

Abstract
There is an endemic cardiomyopathy currently occurring in China, termed, Keshan disease (KD). The authors previously compared mitochondrial-associated gene expression profiles of peripheral blood mononuclear cells (PBMCs) derived from KD patients and normal controls, using mitochondria-focused cDNA microarray technology. The results detected an upregulation of the enzyme-associated CYP1A1 gene, (ratios ≥2.0). The aryl hydrocarbon receptor (AhR) regulates the expression of numerous cytochrome P450 (CYP) genes including members of the CYP1 family; CYP1A1 and CYP1A2. Several previous studies have suggested roles for the aryl hydrocarbon receptor (AhR) and the genes that it regulates. An example involves cytochrome P4501A1 (CYP1A1), in the pathogenesis of heart failure, cardiac hypertrophy and other cardiomyopathies. Mitochondria comprise ~30% of the intracellular volume in mammalian cardiomyocytes, and subtle alterations in mitochondria can markedly influence cardiomyopathies. The present study investigated alterations in the activity and functions of mitochondria following AhR-induced overexpression of CYP1A1. AC16 cells were treated with the CYP1A1 inducer 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and cytotoxicity was then evaluated in MTT assays. Reverse transcription-quantitative polymerase chain reactions, western blot analysis and 7-ethoxyresorufin O-deacylase assays were performed to analyze the mRNA and protein levels, and the enzymatic activity of CYP1A1. Mitochondrial activity and mass were analyzed using an inverted fluorescence microscope and a fluorescence microplate reader. Reactive oxygen species (ROS) activity was analyzed using flow cytometry. The results of the current study demonstrated that TCDD gradually increased mRNA and protein levels of AhR and CYP1A1, in addition to the enzymatic activity. Mitochondrial activity and the quality of mitochondrial membranes were also significantly attenuated, and mitochondrial ROS levels were elevated in the TCDD-induced cardiomyocytes. The results indicate the involvement of the AhR/CYP1A1 signaling pathway in the mechanism of action of TCDD in human cardiomyocytes. The present findings may provide an explanation for myocardial injuries caused by polycyclic aromatic hydrocarbons. The authors conclude that exposure to TCDD results in regulatory alteration to the expression of detoxification genes that ultimately affect the metabolic activation and function of cardiomyocytes.
Keywords: cardiomyocytes, aryl hydrocarbon receptor, CYP1A1, mitochondria
Introduction
The aryl hydrocarbon receptor (AhR) is a cytosolic receptor and ligand-activated transcriptional factor. In the absence of a ligand, cytoplasmic AhR is present in a primarily dormant state in an inactive complex that contains two 90-kDa heat-shock proteins (HSP90), hepatitis B virus X-associated protein, p23 and the AhR inhibitory protein (1) It has been hypothesized that HSP90 maintains AhR in a conformation that is capable of high-affinity ligand binding to prevents its nuclear translocation (2). AhR ligands consist primarily of polycyclic aromatic hydrocarbons (PAHs) (3). Upon binding a ligand, AhR is activated. AhR is able to dissociate from HSP90, translocate into the nucleus and form a heterodimer with the closely associated nuclear protein aryl hydrocarbon receptor nuclear translocator (ARNT) (4). The AhR-ARNT heterodimer creates a specific DNA recognition sequence, GCGTG, within a responsive element, known as the xenobiotic responsive element (XRE). XREs are located in the promoter regions of a number of receptor-regulated genes, including CYP1A1, CYP1A2, CYP1B, and CYP2E1 (1). CYP1A1 is of particular interest because it is the most active of the group of cytochrome P450s that metabolize ligands of AhR, PAHs, into reactive species.
Cardiac CYP1A1 transcription was significantly induced by treatment with AhR ligands, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), benzo(a)pyrene (BaP), 3,3,4,4,5-pentachlorobiphenyl (PCB 126), β-naphthoflavone (bNF) and 3-methylcholanthrene, in a variety of mammalian species (5,6). Previous studies investigating the expression of constitutive CYP1A1 mRNA have demonstrated that TCDD and BaP are the most potent inducers of CYP1A1 expression (3). It has been suggested that CYP1A1 mRNA can be superinduced by treatment with TCDD, which is the most potent AhR ligand described (7). TCDD is a pervasive environmental contaminant that induces hepatic and myocardial oxidative stress (8,9).
CYP1A1 belongs to the cytochrome P450 family (family 1, subfamily A, polypeptide 1). CYP1A1 was initially thought to be localized only to the endoplasmic reticulum, until a mitochondrial form of CYP1A1 was characterized in the livers of rats that were pre-treated with bNF and, more recently, on inner mitochondrial membranes (10). CYP1A1 has been demonstrated to produce elevated levels of reactive oxygen species (ROS) in fish and rodent mitochondria and microsomes when challenged with the PAH compounds 3,3′,4,4′-tetrachlorobiphenyl (11) and PCB 126 (12). It has been previously indicated that the CYP1A1 metabolism serves a major role in detoxifying foreign chemicals and metabolic activation, which leads to oxidative damage (3). A previous study established a link between CYP1A1 and oxidative stress (8). However, little is currently understood regarding the mechanisms underlying mitochondrial oxidative stress in human cardiomyocytes.
