

Abstract
In recent years, allosteric modulation of 7 transmembrane spanning receptors (7TMRs) has become a highly productive and exciting field of receptor pharmacology and drug discovery efforts. Positive and negative allosteric modulators (PAMs and NAMs, respectively) present a number of pharmacological and therapeutic advantages over conventional orthosteric ligands, including improved receptor-subtype selectivity, a lower propensity to induce receptor desensitization, the preservation of endogenous temporal and spatial activation of receptors, greater chemical flexibility for optimization of drug metabolism and pharmacokinetic parameters, and saturability of effect at target receptors, thus improving safety concerns and risk of overdose. Additionally, the relatively new concept of allosteric modulator-mediated receptor signal bias opens up a number of intriguing possibilities for PAMs, NAMs, and allosteric agonists, including the potential to selectively activate therapeutically beneficial signaling cascades, which could yield a superior tissue selectivity and side effect profile of allosteric modulators. However, there are a number of considerations and caveats that must be addressed when screening for and characterizing the properties of 7TMR allosteric modulators. Mode of pharmacology, methodology used to monitor receptor activity, detection of appropriate downstream analytes, selection of orthosteric probe, and assay time-course must all be considered when implementing any high-throughput screening campaign or when characterizing the properties of active compounds. Yet compared to conventional agonist/antagonist drug discovery programs, these elements of assay design are often a great deal more complicated when working with 7TMRs allosteric modulators. Moreover, for classical pharmacological methodologies and analyses, like radioligand binding and the assessment of compound affinity, the properties of allosteric modulators yield data that are more nuanced than orthosteric ligand–receptor interactions. In this review, we discuss the current methodologies being used to identify and characterize allosteric modulators, lending insight into the approaches that have been most successful in accurately and robustly identifying hit compounds. New label-free technologies capable of detecting phenotypic cellular changes in response to receptor activation are powerful tools well suited for assessing subtle or potentially masked cellular responses to allosteric modulation of 7TMRs. Allosteric modulator-induced receptor signal bias and the assay systems available to probe the various downstream signaling outcomes of receptor activation are also discussed.
1. INTRODUCTION
7 Transmembrane spanning receptors (7TMRs) are proteins that represent the majority of drug targets to date.1–4 These membrane proteins communicate information received via extracellular signals, such as hormones, sensory stimuli, and neurotransmitters, to intracellular components by regulating signal transduction via heterotrimeric G-proteins and G-protein independent pathways (e.g., β-arrestins).5,6 7TMR proteins share a similar topology in that they possess an extracellular amino-terminus, seven transmembrane helices connected via three extracellular and three intracellular loops, and an intracellular carboxy-terminal tail.5,6 Based on sequence homology and functional roles, 7TMRs are commonly divided into six main families,7 A through F, with Families A–C being the most commonly studied. These receptors are distinguished by variation in their amino acid sequences and binding sites for endogenous ligands. For example, Family A 7TMRs contain a short N-terminus and generally interact with ligands via the 7 transmembrane spanning region. In contrast, Family B receptors are characterized by a longer extracellular domain, and ligands for these receptors bind within this region as well as the extracellular loop areas.8 Finally, Family C receptors interact with ligands via a large N-terminal region, termed the Venus flytrap domain, which is a bilobed structure that “closes” when agonists bind.9
Endogenous ligands interact with their receptor partner via a site that is commonly termed the “orthosteric” site. The orthosteric ligands for Family A and C are generally small, and characterized by ligands such as neurotransmitters, hormones, and ions. In contrast, the orthosteric ligands for Family B 7TMRs are peptides, such as glucagon-like peptide (GLP-1). Within the 7TMR superfamily, there are smaller groups of related proteins that interact with the same endogenous ligand. Examples of these subfamilies include muscarinic and serotonergic receptors for Family A, the corticotropin and GLP receptors for Family B, and the taste and metabotropic glutamate receptors for Family C.7 In many cases of highly conserved orthosteric binding sites, it has been very difficult to develop ligands that are specific for a given receptor. For example, in the case of the five muscarinic receptors, this lack of subtype selectivity has made it extremely difficult to probe the therapeutic potential of activating the predominantly CNS-expressed M1, M4, and M5 receptors due to simultaneous activation of peripherally expressed M2 and M3 subtypes, resulting in severe side effects.
Currently, the majority of FDA-approved drugs that act at 7TMRs bind at the orthosteric site and regulate receptor function by directly stimulating a receptor response (agonist), blocking constitutive activity of the receptor (inverse agonism), or competing with binding of the native agonist (competitive antagonism). Orthosteric pharmacology is expected for most 7TMR marketed drugs, as the majority of these compounds were identified by employing radioligand binding assays targeting the orthosteric site. While there are certainly many therapeutically relevant 7TMR ligands that interact via the orthosteric site (e.g., haloperidol and other D2 dopamine receptor antagonists for the treatment of psychosis or β-adrenergic receptor agonists and antagonists for the control of heart rate and blood pressure), the lack of receptor selectivity inherent to many orthosteric ligands has, for some time, thwarted exploration of the therapeutic potential of many of the 7TMRs.
Roadblocks in orthosteric ligand development have prompted the approach of exploring compounds that interact with 7TMRs at “allosteric,” or “other” binding sites on the receptor to modulate receptor activity.10,11 Binding of an allosteric modulator to a topographically distinct site induces conformational changes of the protein that can modify receptor activity in, most simply, a positive, negative, or neutral direction. Mechanistically, modulators can affect affinity of the orthosteric ligand, the efficacy of the orthosteric ligand, both, or bind to the receptor and not alter orthosteric agonist activity.11 A modulator that changes the affinity of an orthosteric agonist does so by changing the association or dissociation rate (or both) of the orthosteric ligand. For efficacy modulation, the conformational change in the 7TMR upon allosteric ligand binding leads to a change in signaling capacity and thereby either facilitates or inhibits receptor coupling to downstream effectors. The functional potency of an allosteric ligand is a combination of its affinity for its allosteric site as well as the degree of cooperativity exerted with the orthosteric ligand. It is possible for a modulator to possess very weak affinity for its own binding site but induce strong functional responses due to high cooperativity; in contrast, some compounds may possess high affinity but exert only weak cooperativity with the orthosteric binding site.
Allosteric modulators of 7TMRs fall into several categories. Some compounds directly activate a receptor via an allosteric site; these compounds are termed allosteric agonists. Positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) are compounds that, respectively, increase or decrease the affinity or efficacy of an orthosteric ligand. In addition to PAMs and NAMs, there are also silent allosteric modulators (SAMs or neutral allosteric ligands) that block the activity of PAMs and NAMs but do not affect the response to the orthosteric agonist. SAMs have no effect on orthosteric ligand responses, but they can block the activity of PAMs and NAMs and are quite useful tool compounds to probe receptor function (discussed in more detail below). It is also possible for allosteric compounds to act as “partial antagonists,” a mode of pharmacology that is impossible for orthosteric antagonists, as orthosteric ligands block agonist activity by a competitive binding interaction. “Partial” antagonists, in contrast, are compounds that can fully occupy their allosteric binding site but induce very weak, yet saturable, negative cooperativity with the orthosteric ligand.
