

Abstract
Background
People with fragile X syndrome (FXS) often have deficits in social behavior, and a substantial portion meet criteria for autism spectrum disorder. Though the genetic cause of FXS is known to be due to the silencing of FMR1, and the Fmr1 null mouse model representing this lesion has been extensively studied, the contributions of this gene and its protein product, FMRP, to social behavior are not well understood.
Methods
Fmr1 null mice and wildtype littermates were exposed to a social or non-social stimulus. In one experiment, subjects were assessed for expression of the inducible transcription factor c-Fos in response to the stimulus, to detect brain regions with social-specific activity. In a separate experiment, tissue was taken from those brain regions showing differential activity, and RNA sequencing was performed.
Results
Immunohistochemistry revealed a significantly greater number of c-Fos-positive cells in the lateral amygdala and medial amygdala in the brains of mice exposed to a social stimulus, compared to a non-social stimulus. In the prelimbic cortex, there was no significant effect of social stimulus; although the number of c-Fos-positive cells was lower in the social condition compared to the non-social condition, and negatively correlated with c-Fos in the amygdala. RNA sequencing revealed differentially expressed genes enriched for molecules known to interact with FMRP and also for autism-related genes identified in the Simons Foundation Autism Research Initiative gene database. Ingenuity Pathway Analysis detected enrichment of differentially expressed genes in networks and pathways related to neuronal development, intracellular signaling, and inflammatory response.
Conclusions
Using the Fmr1 null mouse model of fragile X syndrome, we have identified brain regions, gene networks, and molecular pathways responsive to a social stimulus. These findings, and future experiments following up on the role of specific gene networks, may shed light on the neural mechanisms underlying dysregulated social behaviors in fragile X syndrome and more broadly.
Electronic supplementary material
The online version of this article (doi:10.1186/s13229-017-0148-6) contains supplementary material, which is available to authorized users.
Keywords: Fragile X syndrome, Autism spectrum disorder, Social behavior, RNA sequencing, Amygdala, Prefrontal cortex
Background
Social impairment is one of the two core symptom domains in autism spectrum disorder (ASD) [1], but we have limited understanding of the mechanisms underlying this impairment. One way to probe the molecular underpinnings of social difficulties in ASD is to study single gene syndromes that frequently include prominent ASD symptoms, such as fragile X syndrome (FXS), which is found in about 1% of children with ASD [2–4]. Between one quarter and two thirds of children with FXS meet criteria for ASD, depending upon the study [5–15]. In some but not all studies, the average child with FXS and autism spectrum disorder (FXS-ASD) has fewer ritualistic behaviors and perhaps fewer social deficits than the average child with autism spectrum disorder without FXS [10, 16–20], but these differences are not large enough to identify a behaviorally defined subgroup without genetic testing.
FXS is caused by expansion of a trinucleotide repeat in the 5′ untranslated region of the FMR1 gene leading to gene hypermethylation and transcriptional silencing. Fmr1 null mice have been a primary tool to investigate the cellular, synaptic, and molecular changes underlying FXS [21]. These mice display behavioral, structural, and molecular abnormalities that mirror some of those seen in FXS [22]. While the preponderance of evidence suggests that Fmr1 null mice do have changes in social behavior compared to wildtype controls, results have been inconsistent across labs and appear to be dependent on the inbred strain background of the mice being studied, with abnormalities more frequently reported on an FVB/129S mixed background [22–25]. The most common finding is a loss of preference for a novel mouse over a familiar mouse, but some laboratories have also reported that Fmr1 mice fail to prefer a novel mouse over a novel object, show diminished free social interactions, or are more likely to back out of the tube test for dominance [23–43]. In addition to abnormal social behavior, Fmr1 null mice have been reported to show several other behavioral deficits, including hyperactivity, attention deficits, learning deficits, and abnormal reactions to sensory stimuli [25, 44–47], although these behavioral changes have also been inconsistent across labs.
Many studies have focused on the molecular consequences of FMR1 loss with the goal of developing an effective treatment for FXS. Considerable data support a critical role for the gene’s protein product, FMRP, downstream of group 1 metabotropic glutamate receptor (mGluR) signaling, leading to the mGluR theory of fragile X [48]. Although improvement of behavioral and synaptic abnormalities was observed in animal models following treatment with mGluR5 negative allosteric modulators [49, 50], clinical trials have not yet yielded benefit in adolescents or adults with FXS [51]. Other potential treatments have also been studied, including lithium [52], minocycline [53], and arbaclofen [54], but no treatment has yet shown consistent improvement in behavior in the human population.
Despite extensive characterization of the Fmr1 null mouse, the role of FMRP in mediating social behavior remains relatively unknown. The aim of the current investigation was to use the Fmr1 (FVB/129) null mouse as a tool to further investigate molecular changes that may impact social behavior. We designed a series of experiments to understand the brain areas involved in response to social stimuli in these animals and to identify changes in levels of RNA expression that may identify molecular pathways altered in this model. Beyond insight into potential mechanisms underlying altered social behavior in the Fmr1 null mouse, we hoped to use this well-studied model as a proof-of-concept of this use of social stimuli to identify genes and pathways that can be cross-validated with other data from FXS and ASD.
Methods
Animals
Male Fmr1 null mice [55] (Fmr1 null, FVB.129P2-Pde6b + Tyrc-ch Fmr1tm1Cgr/J; stock #4624) and wildtype mice (WT, FVB.129P2-Pde6b + Tyrc-ch/AntJ; stock #4828) were purchased from Jackson Laboratories. These wildtype mice were generated from the Fmr1 line and are considered the standard control for Fmr1 null mice, as have been used in behavioral and molecular studies [56, 57]. Mice were housed two to five to a cage in the Vanderbilt Neurobehavioral core with a 12-h light/dark cycle and food and water available ad libitum. All mice were between 8 and 14 weeks of age for all experiments. Behavioral experiments commenced following a habituation period of at least 1 week at the Vanderbilt Neurobehavioral core.
Home cage social exposure for immunohistochemistry
We first tested the classic 3-chamber sociability apparatus, as implemented by the Crawley lab [58], for social exposures in wildtype C57BL/6J mice. During these experiments, the social exposure was conducted in a single chamber containing a stimulus mouse in an inverted pencil cup. The non-social exposure was conducted in a single chamber containing only an inverted pencil cup. Using this approach, we found widespread c-Fos activation that did not easily separate between the social and non-social condition (data not shown), similar to a recent study performed in juvenile C57BL/6 mice [59] which found only one brain area (basolateral amygdala) within which c-Fos activation was differentiated between social and non-social groups. We therefore modified this task to optimize the detection of social-specific c-Fos activation in a separate set of C57BL/6J mice. First, each mouse was tested within a standard housing cage to prevent a ceiling effect of c-Fos activation from the novel, open 3-chamber apparatus. Second, in order to control for exposure to novelty, mice were habituated to all components of the stimulus (cage, pencil cup) except for the social stimulus, which was controlled for using a toy horse (Playmobil) as a novel, non-social stimulus. After initial experiments in C57BL/6J animals, Fmr1 null and wildtype mice (n = 16, four per stimulus per genotype) were used in the behavioral experiments described here.
To habituate mice to the pencil cup stimulus, each mouse was individually separated into a novel cage that contained an empty pencil cup 3 days prior to the behavioral experiments (see Fig. 1a). After 3 days of habituation, each group of four mice (a “quad”), composed of two wildtype mice and two mutant mice, with each mouse separately housed, was placed in a room within the Vanderbilt Neurobehavioral core. The mice were allowed to habituate to the room for 1 h. A social stimulus, a novel mouse, was then placed in the pencil cups of one wildtype and one mutant cage, and a non-social stimulus, a toy horse (Playmobil), was placed in the pencil cups of the remaining wildtype and mutant cage (Fig. 1b). The exposure period lasted 30 min.
Fig. 1.