In the present study, it was investigated whether the AhR-induced overexpression of CYP1A1 causes mitochondrial oxidative stress-related effects in the human cardiac-derived AC16 cell line. The present study may aid to determine the involvement of AhR and its downstream gene product, CYP1A1, in the mechanisms underlying dioxin-induced subacute levels of mitochondrial oxidative stress.
Materials and methods
Chemicals
The reagent, TCDD, was purchased from AccuStandard, Inc. (New Haven, CT, USA). A pre-calculated volume of TCDD (10 µl) was first dissolved in dimethyl sulfoxide (DMSO; 300.5 µl). A concentration of 100 nM TCDD stock liquid of TCDD was obtained. The appropriate stock solutions of TCDD were added directly to the culture media to achieve a final concentration of 0.1, 1, 5 and 10 nM of TCDD. The concentration of DMSO was not allowed to exceed 0.05% (v/v). Dulbecco's modified Eagle's medium (DMEM)/F-12 was purchased from Beijing Aipoo HuaMei Biotechnology Co., Ltd. (Beijing, China). Fetal bovine serum (FBS) was purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). A High-Capacity cDNA Reverse Transcription kit and SYBR-Green Super Mix were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The reverse transcription-quantitative polymerase chain reaction (RT-qPCR) primers were synthesized by AuGCT DNA-SNY Biotechnology, Inc. (Beijing, China). Rabbit anti-human CYP1A1 (catalog no. sc-39397)/AhR (catalog no. sc-0816R) polyclonal antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell cultures and treatments
The AC16 human myocardial cell line (Land Biology, Inc., Guangzhou, China) was cultured in standard DMEM that was supplemented with 10% FBS and 1% penicillin-streptomycin (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) in 25 cm3 plastic culture flasks. The cells were grown in the culture flasks at 37°C in a 5% CO2 humidified incubator. The appropriate stock solutions of TCDD were added directly to the culture media to achieve a final concentration of 0.1 nM or 10 nM of TCDD. To measure the effects of TCDD on cell viability and cardiomyocyte mitochondrial membrane oxidative injury, cells were grown at a density of 7.5×104 cells/well in 96-well tissue culture plates. For the analyses used to determine mRNA and protein levels and the intracellular production of oxidants, the cardiomyocytes were seeded into 6-well plates at a density of 1.5×106 cells/well. To measure mitochondrial activity, the cells were cultured in 24-well tissue culture plates at a density of 1.5×105 cells/well.
The effect of TCDD on cell viability
The cytotoxic effects of TCDD on AC16 myocardial cell viability were determined by measuring the capacity of the reducing enzymes present in viable cells to MTT to formazan crystals. Briefly, cardiomyocytes were treated with various concentrations of TCDD (0, 0.1, 1, 5 or 10 nM) for 24 h. A volume of 20 µl MTT solution (5 mg/ml in PBS) was then added, and the cells were incubated for 4 h at 37°C. The supernatants were then aspirated, and 150 µl DMSO was added to each well. The plates were shaken for 10 min to thoroughly dissolve the formazan crystals. The absorbance at 570 nm was measured for each well using a microplate reader (Infinite™ M2000; Tecan, Männedorf, Switzerland).
RNA extraction and RT-qPCR
Following incubation of the AC16 cardiomyocytes with various concentrations of TCDD (0, 0.1, 1, 5 or 10 nM) for 24 h, total RNA was extracted using an E.Z.N.A Total RNA kit I (R6834-01; Omega Bio-Tek, Inc., Norcross, GA, USA) according to the manufacturer's instructions. The concentration and purity of the total RNA was evaluated by measuring the 260/280 nm absorbance ratio using a Thermo Nanodrop 2000 (NanoDrop; Thermo Fisher Scientific, Inc., Wilmington, DE, USA). To obtain cDNA, total RNA (0.5 µg) was reverse transcribed using a PrimeScript™ RT reagent kit (DRR037A; Takara Bio, Inc., Otsu, Japan), three times. RT-qPCR was then performed using a Bio-Rad IQ5 PCR Detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with SYBR Premix Ex Taq™ II (RR820A; Takara Bio Inc.) according to the manufacturer's instructions. The following specific primer sequences were used: CYP1A1 forward, 5′-CCTCCTCAACCTCCTGCTAC-3′ and reverse, 5′-AAGCAAATGGCACAGAYGAC-3′; CYP2C19 forward, 5′-GGTCCTTGTGCTCTGTCTCT-3′ and reverse, 5′-CATATCCATGCAGCACCACC-3′; GAPDH forward, 5′-TGCACCACCAACTGCTTAGC-3′ and reverse, 5′-GGCATGGACTGTGGTCATGAG-3′.