In addition to improvements in target selectivity, allosteric modulators of 7TMRs and other proteins have a number of potential advantages in terms of drug development. Many compounds that interact via orthosteric sites are structural mimics of the endogenous ligand and are often small, polar amino acids, peptides, or more complex ligands that are not easily optimized in terms of human drug metabolism and pharmacokinetic (DMPK) parameters. In contrast, allosteric ligands are often small molecules with structures that are much more amenable to chemical optimization for drug candidates. Additionally, since the effects of an allosteric modulator are saturable, once allosteric sites are occupied, no additional response is observed. This ceiling effect greatly improves the potential safety of an allosteric modulator and has been used with clinical success in manipulation of other protein classes, such as the γ-aminobutyric acid A (GABAA) chloride channel.12 In this case, direct activation of the GABAA channel results in severe toxicity or death; however, the use of benzodiazepines, which potentiate the activity of GABA at these channels, results in a safe and effective clinical profile. Additionally, as orthosteric agonists may chronically activate a receptor, true PAMs require the presence of the endogenous ligand for activity and therefore maintain temporal and spatial activity of the endogenous agonist. It should be noted that a potential disadvantage of PAMs is the requirement for agonist; in neurodegenerative diseases such as Alzheimer’s disease, for example, endogenous levels of acetylcholine must remain high enough for potentiation to occur. In this situation, a compound with allosteric agonist, or ago-PAM, activity may actually be preferred; the final profile of a modulator may differ depending upon the disease being targeted.
2. GENERAL CONSIDERATIONS FOR ASSAYS DESIGNED TO IDENTIFY AND CHARACTERIZE ALLOSTERIC MODULATORS
Due to potential advantages in selectivity, DMPK properties, saturable efficacy, and chemical tractability, recent years have seen an explosion in the development of allosteric modulators for 7TMRs. This is, in large part, due to the development of functional assays that permit identification of ligands that modulate a receptor without regard to binding site (although there are radioligands for some allosteric sites, the majority of de novo “screening” approaches rely on methods that do not require knowledge of the binding site, thereby broadening the search for compounds that modulate agonist activity via diverse mechanisms).
The majority of currently used functional methods for detecting the activity of allosteric modulators fall into two broad categories, kinetic assays and endpoint assays. Many of the considerations needed to move from a focus on orthosteric to allosteric ligands are similar for these two assay types. For example, one issue of critical importance is the choice of agonist used to profile the activity of allosteric compounds. While it is often assumed that compounds that compete with orthosteric ligands are binding to the exact same site on the receptor protein, and are, therefore, also orthosteric, this idea is somewhat naïve. Different “orthosteric” ligands may contact distinct amino acids or engender specific receptor conformations that affect the ability of an allosteric ligand to cooperatively modulate that agonist. There are now clear examples of allosteric modulators producing drastically different levels of cooperativity in the presence of distinct orthosteric agonists. This phenomenon has been termed “probe dependence”13 and has implications for both the functional characterization of allosteric modulators and the determination of allosteric ligand structure–activity relationships (SARs). In extreme cases, probe dependence can result in a complete alteration in the mode of pharmacology or a loss of selectivity of an allosteric ligand. This has been shown convincingly using compounds with the M4 muscarinic receptor (Refs. 14,15 and Fig. 1.1). As shown in Fig. 1.1, the M4 muscarinic receptor PAM VU0152100 shows varying degrees of positive cooperativity with various orthosteric agonists, ranging from almost no potentiation of the agonist xanomeline, intermediate levels of potentiation for the endogenous ligand, acetylcholine, and strong potentiation of the agonist oxotremorine. Additionally, as previously shown for the M4 PAM LY2033298,14,15 the use of oxotremorine or xanomeline as agonists unmasks PAM LY2033298 activity of VU0152100 at M2 muscarinic receptors. These results suggest that codosing or coapplication of these PAMs with oxotremorine or xanomeline would result in potential off-target activity at M2 receptors, confounding data interpretation. An additional point regarding probe dependence is that there may be differences in probe dependence between species homologues of a given receptor; Suratman et al. showed that LY2033298 exhibits probe-dependent effects between rodent and human M4 receptors.16 Finally, there are many 7TMRs that have more than one endogenous orthosteric agonist. For example, the GLP-1 receptor responds to at least two endogenous peptides, GLP-1(7–36) and oxyntomodulin17 as well as other endogenous ligands, such as flavonoids.18 It has recently been shown that a novel allosteric ligand potentiates the response to oxyntomodulin and not GLP-1(7–36).17 These observations highlight the requirement for careful consideration in the choice of orthosteric ligands to assess the effects of allosteric modulators. For screening purposes, the agonist that is considered to be the primary endogenous orthosteric ligand should be the most likely choice if it is readily available and chemically stable during the assay employed for compound discovery and characterization. “Alternative” agonists should then be employed when more deeply characterizing an allosteric modulator of interest, particularly if coapplication (e.g., in electrophysiology experiments) or codosing (for in vivo assessment) of a distinct agonist/modulator combination is planned.
Figure 1.1.
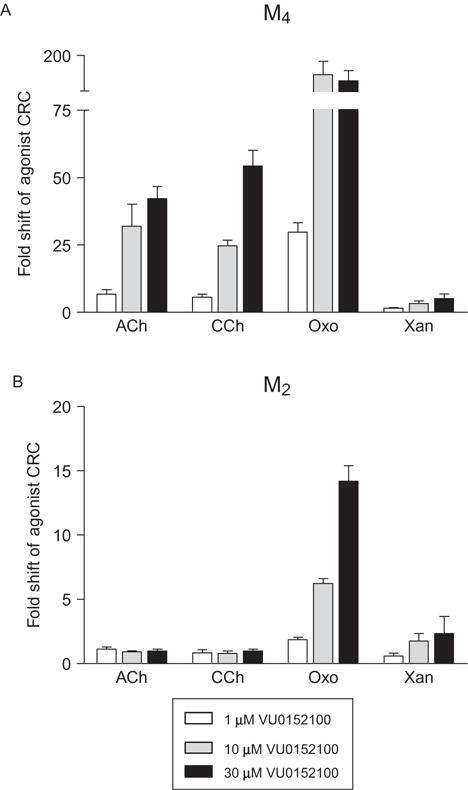
Probe dependence revealed in the presence of the M4 muscarinic acetylcholine receptor PAM, VU0152100, and various orthosteric agonists. (A) Varying concentrations of VU0152100 (1 μM (white bars), 10 μM (gray bars), and 30 μM (black bars)) were applied 2.5 min prior to the addition of increasing concentrations of the orthosteric agonists acetylcholine (ACh), carbachol (CCh), oxotremorine (Oxo), or xanomeline (Xan) and calcium mobilization was measured in cells expressing the human M4 receptor and the chimeric G-protein Gqi5. The potentiation activity of VU0152100 is clearly different in the presence of agonists other than the endogenous agonist, ACh, ranging from robust potentiation in the presence of Oxo versus almost no potentiation in the presence of Xan. (B) The selectivity profile of VU0152100 is altered in the presence of different orthosteric agonists, with the Oxo+VU0152100 and Xan+VU0152100 combinations showing potentiation at the M2 receptor in calcium mobilization assay using cells coexpressing the human M2 receptor and Gqi5. Data represent three independent experiments performed in triplicate (C.M. Niswender, previously unpublished data). Similar findings were previously reported in Refs. 14,15.
