

Abstract
Reactive microglia are present in lesions of myelin-associated white matter disorders resulting in injuries to oligodendrocyte precursor cells (OPCs). Therefore, protection of OPCs from injury due to excessive activation of microglia is important in treating these diseases. Heat shock protein 60 (HSP60) has been demonstrated to be released extracellularly in the failing heart upon stress or injury. However, the role of HSP60 in the central nervous system and whether it participates in the toxic effects of microglia on OPCs remains unclear. The present study used the co-culture, cell death assays, binding assays, immunochemistry, western blot and ELISA. HSP60 was demonstrated to be released extracellularly by LPS-activated microglia and to bind to OPCs, triggering OPC apoptosis. When pretreated with toll-like receptor (TLR) 4 blocking antibody, the viability of OPCs increased, while the expression of nuclear factor κB (NFκB), caspase 3 and the release of proinflammatory cytokines triggered by HSP60 decreased. These results suggest that HSP60 released by microglia may mediate OPC apoptosis through binding to TLR4 on the surface of OPCs and subsequently activating the TLR4-NFκB signaling pathway. HSP60 may, therefore, serve as a potential target for treatment of myelin-associated neurodegenerative diseases that are accompanied by microglia activation.
Keywords: heat shock protein 60, oligodendrocyte precursor cell, microglia, toll-like receptor 4
Introduction
The activation of microglia, the primary immune cells of the central nervous system (CNS), occurs in almost all neurological disorders (1). Activated microglia undergo marked morphological changes and secrete various proinflammatory and neurotoxic factors, including interleukin (IL)-1β and tumor necrosis factor (TNF)-α, which contribute to neurodegeneration (2,3). Excessive microglial activation is toxic not only to neurons however also to other glial cells, such as oligodendrocytes (OL). Differentiation of oligodendrocyte precursor cells (OPCs) into OL is a crucial step in the formation of myelin in the CNS (4). Reactive microglia are present in lesions of myelin-associated white matter disorders, such as periventricular leukomalacia and multiple sclerosis, resulting in injuries to OPCs and leading to myelin defects via excessive production of oxidative stress and cytokines (5). Due to the importance of microglial activation in demyelination, the mechanism by which activated microglia affect the survival of OPCs was examined in the present study.
Heat shock protein 60 (HSP60) is a stress chaperone protein. HSP60 has dual functions in cell apoptosis: Under physiological conditions, HSP60 protects cells from apoptosis, whereas under stress conditions, HSP60 translocates to the plasma membrane, is released extracellularly and becomes toxic by targeting self-reactive T cells (6). The role of HSP60 in the myocardium has been previously reported (7–12), however, the role of HSP60 in the CNS remains to be fully elucidated. Previous studies have demonstrated that HSP60 is involved in the neuroprotection properties of Lycium barbarum polysaccharides (13), naloxone (14) and dextromorphan (15) by activation of Toll like receptor (TLR) 4 on microglia, for which HSP60 is a ligand (16). Notably, OPCs have been reported to express functional TLR4 (17,18).
In the present study, the hypothesis that extracellular HSP60, released by LPS-activated microglia, may bind to TLR4 on OPCs and induce OPC apoptosis was examined. The results demonstrated that OPC apoptosis was decreased by treatment with a TLR4 blocking antibody. These observations indicate that HSP60, released during microglia activation, may display a paracrine effect on neighboring OPCs leading to OPC cell death. The present study is, to the best of our knowledge, the first report of HSP60 exhibiting a toxic effect on OPCs. The current study may provide a potential target for treatment of myelin-associated neurodegenerative diseases that are accompanied by microglia activation.