Protein extraction and western blot analysis
Cardiomyocytes were pre-treated with various concentrations of TCDD (0, 0.1, 1, 5 or 10 nM) for 24 h. The cells were then rinsed with ice-cold PBS, and the cytoplasmic and nuclear proteins were lysed using radioimmunoprecipitation buffer (HEART Biological Technology Co., Ltd., Xi'an, China). A total of 25 µg protein was separated from each treatment group using 12% SDS-PAGE, and the separated proteins were then electrophoretically transferred onto polyvinylidene difluoride membranes. The membranes were blocked in 0.75 g skim milk dissolved in 15 ml tris-buffered saline with Tween-20 (TBST) and probed with primary polyclonal rabbit anti-human antibodies against CYP1A1 (catalog no. sc-39397; 1:500), AhR (catalog no. sc-166586; 1:800) and β-actin (catalog no. sc-130656; 1:400; Santa Cruz Biotechnology, Inc.) at 4°C overnight. The primary antibodies were then discarded and the membranes were washed three times with TBST and incubated with the appropriate horseradish peroxidase-conjugated goat anti-rabbit IgG (catalog no. A0216; 1:400; Beyotime Institute of Biotechnology, Haimen, China) for 2 h with shaking at room temperature. The bands were visualized using enhanced chemiluminescence according to the manufacturer's instructions(EMD Millipore, Billerica, MA, USA).
Determination of CYP1A1 enzymatic activity
The activity of 7-ethoxyresorufin O-deacylase (EROD) was used as a diagnostic marker of CYP1A1 enzymatic activity. The 7-ethoxyresorufin generated resorufin when it was catalyzed by EROD. The wavelengths used to analyze excitation and emission (λex, 488 nm and λem, 505 nm) were 530 and 590 nm, respectively. The rate of the change in fluorescence was recorded prior to the experiment and after the addition of a known quantity of resorufin using a microplate reader.
Effect of TCDD on intracellular ROS production
To determine whether TCDD-induced CYP1A1 overexpression resulted in an increase in intra-cellular ROS production, intracellular oxidant production was analyzed using the non-fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Beyotime Institute of Biotechnology). Cardiomyocytes were seeded into 6-well plates at a density of 1.5×106 cells/well and treated with various concentrations of TCDD. The cells were incubated with diluted DCFH-DA at 37°C for 20 min and reverse-blended every 3–5 mins. The cells were then washed three times with serum-free cell culture medium, and the DCF fluorescence distribution was detected in 2×107 cells using flow cytometry.
Detection of living cellular mitochondrial activity
The mitochondrial activity of cardiomyocytes was analyzed using the fluorescent probe Mito Tracker RED staining assay kit (Genmed Scientifics, Inc., Wilmington, DE, USA) according to the manufacturer's instructions. Mito Tracker RED is a type of fluorescent dye that contains chlorine methyl thiol groups, which freely travel through the cell membrane and selectively accumulate in mitochondria. Cardiomyocytes were seeded in 24-well plates, and the cell culture fluid was removed when confluence reached 70%. The medium was then preheated to 37°C (500 µl/well) and added to each well. Following this, solutions of the liquid Reagents C and D (20:1; 1 µl/well) were added, and the cells were incubated at 37°C for 20 min. Finally, fluorescence intensity was analyzed using an inverted fluorescence microscope.
Fluorometric assays for detecting mitochondrial membrane damage/oxidation
Cardiolipin is one of the main components in mitochondrial membranes (13). The hydrophobic fluorescence dye 10-N-nonyl acrydine orange (NAO) is a stain that binds specifically to mitochondrial cardiolipin and then emits fluorescence. The Mitochondria Damage Testing kit (Genmed Scientifics, Inc.) was used according to the manufacturer's instructions to directly assess the degree of damage and oxidation in the mitochondrial membrane. Briefly, the cells were grown in 96-well plates. The culture medium, which contained various concentrations of TCDD, was removed when the cells covered the bottom of the well, and the cells were then washed with 100 µl reagent A. After the solution containing reagent A was thoroughly drained, the cells were then incubated at 37°C for 20 min using 50 µl/well of reagent B and reagent C (ratio, 1:99). Finally, the cells were washed with reagent A again. The relative fluorescence units were measured using a microplate reader (λex 488 nm and λem 505 nm).
Statistical analysis
Statistical analysis was performed using SPSS software (version, 18.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of TCDD on cardiomyocyte viability
An MTT assay was performed to determine which optimal concentrations to use in the following studies. As demonstrated in Fig. 1, when TCDD was applied at 0.1–10 nM, it demonstrated no significant cytotoxicity towards cardiomyocytes (P>0.05). The percentage of surviving cells was calculated for each concentration group by dividing the difference between the optical density (OD) values of the intervention group and the blank group by the difference between the OD values of the negative control group and the blank group.
Figure 1.