An additional point is warranted regarding differential signaling that may be induced by allosteric modulators. As receptor conformations may be unique when a 7TMR interacts with its endogenous agonist in the absence versus the presence of an allosteric modulator, the presence of the modulator may change the efficacy or types of signaling cascades that are engaged when the modulator is present. Additionally, the modulator may induce effects on its own that are unique from those generated when the receptor is engaged by its endogenous agonist. This concept has been termed functional selectivity, biased agonism, ligand-directed trafficking of stimulus, or stimulus bias.19–23
Historically, the study of 7TMR pharmacology has been dominated by radioligand binding and the measurement of a single downstream second messenger (i.e., the stimulation or inhibition of cyclic adenosine monophosphate (cAMP) accumulation for Gs and Gi/o receptors, respectively, and phosphatidylinositol (PI) hydrolysis for Gq-coupled receptors).6 With the discovery of the many other members of the heterotrimeric G-protein family, the identification of arrestin-mediated 7TMR signaling pathways, and a greater appreciation for the various cellular responses 7TMRs can mediate, the identification of stimulus bias has come to the forefront of 7TMR drug discovery.10,24 The theoretical framework that supports the development of biased ligands is that certain signaling cascades may mediate greater therapeutic benefits than other signaling pathways or that some signaling pathways might be deleterious and cause unwanted side effects. This is clearly exemplified by the action of carvedilol and its superior properties as a therapeutic for the treatment of congestive heart failure.25 Additionally, receptors transcribed from the same gene may be endogenously coupled to different signaling pathways in different cell types of the body, and the development of ligands that stimulate one pathway preferentially over another can provide for greater tissue specificity of drug effects.
Allosteric modulators have clearly been shown to bias the signaling of 7TMRs.21,26,27 Biased PAMs or NAMs might be advantageous therapeutics if they respectively potentiate or inhibit the response of the endogenous agonist for one signaling cascade versus another. A particularly intriguing concept is the identification of allosteric modulators that may switch receptor signaling to pathways that the endogenous agonist normally does not signal through (e.g., switching the signaling of a Gs-coupled receptor to Gi/o). The range of responses that allosteric modulators could affect is immense and opens up 7TMR pharmacology to even greater diversity. Thus, it is prudent that the in-depth characterization of allosteric modulators be performed in more than one assay system and that these assays sample a diverse range of potential receptor responses, even those that are not normally mediated by the receptor of interest. Figure 1.2 demonstrates a situation where a hypothetical compound (compound “B”) appears to be inactive in a typical assay for Gs-coupled receptors (i.e., the accumulation of cAMP) but shows robust potentiation of β-arrestin recruitment. In this situation, if only cAMP was measured, an experimenter who finds both compounds “A” and “B” to be active in vivo may erroneously conclude that the effect is not specific to the receptor of interest. Yet, with knowledge of other signaling pathways, concluding that the effect is nonspecific would be premature; moreover, these data would suggest a role for β-arrestin in mediating the effects observed in vivo, which may significantly impact drug discovery efforts going forward. With greater understanding of a new compound’s properties in vitro, the more accurately in vivo pharmacodynamic properties can be predicted and the more rapidly compounds can be optimized in vitro and advanced through drug development programs.
Figure 1.2.
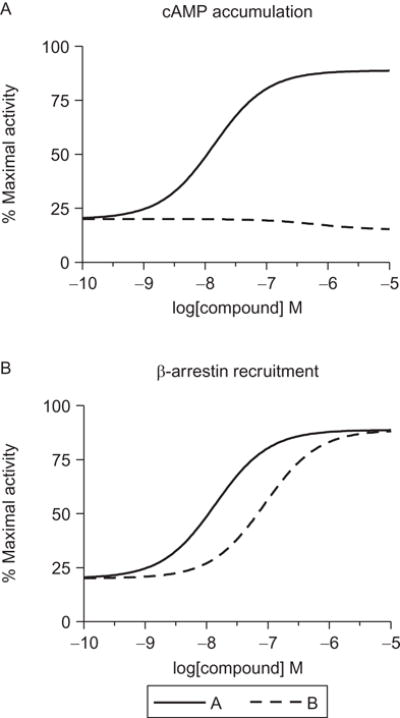
Allosteric ligands can show a range of efficacies as modulators of agonist activity. Displayed are CRCs for hypothetical compounds “A” and “B” that act at a Gs-coupled receptor. (A) One of the standard assays for measuring activity of Gs-coupled receptors is the accumulation of cAMP, and for these compounds, A appears to be a robust PAM at this receptor while compound B appears to be essentially inactive. However, in a β-arrestin recruitment assay (B), both compounds robustly potentiate the agonist’s recruitment of β-arrestin to the receptor, indicating that compound B is an example of a β-arrestin-biased PAM. Having both datasets in hand allows for a more complete understanding of the pharmacology of individual compounds.
In addition to the interaction of orthosteric and allosteric ligands in mediating preference for certain signaling pathways, there are also contributions of cellular proteins (G-proteins, β-arrestins, kinases, etc.) that can induce signal pathway-dependent modulation. These signaling proteins can also be considered “allosteric” regulators of orthosteric agonist function. This phenomenon has been termed “context dependence,” indicating that the cellular environment can impact receptor, and compound-induced, pharmacology. This case has been demonstrated for the metabotropic glutamate receptor 7 antagonist MMPIP, which exhibits strikingly different efficacies and potencies when the receptor is expressed in distinct cell backgrounds, and this appears to translate to native tissues.28 Additionally, the level of receptor expression can dictate the in vitro pharmacological profile of a compound, with compounds appearing as ago-PAMs when receptor expression is high and pure PAMs when receptor expression is decreased.29 Therefore, employment of assays in a variety of cell backgrounds and with differential levels of receptor expression may be helpful in translating the in vitro profiles of allosteric modulators to the results observed in native tissues or in vivo models.
In addition to issues regarding the choice of agonist and the recognition of the potential for functionally selective signal transduction and pharmacological profiles, experimental design to assess the activity of allosteric modulators is also influenced by the goals of each particular experiment. For example, some studies are designed to screen large numbers of compounds for which pharmacology is, yet, unknown. Other experiments are required to provide a more quantitative aspect of allosteric interaction at the receptor. In general, experiments designed to detect PAM activity are typically designed such that the observed response to a low concentration of orthosteric agonist (e.g., a concentration eliciting a response that is 10–20% of the maximal agonist response, EC10 or EC20) is enhanced. In contrast, for NAMs, the dampening effect on a submaximal concentration of orthosteric agonist (e.g., EC80 or EC90) is measured. By definition, true neutral or “silent” allosteric modulators do not affect the response to the endogenous ligand; therefore, experiments designed to detect their activity must include the presence of a PAM or NAM that interacts at the same site on the receptor. These neutral modulators serve as highly useful tools when characterizing PAMs and NAMs at the site of interaction on the receptor, discussed in detail below, and may serve as valuable controls when investigating the effect of target engagement during native tissue or in vivo animal experiments.
3. GENERAL WORKFLOW USED IN IDENTIFYING AND CHARACTERIZING ALLOSTERIC MODULATORS
3.1. High-throughput screening
Most HTS campaigns designed to search for allosteric modulators are focused on the identification of PAMs, NAMs, or both. For HTS purposes, the design of the functional assay typically assesses compound activity at a single concentration (e.g., 10 μM), although some users of 1536-well technology may screen using multiple concentrations of a ligand to quickly understand if the effects are concentration dependent. As the hallmark of a strong HTS campaign is low variability and a robust signal window from day-to-day, signal windows (e.g., EC20 vs. ECmax response for PAMs or vehicle vs. EC80 response for NAMs) must be validated over multiple days during assay development. When searching for both PAMs and NAMs in a single HTS campaign, investigators may consider using an EC50 concentration of agonist; this may allow a wide enough signal window to detect both PAMs and NAMs but would also need to be experimentally validated. Additional parameters to be optimized include the timing of addition of the compound relative to the agonist. For example, coaddition may prevent receptor desensitization that can occur during the experiment. In contrast, ligands with slow association rates require more time to occupy their binding site and so some consideration should be given to allowing the receptor to incubate with allosteric compounds prior to addition of agonist. If reference compounds are available, these parameters can be more easily evaluated; often, however, such decisions are made by taking a conservative approach (i.e., a short preincubation period may be a good compromise).