Materials and methods
Chemicals
LPS was purchased from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany). Antibodies against GAPDH (cat no. ab181602; 1:2,000) and nuclear factor κB (NFκB; cat no. ab31481; 1:1,000) were from Abcam (Cambridge, MA, USA). Recombinant human (rh) HSP60 (cat no. ESP540), anti-HSP60 antibody (cat no. API-SPA-901; 1:1,000) and the HSP60 ELISA kit (cat no. ESK-600) were obtained from Stressgen Biotechnologies Co. (San Diego, CA, USA). Caspase-3 (cat. no. 9665; 1:2,000), TLR4 (cat. no. 2219; 1:1,000), myeloid differentiation primary response 88 (MyD88; cat. no. 4283, 1:1,000) antibodies were from Cell Signaling Technologies, Inc. (Danvers, MA, USA). Anti-O4 antibody was obtained from Merck Millipore (Darmstadt, Germany; cat no. MAB345; 1:200) The Live/Dead Assay kit was from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The proteinase inhibitor cocktails were from Merck & Co., Inc. (Whitehouse Station, USA). IL-6 (cat. no. 85-BMS625), IL-1β (cat no. 85-BMS6002) and TNF-α ELISA kits (cat. no. 85-BMS622) were from eBioscience, Inc. (San Diego, CA, USA). Caspase 3 activity assay kit was from Promega Corporation (Madison, WI, USA; cat. no. G809). The cell death assay (CDD) kit was from Roche Applied Science (Pleasanton, CA, USA; cat no. 11684795910). Bicinchoninic acid (BCA) and enhanced chemiluminescence (ECL) kits were from Pierce (Thermo Fisher Scientific, Inc.). Dulbecco's modified Eagle's medium (DMEM), ProLong Gold Antifade reagent with DAPI (cat. no. P36931), and fetal bovine serum (FBS) were from Gibco (Thermo Fisher Scientific, Inc.). Alexa Fluor 647 Protein Labeling kit (cat no. A20173), neurobasal medium (cat no. 10888022) and B27 (cat no. 17504044) were from Thermo Fisher Scientific, Inc. Recombinant platelet-derived growth factor (PDGF)-AA (cat no. AF-100-13A) and TNF-α (cat no. 315-01A) were bought from PeproTech, Inc. (Rocky Hill, NJ, USA). All other chemicals were purchased from ZSGB-BIO (Shanghai, China), unless otherwise stated.
Primary culture of microglia and OPCs
Purified microglia and OPCs were isolated as previously described with some modifications (19). Mixed cortical glia cell cultures were generated from 10 newborn P1 (weight, 5–7 g) male Sprague Dawley rats (Animal Center of Ningxia Medical University, Yinchuan, China) and maintained in DMEM with 20% FBS for 10 days in 75 cm2 flasks at 37°C and 95% O2/5% CO2. The experiments were approved by the Ethics Committee of Ningxia Medical University and reviewed by the Institutional Review Board of Ningxia Medical University. The culture medium was replaced every 3 days. For OPC collection, cultures were first shaken for 1–2 h at 200 rpm. The microglia cells were detached away from the attached cells and the suspension cells were collected, cultured in fresh flasks with 10% FBS in DMEM. The previous flasks were incubated in fresh medium in 95% O2/5% CO2 at 37°C for 4 h, and then shaken at 260 rpm at 37°C for 16–18 h. The detached cell suspension was seeded and allowed to adhere in uncoated Petri dishes for 1 h at 37°C with 95% O2/5% CO2, which allowed the microglia and astrocytes to become firmly attached, whereas OPCs were loosely attached and could be collected by gently shaking the dishes. Collected OPCs in the medium were then replated at 5,000–50,000 cells/cm2 onto poly-D-lysine-coated plates, dishes or coverslips. OPCs were grown in Neurobasal medium supplemented with 2% B27. To expand the OPCs and to keep them undifferentiated, the culture medium was supplemented with PDGF-AA (10 nM). Microglia cells were cultured in DMEM supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 g/ml). Cultures were maintained at 37°C in a humidified incubator gassed with 95% O2 and 5% CO2.