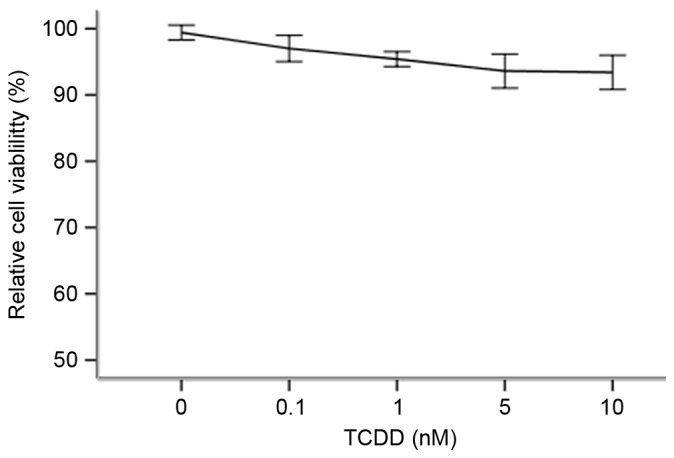
Effects of TCDD on cell viability. Cells were treated with various concentrations of TCDD (0, 0.1, 1, 5 and 10 nM) for 24 h and then analyzed using MTT assays. Treating cells with TCDD produced no adverse effects on cell viability when the treated cells were compared with control cells that were incubated with only culture medium (100% viability). P>0.05 vs. control. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
The induction of CYP1A1 and AhR gene expression by TCDD
To investigate whether TCDD increases the transcription of CYP1A1 mRNA by regulating the expression of the transcription regulator AhR, AC16 cells were treated with increasing concentrations of TCDD (0.1–10 nM) for 24 h. The ratio of absorbance at 260/280 of the total RNA of each group was 1.94–2.02 and was maintained at approximately 2.0, as measured using a Thermo Nanodrop 2000. The RNA samples had sufficient purity. The expression of the CYP1A1 and AhR mRNAs was measured using RT-qPCR. As presented in Fig. 2, TCDD significantly increased the expression of the CYP1A1 and AhR mRNAs in a dose-dependent manner.
Figure 2.
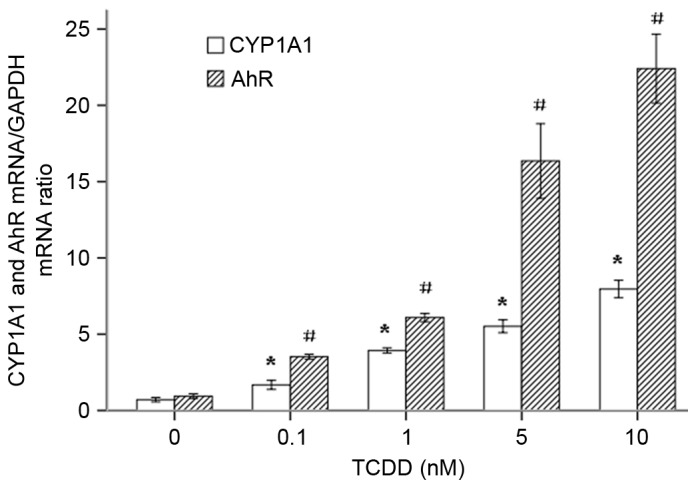
Relative gene expression ratios for the CYP1A1 and AhR mRNAs in TCDD-treated AC16 cardiomyocytes. The cells were pre-treated with various concentrations of TCDD for 24 h. P<0.05 for the expression level of CYP1A1 in each treatment group when compared with the cells that were incubated with only culture medium (*P<0.05 vs. untreated cells); #P<0.05 for the expression level of AhR in each treatment group when compared with the cells that were incubated with culture medium alone (#P<0.05 vs. untreated cells). ArH, aryl hydrocarbon receptor; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
The effects of TCDD on CYP1A1 and AhR protein expression and enzymatic activity
To determine whether the TCDD-induced expression of the CYP1A1 and AhR mRNAs in AC16 cells is translated into an increase in the level of functional proteins, western blot analyses were performed. The results (Fig. 3A) suggested that 24 h of treatment with various concentrations of TCDD significantly increased the protein expression levels of CYP1A1 and AhR, which was consistent with the observed changes in gene expression. ImageJ software (version, 1.43, National Institutes of Health, Bethesda, MD, USA) was used to calculate and analyze gray values to assess the presence of comparable amounts of protein in each lane, as presented in Fig. 3B. The EROD activity observed in each of the different groups is presented in Fig. 4. AC16 cardiomyocytes that were incubated for 24 h displayed a distinct concentration-dependent elevation in CYP1A1 enzymatic activity.
Figure 3.

Effects of TCDD on the protein expression of CYP1A1 and AhR in AC16 cardiomyocytes. Cells were incubated for 24 h with various concentrations of TCDD (0, 0.1, 1, 5 and 10 nM). (A) The proteins (25 µg) were separated using 10% SDS-PAGE. The protein levels of CYP1A1 and AhR were determined using western blot analysis. β-actin was used as the loading control. (B) The bar graph represents the relative normalized levels of the CYP1A1 and AhR proteins. The results are expressed as a percentage of the control values, which were determined to represent 100%. P<0.05 for the expression of CYP1A1 in each treatment group compared to the expression observed in the untreated cells (*P<0.05 vs. untreated cells). #P<0.05 for the expression of AhR in each treatment group compared with the expression observed in the cells that were incubated with only culture medium (#P<0.05 vs. untreated cells). TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; ArH, aryl hydrocarbon receptor.