Common screening platforms used for the search for allosteric ligands of 7TMRs are those that allow for multiple additions of compounds and agonists to a single well, enabling the detection of multiple modes of pharmacology within one experiment. While not always practical, many HTS-based assays for 7TMRs rely on kinetic plate reader technology (e.g., fluorometric imaging plate reader (FLIPR, Molecular Devices) or functional drug screening systems (FDSS, Hamamatsu)). It is possible to use these types of systems to perform multiadd assays, where compound can be added first, followed by a low (EC20) and then a high (EC80) concentration of orthosteric agonist.30 Figure 1.3 demonstrates an example of a triple-add primary screening experimental design. In this experiment, glutamate responses in metabotropic glutamate receptor 5 (mGlu5) expressing cells were assessed using a calcium mobilization assay (assay details and considerations are discussed in more detail in Section 5.2). A baseline read was taken for 3 s, followed by compound addition. An EC20 concentration of agonist was added at 144 s, and then an EC80 concentration at 241 s (solid line, panels A–C). In panel A, the vehicle response is compared to the response observed in the presence of the PAM VU0360172 (dotted line), a pure PAM. This compound elicits no effect alone (compound addition) but potentiates the EC20 glutamate response. In panel B, cells expressing a much higher density of mGlu (Bmax = 25.2 vs. 7.8 fmol/105 cells29) were employed for compound profiling; in this situation, VU0360172 induces a response alone in addition to potentiation of the EC20 response and would be categorized as an “ago-PAM.” In panel C, the mGlu5 NAM VU0405395 induces reductions in both the EC20 and EC80 responses.
Figure 1.3.
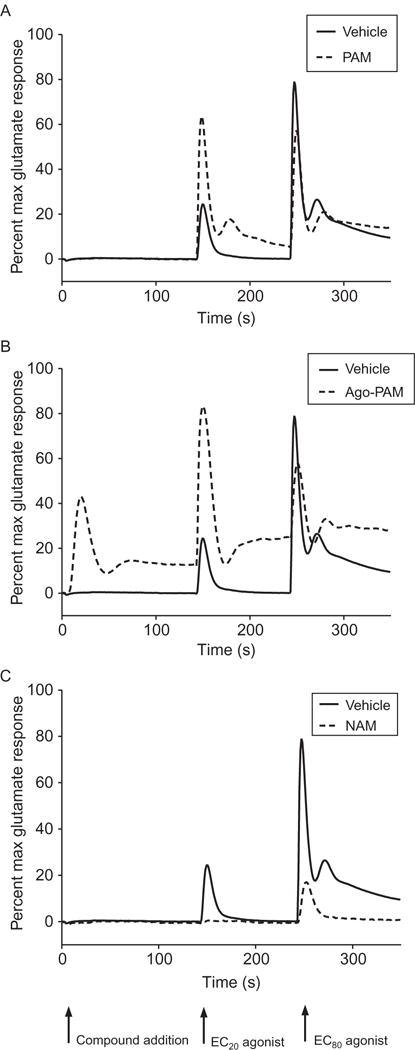
A triple-addition protocol allows for the detection of multiple modes of pharmacology in a single experiment. Calcium assay triple-add traces are shown for an mGlu5 PAM (A), Ago-PAM (B), and NAM (C). The protocol consists of the addition of test compound followed by EC20 and EC80 concentrations of orthosteric agonist to detect agonist, PAM, and NAM activity, respectively. Data shown are expressed as percent of the response of a maximally effective glutamate concentration. (A) The mGlu5 PAM VU0360172 was added to cells expressing a low density of mGlu5 receptors and traces were compared to those of vehicle (vehicle, solid line; VU0360172, dotted line). (B) VU0360172 was added to cells expressing a high level of mGlu5, invoking an Ago-PAM response (note response observed in the “compound addition” window). (C) The mGlu5 NAM, VU0405395, was added to the same cell line as that in panel (A) and results in inhibition of both the EC20 and EC80 peaks. Each data set is from a single representative experiment (P.N. Vinson, previously unpublished data).
It should be noted that different receptors may require distinct differences in the timing of addition of ligands when such a strategy is employed. Addition of vehicle, generally DMSO, instead of test compound for a sample of wells is required for proper comparison and statistical calculations required for “hit” determination. In addition, a set of wells should be set aside for the addition of a maximally effective agonist concentration, allowing for response normalization. Criteria for hit selection may vary, but reliability is dependent on the signal window and variability of data for the high and low responses; screening statistics are generally performed either within or across plates for a given run, with acceptable signal windows generating Z′ values>0.5 with this parameter represented by Eq. 1.1)31
| [1.1] |
where control (a) and control (b) are the two boundaries within which the experimental response is being monitored, for example, the EC20 and ECmax responses for a PAM assay. These types of statistics provide confidence in the selection of hits for follow-up experiments.
3.2. Potency determinations
Primary screening follow-up or characterization during more advanced hit optimization usually requires the generation of CRCs for (1) confirmation of a concentration-dependent response and (2) the determination of in vitro potency and efficacy of the compound of interest. For allosteric modulators, these studies are generally performed by adding increasing concentrations of the allosteric ligand in the presence of a constant amount of agonist (e.g., either a low, EC20, or high, EC80, concentration of agonist for PAMs and NAMs, respectively). It is important to note that the parameters of the allosteric modulator response that are observed can be dramatically influenced by the agonist concentration (Fig. 1.4); therefore, it is important to maintain a tight tolerance on the range of responses observed with agonist alone. For example, one may place a range of 10–30% for a nominal EC20 addition of agonist and require that the agonist response be maintained within this range for appropriate comparison across datasets, especially for SAR interpretation. As can be seen in Fig. 1.4A, CRCs generated for a PAM of the metabotropic glutamate receptor 4 (mGlu4) PAM, VU0400195, in the presence of increasing concentrations of the orthosteric agonist glutamate demonstrate dramatic differences in potency depending upon the glutamate concentration. For example, in the presence of a glutamate concentration that elicits no apparent effect alone (330 nM, −6.5, open triangles), VU0400195 weakly potentiates the receptor with a potency of 5 μM. Raising the glutamate concentration to 10 μM (−5, inverted triangles), a concentration that elicits a response that is approximately 80% of the glutamate maximal response, results in a leftward shift of the VU0400195 potency to approximately 120 nM, an almost 60-fold change.
Figure 1.4.