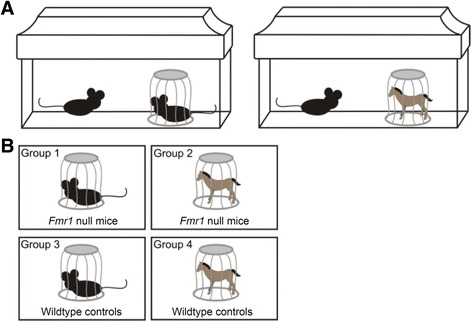
Experimental conditions and groups. a Mice were placed in a cage with an empty pencil cup 3 days prior to testing. At the time of the test, a social stimulus (left) or non-social stimulus (right) was placed in the pencil cup for 30 min. b There were four testing groups: Fmr1 KO with social or non-social stimulus exposure, or wildtype controls with social or non-social stimulus exposure
Tissue harvest for immunohistochemistry
Tissue harvest was conducted 2 h after the conclusion of the behavioral exposure period to allow for expression of c-Fos. As previously described [60], each mouse was deeply anesthetized with pentobarbital (Nembutal, Lundbeck, Deerfield, IL) and then intracardially perfused with 4% paraformaldehyde (ph~7.4). Whole brains were harvested and stored in 4% paraformaldehyde for 24 h and then in 30% sucrose for at least 24 h. Each brain was sectioned on a sliding microtome into 40 μm sections. Sections were stored in a freezing solution (PBS, ethylene glycol, and glycerol) until staining.
Immunohistochemistry
Initial staining was performed in C57BL/6J mice following the behavioral and tissue harvest methods described above. Multiple brain areas including the orbital cortex, frontal association area, prelimbic cortex, cingulate cortex, insular cortex, accumbens core, accumbens shell, caudate putamen, peduncular cortex, thalamic nuclei, hypothalamic nuclei, medial amygdala, basolateral amygdala, CA1 of the hippocampus, CA2 of the hippocampus, CA3 of the hippocampus, central amygdaloid nucleus, oriens layer of hippocampus, lateral amygdaloid nucleus, zona incerta, parietal association area, subthalamic nucleus, enterohinal cortex, mammullary nuclei, and perihinal cortex were examined for differential expression of c-Fos by counting stained cells. In these mice, we found that the areas with the most significant differential c-Fos staining in the social versus novel object comparison were the medial amygdala and lateral amygdala (Fig. 2). These brain areas were then selected as target areas for staining and cell counting in the Fmr1 null and wildtype control mice. Early in the cell counting procedure for Fmr1 mice, we visually scanned regions listed above and observed what appeared to be differential expression of c-Fos within the prelimbic cortex (PLC) in wildtype control animals. Because of these expression differences and the functional relationship between the amygdala and prefrontal cortex, c-Fos expression within the PLC was included in Fmr1 analyses.
Fig. 2.
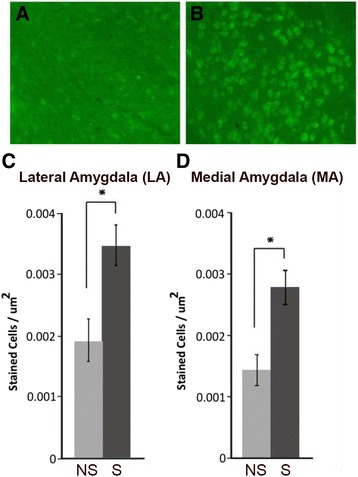
c-Fos staining in C57BL/6J mice. The number of cells was greater in the social (S) condition (b) compared to the non-social (NS) condition (a) in the lateral/basolateral amygdala (c), and the medial amygdala (d). *p < 0.05
Brain sections containing the medial and lateral amygdala and prefrontal cortex were stained for c-Fos expression in the Fmr1 null and wildtype control animals with each genotype and stimulus group represented evenly in each batch of staining. As described previously [60], brain sections were washed in 0.1 M phosphate buffer (PB, pH~7.8) for 20 min, 0.05 M Tris-buffered saline (TBS, pH~7.6) for 20 min, and 0.05 M tris-buffered saline with 0.4% triton-x (TBST, pH~7.6) for 20 min. Sections were then placed in a c-Fos rabbit anti-mouse primary antibody (1:2000) in TBST for 60 h at 6 °C. Four washes in TBST for 15 min preceded a donkey anti-rabbit secondary antibody (1:200) in TBST for 2 h. Sections were then washed in TBST and TBS for 15 min each and washed twice in tris-buffer (TB, pH~8.2) for 15 min. Brain sections were then mounted on slides and covered. Using a Zeiss Axio Imager.M2 microscope and AxioVision 4.8 software, multiple 5× pictures of each brain section were acquired, stitched together, and converted to a single image per slice (Carl Zeiss, Inc., Thornwood, NY, USA). An anterior/posterior (AP) coordinate was assigned to each slice according to the Franklin and Paxinos mouse atlas [61]. Brain sections containing areas of interest were matched across the quad according to AP coordinate labels and 20× pictures were taken for cell counting.
Cell counting
We used the Franklin and Paxinos mouse atlas [61] to select regions for cell counting. Following the methods of Hale and colleagues [62], we selected ten rostrocaudal levels ranging from −0.94 to −2.06 mm from bregma for counting c-Fos stained cells in the medial and lateral/basolateral amygdala. All medial amygdala (MA) cell counts were within the anatomical boundaries of the medial amygdala nucleus anterior dorsal, the medial amygdala nucleus anteroventral, the medial amygdala nucleus anterior, the medial amygdala nucleus posterodorsal, and the medial amygdala nucleus posteroventral. All lateral/basolateral amygdala (LA) cell counts were within the anatomical boundaries of the lateral amygdaloid nucleus, the basolateral amygdaloid nucleus anterior, the lateral amygdaloid nucleus dorsolateral, the lateral amygdaloid nucleus ventrolateral, the basolateral amygdaloid nucleus posterior, and the lateral amygdaloid nucleus ventromedial. For cell counts within the prefrontal cortex, we used 11 rostrocaudal levels ranging from 2.68 to 1.54 mm from bregma and used the anatomical borders of the prelimbic area (PLC; Fig. 3). In all areas, cells were counted from the left and right hemispheres and averaged. The area of each selected region was also measured with NIH ImageJ software [63] to determine number of stained cells per square micrometer. The number of stained cells per square micrometer was then averaged across sections for each mouse (n = 4–5 per group) and compared across genotypes and conditions by a two-way ANOVA. The number of c-Fos-positive cells was also assessed for correlation between brain regions using a Pearson’s correlation.
Fig. 3.
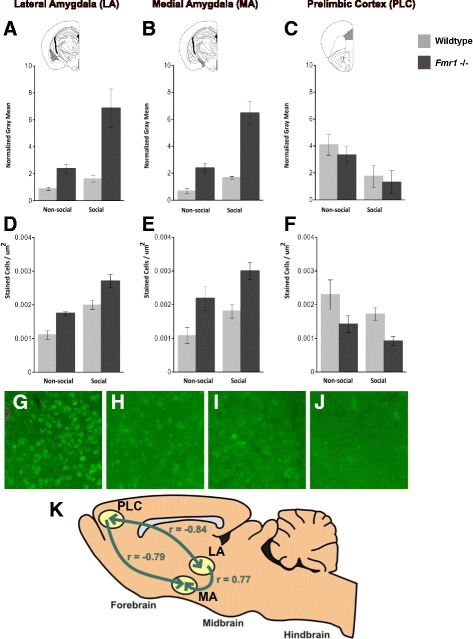
c-Fos staining in Fmr1 mice. The intensity of c-Fos staining (a–c) and the number of c-Fos-positive cells (d–f) in the lateral amygdala (a, d), medial amygdala (b, e), and prelimbic cortex (c, f). Insets show anatomical regions assessed shaded in gray. Representative staining for c-Fos in the PLC of (g) wildtype mouse exposed to a non-social stimulus, (h) wildtype mouse exposed to a social stimulus, (l) Fmr1 KO mouse exposed to a non-social stimulus, and (j) Fmr1 KO mouse exposed to a social stimulus. The number of c-Fos-positive cells was compared between brain regions for each subject. There was a significant positive correlation between LA and MA, and a significant negative correlation between each amygdalar region and the PLC (k)
Measurement of intensity
To judge the intensity of the c-Fos staining, we followed procedures previously described by Boushell and colleagues [64]. We used ImageJ to select a rectangular region within the anatomical boundaries described above for each area. Each image was converted to a 16-bit grayscale image. We then measured the mean gray of the selected area as well as the mean gray of the background (area between stained cells). The background mean gray was subtracted from the mean gray of the selected region, and the resulting normalized mean gray was used as an indication of staining intensity. Normalized mean gray was averaged across sections for each mouse (n = 4 per group) and compared across genotypes and conditions by a two-way ANOVA.