Coculture of microglia and OPCs
For coculture experiments, microglia cells were seeded at density 1,000–50,000 cells/cm2 onto Transwell inserts (Millipore; Merck KGaA), and OPCs were also seeded at a density 1,000–50,000 cells/cm2 on coverslips in a dish separately for 24 h. Then microglia cells were treated with 1 µg/ml LPS for 4 h and no LPS treatment group as control. The medium was discarded and the Transwell with the microglia was moved on top of the dish, thus sharing medium (DMEM/10% FBS) with the OPCs for 16 h. The Transwell was then removed and OPCs were fixed with 4% paraformaldehyde in PBS for 15 min, washed 3 times with PBS, and permeabilized with 0.5% Triton X-100 in PBS for 20 min. Cells were washed 3 times again with PBS and blocked with 5% BSA for 1 h at 37°C. Cells were then incubated with anti-O4 mouse antibody in PBS overnight at 4°C. Following washing with PBS, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (cat no. GTX77318; dilution, 1:1,000; GeneTex, Inc., Irvine, CA, USA) for 1 h at 37°C, washed with PBS and mounted using ProLong Gold Antifade reagent with DAPI. Cells were visualized using an Eclipse 660 fluorescent microscope (Nikon Corporation, Tokyo, Japan). The images were analyzed using 5 randomly selected fields of view and quantified using ImageJ version 1.48u (imagej.nih.gov/ij/).
Binding assay
rhHSP60 was labeled with Alexa Fluor 647 using a protein labeling kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. For live staining, OPCs were incubated with Alexa 647-conjugated rhHSP60 at 4°C for 30 min. Cells were visualized using an Eclipse 660 fluorescent microscope and a Spot cooled CCD (both obtained from Nikon Corporation, Tokyo, Japan).
Apoptosis
OPCs were treated with 5 µg/ml rhHSP60 or 10 ng/ml TNF-α for 16 h at 37°C. Caspase 3 activity was measured using a kit. DNA fragmentation was measured using the CDD assay, as per the manufacturer's instructions.
Blocking antibody
Following 30 min of pre-incubation with the TLR4 blocking antibody (20 µg/ml), cells were treated with rhHSP60, TNF-α or LPS for 16 h, prior to experiments.
ELISA
The levels of IL-6, IL-1β, HSP60 and TNF-α in culture medium were quantified with commercial ELISA kits, according to the manufacturer's directions. Absorbance was determined at 450 nm using a microplate reader.
Western blotting
Cells were washed with PBS 3 times and lysed for 15 min at 4°C with radioimmunoprecipitation assay buffer containing 1% NP40, 150 mM NaCl, 50 mM Tris-HCl, a protease inhibitor mixture, 50 mM NaF and 1 mM Na3VO4 for phosphatase inhibition. Then the cell lysates were centrifuged at 1,200 × g for 10 min at 4°C and used for western blotting. The protein concentration was determined by BCA kit, according to the manufacturer's instructions. Equal quantities of protein (10 µg per well) were loaded and ran on a 10% SDS-polyacrylamide gels and then transferred to a polyvinylidene fluoride membrane. Membranes were blocked with 5% nonfat dry milk in 0.1% Tween-20-Tris buffered saline (TBST; pH 7.4) overnight at 4°C and incubated with primary antibodies in TBS/0.1% Tween-20 (TBST) overnight at 4°C. Following washing in milk-TBST, blots were incubated with horseradish peroxidase-conjugated secondary antibodies goat anti-rabbit (cat. no. ZB-2301; 1:5,000) and anti-mouse IgG (cat. no. ZB-2305; 1:5,000; ZSGB-BIO, Beijing, China). The target proteins were detected by enhanced chemiluminescence (ECL) detection system and X-ray films.
Statistical analysis
Statistical differences were determined using one-way analysis of variance followed by a Holm-Sidak test. Data in the text and figures are presented as the mean ± standard error. Experiments were repeated 3 to 6 times. P<0.05 was considered to indicate a statistically significant difference.
Results
Coculture of activated microglia with OPCs results in OPC death
In order to observe the effects of activated microglia on OPCs, the two cell types were cocultured. Microglia were activated with 1 µg/ml LPS for 4 h in a transwell, then the transwell was moved on top of a dish where OPCs were cultured, so that the two cell types shared medium. Following 16 h of coculture, OPCs were stained for expression of the O4 oligodendrocyte-specific marker, visualized by a FITC-conjugated secondary antibody and observed under a fluorescent microscope. The results demonstrated that the number of live OPCs decreased significantly following coculture with activated microglia compared with co-culture control with non LPS-activated microglia (Fig. 1), indicating that activated microglia were toxic to neighboring OPCs. Based on this result, the molecular mechanism of microglia-induced OPC cell death was explored.
