocol “standard dynamics cascade” was utilized, and following the assignment of constraints, calculations were performed holding the protein outside the binding pocket rigid, by taking into consideration recommendations made for this type of calculation.57 We selected a suitable variant to efficiently analyze the MD trajectories, as solvent effects were taken into account with a relatively fast variant of implicit treatment (distance-dependent dielectric constants; dielectric constant = 4). The frequency of snapshots collected during the MD simulations was increased from 100 fs (default) to 2 ps, while other parameters were not adjusted (target temperature = 300 K; time step = 1 fs). Afterward, the evaluation was accomplished by employing the protocol “analyze trajectory”, which involved appropriate monitoring tools (i.e., intermolecular H-bonds and H-bonds).
Glossary
Abbreviations
- 14-BM
14-benzyloxymetopon
- 14-EM
14-ethoxymetopon
- 14-OBO
14-O-benzyloxymorphone
- 14-OEO
14-O-ethyloxymorphone
- 14-OMO
14-O-methyloxymorphone
- 14-MM
14-methoxymetopon
- BOMO
5-benzyl-14-o-methyloxymorphone
- GPCR
G protein-coupled receptor
- Ki
inhibition constant
- MD
molecular dynamics
- OM
oxymorphone
- OR
opioid receptor
- PPOM
14-phenylpropoxymetopon
- SAR
structure–activity relationship
- TM
transmembrane
Author Contributions
M.S. and H.S. designed the study. S.M.N. conceived and performed the experiments. All of the authors analyzed the data and discussed the results. The manuscript was written through contributions of all of the authors. All of the authors read and approved the final manuscript.
This work was partly supported by grants (all to M.S.) from the Austrian Science Fund (FWF) (TRP19-B18), the Tyrolean Research Fund (TWF) (UNI-0404/1596), the Förderungsbeiträge Aktion D. Swarovski KG 2014, and the Bilateral Cooperation Program Austria-France “Amadée” (FR 12/2016).
The authors declare no competing financial interest.
References
- Pasternak G. W.; Pan Y. X. (2013) Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 65, 1257–1317. 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst S.; Hosztafi S. (2008) The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 95, 3–44. 10.1556/APhysiol.95.2008.1.1. [DOI] [PubMed] [Google Scholar]
- Schmidhammer H.; Spetea M. (2010) Synthesis of 14-alkoxymorphinans and their pharmacological activities. Top. Curr. Chem. 299, 63–91. 10.1007/128_2010_77. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Asim M. F.; Wolber G.; Schmidhammer H. (2013) The μ opioid receptor and ligands acting at the μ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 19, 7415–7434. 10.2174/13816128113199990362. [DOI] [PubMed] [Google Scholar]
- Benyamin R.; Trescot A. M.; Datta S.; Buenaventura R.; Adlaka R.; Sehgal N.; Glaser S. E.; Vallejo R. (2008) Opioid complications and side effects. Pain Physician 11, 105–120. [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spetea M.; Schmidhammer H. (2012) Recent advances in the development of 14-alkoxy substituted morphinans as potent and safer opioid analgesics. Curr. Med. Chem. 19, 2442–2457. 10.2174/092986712800269308. [DOI] [PubMed] [Google Scholar]
- Schmidhammer H.; Spetea M. (2012) Development of 5-substituted n-methylmorphinan-6-ones as potent opioid analgesics with improved side-effect profile. Int. J. Med. Chem. 2012, 1–12. 10.1155/2012/208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke R. M.; Brown A. J. H.; Marshall F. H.; Mason J. S. (2015) Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discovery Today 20, 1355–1364. 10.1016/j.drudis.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A. (2015) New paradigms in GPCR drug discovery. Biochem. Pharmacol. 98, 541–555. 10.1016/j.bcp.2015.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobb F. M.; Negri A.; Beuming T.; Sherman W. (2016) Molecular dynamics techniques for modeling G protein-coupled receptors. Curr. Opin. Pharmacol. 30, 69–75. 10.1016/j.coph.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Tesmer J. J. (2016) Hitchhiking on the heptahelical highway: structure and function of 7TM receptor complexes. Nat. Rev. Mol. Cell Biol. 17, 439–450. 10.1038/nrm.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski B.; Latek D.; Yuan S.; Ghoshdastider U.; Debinski A.; Filipek S. (2012) Action of molecular switches in GPCRs – theoretical and experimental studies. Curr. Med. Chem. 19, 1090–1109. 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhoff K. J.; Shukla D.; Lawrenz M.; Bowman G. R.; Konerding D. E.; Belov D.; Altman R. B.; Pande V. S. (2014) Cloud-based simulations on google exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 6, 15–21. 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T. W.; Frimurer T. M.; Holst B.; Rosenkilde M. M.; Elling C. E. (2006) Molecular mechanism of 7TM receptor activation - a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519. 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Bruno A.; Costantino G. (2012) Molecular dynamics simulations of G protein-coupled receptors. Mol. Inf. 31, 222–230. 10.1002/minf.201100138. [DOI] [PubMed] [Google Scholar]