RNA sequencing
A separate group of Fmr1 null and wildtype mice (n = 36, 9 per stimulus per genotype) was exposed to the same social stimulus procedure described above for IHC, followed by a 10-h wait period to allow for resulting changes in RNA transcription or processing. Each brain was harvested immediately after rapid decapitation and sectioned in a brain matrix. A 1-mm Palkovits punch was used to collect tissue samples from lateral amygdala and medial amygdala. Samples were obtained from the prefrontal cortex (PFC, including PLC) by razor blade dissection. Samples were then stored at−80 °C prior to total RNA extraction using the Qiagen RNeasy mini kit (Qiagen) according to manufacturer’s protocol. RNA concentration and purity were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). The Agilent Bioanalyzer 2100 (Agilent Technologies) was used to confirm RNA integrity (RIN greater than 7.0). To reduce variability, tissue samples were pooled across groups with three brain samples per pool and three pools per group. Samples were diluted to result in consistent RNA concentrations across samples within each pool. Following strand-specific library preparation, paired-end sequencing with 30 M reads per library was performed using Illumina HiSeq 2000.
Differential expression analyses
Reads were mapped to the mouse genome mm10 using TopHat−2.0.10 [65]. The mapping quality for these samples was high, with 89% or more of the reads mapped to the genome. Then, following a standard data analysis protocol [66], we counted the number of reads that fell into annotated genes by samtools−0.1.19 [67] and HTSeq-0.5.4p5 [68] and analyzed the correlation between samples based on normalized counts. The correlation between biological replicates was high for samples from LA and PFC (Additional File 1). For samples from MA, we chose the two replicates with the best correlation from each group. Finally, we performed count-based differential expression analysis using edgeR_3.4.2 [69], which implements general differential analyses based on the negative binomial model. Differential expression analyses were performed for (1) social versus non-social in wildtype, (2) social versus non-social in mutant, (3) mutant versus wildtype in non-social, (4) mutant versus wildtype in social, and (5) the interaction across genotype and condition, which was considered the primary analysis.
Analyses of differentially expressed genes
Differentially expressed genes (DEGs) from each comparison in each brain region were compared against lists of FMRP target genes [70] and validated autism candidate genes from the Simons Foundation Autism Research Initiative (SFARI) gene database (gene.sfari.org). DEG lists were considered significantly enriched for FMRP or ASD-related genes when a chi-square test revealed overlap at a higher level than chance (p < 0.05). Enrichment of differentially expressed genes in specific networks and pathways was discovered using Ingenuity Pathway Analysis (Qiagen), using differentially expressed genes at the p < 0.05 alpha level resulting from the comparisons between genotypes and conditions from each brain region. Pathways were considered significantly enriched for differentially expressed genes when p < 0.05, with the Benjamini-Hochberg correction for multiple testing applied. Upstream regulators were also assessed using Ingenuity Pathway Analysis, identified by significant enrichment of DEGs in the network of molecules known to be affected by a given regulator (p < 0.05).
Results
Immunohistochemistry intensity measurements
Immunohistochemistry (IHC) staining intensity analyses for c-Fos revealed significant genotype × stimulus interaction effects for the LA (F(1,12) = 6.48, p = 0.026), and MA (F(1,12) = 10.71, p = 0.007) but not the PLC (F(1, 12) = 0.044, p = 0.838; Fig. 3a–c). Bonferroni post hoc analyses for staining intensity measurements indicated that the social mutant group displayed significantly greater staining intensity than all other groups in both the LA (social mutant group compared to non-social wildtype group, p = 0.001; social wildtype group, p = 0.002; and non-social mutant group, p = 0.006) and the MA (social mutant group copmared to non-social wildtype group, p < 0.001; social wildtype group, p < 0.001; and non-social mutant group, p < 0.001). Main effects for the social stimulus eliciting greater c-Fos intensity were seen across both wildtype and Fmr1 null mice in the LA (F(1,12) = 12.64, p = 0.004) and MA (F(1,12) = 28.94, p < 0.001), while main effects for the social stimulus eliciting less c-Fos intensity were seen in the PLC (F(1,12) = 8.11, p = 0.015). Genotype main effects were also seen, with overall higher staining intensity in the Fmr1 null mice compared to wildtype mice in the LA (F(1,12) = 20.97, p = 0.001) and MA (F(1,12) = 47.75, p < 0.001).
Immunohistochemistry and cell counts
In contrast to the c-Fos intensity measurements, cell count analyses did not demonstrate interaction effects in any of the brain areas (LA, F(1,12) = 0.085, p = 0.776; MA, F(1,12) = 0.03, p = 0.868; PLC, F(1,12) = 0.018, p = 0.896; Fig. 3d–f). The social stimulus elicited c-Fos expression in a greater number of cells than the non-social stimulus across genotypes in the LA (F(1,12) = 44.05, p < 0.001) and the MA (F(1,12) = 8.23, p = 0.014). A trend toward a significant effect of social stimulus in the opposite direction was observed in the PLC (F(1,12) = 3.70, p = 0.078). Genotype also had a significant effect, with Fmr1 null mice showing a greater number of c-Fos positive cells in the LA (F(1,12) = 23.73, p = 0.001) and the MA (F(1,12) = 18.43, p = 0.001), with fewer c-Fos positive cells in the PLC (F(1,12) = 8.97, p = 0.011).
Within individual animals, the numbers of c-Fos-positive cells in the LA and MA were highly correlated (r = 0.77, df = 10, p = 0.003). In contrast, cell counts were strongly negatively correlated between the PLC and LA (r = −0.84, df = 10, p = 0.0007) and between the PLC and MA (r = −0.79, df = 10, p = 0.002; Fig. 3k).
Differentially expressed genes identified by RNA sequencing
RNA integrity was confirmed by RIN assessment (mean ± SEM = 9.07 ± 0.13) prior to RNAseq analysis. Differentially expressed genes (DEGs) from each comparison in each brain region (Additional files 2, 3, and 4) were compared against lists of FMRP target genes [70]. In these comparisons, the Fmr1 null versus wildtype in the non-social condition would be expected to be the closest representation of gene expression differences driven by genotype alone. This comparison showed a significant enrichment of FMRP target genes among DEGs in the LA (p < 0.0001) and the PFC (p < 0.0001) but not the MA (p = 0.69). Little signal was seen for DEGs enriched for FMRP target genes in the genotype × stimulus interaction analyses, with a nominally significant enrichment (p = 0.045) in the LA but not significant enrichment in MA or PFC. In the comparisons of wildtype mice in the social condition and non-social condition, FMRP target genes were enriched above the level of chance in the MA and PFC (p < 0.05) but not the LA; whereas these genes were not enriched in the comparisons of mutant mice in the social and non-social condition (Fig. 4a).
Fig. 4.
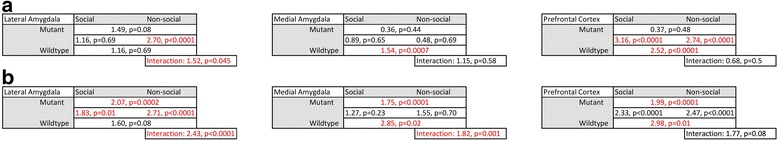
Overlap of differentially expressed genes from Fmr1 mice with FXS- and ASD-associated genes. Chi-square tests contrasted the list of differentially expressed genes from each comparison (social compared to non-social in mutants, top line; social compared to non-social in wildtype, bottom center; mutant compared to wildtype in social conditions, left; mutant compared to wildtype in non-social conditions, right; interaction between genotype and stimulus, bottom right) with a known FMRP targets and b the list of SFARI candidate genes for ASD. Chi-square and p values are listed, and contrasts with significant overlap are highlighted in red text
DEGs were also compared against validated autism candidate genes from the Simons Foundation Autism Research Initiative (SFARI) gene database (gene.sfari.org). In these analyses, the initial focus was the interaction analyses to identify genes that were differentially affected by the social stimulus in wildtype versus Fmr1 null mice, which revealed significant enrichment for SFARI genes in the LA (p < 0.0001) and MA (p = 0.001) and a trend in the PFC (p = 0.08). Almost all social versus non-social comparisons within genotype also showed a significant enrichment, suggesting that ASD candidate genes are differentially affected by a social stimulus in these brain regions. Genotype comparisons in both the non-social and social conditions were enriched for SFARI genes in the LA and PFC but not the MA (Fig. 4b).
To assess common DEGs across comparisons within each brain region, we evaluated the overlap between gene sets resulting from comparisons of wildtype mice in the social and non-social conditions, the comparisons of mutant and wildtype mice in the non-social condition, and the interaction comparisons (Fig. 5; Additional File 5). In the LA, 83 genes were differentially expressed in all three comparisons (p < 0.05). In the MA, 34 genes were differentially expressed in all three comparisons (p < 0.05). In the PFC, 26 genes were differentially expressed in all three comparisons (p < 0.05). To assess commonalities in DEGs across brain regions, we examined the overlap between interaction comparisons. Three genes, Tyrosine hydroxylase (Th), Kringle Containing Transmembrane Protein 1 (Kremen1), and Epsin 3 (Epn3), were found to be significant DEGs in all three interaction comparisons (p < 0.05; Fig. 6). The most common DEGs across comparisons and brain regions are shown in Additional File 6.