Figure 4.
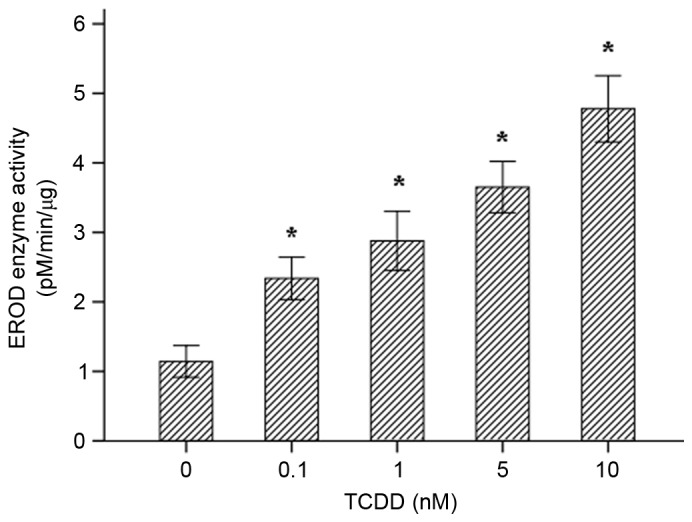
CYP1A1 activity was measured in intact living cells using 96-well cell culture plates and 7-ethoxyresorufin as the substrate. The effects of TCDD treatment on the EROD activity of CYP1A1 in AC16 cells (mean ± standard deviation, n=3) are presented. *P<0.05 vs. control group. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; EROD, 7-ethoxyresorufin O-deacylase.
ROS generation in cultured cardiomyocytes stimulated with TCDD
Exposure to TCDD induced a significant increase in the generation of ROS for concentration of 1, 5 and 10 nM TCDD (Fig. 5, P<0.05). All values are presented relative to the control cells (exposed to DMSO (100%). Treatment with 0.1 nM TCDD did not cause a significant change in ROS production (P>0.05).
Figure 5.
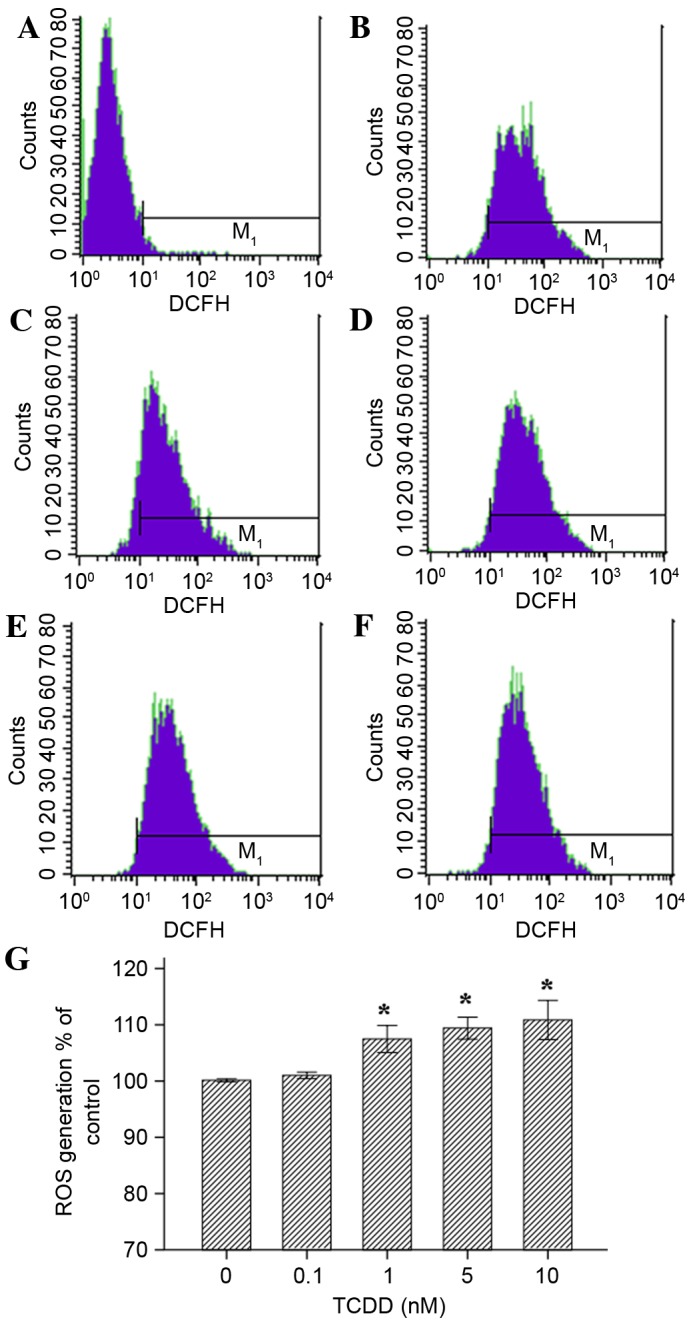
Effects of TCDD on the generation of ROS in AC16 cardiomyocytes that were pre-treated with TCDD for 24 h. Intracellular ROS levels were estimated using flow cytometry with the probe DCFH-DA, which is oxidized to form DCF in the presence of ROS. The cells were treated as follows: (A) Blank group (non-probe); (B) control (concentration=0 nM); (C) intervention group, (0.1 nM); (D) intervention groups (1 nM); (E intervention group (5 nM); and (F) intervention group (10 nM). (G) A guidance line has been drawn through the histograms being compared. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; ROS, reactive oxygen species; DCFH-DA, 2′,7′-dichlorofluorescein diacetate; DCF, dichlorofluorescein.