- Cui X.; Yeliseev A.; Liu R. (2013) Ligand interaction, binding site and G protein activation of the mu opioid receptor. Eur. J. Pharmacol. 702, 309–315. 10.1016/j.ejphar.2013.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaserer T.; Lantero A.; Schmidhammer H.; Spetea M.; Schuster D. (2016) μ Opioid receptor: novel antagonists and structural modeling. Sci. Rep. 6, 21548. 10.1038/srep21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hübner H.; Huang X.-P.; Sassano M. F.; Giguère P. M.; Löber S.; Da D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. (2016) Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel A. C.; Gassaway M. M.; Kapoor A.; Váradi A.; Majumdar S.; Filizola M.; Javitch J. A.; Sames D. (2016) Synthetic and receptor signaling explorations of the Mitragyna alkaloids: Mitragynine as an atypical molecular framework for opioid receptor modulators. J. Am. Chem. Soc. 138, 6754–6764. 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhammer H.; Aeppli L.; Atwell L.; Fritsch F.; Jacobson A. E.; Nebuchla M.; Sperk G. (1984) Synthesis and biological evaluation of 14-alkoxymorphinans. 1. Highly potent opioid agonists in the series of (−)-14-methoxy-N-methylmorphinan-6-ones. J. Med. Chem. 27, 1575–1579. 10.1021/jm00378a009. [DOI] [PubMed] [Google Scholar]
- Lattanzi R.; Spetea M.; Schüllner F.; Rief S. B.; Krassnig R.; Negri L.; Schmidhammer H. (2005) Synthesis and biological evaluation of 14-alkoxymorphinans. 22. Influence of the 14-alkoxy group and the substitution in position 5 in 14-alkoxymorphinan-6-ones on in vitro and in vivo activities. J. Med. Chem. 48, 3372–3378. 10.1021/jm040894o. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Bohotin C. R.; Asim M. F.; Stübegger K.; Schmidhammer H. (2010) In vitro and in vivo pharmacological profile of the 5-benzyl analogue of 14-methoxymetopon, a novel μ opioid analgesic with reduced propensity to alter motor function. Eur. J. Pharm. Sci. 41, 125–135. 10.1016/j.ejps.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhammer H.; Krassnig R. (1990) Synthesis and biological evaluation of 14-alkoxymorphinans. 6. 14-O-Ethyloxymorphone (= (−)-4,5α-epoxy-14-ethoxy-3-hydroxy-N-methylmorphinan-6-one). Sci. Pharm. 58, 255–257. [Google Scholar]
- Schmidhammer H.; Schratz A.; Mitterdorfer J. (1990) Synthesis and biological evaluation of 14-alkoxymorphinans. 8. 14-Methoxymetopon, an extremely potent opioid agonist. Helv. Chim. Acta 73, 1784–1787. 10.1002/hlca.19900730623. [DOI] [Google Scholar]
- Fürst S.; Búzás B.; Friedmann T.; Schmidhammer H.; Borsodi A. (1993) Highly potent novel opioid receptor agonist in the 14-alkoxymetopon series. Eur. J. Pharmacol. 236, 209–215. 10.1016/0014-2999(93)90591-5. [DOI] [PubMed] [Google Scholar]
- Freye E.; Schmidhammer H.; Latasch L. (2000) 14-Methoxymetopon, a potent opioid, induces no respiratory depression, less sedation, and less bradycardia than sufentanil in the dog. Anesth. Analg. 90, 1359–1364. 10.1097/00000539-200006000-00018. [DOI] [PubMed] [Google Scholar]
- King M. A.; Su W.; Nielan C.; Chang A. H.; Schütz J.; Schmidhammer H.; Pasternak G. W. (2003) 14-Methoxymetopon, a very potent μ-opioid analgesic with an unusual pharmacological profile. Eur. J. Pharmacol. 459, 203–209. 10.1016/S0014-2999(02)02821-2. [DOI] [PubMed] [Google Scholar]
- Bileviciute-Ljungar I.; Spetea M.; Guo Y.; Schütz J.; Windisch P.; Schmidhammer H. (2006) Peripherally mediated antinociception of the μ-opioid receptor agonist 2-[(4,5α-epoxy-3-hydroxy-14β-methoxy-17-methylmorphinan-6β-yl)amino]acetic acid (HS-731) after subcutaneous and oral administration in rats with carrageenan-induced hindpaw inflammation. J. Pharmacol. Exp. Ther. 317, 220–227. 10.1124/jpet.105.096032. [DOI] [PubMed] [Google Scholar]
- Király K. P.; Riba P.; D’Addario C.; Di Benedetto M.; Landuzzi D.; Candeletti S.; Romualdi P.; Fürst S. (2006) Alterations in prodynorphin gene expression and dynorphin levels in different brain regions after chronic administration of 14-methoxymetopon and oxycodone-6-oxime. Brain Res. Bull. 70, 233–239. 10.1016/j.brainresbull.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Schütz J.; Spetea M.; Koch M.; Aceto M. D.; Harris L. S.; Coop A.; Schmidhammer H. (2003) Synthesis and biological evaluation of 14-alkoxymorphinans. 20. 14-Phenylpropoxymetopon: an extremely powerful analgesic. J. Med. Chem. 46, 4182–4187. 10.1021/jm030878b. [DOI] [PubMed] [Google Scholar]