Fig. 5.
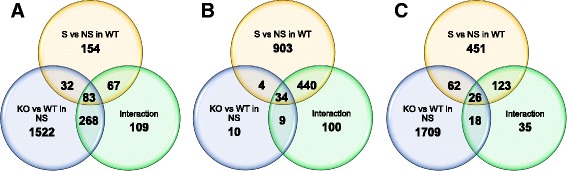
Overlap of differentially expressed genes in baseline comparisons for each brain region. Differentially expressed gene lists (p < 0.05) from each baseline comparison (mutants compared to wildtype in non-social conditions in blue, social compared to non-social stimulus in wildtype mice in yellow) were compared with the list from the interaction analysis of genotype and stimulus variables (green) to detect overlap and focus on genes of interest. The number of overlapping genes is shown in corresponding regions of the Venn diagrams for LA (a), MA (b), and PFC (c)
Fig. 6.
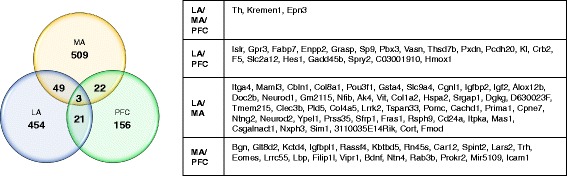
Overlap of differentially expressed genes in LA, MA, and PFC. Differentially expressed gene lists (p < 0.05) from the interaction analysis of genotype and stimulus variables were compared from each brain region (LA in blue, MA in yellow, PFC in green). The number of overlapping genes is shown in corresponding regions of the Venn diagram, and the list of overlapping genes for each comparison is shown in the adjoining table
Pathway analyses
A number of canonical pathways exhibited overrepresentation of molecular components within the DEGs (Additional File 7). Within the LA and MA interaction comparisons, enrichment was observed for pathways corresponding to neuronal development (i.e., Axonal guidance signaling p = 0.0005 and p = 3.24 ×10−06, respectively) and intracellular signaling (i.e., AMPK signaling in LA p = 0.0032; calcium signaling in MA p = 0.0002). Within the PFC interaction comparison, enrichment was seen for inflammatory response (i.e., IL-6 signaling p = 0.0035) and intracellular signaling pathways (i.e., TR/RR activation p = 0.0009). However, in the LA and PFC, no pathways remained significant when the B-H correction for multiple testing was applied.
Network analyses yielded clusters of DEGs whose corresponding proteins are known to interact (Figs. 7, 8, and 9). Enrichment for differentially expressed genes was found in networks associated with nervous system development and function in the LA interaction comparisons, organismal and nervous system development in the MA interaction comparisons, and oxidative stress and developmental disorders in the PFC interaction comparisons. Finally, interaction DEGs were enriched for genes known to be affected by endogenous or exogenous regulators, including beta-estradiol and transforming growth factor beta1, in all three brain regions (3.86 ×10-17 ≤ 5.60 ×10-08; Additional File 8).
Fig. 7.
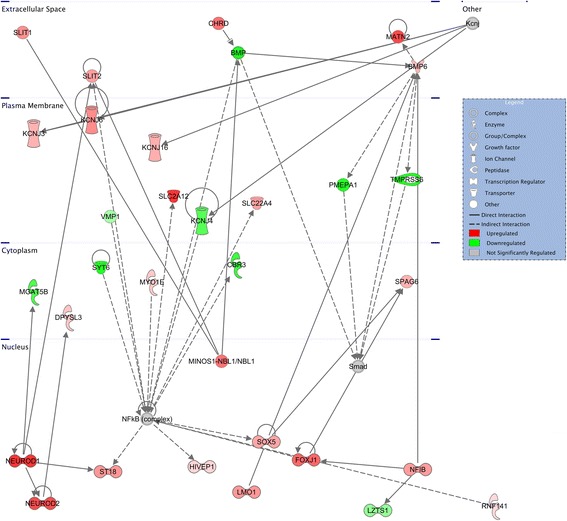
Top network enriched for differentially expressed genes in LA. The network with the greatest enrichment of differentially expressed genes from the interaction comparison of genotypes and stimuli from the lateral amygdala centers on the hub molecule NFkB, which was itself not significantly regulated
Fig. 8.
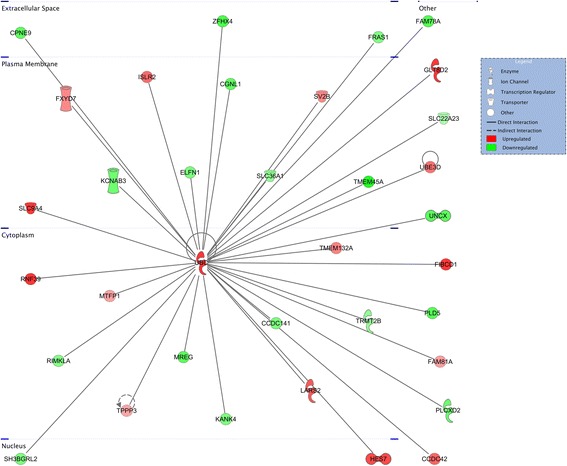
Top network enriched for differentially expressed genes in MA. The network with greatest enrichment of differentially expressed genes from the interaction comparison of genotypes and stimuli from the medial amygdala centers on the hub molecule ubiquitin
Fig. 9.
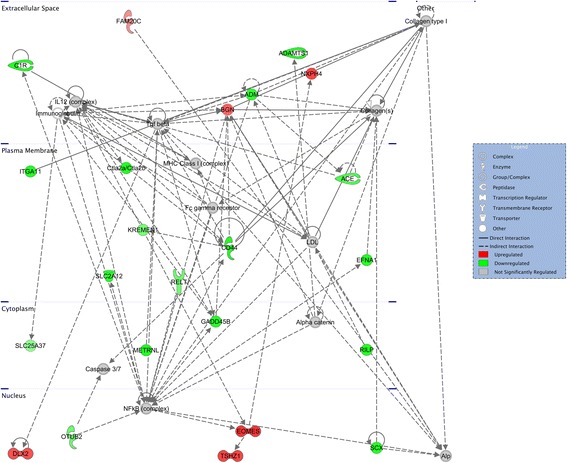
Top network enriched for differentially expressed genes in PFC. The network with the greatest enrichment of differentially expressed genes from the interaction comparison of genotypes and stimuli from the prefrontal cortex centers on the hub molecule NFkB, which was itself not significantly regulated
Discussion
Using the Fmr1 null mouse as a model of one of the most common ASD-associated syndromes, we were able to identify changes in immediate early gene activation and gene expression following the presentation of a novel mouse, in comparison to a novel object. This may provide insights into pathways and networks underlying altered social response in the Fmr1 null mice and potentially in people with FXS. Further, the strong overlap with SFARI genes suggests that a social stimulus can be used to provoke changes in gene expression that correspond with ASD-relevant brain regions and molecular pathways within a genetic mouse model of ASD.
Our initial immunohistochemistry in wildtype C57BL/6J mice revealed the LA and MA as having the greatest degree of differential activation, as evidenced by c-Fos levels, in response to a social stimulus compared to a non-social stimulus, while similar findings in Fmr1 null mice also showed differential activation in the PLC. These findings are in line with recent work by Ferri and colleagues [59], who demonstrated greater levels of immediate early gene products c-Fos and Egr-1 in the BLA of juvenile C57BL/6J mice in response to a social stimulus, as compared with either a non-social stimulus or no stimulus exposure (home cage controls). In contrast, however, they did not observe an increase in c-Fos in response to a social stimulus compared to non-social stimulus in the MA; both the social and non-social stimulus conditions were elevated above the level of home cage controls for the MA. They propose that the MA is more likely to be responsive to various social stimuli after adolescence, which could explain the difference in findings between our studies. In addition, they found only a trend toward a significant difference in c-Fos cell counts in response to stimuli in the PLC, as in our results, though we also found a significant effect of stimulus on staining intensity, and the direction of the effect was the same in both studies, with the social stimulus exposure corresponding to lower c-Fos levels than non-stimulus exposure in the PLC.