Figure 1.
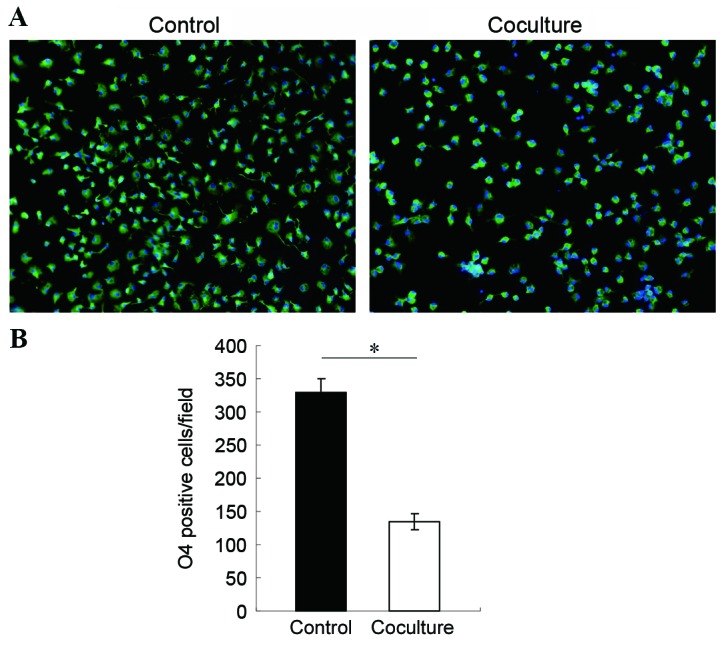
Coculture of activated microglia with OPCs results in OPC death. Microglia cells were activated with LPS for 4 h, then cocultured with OPCs for 16 h, non-activated microglia cocultured with OPCs as control. OPCs were stained with anti-O4 antibody and FITC-conjugated secondary antibody (original magnification, ×20). (A) Representative images of OPCs stained for O4 expression (green, O4; blue, nuclei using DAPI). (B) Quantification of O4-positive cells in control untreated cells and OPC cells cocultured with LPS-activated microglia. *P<0.05 vs. control. OPCs, oligodendrocyte progenitor cells; LPS, lipopolysaccharide; O4, immunoglobulin kappa variable 1D-37; FITC, fluorescein isothiocyanate.
Expression and release of HSP60 in activated microglia
To test the hypothesis that HSP60 may be important in microglia-mediated OPC death, HSP60 expression levels in activated microglia were examined. Microglia cells were treated with 1 µg/ml LPS for 4 h, then the protein lysate was analyzed by western blotting for HSP60 expression levels. Compared with untreated control cells, HSP60 protein expression levels increased significantly in LPS-treated microglia (Fig. 1A). In order to examine the levels of secreted extracellular HSP60, ELISA assay was performed in samples of the culture media from LPS-treated and untreated microglia cells. The results demonstrated that HSP60 levels in the culture medium also significantly increased in LPS-treated microglia, compared with untreated control cells (Fig. 2B). These results indicated that HSP60 was highly expressed by activated microglia and was released extracellularly. Therefore, HSP60 might be the toxic factor released by microglia that caused the OPC death.
Figure 2.
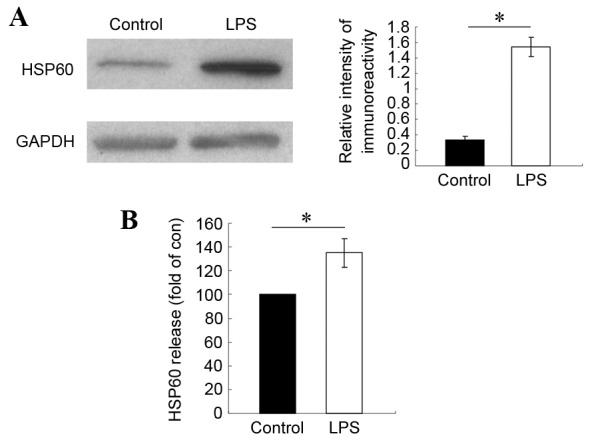
Expression and release of HSP60 increases significantly in microglia following LPS activation. Microglia cells were treated with 1 µg/ml LPS for 4 h. (A) HSP60 protein levels were measured by western blotting. GAPDH was used as a loading control and HSP60 signal is reported as a relative ratio to GAPDH. (B) Extracellular levels of HSP60 in culture medium were measured by ELISA, and presented as fold ratio relative to control. Results are representative of 3 independent experiments. *P<0.05 vs. control. HSP60, heat shock protein 60; LPS, lipopolysaccharide.