TCDD reduced mitochondrial activity
The changes in mitochondrial activity that were associated with TCDD were assessed qualitatively by staining the mitochondria with a Mito Tracker RED probe. Mito Tracker RED is the reduced form of a red fluorescent dye (λex=579 nm and λem=599 nm), which does not fluoresce on its own. Following incubation with Mito Tracker RED, the probe penetrates the cell membrane via passive transport, and the oxidized fluorescence probe inside the cells selectively gathers on the activated mitochondria. The results obtained using an inverted fluorescence microscope demonstrated that TCDD induced changes in mitochondrial activity that resulted in the loss of red fluorescence (Fig. 6).
Figure 6.

Effects of TCDD on mitochondrial activity in AC16 cells. The mitochondria of living cells were stained using Mito Tracker RED. Digital images of each well were obtained using inverted fluorescence microscopy at ×40 magnification. (A), (B), (C), (D) and (E) represent 0, 0.1, 1, 5 and 10 nM of TCDD, respectively. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
The effect of TCDD on mitochondrial mass in cardiomyocytes
NAO is a fluorescent probe that is widely used to determine mitochondrial mass in living cells. When the mitochondrial membrane quality is damage or oxidized, the NAO dye combines with membrane cardiolipin and is reduced, resulting in a decrease in fluorescence intensity. The results of the assays indicated that as TCDD concentration increased, the quality of the mitochondrial membrane decreased for concentrations of 1, 5 and 10 nM (Fig. 7, P<0.05).
Figure 7.
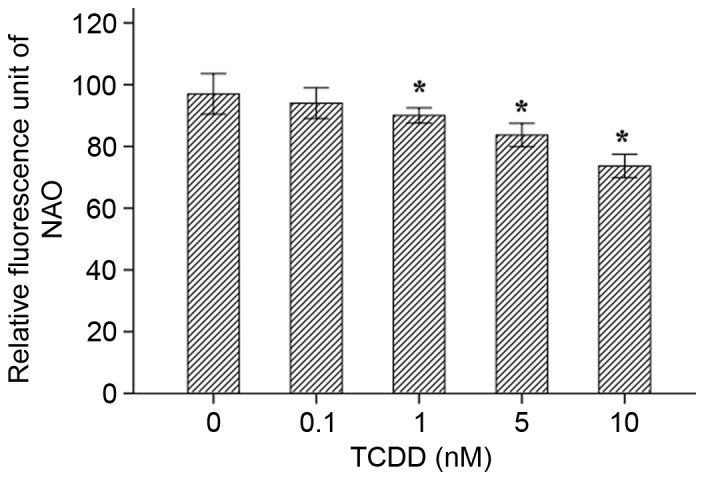
Mitochondria mass was measured in living cells using 10-N-nonyl acridine orange as a substrate. Fluorescence intensities were assessed using a microplate reader. *P<0.05 vs. Control group (concentration=0 nM). TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Discussion
Previous studies demonstrated that CYP1A1 was constitutively expressed and inducible in H9c2 cells and that the cardiotoxic effects of AhR ligands included structural malformations, ventricular hypertrophy and abnormal myocyte contractility in several in vivo models (7,9,14). In the present study, five different concentrations of the AhR ligand TCDD were applied to AC16 cardiac cells. The results demonstrated that this myocardial cell line may constitutively express the CYP1A1 mRNA and protein, leading to its enzyme activity. In addition, the process of inducing gene expression of CYP1A1 was accompanied by a decline in mitochondrial activity and quality and an increase in mitochondrial ROS.