In the Fmr1 null animals, we found hyperactivation of the amygdala of Fmr1 null animals following the presentation of a novel mouse. These findings suggest that the activation is dysregulated by the deletion of Fmr1 and support human imaging studies that associate the amygdala with social response in FXS. For example, Watson and colleagues [71] noted increased activation of the left amygdala in response to successive eye gaze exposures in children with FXS compared to controls. Structural imaging suggests a reduction in amygdala volume in FXS [72, 73], and Hoeft and colleagues [74] found that decreased amygdala volume in FXS patients is correlated with altered activation patterns. Recent imaging results in the Fmr1 null mouse model on the FVB background showed no significant change in the frontal cortex or amygdala volumes, with quite subtle changes overall including primarily white matter, as well as a small increase in ventricle size; although the resolution of MRI for individual brain structures is substantially reduced simply as a result of mouse brain size [75, 76]. The hypoactivation of the PLC in response to the social stimulus was significant for c-Fos intensity but not cell count, but the direction of the effects observed in both measures corresponds with human imaging findings. An fMRI study in children with FXS revealed decreased PFC activation in response to direct eye gaze in comparison to age-matched, typically developing children [71]. Decreased gray matter in the mPFC has also been reported in children with FXS [74].
Our immediate early gene findings also overlap with the broader literature on neural mechanisms of social behavior. For instance, a wide array of data support a role for the medial amygdala in social recognition [77], aggression [78, 79], and mating behavior [80] in mice. The positive correlation between lateral and medial amygdala c-Fos levels in response to a social stimulus align with recent work demonstrating the importance of the intra-amygdalar pathway in processing social behavior and formulating an appropriate response in the context of social fear learning [81]. The high, inverse correlation between amygdala and PLC immediate early gene activation aligns with previous literature suggesting that these two regions communicate during social behavior. The involvement of prefrontal-limbic circuitry, which includes both PFC and the amygdala, has been suggested in several social behaviors in humans including social anxiety [82], social phobia [83], deception [84], social cooperation [85], and empathy [86]. Swartz and colleagues [87] also found a decrease in connectivity between the PFC and amygdala while viewing sad faces as measured by functional magnetic resonance imaging (fMRI). Together with the connectivity and correlational findings between the PLC and amygdala, the findings of lower activation of PLC and higher activation of the amygdala activity in response to a social stimulus suggests dysregulation of at least one node within this circuit in Fmr1 null mice. This dysregulation in response to a social stimulus may be involved in the differences in social behavior observed in this mouse model of FXS.
The patterns of differentially expressed genes in the LA, MA, and PFC also show convergence with previous data. It is reassuring that this convergence includes highly significant overlap for genes bound by FMRP. Further, it is interesting that this overlap is primarily in the mutant versus wildtype comparison in LA after exposure to the non-social stimulus and in PFC after exposure to either the social or non-social stimulus. The absence of significant overlap with FMRP-bound genes in the genotype comparison in the MA is surprising and could indicate that this is a brain region where FMRP plays less of a direct role in regulating gene expression. Previous work in Fmr1 null mice has identified changes in electrophysiology and dendritic spine morphology in the basolateral amygdala and the medial prefrontal cortex [88–92]; whereas previous studies have not examined the MA in these animals. These data suggest that the MA could be a brain region that lacks substantial baseline changes due to the direct effects of Fmr1 deletion but may still show dysregulated activity within a broader circuit, but further work is necessary to examine this possibility. Interestingly, in addition to genotype-driven differences, we also saw a significant enrichment of FMRP-bound genes in the social versus non-social comparison in wildtype animals in both the MA and the PFC, although we did not observe a significant interaction between stimulus and genotype. This suggests that FMRP-bound genes may play an important role in response to a social stimulus, aligning with previous data implicating FMRP as a central hub that regulates genes implicated in ASD [93].
In addition to FMRP-bound genes, which could be expected to show differences in Fmr1 null mice, we also observed substantial overlap of DEGs with genes implicated in ASD. Our primary analysis for this comparison was the genotype × condition interaction, which showed a significant enrichment for SFARI ASD risk genes for the LA and MA, but only a trend for the PFC. This suggests that a social stimulus may be useful to focus further study on DEGs that are particularly relevant to ASD. Interestingly, Th, encoding tyrosine hydroxylase, was one of three genes that was differentially expressed in the genotype × stimulus interaction in all three brain regions. Tyrosine hydroxylase is the rate-limiting enzyme in dopamine synthesis, and the dopamine system has been implicated in FXS, in ASD, and in social response in general. For example, loss of FMRP alters signaling downstream of the dopamine D1 receptor [94]. Recent work also shows increased dopamine levels in the striatum of Fmr1 null mice [27]. De novo and inherited single nucleotide variants have been detected in a number of genes in the dopamine system in ASD. Elegant work using optogenetic and pharmacogenetic approaches has demonstrated that dopaminergic signaling is critical for a number of social functions, including social approach, social reward, and response to social deprivation [95–99]. Beyond the significant interactions, the social versus non-social contrast showed highly significant overlap with the SFARI gene set for Fmr1 null animals in all three brain regions, further supporting the use of a social stimulus to reveal ASD-relevant DEGs in a genetic mouse model.
Network analysis also pointed toward convergence with previous data, with DEGs clustering in networks that are associated with nervous system development. In addition to further validating the model and social stimulus procedure as producing gene expression differences in pathways relevant for ASD, the analyses may provide focus for future research and potential treatments. By identifying networks in specific brain regions that are differentially regulated in Fmr1 null mice, we can consider different ways to probe or manipulate the pathway to potentially identify a time during development or adulthood in which perturbation of the dysregulated pathway may rescue specific phenotypes. Additionally, the networks identified may provide specific molecules to consider targeting for treatment, including non-DEGs such as NFκB that occupy a position as molecular hubs interacting with multiple DEGs (Figs. 7 and 9). Finally, the upstream regulator analyses may highlight potential treatments that could reverse the observed pattern of differential gene expression, such as beta-estradiol, which was recently shown to rescue behavioral phenotypes in zebrafish lacking Cntnap2, an ASD-risk gene [100]. Most importantly, these pathway and network findings direct us to specific aspects of development, for example axonal guidance signaling or immune response pathways that will be beneficial to examine in Fmr1 null mice, particularly in relation to their social behavior.
These findings also have some limitations, including the moderately high degree of variability within the RNA sequencing data. Of the three brain areas, biological replicates had a high degree of convergence with the exception of one sample within the medial amygdala that we excluded from all analyses. Additionally, our RNA sequencing approach was focused primarily on pathway and network analyses, with few individual genes reaching significance after correction for multiple testing. We set an α level of 0.05 as a threshold for these analyses to balance a desire for specificity with a desire to eliminate false negatives, which could obscure convergence across multiple DEGs. Adjusting the threshold in either direction could yield different findings. Similarly, application of the Benjamini-Hochberg correction at the level of 0.05 for canonical pathway analysis resulted in no enriched pathways in the LA and PFC passing the threshold for significance. These individual DEGs and pathways are therefore exploratory and should be replicated before targeting specific, individual genes.
Another factor to consider is the introduction of the stimulus into the homecage. We minimized the potential for aggression by only housing the test animal in the new cage for 3 days, and containing the stimulus in the pencil cup, but it is possible that the reaction to the stimulus was influenced by territoriality in a way that might be lessened (or increased) by other circumstances.
Finally, the statistical differences between measurements of c-Fos intensity and cell counts should be considered. The effect of the interaction between genotype and stimulus was only significant in c-Fos intensity, although virtually all findings were in the same relative directions between analyses. The significant interaction effect on intensity was due to a larger effect size, and it is possible that this measure is more sensitive to biological variation than cell count. For example, the level of c-Fos expression may vary between cells, leading to cell-specific transcriptional responses. Intensity measurement has the added advantage of being more objective, not requiring an experimenter to apply a threshold as in the cell count. The meaning of each measurement and resulting findings should be considered, but one is not necessarily preferable to the other.
Conclusions
The findings presented here align with previous research demonstrating the role of the amygdala and the PFC in social behavior in mice and validate the use of the Fmr1 null mouse for investigating circuitry contributing to social behavior. The use of this well-studied genetic mouse model provides the opportunity to look for overlap with previous findings, potentially serving as a proof of concept for this experimental design focus on the response to a social stimulus. The differential neuronal activation patterns and differential gene expression patterns elicited by a novel social stimulus did intersect with reported neuroimaging findings in fragile X syndrome as well as previous molecular findings in the Fmr1 null mouse. This approach may therefore prove useful in exploring the neural substrates of social behavior and for identifying molecular pathways contributing to abnormal social behaviors in other mouse models.