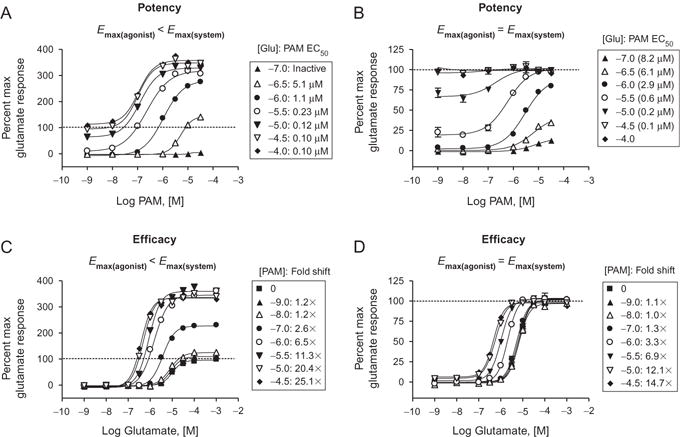
Profiles of allosteric modulators in various assays and coupling scenarios. (A and B) Potency data for the mGlu4 PAM VU0400195 in the presence of different concentrations of the orthosteric agonist glutamate are shown for two assay systems. In a Gqi5-mediated calcium assay (A), a maximal concentration of glutamate does not induce the maximal effect possible within the system (Emax(agonist) <Emax(system)), shown as a dotted line for the maximal glutamate alone response. In a different assay system, mGlu4-mediated activation of G protein inwardly rectifying potassium channels (B), glutamate induces a maximal response alone that cannot be further potentiated by PAMs (Emax(agonist) =Emax(system)); again, dotted line indicated the maximal glutamate response. Data for both assay systems reveal that there is a substantial leftward shift in the potency of the PAM at various glutamate concentrations. In the case of VU0400195, glutamate concentrations that induce no apparent effects alone can be potentiated by this PAM. For example, addition of 330 nM glutamate does not induce an apparent response in either calcium or GIRK assays, yet a concentration-dependent effect is observed in the presence of VU0400195 with a potency of 5–6 μM (white triangles). The presence of increasing concentrations of glutamate results in a dramatic 60-fold left shift in VU0400195 potency (e.g., addition of VU0400195+10 μM glutamate shifts the potency of VU0400195 to 0.1 μM, closed inverted triangles). Therefore, determination of PAM potencies can be dramatically affected by the concentration of orthosteric agonist used in the assay. (C and D). These efficacy experiments again show the ability of a PAM to increase the maximal response in one assay (C), but not in another (D). In both assays, PAM activity can also be observed as a leftward shift of the agonist concentration–response. Data are representative of three independent experiments performed in triplicate (C.M. Niswender, previously unpublished data).
Another issue that should be noted relates to the assay system chosen and the ability of the orthosteric agonist to induce a maximal response alone. This “%max” response is often tracked as it is a measure of compound efficacy, although it should be noted that the true efficacy of a compound may be underestimated due to a ceiling effect. Depending upon the assay, the orthosteric agonist may be able to elicit a response that is equivalent to the maximal response possible within that system. However, an orthosteric agonist may elicit a response that is significantly less than the maximal response capable of being achieved within that system or assay. An example of this is shown in Fig. 1.4. When mGlu4 PAM activity is assessed in one assay, the response achieved in the presence of increasing concentrations of PAM is well above the “max” response elicited by glutamate alone (panels A and C, dotted line). In contrast, when assessed using another assay (panels B and D), the maximal response observed in the presence of PAM is the same as the maximal response elicited by glutamate.
Potency determinations are often used to support chemical optimization of allosteric modulators and in interpreting SAR. One interesting observation for multiple classes of allosteric ligands is the ability of ligands to “switch” modes of pharmacology with rather simple chemical modifications. For example, subtle changes to an mGlu5 PAM scaffold that includes the compound DFB generates compounds ranging from NAM, PAM, to SAM activity.32 Within another mGlu5 scaffold, a “partial antagonist” was initially identified via an HTS program.33 This compound maximally inhibited 71% of the glutamate response with an mGlu5 IC50=486 nM. Introduction of a 3′-methyl group transformed this partial antagonist into a very potent full NAM (mGlu5 IC50=7.5 nM). When this methyl group was moved just one position, the resulting compound was a PAM with an EC50 of 3.3 μM; further optimization within this series of PAMs eventually resulted in the development of an mGlu5 PAM with good in vivo activity. These results highlight the advantages of testing for multiple modes of pharmacology when supporting an allosteric modulator chemistry optimization effort.
3.3. Efficacy determinations
As stated in Section 3.2, a measure of a modulator’s efficacy may be obtained from a CRC of the modulator in the presence of a constant concentration of agonist. However, this approach is limiting in that it is but a snapshot of the “strength” of an allosteric modulator. By measuring the shift of a CRC curve of the agonist in the presence of either a maximally effective concentration of modulator or a range of modulator concentrations (Fig. 1.4), one can measure the change in the EC50 of the agonist and express the change between the EC50 in the absence of modulator and presence of modulator. By expressing these two values as a ratio (EC50 in the absence of modulator/EC50 in the presence of modulator), the resulting “fold shift” gives a quantitative expression of the modulator’s efficacy with PAMs giving a fold shift of greater than 1 and NAMs a fold shift significantly less than 1. It should be noted that fold-shift ratios are normally distributed on a logarithmic scale, and interpretation of a PAM’s or NAM’s efficacy should take this into account. This approach can be applied using most functional assays where an effect of allosteric modulation is observed on the agonist response. Due to the noncompetitive nature between allosteric modulators and the orthosteric agonist, the agonist curves in the presence of NAMs can exhibit a decrease in the maximum response and/or a limited rightward shift (Fig. 1.5A), whereas competitive antagonists will shift the CRC in an parallel rightward fashion (Fig. 1.5B). These types of experiments can assist in confirming that a compound is acting via an allosteric mechanism of action and does not compete with the orthosteric site. Additionally, in certain assays, this type of strategy may allow for the detection of activity of the compound on its own, suggestive of allosteric agonism in that assay system (Fig. 1.5C).
Figure 1.5.
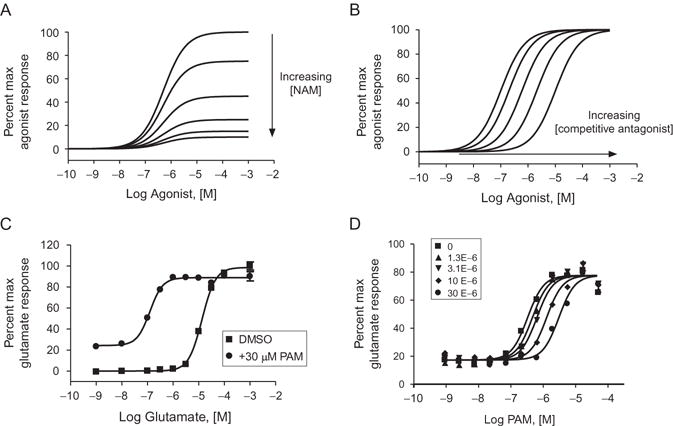
Examples of allosteric modulator profiles and use of SAMs as tools to determine ligand binding sites. (A) Simulation of the right-shift and depression of the Emax that is observed when increasing concentrations of a high efficacy NAM are applied with an agonist CRC. Note that the effect is saturable. (B) In comparison, this panel shows a simulation of the behavior of an antagonist that is competitive with the agonist. Note that the effect of the antagonist is surmountable as demonstrated by the return to maximal response by the agonist. (C) Example of a baseline effect observed in a fold-shift assay by a PAM that imparts a response independent of orthosteric agonist (DMSO control CRC curve, squares; curve in the presence of an ago-PAM, circles; note the increase in the response in the presence of low concentration of agonist. C.M. Niswender, previously unpublished data). (D) The effect of increasing concentrations of a neutral allosteric ligand (5-MPEP) on a PAM (VU0404211) CRC in a Ca+2 assay. 5-MPEP is known to interact at the MPEP site on mGlu5 and may be used as a probe to determine if a PAM is competitive at the same site. Note the features of competitive behavior of the data that can be fit to the Gaddum/Schild equation to test this hypothesis (P.N. Vinson, previously unpublished data).