The AC16 cell line is a commercially available myogenic cell line that is derived from human heart cells. TCDD is a congener, which is the most well studied in a family of halogenated aromatic hydrocarbons known as dioxins (15). Due to their high lipophilicity and long half-life, dioxins persist in the environment for long periods of time and accumulate in biological systems. Dioxins additionally induce a wide range of toxic effects in the heart, including impairment of cardiomyocyte differentiation and induction of hypertrophy and heart failure (7,16,17). Furthermore, AhR ligands have been reported to alter arrangement of the mitochondria, resulting in twisted and disrupted cristae, in addition to altering arrangement of sarcomeres (16). AhR ligands can cause the misalignment or disarrangement of the myofibrillar organization in cardiomyocytes, which are derived from embryonic stem cells (16). Mitochondria are important organelles that are involved in metabolism and apoptosis, and they serve important roles in cardiomyopathies (18). In the present study, two commonly used fluorescent dyes were used to analyze mitochondrial activity and mass in living cells. The results indicated that, following incubation with TCDD, the red fluorescence intensity in cardiomyocytes was significantly weaker than the intensity observed in the control group, indicating that TCDD affects mitochondrial activity.
The transcriptional regulation of the CYP1A1 gene is controlled by the soluble intracellular receptor protein, AhR, and this process serves a major role in cardiovascular diseases, due to the fact that these two genes are inducible by PAHs. Generally, the induction of CYP1A1 is considered to be cardiotoxic as it leads to the generation of ROS (19), DNA adducts and endogenous arachidonic acid metabolites (1). CYP1A1 is known to metabolize arachidonic acid into different regioisomers of hydroxyeicosatetraenoic acids (HETEs) (7). A previous study indicated that HETEs and possibly other CYP ω-hydroxylation products serve deleterious roles in cardiac injuries (20). Furthermore, inhibiting ω-hydroxylase has been reported to reduce the size of infarcts (21). Whether this represented a direct effect by AhR ligands on cardiotoxicity requires further elucidation. To the best of our knowledge, no previous studies have demonstrated that the AC16 human heart cell line can be induced to express CYP1A1 mRNA, protein and enzymatic activity. The present experiments, which were performed in TCDD-treated AC16 cells, indicated that CYP1A1 was significantly expressed and inducible in these cells. Because AhR is a transcriptional regulator of CYP1A1, the expression levels of AhR mRNA and protein were also assessed. As predicted, the results confirmed that in cardiomyocytes, treatment with TCDD resulted in markedly higher levels of AhR expression than were observed in the blank control group. The regulation of CYP1A1 gene expression involves the activation of a cytosolic transcriptional factor, AhR, which is the first step in a series of molecular events that promote the transcription and translation of CYP1A1 (1,22).
In the present study, ROS levels were increased and the quality of mitochondrial membranes was reduced in TCDD-treated cardiomyocytes. A previous study (23) demonstrated that an increase in intracellular ROS involved numerous signaling pathways and predisposed cells to damage. In addition, ROS can lead to thiol oxidation and the inhibition of ATP synthesis and translocation of adenine nucleotides (24). ROS can also cause the peroxidation of unsaturated fatty acids in membrane phospholipids, particularly cardiolipin, on the inner mitochondrial membrane, which leads to the further inhibition of respiratory chain activity (25). The initiation of the ROS-dependent opening of mitochondrial permeability transition pores has been specifically implicated in cardiomyocyte cell death (26). Taken together, the observations made in the current study suggest a previously unsuspected connection between ROS and TCDD-induced mitochondrial dysfunction.
In conclusion, the present study demonstrates that AC16 cardiomyocytes respond to TCDD by increasing CYP1A1 activity and increasing the mRNA and protein expression levels of CYP1A1 and AhR. The observations support the involvement of the AhR/CYP1A1 signaling pathway in TCDD toxicity in AC16 cells. Due to the fact that CYP1A1 is involved in xenobiotic metabolism and detoxification in humans, malfunctions in this enzyme caused by TCDD disrupting the normal metabolism and detoxification machinery are suggested to lead to increased susceptibility of the heart to environmental and other stressors, in addition to increased risk of cardiomyopathy.
Acknowledgements
The present study was supported by the National Natural Scientific Foundation of China (grant nos. 81273008 and 30872192).