Additional files
Correlations between biological replicates. The correlations of mapped reads between biological replicates using normalized counts per million for all reads were high for all comparisons in the lateral amygdala (LA) and prefrontal cortex (PFC), but less strong in some replicates in the medial amygdala (MA). MT mutant; WT wildtype; NS non-social; S social. Red p < 0.001; white p > 0.001. (PDF 144 kb)
Differentially expressed genes in lateral amygdala (XLSX 5388 kb)
Differentially expressed genes in medial amygdala (XLSX 5377 kb)
Differentially expressed genes in the prefrontal cortex (XLSX 5258 kb)
Overlapping genes of comparisons in each brain region. The differentially expressed genes (DEGs) that overlapped in specific comparisons are listed. The first column indicates DEGs significantly regulated in both the mutant versus wildtype comparison in the non-social condition (MUvsWTinNS) and in the social versus non-social conditions in the wildtype animals (SvsNSinWT). The second column indicates DEGs significantly regulated in both the MUvsWTinNS condition and in the interaction between genotype and stimulus exposure. The third column indicates DEGs significantly regulated in both the SvsNSinWT condition and in the interaction between genotype and stimulus exposure. Overlap in all three comparisons indicates DEGs that were significantly regulated in the MUvsWTinNS, SvsNSinWT, and interaction analyses. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 37 kb)
Most commonly differentially expressed genes across all comparisons. The genes that had the most frequent differential expression in analyses of all comparisons in all brain regions are listed, showing where they had significant differences in expression level for a given comparison (X) in each brain region. M mutant; W wildtype; NS non-social; S social. (PDF 270 kb)
Canonical pathway results for RNAseq data. Canonical pathway analysis revealed pathways with significant enrichment of genes that were differentially expressed in each comparison. The genotype and stimulus condition comparison is shown on the left, with the name of the top 10 enriched pathways, the number of differentially expressed genes that overlap with each pathway, and the adjusted p value shown from left to right. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 18 kb)
Upstream Regulator results for RNAseq data. Upstream regulator analysis revealed regulatory molecules known to affect a certain network of molecules, overlapping with genes that were differentially expressed in each comparison. The genotype and stimulus condition comparison is shown on the left, with the name of the top 10 upstream regulators, the predicted direction of activation or inhibition based on the up/down-regulation of differentially expressed genes that overlap with the affected network, and the adjusted p value shown from left to right. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 12 kb)
Acknowledgements
Studies were performed at VANTAGE and Mouse Neurobehavior Lab facilities at Vanderbilt University.
Funding
The study was funded by MH094604 to JV-V, and training grants MH065215 to TDR, and MH016434 to AMJA, which did not play a role in the design or execution of the study.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
TDR designed and performed the animal studies, immunohistochemistry, RNA preparation, RNA sequencing statistical analysis, and wrote the manuscript. AMJA performed the Ingenuity Pathway Analysis and wrote the manuscript. TMK performed immunohistochemistry and contributed to experimental design. CGF performed tissue harvest and RNA preparation. BZ and JW performed RNA sequencing and analysis. JVV designed the experiments, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
Dr. Veenstra-VanderWeele has consulted or served on advisory boards for Roche, Novartis, and SynapDx. He has received research funding from Roche, Novartis, SynapDx, Seaside Therapeutics, and Forest. The other authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee (protocol M/11/101).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ANOVA
Analysis of variance
- ASD
Autism spectrum disorder
- FMR1
Fragile X mental retardation 1 gene
- FMRP
Fragile X mental retardation protein
- FXS
Fragile X syndrome
- GABA
Gamma-amino butyric acid
- ID
Intellectual disability
- IHC
Immunohistochemistry
- IPA
Ingenuity pathway analysis
- LA
Lateral amygdala
- MA
Medial amygdala
- mGluR
Metabotropic glutamate receptor
- PBS
Phosphate-buffered saline
- PFC
Prefrontal cortex
- PLC
Prelimbic cortex
- RNAseq
Ribonucleic acid sequencing
- SFARI
Simons Foundation Autism Research Initiative
- TBST
Tris-buffered saline with triton
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13229-017-0148-6) contains supplementary material, which is available to authorized users.
Contributor Information
Tiffany D. Rogers, Email: tiffanydrogers@gmail.com
Allison M. J. Anacker, Email: allison.anacker@gmail.com
Travis M. Kerr, Email: travis.m.kerr@gmail.com
C. Gunnar Forsberg, Email: forsbeca@musc.edu.
Jing Wang, Email: jing.wang.2@bcm.edu.
Bing Zhang, Email: bing.zhang@bcm.edu.
Jeremy Veenstra-VanderWeele, Phone: 646-774-5251, Email: veenstr@nyspi.columbia.edu.
References
- 1.American Psychiatric Association . Diagnostic and statistical manual of mental disorders. 5. Arlington: American Psychiatric Publishing; 2013. [Google Scholar]
- 2.Piven J, Gayle J, Landa R, Wzorek M, Folstein S. The prevalence of fragile X in a sample of autistic individuals diagnosed using a standardized interview. J Am Acad Child Adolesc Psychiatry. 1991;30:825–830. [PubMed] [Google Scholar]
- 3.Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hersh JH, Saul RA, Committee on Genetics Health supervision for children with fragile X syndrome. Pediatrics. 2011;127(5):994–1006. doi: 10.1542/peds.2010-3500. [DOI] [PubMed] [Google Scholar]
- 5.Bailey DB, Jr, Raspa M, Olmsted M, Holiday DB. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. 2008;146A:2060–2069. doi: 10.1002/ajmg.a.32439. [DOI] [PubMed] [Google Scholar]
- 6.Yu TW, Berry-Kravis E. Autism and fragile X syndrome. Semin Neurol. 2014;34:258–265. doi: 10.1055/s-0034-1386764. [DOI] [PubMed] [Google Scholar]
- 7.Lewis P, Abbeduto L, Murphy M, Richmond E, Giles N, Bruno L, Schroeder S. Cognitive, language and social-cognitive skills of individuals with fragile X syndrome with and without autism. J Intellect Disabil Res. 2006;50:532–545. doi: 10.1111/j.1365-2788.2006.00803.x. [DOI] [PubMed] [Google Scholar]
- 8.Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 9.Philofsky A, Hepburn SL, Hayes A, Hagerman R, Rogers SJ. Linguistic and cognitive functioning and autism symptoms in young children with fragile X syndrome. Am J Ment Retard. 2004;109:208–218. doi: 10.1352/0895-8017(2004)109<208:LACFAA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 10.Hall SS, Lightbody AA, Hirt M, Rezvani A, Reiss AL. Autism in fragile X syndrome: a category mistake? J Am Acad Child Adolesc Psychiatry. 2010;49:921–933. doi: 10.1016/j.jaac.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzocco MMM, Knight SJL, Abrams MT, Doheny KF, Baumgardner TL, Freund LS, Davies KE, Reiss AL. Cognitive, behavioral, and neuroanatomical assessment of two unrelated male children expressing FRAXE. Am J Med Genet. 1997;74:73–81. doi: 10.1002/(SICI)1096-8628(19970221)74:1<73::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 12.Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr, Roberts J, Mirrett P. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A:1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- 13.Bailey DB, Jr, Mesibov GB, Hatton DD, Clark RD, Roberts JE, Mayhew L. Autistic behavior in young boys with fragile X syndrome. J Autism Dev Disord. 1998;28:499–508. doi: 10.1023/A:1026048027397. [DOI] [PubMed] [Google Scholar]
- 14.Kau AS, Tierney E, Bukelis I, Stump MH, Kates WR, Trescher WH, Kaufmann WE. Social behavior profile in young males with fragile X syndrome: characteristics and specificity. Am J Med Genet. 2004;126A:9–17. doi: 10.1002/ajmg.a.20218. [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- 16.Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Wolff JJ, Bodfish JW, Hazlett HC, Lightbody AA, Reiss AL, Piven J. Evidence of a distinct behavioral phenotype in young boys with fragile X syndrome and autism. J Am Acad Child Adolesc Psychiatry. 2012;51:1324–1332. doi: 10.1016/j.jaac.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dissanayake C, Bui Q, Bulhak-Paterson D, Huggins R, Loesch DZ. Behavioural and cognitive phenotypes in idiopathic autism versus autism associated with fragile X syndrome. J Child Psychol Psychiatry. 2009;50:290–299. doi: 10.1111/j.1469-7610.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- 19.Rogers SJ, Wehner EA, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008;113:44–53. doi: 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 21.The Dutch-Belgian fragile X consortium. FMR1 knockout mice: a model to study fragile X mental retardation. Cell 1994, 78:23–33. [PubMed]