- Kaczor A. A.; Rutkowska E.; Bartuzi D.; Targowska-Duda K. M.; Matosiuk D.; Selent J. (2016) Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 132, 359–399. 10.1016/bs.mcb.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Kolinski M.; Filipek S. (2008) Molecular dynamics of mu opioid receptor complexes with agonists and antagonists. Open Struct. Biol. J. 2, 8–20. 10.2174/1874199100802010008. [DOI] [Google Scholar]
- Kim J. M.; Altenbach C.; Kono M.; Oprian D. D.; Hubbell W. L.; Khorana H. G. (2004) Structural origins of constitutive activation in rhodopsin: role of the K296/E113 salt bridge. Proc. Natl. Acad. Sci. U. S. A. 101, 12508–12513. 10.1073/pnas.0404519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accelrys Software Inc. (2012) Discovery Studio, release 3.0, Accelrys Inc., San Diego, CA, www.accelrys.com.
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Chessari G.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Nissink J. W.; Taylor R. D.; Taylor R. (2005) Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 48, 6504–6515. 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. (2005) LigandScout: 3-D Pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45, 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. (2016) MOPAC2016, Stewart Computational Chemistry, Colorado Springs, CO; http://OpenMOPAC.net. [Google Scholar]
- Günther S.; Senger C.; Michalsky E.; Goede A.; Preissner R. (2006) Representation of target-bound drugs by computed conformers: implications for conformational libraries. BMC Bioinf. 7, 293. 10.1186/1471-2105-7-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. S.; Feig M.; Salsbury F. R. Jr.; Brooks C. L. 3rd (2003) New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 24, 1348–1356. 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Henriksen G.; Platzer S.; Marton J.; Hauser A.; Berthele A.; Schwaiger M.; Marinelli L.; Lavecchia A.; Novellino E.; Wester H. J. (2005) Syntheses, biological evaluation, and molecular modeling of 18F-labeled 4-anilidopiperidines as mu-opioid receptor imaging agents. J. Med. Chem. 48, 7720–7732. 10.1021/jm0507274. [DOI] [PubMed] [Google Scholar]
- Dosen-Micovic L.; Ivanovic M.; Micovic V. (2006) Steric interactions and the activity of fentanyl analogs at the mu-opioid receptor. Bioorg. Med. Chem. 14, 2887–2895. 10.1016/j.bmc.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. (2009) Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Giangreco I.; Hall R. J.; Korb O.; Mortenson P. N.; Murray C. W. (2011) Docking performance of fragments and drug-like compounds. J. Med. Chem. 54, 5422–5431. 10.1021/jm200558u. [DOI] [PubMed] [Google Scholar]
- Liebeschuetz J. W.; Cole J. C.; Korb O. (2012) Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput.-Aided Mol. Des. 26, 737–748. 10.1007/s10822-012-9551-4. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Sun H.; Yao X.; Li D.; Xu L.; Li Y.; Tian S.; Hou T. (2016) Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 18, 12964–12975. 10.1039/C6CP01555G. [DOI] [PubMed] [Google Scholar]
- Wu G.; Robertson D. H.; Brooks C. L. 3rd; Vieth M. (2003) Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 24, 1549–1562. 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- Brooks B.; Bruccoleri R.; Olafson B.; States D. J.; Swaminathan S.; Karplus M. (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Hwang M. J.; Stockfisch T. P.; Hagler A. T. (1994) Derivation of class ii force fields. 2. Derivation and characterization of a class ii force field, CFF93, for the alkyl functional group and alkane molecules. J. Am. Chem. Soc. 116, 2515–2525. 10.1021/ja00085a036. [DOI] [Google Scholar]
- Stewart J. J. (2013) Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruschel D.; Zagrovic B. (2009) Conformational averaging in structural biology: issues, challenges and computational solutions. Mol. BioSyst. 5, 1606–1616. 10.1039/b917186j. [DOI] [PubMed] [Google Scholar]
- Mortier J.; Rakers C.; Bermudez M.; Murgueitio M. S.; Riniker S.; Wolber G. (2015) The impact of molecular dynamics on drug design: applications for the characterization of ligand-macromolecule complexes. Drug Discovery Today 20, 686–702. 10.1016/j.drudis.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical Integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Mackerell A. D. Jr. (2004) Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 25, 1584–1604. 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]