If a SAM exists and has been demonstrated to interact at a specific site on the receptor (e.g., through radioligand binding competition assays), such a compound may be used as a probe in a functional assay to determine whether or not another modulator is interacting at the same site in a competitive manner. An example of this is the SAM 5-MPEP.34 5-MPEP induces no effect on the response to glutamate alone but completely displaces a radioligand at a site on mGlu5 known to interact with many allosteric ligands (the so-called “MPEP” site). Shown in Fig. 1.5D is an experiment in which increasing concentrations of 5-MPEP were preapplied to cells expressing mGlu5. Approximately 2 min later, a CRC of PAM in the presence of an EC20 concentration of glutamate was added. The parallel rightward shift of the CRC indicates that 5-MPEP interacts competitively with the PAM being profiled, suggesting that this PAM interacts with mGlu5 via the MPEP binding site. Had the PAM in question interacted at an alternate site, the curves would not have shifted in a parallel fashion, but would rather have decreased in maximal response as the concentration of 5-MPEP was increased (e.g., see Ref. 35).
4. DATA ANALYSIS: GENERAL FEATURES FOR ALLOSTERIC INTERACTIONS
While detailed methods are provided in other portions of this book, a brief discussion on analysis of functional data is warranted. The simplest way to examine experimental data with allosteric modulators is to simply fit CRC data or fold shift to a common four-parameter logistic equation:
| [1.2] |
where Y is the measured response, X is the logarithm of ligand concentration, Bottom and Top are the lower and upper bounds of a sigmoidal curve, respectively, EC50 is the concentration of ligand where 50% of maximal response is observed, and HillSlope indicates the Hill coefficient used to describe the steepness of the sigmoidal curve.
However, there are additional pieces of information, such as an estimate of the affinity of the modulator, which cannot be accommodated by this simple equation. While most mass action-based molecular models of allosteric modulation contain too many parameters to be fitted to real data, there are now modified methods that are useful in fitting experimental data using allosteric modulators and provide some quantitative aspects to the interactions of allosteric ligands with their targets, such as an estimate of modulator affinity. This can be done using an “operational” model of allosterism, which can be used to describe cooperativity and allosteric agonist effects.13 The following equation describes the effects of an allosteric modulator on the signaling of an orthosteric agonist:
| [1.3] |
where E is the effect (i.e., measured response); [A] and [B] are the concentrations of orthosteric and allosteric ligand, respectively; KA and KB are the equilibrium dissociation constants of the orthosteric and allosteric ligands, respectively; α describes a cooperativity factor relating to the allosteric effect of each ligand on the other ligand’s binding affinity; β, which ranges from zero to infinity, quantifies the extent to which the allosteric modulator modifies the efficacy of the orthosteric agonist at a given signal pathway; τA and τB relate to the ability of the orthosteric and allosteric ligands, respectively, to promote receptor activation (i.e., direct agonism) and are dictated by the intrinsic efficacy of each ligand, the efficiency of stimulus–response coupling, and the total density of receptors; Em is the maximal possible system response (described as Emax in Fig. 1.3): and n is the slope factor linking occupancy to response.
This model allows an investigator to fit experimental data, and there are three key parameters that can be routinely derived from the model’s application to full concentration–response and curve–shift relationships. These include the KB of the allosteric modulator, which is an estimate of the affinity of the allosteric ligand; a composite cooperativity parameter, αβ, which provides information on the overall allosteric effect on the orthosteric agonist in the chosen functional assay (and, depending on the bias of the modulator for orthosteric agonist affinity vs. efficacy, can be close to the “fold-shift” value); and the modulator efficacy parameter, τB, which provides information on the ability of the allosteric ligand to promote agonism in the absence of orthosteric ligand. The estimate of affinity is particularly important, as many 7TMRs do not have radioligands that correspond to an allosteric site. Affinity estimates can be particularly helpful in determining which compounds might be candidates for radiolabeling or for potential development into positron emission tomography (PET) tracers, tools that are critical for relating occupancy of a target to in vivo efficacy and can help guide clinical development.
5. KINETIC ASSAYS TO MEASURE ALLOSTERIC INTERACTIONS AT 7TMRs
5.1. Fluorescence-based second-messenger assays
Identifying a general method for measuring specific target engagement is a highly desirable feature of an assay used in pharmacological research and drug discovery efforts. The introduction of probes that fluoresce upon binding to ionic species has met that need for many 7TMRs by measuring intracellular ion changes occurring through specific signaling pathways upon ligand binding to the receptor. This process occurs within seconds, enabling the tracking of changes in intracellular ion levels kinetically. Typically, one point corresponding to the maximum response is used to characterize the effect of a compound, but the kinetic readout allows for a more detailed assessment of the dynamics of the cellular response; multiple compound additions per well (e.g., a compound’s agonist and PAM or NAM activity can be determined in a single well) can also be employed. These assays have become critical for HTS, lead optimization, and basic science investigation of 7TMR pharmacology. The application of these methods to allosteric modulators is described below and refers to plate-based formats and fluorescent imaging techniques using instruments such as the Hamamatsu FDSS or Molecular Devices FLIPR. However, lower end fluorescent readers also may be used for lower throughput and resolution.
5.2. Calcium mobilization, general principles
In general, monitoring Ca2+ mobilization (aka “calcium flux” assay) exploits the stimulation of phospholipase C by 7TMRs that couple to the Gq family of heterotrimeric G-proteins and the subsequent inositol triphosphate (IP3)-stimulated release of Ca2+ from the endoplasmic reticulum into the cytoplasm. The binding of calcium to a preloaded fluorescent dye provides a means of monitoring the change in Ca2+ concentration in response to the cellular event. Fortunately, the development of chimeric G-proteins, such as Gqi5, and the identification of promiscuous G-proteins, such as Gα15 and Gα16, that can redirect the signaling of non-Gq-coupled receptors to the phospholipase C (PLC) pathway have enabled the application of this method beyond receptors that naturally couple to Gq.36–38 It should also be noted that, depending upon how the assay is performed, 7TMR-mediated activation of cell surface calcium channels, often via the βγ subunits of the heterotrimeric G-protein, can also contribute to the calcium signal induced via 7TMR activation. If a compound does possess functional selectivity, it is possible that some modulators may induce peculiarities in the calcium trace that may be indicative of differential modulation of these pathways and can be monitored for an early read on potentially functionally selective signaling events. Recently, Blatterman et al. have reported a compound that functionally uncouples the Gβγ pathway from that of Gα39; therefore, the ability to monitor distinct parts of the G-protein signaling cascade could be useful in compound profiling.
Other interesting and, perhaps, informative features may exist within the raw data of a calcium trace, which may be the result of applied modulator. Some of these features include calcium oscillations, effect on a second agonist addition (i.e., in a “triple-add” design (Fig. 1.3), comparing the pharmacology of the EC80 peak to determine if there is a reduction, enhancement, or retention of the response at the control level), and calcium response at a plateau phase of the trace. Although the allosteric CRC curve is traditionally generated from the kinetic trace data by calculating the peak response, one could also extract and plot responses corresponding to a different point on the trace or simply qualitatively describe such features. Knowing the physiological relevance is the challenge; yet, with current data extraction, analysis, and storage techniques, it is possible to track these different features for post hoc comparison to a downstream assay either in vitro or in vivo.