- 22.Kazdoba TM, Leach PT, Silverman JL, Crawley JN. Modeling fragile X syndrome in the FMR1 knockout mouse. Intractable Rare Dis Res. 2014;3:118–133. doi: 10.5582/irdr.2014.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, D’Ercole AJ, Crawley JN, Magnuson TR, Lauder JM. Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 2009;8:129–142. doi: 10.1111/j.1601-183X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pietropaolo S, Guilleminot A, Martin B, D’Amato FR, Crusio WE. Genetic-background modulation of core and variable autistic-like symptoms in FMR1 knock-out mice. PLoS One. 2011;6:e17073. doi: 10.1371/journal.pone.0017073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, Paylor R. Modifying behavioral phenotypes in FMR1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 2011;4:40–56. doi: 10.1002/aur.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the FMR1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 2005;4:420–430. doi: 10.1111/j.1601-183X.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- 27.Sorensen EM, Bertelsen F, Weikop P, Skovborg MM, Banke T, Drasbek KR, Scheel-Kruger J. Hyperactivity and lack of social discrimination in the adolescent FMR1 knockout mouse. Behav Pharmacol. 2015;26:733–740. doi: 10.1097/FBP.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 28.Naviaux JC, Wang L, Li K, Bright AT, Alaynick WA, Williams KR, Powell SB, Naviaux RK. Antipurinergic therapy corrects the autism-like features in the fragile X (FMR1 knockout) mouse model. Mol Autism. 2015;6:1. doi: 10.1186/2040-2392-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sare RM, Levine M, Smith CB: Behavioral phenotype of Fmr1 knock-out mice during active phase in an altered light/dark cycle. eNeuro 2016, 3. [DOI] [PMC free article] [PubMed]
- 30.Qin M, Huang T, Kader M, Krych L, Xia Z, Burlin T, Zeidler Z, Zhao T, Smith CB: R-baclofen reverses a social behavior deficit and elevated protein synthesis in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 2015, 18. [DOI] [PMC free article] [PubMed]
- 31.Oddi D, Subashi E, Middei S, Bellocchio L, Lemaire-Mayo V, Guzman M, Crusio WE, D’Amato FR, Pietropaolo S. Early social enrichment rescues adult behavioral and brain abnormalities in a mouse model of fragile X syndrome. Neuropsychopharmacology. 2015;40:1113–1122. doi: 10.1038/npp.2014.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hebert B, Pietropaolo S, Meme S, Laudier B, Laugeray A, Doisne N, Quartier A, Lefeuvre S, Got L, Cahard D, et al. Rescue of fragile X syndrome phenotypes in FMR1 ko mice by a BKCa channel opener molecule. Orphanet J Rare Dis. 2014;9:124. doi: 10.1186/s13023-014-0124-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci. 2014;34:9867–9879. doi: 10.1523/JNEUROSCI.1162-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gantois I, Pop AS, de Esch CE, Buijsen RA, Pooters T, Gomez-Mancilla B, Gasparini F, Oostra BA, D’Hooge R, Willemsen R. Chronic administration of AFQ056/mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res. 2013;239:72–79. doi: 10.1016/j.bbr.2012.10.059. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 s6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ. Genetic manipulation of step reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 2012;11:586–600. doi: 10.1111/j.1601-183X.2012.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pacey LK, Doss L, Cifelli C, van der Kooy D, Heximer SP, Hampson DR. Genetic deletion of regulator of G-protein signaling 4 (rgs4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol Cell Neurosci. 2011;46:563–572. doi: 10.1016/j.mcn.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Liu ZH, Chuang DM, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol. 2011;14:618–630. doi: 10.1017/S1461145710000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. Gsk3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5:e9706. doi: 10.1371/journal.pone.0009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahlhaus R, El-Husseini A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav Brain Res. 2010;208:96–105. doi: 10.1016/j.bbr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 41.Liu ZH, Smith CB. Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci Lett. 2009;454:62–66. doi: 10.1016/j.neulet.2009.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNaughton CH, Moon J, Strawderman MS, Maclean KN, Evans J, Strupp BJ. Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behav Neurosci. 2008;122:293–300. doi: 10.1037/0735-7044.122.2.293. [DOI] [PubMed] [Google Scholar]
- 43.Mineur YS, Huynh LX, Crusio WE. Social behavior deficits in the Fmr1 mutant mouse. Behav Brain Res. 2006;168:172–175. doi: 10.1016/j.bbr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 44.Moon J, Beaudin AE, Verosky S, Driscoll LL, Weiskopf M, Levitsky DA, Crnic LS, Strupp BJ. Attentional dysfunction, impulsivity, and resistance to change in a mouse model of fragile X syndrome. Behav Neurosci. 2006;120:1367–1379. doi: 10.1037/0735-7044.120.6.1367. [DOI] [PubMed] [Google Scholar]
- 45.Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999;94:185–192. doi: 10.1016/S0306-4522(99)00285-7. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/S0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 47.Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry. 2004;9:417–425. doi: 10.1038/sj.mp.4001432. [DOI] [PubMed] [Google Scholar]
- 48.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 49.de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, Brinkman M, Rerat K, Koumaras B, Zhu L, et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. 2016;8:321ra325. doi: 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- 52.Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, Weiler IJ, Greenough WT. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29:293–302. doi: 10.1097/DBP.0b013e31817dc447. [DOI] [PubMed] [Google Scholar]
- 53.Leigh MJ, Nguyen DV, Mu Y, Winarni TI, Schneider A, Chechi T, Polussa J, Doucet P, Tassone F, Rivera SM, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile X syndrome. J Dev Behav Pediatr. 2013;34:147–155. doi: 10.1097/DBP.0b013e318287cd17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berry-Kravis EM, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K, Mu Y, Nguyen DV, Gonzalez-Heydrich J, Wang PP, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4:152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- 55.Bakker CE, Verheij C, Willemsen R, Vanderhelm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, Oostra BA, Reyniers E, et al. Fmr1 knockout mice––a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- 56.Arnett MT, Herman DH, McGee AW. Deficits in tactile learning in a mouse model of fragile X syndrome. PLoS One. 2014;9:e109116. doi: 10.1371/journal.pone.0109116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vislay RL, Martin BS, Olmos-Serrano JL, Kratovac S, Nelson DL, Corbin JG, Huntsman MM. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J Neurosci. 2013;33:7548–7558. doi: 10.1523/JNEUROSCI.2764-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 59.Ferri SL, Kreibich AS, Torre M, Piccoli CT, Dow H, Pallathra AA, Li H, Bilker WB, Gur RC, Abel T, Brodkin ES: Activation of basolateral amygdala in juvenile C57BL/6J mice during social approach behavior. Neuroscience 2016;335(29):184–94. [DOI] [PMC free article] [PubMed]
- 60.Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, Cohen J, Mannangatti P, Jessen T, Thompson BJ, et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci U S A. 2012;109:5469–5474. doi: 10.1073/pnas.1112345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. 3. New York: Elsevier; 2008. [Google Scholar]
- 62.Hale MW, Johnson PL, Westerman AM, Abrams JK, Shekhar A, Lowry CA. Multiple anxiogenic drugs recruit a parvalbumin-containing subpopulation of gabaergic interneurons in the basolateral amygdala. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1285–1293. doi: 10.1016/j.pnpbp.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to imageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boushell LW, Kaku M, Mochida Y, Bagnell R, Yamauchi M. Immunohistochemical localization of matrixmetalloproteinase-2 in human coronal dentin. Arch Oral Biol. 2008;53:109–116. doi: 10.1016/j.archoralbio.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. Tophat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nature protocols. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing S: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S, Pyl P, Huber W: HTSeq–a python framework to work with high-throughput sequencing data. bioRxiv 2014. [DOI] [PMC free article] [PubMed]