Extracellular rhHSP60 binding to OPCs leads to OPC apoptosis
To examine further the effect of HSP60 on OPCs, OPCs were incubated with fluorescently-labeled rhHSP60. As demonstrated in Fig. 3A, Alexa 647-rhHSP60 bound on the surface of OPCs. Extracellular HSP60 (exHSP60) has been reported to be a ligand of TLR4 (20). Therefore, exHSP60 binding on TLR4 in OPCs might lead to OPC apoptosis. Apoptosis was examined by caspase 3 activity and DNA fragmentation assays following OPCs treatment with rhHSP60 for 16 and 20 h, respectively. TNF-α treatment (10 ng/ml), which causes apoptosis in OPCs, was used as a positive control. Both rhHSP60 and TNF-α treatments resulted in a similar increase of caspase 3 activity in OPCs, compared with untreated cells (Fig. 3B). The CDD assay confirmed this result, with both rhHSP60 and TNF-α significantly triggering increased DNA fragmentation in OPCs, compared with untreated cells (Fig. 3C).
Figure 3.

Extracellular HSP60 binds OPCs and causes apoptosis. (A) OPCs were incubated with Alexa 647-conjugated rhHSP60 (red signal) at 4°C for 30 min, then stained for O4 expression (green signal). Representative images are presented. Magnification, ×20. (B) Caspase 3 activity and (C) DNA fragmentation were measured in OPCs treated with 5 µg/ml rhHSP60 or 10 ng/ml TNF-α and compared with untreated cells (control). *P<0.05 vs. control. HSP60, heat shock protein 60; OPCs, oligodendrocyte progenitor cells; rh, recombinant human; O4, immunoglobulin κ variable 1D-37; OD, optical density.
TLR4 blocking antibody inhibits rhHSP60-mediated caspase-3 expression
Since HSP60 has been proposed to be a ligand for TLR4, the role of TLR4 in HSP60-mediated OPC apoptosis was examined, by pretreating OPCs with a TLR4 blocking antibody. Without the blocking antibody, rhHSP60 and TNF-α treatment triggered a significant increase in caspase 3 expression in OPCs compared with control untreated cells (Fig. 4A). However, pretreatment with 20 µg/ml anti-TLR4 blocking antibody for 30 min prevented the increase in caspase 3 expression mediated by rhHSP60 or TNF-α; no significant difference in caspase 3 expression was observed following treatment with rhHSP60 or TNF-α compared with control untreated cells (Fig. 4B).
Figure 4.
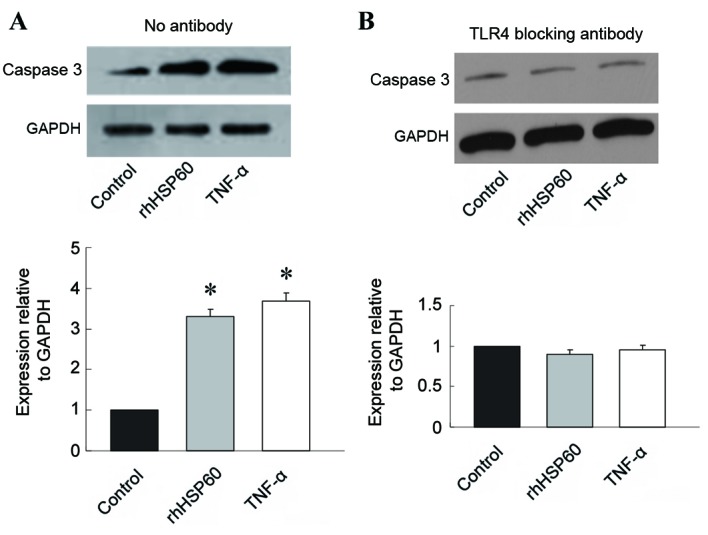
TLR4 blocking antibody inhibits rhHSP60-mediated caspase 3 expression in OPCs. Following 30-min pre-incubation with a TLR4 blocking antibody (20 µg/ml), OPCs were treated with rhHSP60 or TNF-α for 16 h. Cell lysates were then analyzed for caspase 3 protein expression levels by western blotting and quantified relative to GAPDH. (A) No antibody pretreatment. (B) TLR4 blocking antibody pretreatment. *P<0.05 vs. control untreated cells. TLR4, toll-like receptor 4; rhHSP60, recombinant human heat shock protein 60; OPCs, oligodendrocyte progenitor cells; TNF-α, tumor necrosis factor α.