5.3. Thallium flux, general principles
The influx of ions into the cell through ion channels may also be monitored using fluorescent probes. The discovery that thallium may be used as a surrogate for ions,40 such as potassium, allows for the ability to use this ion to monitor the activity of potassium channels such as G-protein-coupled inwardly rectifying potassium channels (GIRKs), human ether-a-go-go, Kv7.4 (KCNQ4), K(ATP) channels, and renal outer medullary potassium channels.40–51 As many Gi/o-coupled receptors modulate potassium channels, predominantly via the Gβγ subunits of the heterotrimeric G-protein, this technique has been used as another kinetic assay to examine the activity of allosteric modulators targeting this class of receptors.43 For cells expressing both Gi/o-coupled receptors and GIRKs, the release of the Gβγ subunits upon 7TMR stimulation activates GIRK heteromers. By bathing the cells in buffer containing thallium and preloading the cells with a thallium-sensitive dye (e.g., BTC, AM, FluoZin™-2, or dyes contained within propriety kits such as FluxOR®, Invitrogen), a readout of receptor activity can be achieved. The kinetics of this ion flux is characteristically different from those of calcium mobilization (Fig. 1.6), and the conditions of the experiment are such that repeated additions of agonist may not be well tolerated, making addition of multiple boluses of thallium problematic. In this case, compound can be added in non-thallium-containing buffer and then the agonist can be coapplied with thallium. While data can be analyzed using stand-alone time points as in calcium mobilization experiments, we have found that measuring the slope within the first few seconds after the thallium+/− agonist addition results in a robust relationship from which to generate CRC data.43 One additional note regarding thallium: because the compound is a heavy metal, handling and disposal must be performed according to local safety guidelines.
Figure 1.6.
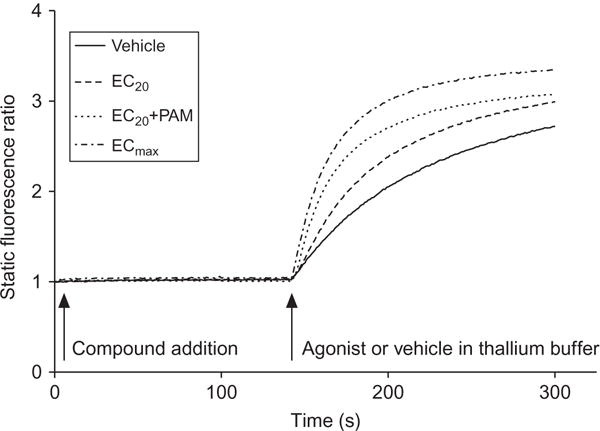
Characteristic traces observed from a thallium flux experiment. Shown are data collected during a thallium flux assay in hM4 muscarinic receptor/GIRK-HEK cell line (P.N. Vinson, previously unpublished data). The M4 PAM, VU0152100, was added to cells that had been incubated with the dye FluoZin TM-2 AM followed by an EC20 concentration of acetylcholine in buffer containing thallium. The response is measured as the slope of the linear portion of the curve after thallium addition. The vehicle response can then be subtracted, followed by normalization to the baseline-corrected ECmax of agonist, to normalize across experiments.
5.4. Fluorescent probe methodology
The process of performing a plate-based fluorescent probe kinetic imaging assay should be optimized for each cell line and agonist and detailed methods may be found in primary publications using this approach as well as reviews.17,43,52–54 In general, the process begins with plating cells expressing the receptor of interest (recombinant or natively expressed) in black-welled, clear-bottomed plates (96, 384, 1536; poly-D-lysine-coated or noncoated as appropriate for adherent cells) at a density allowing for optimal response approximately 24 h later. It is important to know if other, natively expressed, proteins could possibly respond to the agonist used in the assay, resulting in confounding experimental results. One approach to test for off-target effects is to perform a parallel assay in the parental cell line that lacks the receptor of interest (if using a recombinant source) or to use a specific antagonist, if it exists. For the assay itself, the cell medium is typically removed and replaced with a buffer solution such as Hanks balanced salt solution buffered with HEPES. This may be performed manually or by using an automated plate washer. An optimized concentration of dye in buffer is added and incubated for a period of time (usually on a scale of approximately 1 h) to allow penetration of the dye into the cytosolic space. Note that many dyes require a surfactant such as pluronic acid to improve their distribution in an aqueous solution. Most dyes are designed to be activated by ester cleavage by naturally occurring esterases to produce a molecule with high affinity for the ion of interest. An additional consideration to maximize the dye concentration within the cell is to include the anion exchange inhibitor, probenecid, to the buffer used throughout the experiment. The dye-loading step is also amenable to manual or automated (e.g., bulk liquid dispenser) methods. To reduce background fluorescence, the buffer may be exchanged after dye incubation, although there are dye products where this step may not be necessary. We have observed that some assays benefit from the cells having a “settling time” of approximately 10 min after the second buffer exchange, resulting in a more stable response. The experimental readout on the fluorescence instrument begins by the acquisition of baseline followed by the collection of the readout resulting from compound and subsequent agonist additions. Each of these steps should be considered when introducing and optimizing a new assay of this type; any manipulation that occurs could have a detrimental effect on the cells’ response and optimal dye concentration and incubation time and temperature may be different across cell lines. Many of these types of kinetic assays can be performed on lower end fluorescence plate readers with liquid-handling capabilities (e.g., Molecular Devices FlexStations), which lack the advantage of simultaneously adding compound and recording data from all wells on an assay plate, but come at only a fraction of the cost of an FDSS or FLIPR assay system. It should be noted, however, that because these fluorescence plate readers cannot read all wells simultaneously, different wells on the plate will be read at distinct time points after the addition of dye or compound and a drift in the assay signal may be observed that should be taken into account when analyzing data.
5.5. Alternatives to fluorescence technology
An alternative to permeable fluorescent dyes to detect changes in intracellular Ca2+ is the photoprotein aequorin (AEQ).55 This protein, which originates from jellyfish, binds to calcium, resulting in oxidation of its cofactor, coelenterazine, thereby producing CO2 and light emission, the latter of which is detected through luminescence. Aequorin is coexpressed with the receptor of interest (along with, if necessary, a PLC-coupling G-protein), with stable expression in the mitochondria providing a better signal compared to expression in the cytoplasm.56 The assay design consists of adding coelenterazine followed by the test compound(s). Although this platform is gaining popularity for 7TMR Ca2+ signaling detection,57–59 it has been applied only in limited cases for allosteric modulators.60 The main advantage of this assay is the avoidance of fluorescence signal from test compounds, which can significantly confound data analysis where the fluorescence signal is expected to directly correlate with receptor activity, as is done for dye-based Ca2+ assays. It remains to be seen if this approach will be amenable to designs such the triple-add assay due to the AEQ assay typically applying cofactor-loaded cells to a plate of compound (agonist, antagonist, or modulator). Kits for AEQ luminescence assays include the transient transfection BacMam Aequorin Cellular Assay® kit from Invitrogen and stable cell lines produced by PerkinElmer under the product name AequoScreen®.
6. LABEL-FREE TECHNOLOGY
Label-free technology, so named because it does not require isolation of a specific signaling pathway and requires no biochemical techniques, such as the use of dyes or enzymatic reactions for measurement, is becoming increasingly popular for the study of allosteric modulator pharmacology. As these techniques are “unbiased” in terms of the binding site of a compound and in the signaling pathway induced, they represent a way to cast a wide net in terms of the pharmacology elicited by a modulator. Rather than reflecting effects of receptor activation on a single signaling pathway, these assays provide a measure of effects on a composite of different signaling responses to a 7TMR ligand, although certain pathways will dominate the kinetic trace that is measured.