- 69.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steinberg J, Webber C. The roles of FMRP-regulated genes in autism spectrum disorder: single- and multiple-hit genetic etiologies. Am J Hum Genet. 2013;93:825–839. doi: 10.1016/j.ajhg.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watson C, Hoeft F, Garrett AS, Hall SS, Reiss AL. Aberrant brain activation during gaze processing in boys with fragile X syndrome. Arch Gen Psychiatry. 2008;65:1315–1323. doi: 10.1001/archpsyc.65.11.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, Erba HW, Ringel J, Hayashi KM, Patnaik S, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008;63:40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hazlett HC, Poe MD, Lightbody AA, Styner M, MacFall JR, Reiss AL, Piven J. Trajectories of early brain volume development in fragile X syndrome and autism. J Am Acad Child Adolesc Psychiatry. 2012;51:921–933. doi: 10.1016/j.jaac.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoeft F, Walter E, Lightbody AA, Hazlett HC, Chang C, Piven J, Reiss AL. Neuroanatomical differences in toddler boys with fragile X syndrome and idiopathic autism. Arch Gen Psychiatry. 2011;68:295–305. doi: 10.1001/archgenpsychiatry.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M, DiCicco-Bloom E, Lai JK, Foster JA, Penagarikano O, et al. Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry. 2015;20:118–125. doi: 10.1038/mp.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lai JK, Lerch JP, Doering LC, Foster JA, Ellegood J. Regional brain volumes changes in adult male FMR1-KO mouse on the FVB strain. Neuroscience. 2016;318:12–21. doi: 10.1016/j.neuroscience.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 77.Noack J, Murau R, Engelmann M. Consequences of temporary inhibition of the medial amygdala on social recognition memory performance in mice. Front Neurosci. 2015;9:152. doi: 10.3389/fnins.2015.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hong W, Kim DW, Anderson DJ. Antagonistic control of social versus repetitive self-grooming behaviors by separable amygdala neuronal subsets. Cell. 2014;158:1348–1361. doi: 10.1016/j.cell.2014.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Y, He Z, Zhao C, Li L. Medial amygdala lesions modify aggressive behavior and immediate early gene expression in oxytocin and vasopressin neurons during intermale exposure. Behav Brain Res. 2013;245:42–49. doi: 10.1016/j.bbr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 80.Kondo Y. Lesions of the medial amygdala produce severe impairment of copulatory behavior in sexually inexperienced male rats. Physiol Behav. 1992;51:939–943. doi: 10.1016/0031-9384(92)90074-C. [DOI] [PubMed] [Google Scholar]
- 81.Twining RC, Vantrease JE, Love S, Padival M, Rosenkranz JA: An intra-amygdala circuit specifically regulates social fear learning. Nat Neurosci 2017;20:459–69. [DOI] [PMC free article] [PubMed]
- 82.Guyer AE, Lau JY, McClure-Tone EB, Parrish J, Shiffrin ND, Reynolds RC, Chen G, Blair RJ, Leibenluft E, Fox NA, et al. Amygdala and ventrolateral prefrontal cortex function during anticipated peer evaluation in pediatric social anxiety. Arch Gen Psychiatry. 2008;65:1303–1312. doi: 10.1001/archpsyc.65.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Veit R, Flor H, Erb M, Hermann C, Lotze M, Grodd W, Birbaumer N. Brain circuits involved in emotional learning in antisocial behavior and social phobia in humans. Neurosci Lett. 2002;328:233–236. doi: 10.1016/S0304-3940(02)00519-0. [DOI] [PubMed] [Google Scholar]
- 84.Abe N, Suzuki M, Mori E, Itoh M, Fujii T. Deceiving others: distinct neural responses of the prefrontal cortex and amygdala in simple fabrication and deception with social interactions. J Cogn Neurosci. 2007;19:287–295. doi: 10.1162/jocn.2007.19.2.287. [DOI] [PubMed] [Google Scholar]
- 85.Rilling JK, Glenn AL, Jairam MR, Pagnoni G, Goldsmith DR, Elfenbein HA, Lilienfeld SO. Neural correlates of social cooperation and non-cooperation as a function of psychopathy. Biol Psychiatry. 2007;61:1260–1271. doi: 10.1016/j.biopsych.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 86.Spinella M. Prefrontal substrates of empathy: psychometric evidence in a community sample. Biol Psychol. 2005;70:175–181. doi: 10.1016/j.biopsycho.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 87.Swartz JR, Wiggins JL, Carrasco M, Lord C, Monk CS. Amygdala habituation and prefrontal functional connectivity in youth with autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. 2013;52:84–93. doi: 10.1016/j.jaac.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:11591–11596. doi: 10.1073/pnas.1002262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective gabaergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30:9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Testa-Silva G, Loebel A, Giugliano M, de Kock CP, Mansvelder HD, Meredith RM. Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb Cortex. 2012;22:1333–1342. doi: 10.1093/cercor/bhr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tian M, Zeng Y, Hu Y, Yuan X, Liu S, Li J, Lu P, Sun Y, Gao L, Fu D, et al. 7, 8-Dihydroxyflavone induces synapse expression of AMPA Glua1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology. 2015;89:43–53. doi: 10.1016/j.neuropharm.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 92.Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Paul K, Venkitaramani DV, Cox CL. Dampened dopamine-mediated neuromodulation in prefrontal cortex of fragile X mice. J Physiol. 2013;591:1133–1143. doi: 10.1113/jphysiol.2012.241067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anstrom KK, Miczek KA, Budygin EA. Increased phasic dopamine signaling in the mesolimbic pathway during social defeat in rats. Neuroscience. 2009;161:3–12. doi: 10.1016/j.neuroscience.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, et al. Natural neural projection dynamics underlying social behavior. Cell. 2014;157:1535–1551. doi: 10.1016/j.cell.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hall FS, Wilkinson LS, Humby T, Robbins TW. Maternal deprivation of neonatal rats produces enduring changes in dopamine function. Synapse. 1999;32:37–43. doi: 10.1002/(SICI)1098-2396(199904)32:1<37::AID-SYN5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 98.Matthews GA, Nieh EH, Vander Weele CM, Halbert SA, Pradhan RV, Yosafat AS, Glober GF, Izadmehr EM, Thomas RE, Lacy GD, et al. Dorsal raphe dopamine neurons represent the experience of social isolation. Cell. 2016;164:617–631. doi: 10.1016/j.cell.2015.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moy SS, Nonneman RJ, Shafer GO, Nikolova VD, Riddick NV, Agster KL, Baker LK, Knapp DJ. Disruption of social approach by MK-801, amphetamine, and fluoxetine in adolescent C57BL/6J mice. Neurotoxicol Teratol. 2013;36:36–46. doi: 10.1016/j.ntt.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoffman EJ, Turner KJ, Fernandez JM, Cifuentes D, Ghosh M, Ijaz S, Jain RA, Kubo F, Bill BR, Baier H, et al. Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron. 2016;89:725–733. doi: 10.1016/j.neuron.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlations between biological replicates. The correlations of mapped reads between biological replicates using normalized counts per million for all reads were high for all comparisons in the lateral amygdala (LA) and prefrontal cortex (PFC), but less strong in some replicates in the medial amygdala (MA). MT mutant; WT wildtype; NS non-social; S social. Red p < 0.001; white p > 0.001. (PDF 144 kb)
Differentially expressed genes in lateral amygdala (XLSX 5388 kb)
Differentially expressed genes in medial amygdala (XLSX 5377 kb)
Differentially expressed genes in the prefrontal cortex (XLSX 5258 kb)
Overlapping genes of comparisons in each brain region. The differentially expressed genes (DEGs) that overlapped in specific comparisons are listed. The first column indicates DEGs significantly regulated in both the mutant versus wildtype comparison in the non-social condition (MUvsWTinNS) and in the social versus non-social conditions in the wildtype animals (SvsNSinWT). The second column indicates DEGs significantly regulated in both the MUvsWTinNS condition and in the interaction between genotype and stimulus exposure. The third column indicates DEGs significantly regulated in both the SvsNSinWT condition and in the interaction between genotype and stimulus exposure. Overlap in all three comparisons indicates DEGs that were significantly regulated in the MUvsWTinNS, SvsNSinWT, and interaction analyses. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 37 kb)
Most commonly differentially expressed genes across all comparisons. The genes that had the most frequent differential expression in analyses of all comparisons in all brain regions are listed, showing where they had significant differences in expression level for a given comparison (X) in each brain region. M mutant; W wildtype; NS non-social; S social. (PDF 270 kb)
Canonical pathway results for RNAseq data. Canonical pathway analysis revealed pathways with significant enrichment of genes that were differentially expressed in each comparison. The genotype and stimulus condition comparison is shown on the left, with the name of the top 10 enriched pathways, the number of differentially expressed genes that overlap with each pathway, and the adjusted p value shown from left to right. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 18 kb)
Upstream Regulator results for RNAseq data. Upstream regulator analysis revealed regulatory molecules known to affect a certain network of molecules, overlapping with genes that were differentially expressed in each comparison. The genotype and stimulus condition comparison is shown on the left, with the name of the top 10 upstream regulators, the predicted direction of activation or inhibition based on the up/down-regulation of differentially expressed genes that overlap with the affected network, and the adjusted p value shown from left to right. Analyses from each brain region are shown on separate sheets, accessed by the tabs at the bottom. (XLSX 12 kb)
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.