TLR4 blocking antibody reverses rhHSP60-mediated MyD88, NFκ B and cytokine overexpression in OPCs. To further investigate the signaling pathway leading to rhHSP60-mediated apoptosis, the expression of downstream signaling factors MyD88 and NFκB was examined by western blotting. Protein expression levels of MyD88 and the NFκB subunit p65 significantly increased by rhHSP60 treatment for 16 h in OPCs, compared with untreated control cells (Fig. 5A). This effect was, however, significantly reduced by pretreatment with the TLR4 blocking antibody (Fig. 5A). In addition, release in the medium of the proinflammatory cytokines IL-6 and TNF-α was significantly stimulated by rhHSP60 treatment in OPCs compared with untreated control cells (Fig. 5B), however this effect was also suppressed by pretreatment with the TLR4 blocking antibody (Fig. 5B). These results indicated that HSP60 may act on OPCs through the TLR4-MyD88-NFκB signaling pathway.
Figure 5.

TLR4 blocking antibody inhibits the rhHSP60-mediated MyD88 and NFκB overexpression and cytokine production. Following 30 min of preincubation with a TLR4 blocking antibody, OPCs were treated with rhHSP60 for 16 h. (A) Protein expression levels for MyD88 and the NFκB subunit p65 were measured by western blotting in untreated cells (control), cells treated with rhHSP60 and cells treated with hHSP60 + TLR4 blocking antibody (TLR4 AB). Representative images and quantification are presented relative to the GAPDH loading control. (B) Proinflammatory cytokines IL-6 and TNF-α levels were measured by ELISA in the media of untreated cells (control), cells treated with rhHSP60 and cells treated with hHSP60 + TLR4 blocking antibody (TLR4 AB). *P<0.05 vs. control untreated cells; #P<0.05 vs. rhHSP60. TLR4, toll-like receptor 4; rhHSP60, recombinant human heat shock protein 60; MyD88, myeloid differentiation primary response 88; NFκB, nuclear factor κB; OPCs, oligodendrocyte progenitor cells; IL-6, interleukin 6; TNF-α, tumor necrosis factor α.
Discussion
The present study demonstrated that activated microglia released HSP60. ExHSP60 then bound to OPCs, which resulted in caspase 3 activation and increased DNA cleavage in OPCs, both markers of induction of apoptosis. TRL4 signaling was demonstrated to be essential for the HSP60-mediated caspase 3 activation and release of proinflammatory cytokines. Finally, HSP60 was demonstrated to upregulate the MyD88-NFκB signaling pathway, through TLR4. Thus, the present study suggested that activated microglia released HSP60 resulting in OPC apoptosis, presumably through the TLR4-MyD88-NFκB signaling pathway.
A number of studies have established that activated microglia influence the survival of neurons (21). Studies examining the effect of microglia on OPC survival have yielded conflicting results. Filipovic and Zecevic (22) reported that activated microglia are beneficial for OPC proliferation, while Miller et al (23) reported that activated microglia have divergent effects on OPCs and mature oligodendrocytes, reducing OPC survival and increasing mature oligodendrocyte survival. Additional studies have reported that microglia are toxic to OPC in vivo and in vitro (18,24,25), which is consistent with the current findings. In the present study, it was demonstrated that the number of live OPCs decreased significantly following coculture with LPS-activated microglia.