References
- 1.Korashy HM, El-Kadi AO. The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab Rev. 2006;38:411–450. doi: 10.1080/03602530600632063. [DOI] [PubMed] [Google Scholar]
- 2.Kann S, Huang MY, Estes C, Reichard JF, Sartor MA, Xia Y, Puga A. Arsenite-induced aryl hydrocarbon receptor nuclear translocation results in additive induction of phase I genes and synergistic induction of phase II genes. Mol Pharmacol. 2005;68:336–346. doi: 10.1124/mol.105.011841. [DOI] [PubMed] [Google Scholar]
- 3.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 4.Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 2005;338:311–317. doi: 10.1016/j.bbrc.2005.08.162. [DOI] [PubMed] [Google Scholar]
- 5.Thum T, Borlak J. Testosterone, cytochrome P450 and cardiac hypertrophy. FASEB J. 2002;16:1537–1549. doi: 10.1096/fj.02-0138com. [DOI] [PubMed] [Google Scholar]
- 6.Granberg AL, Brunström B, Brandt I. Cytochrome P450-dependent binding of 7,12-dimethylbenz[a]anthracene (DMBA) and benzo[a]pyrene (B[a]P) in murine heart, lung and liver endothelial cells. Arch Toxicol. 2000;74:593–601. doi: 10.1007/s002040000171. [DOI] [PubMed] [Google Scholar]
- 7.Zordoky BN, El-Kadi AO. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and beta-naphthoflavone induce cellular hypertrophy in H9c2 cells by an aryl hydrocarbon receptor-dependant mechanism. Toxicol In Vitro. 2010;24:863–871. doi: 10.1016/j.tiv.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radic Biol Med. 2002;33:1268–1278. doi: 10.1016/S0891-5849(02)01014-6. [DOI] [PubMed] [Google Scholar]
- 9.Aboutabl ME, El-Kadi AO. Constitutive expression and inducibility of CYP1A1 in the H9c2 rat cardiomyoblast cells. Toxicol In Vitro. 2007;21:1686–1691. doi: 10.1016/j.tiv.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Dong H, Dalton TP, Miller ML, Chen Y, Uno S, Shi Z, Shertzer HG, Bansal S, Avadhani NG, Nebert DW. Knock-in mouse lines expressing either mitochondrial or microsomal CYP1A1: Differing responses to dietary benzo[a]pyrene as proof of principle. Mol Pharmacol. 2009;75:555–567. doi: 10.1124/mol.108.051888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlezinger JJ, White RD, Stegeman JJ. Oxidative inactivation of cytochrome P-450 1A (CYP1A) stimulated by 3,3′, 4,4′-tetrachlorobiphenyl: Production of reactive oxygen by vertebrate CYP1As. Mol Pharmacol. 1999;56:588–597. doi: 10.1124/mol.56.3.588. [DOI] [PubMed] [Google Scholar]
- 12.Schlezinger JJ, Stegeman JJ. Induction and suppression of cytochrome P450 1A by 3,3′,4,4′,5-pentachlorobiphenyl and its relationship to oxidative stress in the marine fish scup (Stenotomus chrysops) Aquat Toxicol. 2001;52:101–115. doi: 10.1016/S0166-445X(00)00141-7. [DOI] [PubMed] [Google Scholar]
- 13.Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heid SE, Walker MK, Swanson HI. Correlation of cardiotoxicity mediated by halogenated aromatic hydrocarbons to aryl hydrocarbon receptor activation. Toxicol Sci. 2001;61:187–196. doi: 10.1093/toxsci/61.1.187. [DOI] [PubMed] [Google Scholar]
- 15.De Abrew KN, Thomas-Virnig CL, Rasmussen CA, Bolterstein EA, Schlosser SJ, Allen-Hoffmann BL. TCDD induces dermal accumulation of keratinocyte-derived matrix metalloproteinase-10 in an organotypic model of human skin. Toxicol Appl Pharmacol. 2014;276:171–178. doi: 10.1016/j.taap.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neri T, Merico V, Fiordaliso F, Salio M, Rebuzzini P, Sacchi L, Bellazzi R, Redi CA, Zuccotti M, Garagna S. The differentiation of cardiomyocytes from mouse embryonic stem cells is altered by dioxin. Toxicol Lett. 2011;202:226–236. doi: 10.1016/j.toxlet.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Zordoky BN, El-Kadi AO. Modulation of cardiac and hepatic cytochrome P450 enzymes during heart failure. Curr Drug Metab. 2008;9:122–128. doi: 10.2174/138920008783571792. [DOI] [PubMed] [Google Scholar]
- 18.Gao S, Li H, Cai Y, Ye JT, Liu ZP, Lu J, Huang XY, Feng XJ, Gao H, Chen SR, et al. Mitochondrial binding of α-enolase stabilizes mitochondrial membrane: Its role in doxorubicin-induced cardiomyocyte apoptosis. Arch Biochem Biophys. 2014;542:46–55. doi: 10.1016/j.abb.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Hunter AL, Bai N, Laher I, Granville DJ. Cytochrome p450 2C inhibition reduces post-ischemic vascular dysfunction. Vascul Pharmacol. 2005;43:213–219. doi: 10.1016/j.vph.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Granville DJ, Gottlieb RA. Having a heart attack? Avoid the ‘HETE’! Am J Physiol Heart Circ Physiol. 2006;291:H485–H487. doi: 10.1152/ajpheart.00241.2006. [DOI] [PubMed] [Google Scholar]
- 21.Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ. Inhibition of cytochrome P450omega-hydroxylase: A novel endogenous cardioprotective pathway. Circ Res. 2004;95:e65–e71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- 22.Denison MS, Fisher JM, Whitlock JP., Jr Protein-DNA interactions at recognition sites for the dioxin-Ah receptor complex. J Biol Chem. 1989;264:16478–16482. [PubMed] [Google Scholar]
- 23.Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc Res. 2006;70:191–199. doi: 10.1016/j.cardiores.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L, Zhang S, Li K, Yu Z. Lycopene protects against hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial dysfunction in primary neonatal mouse cardiomyocytes. PLoS One. 2012;7:e50778. doi: 10.1371/journal.pone.0050778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 26.Papanicolaou KN, Ngoh GA, Dabkowski ER, O'Connell KA, Ribeiro RF, Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]