Activated microglia release various factors that are important in microglia-induced OPC apoptosis (26). HSP60 is primarily a mitochondrial protein, however it has been demonstrated that, upon stress or injury in the heart, HSP60 translocates to the plasma membrane and gets released extracellularly (12). A previous study from our group also demonstrated that HSP60 is highly expressed and released by microglia (13). ExHSP60 has been demonstrated to be a ligand for TLR4 in the immune system (16), however this remains unclear for other cell types. Tian et al (8) reported that extracellular HSP60 induces inflammation through upregulation and activation of TLRs in cardiomyocytes. Taylor et al (17) demonstrated that active TLR4 is present on OPCs. Therefore, in order to examine whether HSP60 can be bound by OPCs, a binding experiment was performed by labeling HSP60 with Alexa Fluor 647 and incubating OPCs with the Alexa647-rhHSP60. The results demonstrated that exHSP60 bound on the surface of OPCs, leading to caspase 3 activation and increased DNA fragmentation compared with untreated cells.
Lehnardt et al (18) demonstrated that HSP60 alone had no effect on neuronal survival. By contrast, when purified microglia were added to neuronal cultures, severe loss of neurons was observed in the presence of HSP60 (18). As a control, the addition of purified microglia from mice with a tlr4 loss-of-function mutation to wild-type neurons had no effect on neuronal survival following HSP60 treatment, confirming the requirement for intact TLR4 signaling (18). Within the TLR4 signaling pathway, the adapter protein MyD88 is an important activator of NFκB. Activation of NFкB is important in the inflammatory response initiated by microglia, as transcription of many proinflammatory genes is mediated by NFкB (27). NFκB has also been reported to initiate transcription of the HSP60 stress protein gene, which elicits a potent proinflammatory response in innate immune cells (11). To determine whether HSP60 affects the TLR4-MyD88-NFκB pathway in OPCs, expression levels of MyD88 and NFκB in OPCs were analyzed following HSP60 treatment. As expected, MyD88 and NFκB subunit p65 protein expression levels in OPCs increased significantly following HSP60 treatment, compared with untreated cells. Activation of TLR4 was necessary for the HSP60-mediated effects on OPCs; pretreatment with a TLR4 blocking antibody, significantly inhibited the rhHSP60-mediated caspase 3 activation, in addition to attenuating the expression of MyD88 and NFκB and the release of the proinflammatory cytokines IL-6 and TNF-α. These observations suggest that HSP60 acts on OPCs through the TLR4-MyD88-NFκB signaling pathway.
Acknowledgements
The present study was supported by grants from Ningxia Natural Science Foundation (grant no. NZ14057), National Natural Science Foundation of China (grant nos. 81460182, 31460257, 81571098 and 31260243), and Ningixa 13th Plan of five-year Major Scientific Program (grant no. 2016BZ 07).
References
- 1.Hanisch UK, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 2.McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol. 2001;169:219–230. doi: 10.1006/exnr.2001.7688. [DOI] [PubMed] [Google Scholar]
- 3.Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: Involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- 4.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-K. [DOI] [PubMed] [Google Scholar]
- 5.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 6.Kim SC, Stice JP, Chen L, Jung JS, Gupta S, Wang Y, Baumgarten G, Trial J, Knowlton AA. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res. 2009;105:1186–1195. doi: 10.1161/CIRCRESAHA.109.209643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta S, Knowlton AA. HSP60, Bax, apoptosis and the heart. J Cell Mol Med. 2005;9:51–58. doi: 10.1111/j.1582-4934.2005.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian J, Guo X, Liu XM, Liu L, Weng QF, Dong SJ, Knowlton AA, Yuan WJ, Lin L. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc Res. 2013;98:391–401. doi: 10.1093/cvr/cvt047. [DOI] [PubMed] [Google Scholar]
- 9.Knowlton AA, Gupta S. HSP60, Bax, and cardiac apoptosis. Cardiovasc Toxicol. 2003;3:263–268. doi: 10.1385/CT:3:3:263. [DOI] [PubMed] [Google Scholar]
- 10.Kirchhoff SR, Gupta S, Knowlton AA. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105:2899–2904. doi: 10.1161/01.CIR.0000019403.35847.23. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Chen L, Hagiwara N, Knowlton AA. Regulation of heat shock protein 60 and 72 expression in the failing heart. J Mol Cell Cardiol. 2010;48:360–366. doi: 10.1016/j.yjmcc.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, Torre-Amione G, Knowlton AA. HSP60 in heart failure: Abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:H2238–H2247. doi: 10.1152/ajpheart.00740.2007. [DOI] [PubMed] [Google Scholar]
- 13.Teng P, Li Y, Cheng W, Zhou L, Shen Y, Wang Y. Neuroprotective effects of Lycium barbarum polysaccharides in lipopolysaccharide-induced BV2 microglia cells. Mol Med Rep. 2013;7:1977–1981. doi: 10.3892/mmr.2013.1442. [DOI] [PubMed] [Google Scholar]
- 14.Cheng W, Hou X, Zhang N, Ma J, Ding F, Li F, Miao Z, Zhang Y, Qi Q, Li G, et al. HSP60 is involved in the neuroprotective effects of naloxone. Mol Med Rep. 2014;10:2172–2176. doi: 10.3892/mmr.2014.2411. [DOI] [PubMed] [Google Scholar]
- 15.Cheng W, Li Y, Hou X, Bai B, Li F, Ding F, Ma J, Zhang N, Shen Y, Wang Y. Determining the neuroprotective effects of dextromethorphan in lipopolysaccharide-stimulated BV2 microglia. Mol Med Rep. 2015;11:1132–1138. doi: 10.3892/mmr.2014.2794. [DOI] [PubMed] [Google Scholar]
- 16.Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: Heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 17.Taylor DL, Pirianov G, Holland S, McGinnity CJ, Norman AL, Reali C, Diemel LT, Gveric D, Yeung D, Mehmet H. Attenuation of proliferation in oligodendrocyte precursor cells by activated microglia. J Neurosci Res. 2010;88:1632–1644. doi: 10.1002/jnr.22335. [DOI] [PubMed] [Google Scholar]
- 18.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z, Watanabe M, Nishiyama A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J Neurosci Methods. 2005;149:50–56. doi: 10.1016/j.jneumeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberger K, Dembny P, Derkow K, Engel O, Krüger C, Wolf SA, Kettenmann H, Schott E, Meisel A, Lehnardt S. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol Neurodegener. 2015;10:5. doi: 10.1186/s13024-015-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 22.Filipović R, Zecević N. 2005. Interaction between microglia and oligodendrocyte cell progenitors involves Golli proteins. Ann N Y Acad Sci. 2005;1048:166–174. doi: 10.1196/annals.1342.015. [DOI] [PubMed] [Google Scholar]
- 23.Miller BA, Crum JM, Tovar CA, Ferguson AR, Bresnahan JC, Beattie MS. Developmental stage of oligodendrocytes determines their response to activated microglia in vitro. J Neuroinflamm. 2007;4:28. doi: 10.1186/1742-2094-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang Y, Cai Z, Rhodes PG. Effects of lipopolysaccharide on oligodendrocyte progenitor cells are mediated by astrocytes and microglia. J Neurosci Res. 2000;62:510–520. doi: 10.1002/1097-4547(20001115)62:4<510::AID-JNR5>3.3.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Fan XL, Zhao Y, Luo GR, Li XP, Li R, Le WD. Estrogen provides neuroprotection against activated microglia-induced dopaminergic neuronal injury through both estrogen receptor-alpha and estrogen receptor-beta in microglia. J Neurosci Res. 2005;81:653–665. doi: 10.1002/jnr.20583. [DOI] [PubMed] [Google Scholar]
- 26.Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S, Schwartz M. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khasnavis S, Jana A, Roy A, Mazumder M, Bhushan B, Wood T, Ghosh S, Watson R, Pahan K. Suppression of nuclear factor-κB activation and inflammation in microglia by physically modified saline. J Biol Chem. 2012;287:29529–29542. doi: 10.1074/jbc.M111.338012. [DOI] [PMC free article] [PubMed] [Google Scholar]