While discussed in detail in another chapter in this volume, there are two main principles currently driving label-free techniques monitoring 7TMR activity in whole cells. The first of these is the cellular impedance technique, based on Ohm’s law of voltage (V)=current (I) × resistance (Z). In this method, cells are layered onto a plate and a current is applied. The current can flow either between the cells or through them, resulting in a certain level of resistance, or impedance. When drugs are applied, the cells change shape due to intracellular cytoskeletal dynamics, resulting in a change in the impedance value from baseline. Examples of this technology are marketed in 96- and 384-well format as the CellKey® system from MSD Analytical Technologies and as the xCELLignce® from Roche in partnership with ACEA Biosciences, Inc. Impedance technology can detect signals from various 7TMRs with distinct “signatures” indicative of Gs versus Gq versus Gi/o-coupled receptors.61–65
An alternate technique to impedance technology involves dynamic mass distribution, or DMR; several readers are now available in 96-, 384-, and 1536-well format marketed as the Epic® (Corning, Incorporated) and the EnSpire® (PerkinElmer), which is based on the Epic® technology. This method relies on plating cells containing the receptor of interest onto a plate in which individual wells contain a resonance waveguide grating. Initial baseline readings are taken by illuminating the plate with a broadband light source and measuring the wavelength of the refracted light (Fig. 1.7A). After cell stimulation, mass redistribution of intracellular components occurs, resulting in a shift in the refracted wavelength. Responses are measured as a picometer wavelength shift from the initial recorded wavelength and can occur in either a positive or negative direction. These types of assays are proving useful for the study of 7TMR pharmacology.28,39,65–82
Figure 1.7.
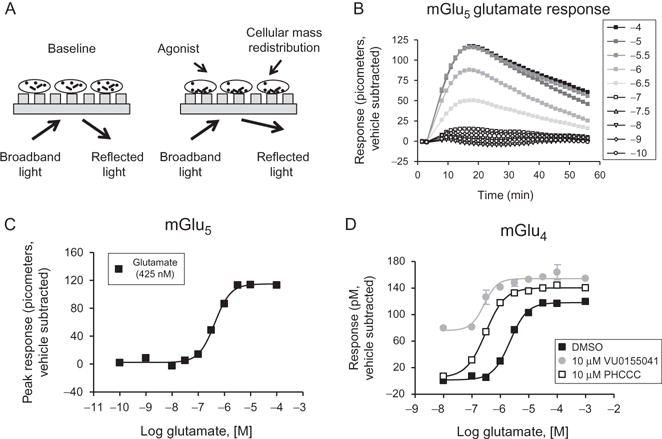
Principle of Epic® technology and examples of glutamate receptor pharmacology assessed using this method. (A) Dynamic mass redistribution principle. Cells are plated onto plates containing a resonance waveguide grating. Broadband light is shone onto the bottom of the place and then reflected. After agonist treatment, mass distribution of intra-cellular components occurs,resulting in a shift in the wavelength of the reflected light measured in picometers. (B) Example Epic® traces observed after the application of the agonist glutamate to cells expressing mGlu5 after subtraction of vehicle responses. (C) The peak responses from panel (B) are plotted as a glutamate CRC. (D) Responses of glutamate alone and glutamate in the presence of two mGlu4 PAMs. In this case, PHCCC acts as a pure PAM, with no agonist activity as noted in other in vitro assays. In contrast, VU0155041 shows agonist activity in other in vitro settings (panel (C) of Fig. 1.5 shows data in the absence and presence of VU0155041 in a thallium flux assay); note the raised baseline in the Epic® data shown here. (C.M. Niswender, previously unpublished data).
Using Corning Epic® technology, we have generated preliminary data profiling the signatures of some of the metabotropic glutamate receptors, plus and minus allosteric modulators. Shown in Fig. 1.7B are traces generated using the Epic® with HEK cells expressing mGlu5. These traces have been normalized to the response that occurs in the presence of vehicle (a downward deflection of the traces). The maximum response induced in each of these traces can then be converted to a CRC (Fig. 1.7C), which results in a calculated potency for glutamate at mGlu5 of approximately 425 nM, well within the range observed with this cell line in our laboratories in calcium mobilization assays.29,34,53 In Fig. 1.7D is shown an experiment with cells expressing the related mGlu4 receptor in which receptor activity has been measured in the presence and absence of the allosteric modulators PHCCC and VU0155041.83–85 These compounds differ in that PHCCC functions as a “pure” potentiator, with no agonist activity observed in multiple in vitro assays and in slice physiology, whereas VU0155041 exhibits ago-PAM activity in vitro.83–85 As can be seen in Fig. 1.7D, the agonist activity of VU0155041 is readily observed using Epic® technology (note the raised baseline similar to that observed in thallium flux assays, Fig. 1.5C) while PHCCC behaves as a pure PAM.
There are a number of advantages to the use of label-free technology. For example, no knowledge of the binding site is required, and functional readouts can be quite diverse, which may be useful for examining the activity of alternate signaling pathways as well as the pharmacology of “orphan” receptors for which signaling pathways and ligands may not yet have been identified. Both types of assays are capable of monitoring signaling induced by endogenously expressed, rather than overexpressed, receptors within cells. This allows for the ability to measure receptor activity in a cell background in which the receptor would normally be expressed, as well as assessing receptor modulation at potentially more physiological levels of expression. In terms of assessing functional selectivity, label-free technology adds an additional tool to enable an early read on compound profiles and potential differences in signal transduction. When performed in kinetic mode, a wealth of information is present in the traces, and “slices” of receptor activity can be taken at different time points, which may reflect distinct signaling events. The ability to read a plate and then return after some time has passed and take additional measurements may also allow for monitoring of very late receptor-mediated events, such as internalization or effects on new protein synthesis.
While label-free techniques have many advantages, there are a few points to consider. While the signals emanating from a receptor may represent a treasure trove of information regarding signaling, it can also be quite complex to tease out the contributions of various signaling events without systematic, detailed follow-up experiments. When used as a platform for HTS, it should be noted that the number of hits may escalate exponentially, as the systems are able of detecting any event that causes impedance or DMR changes, regardless of the specificity for the receptor being interrogated; carefully planned subsequent experiments (untransfected parental cells, alternate cell lines expressing the receptor of interest) are then required to understand which hits are desirable for further study. An additional factor is cost. Aside from the cost of the reader itself, plates containing the appropriate reagents to measure receptor-mediated signals are required and may be cost limiting for some investigators. Nevertheless, as label-free technology continues to evolve, it is anticipated to bring new opportunities for the study of receptor pharmacology, including that induced by allosteric modulators.
7. ENDPOINT ASSAYS
There are a number of compelling reasons for investigators to choose an endpoint assay for allosteric compound screening and characterization. The endpoint assays available for 7TMRs are numerous and diverse, and in many cases, these assays may be the only viable options for studying multiple signaling pathways for a particular receptor with reasonable throughput.86 Many endpoint assays can be multiplexed with each other so long as the readout for one signaling pathway does not interfere with the readout of another (e.g., multiplexed luminescent and fluorescent readouts,87 fluorescent readouts with nonoverlapping spectral windows). Additionally, many endpoint assays can be run using cells or tissue subsequent to a kinetic assay (e.g., kinetic readout of receptor-induced Ca2+ mobilization followed by the detection of IP3 levels). These multiplexing options allow for multiple signaling pathways to be studied and can help in the assessment of allosteric modulator-induced signal bias. Some signaling pathways may be more sensitive to putative agonist or inverse agonist properties of allosteric compounds and with these multiplexing options, a compound’s agonist or inverse agonist effects may be more readily uncovered.16 Moreover, running kinetic assays for 7TMRs may not be feasible for all laboratories as they might require hardware that is not widely available (e.g., robotic and liquid-handling systems, FDSS or FLIPR systems, plate readers equipped with evanescent waveguide sensors), while many endpoint assays are designed to produce robust readouts detectable on fairly common equipment (e.g., luminometers, spectrophotometers) and can be hand-pipetted if necessary.
There are, however, important caveats that must be considered when working with allosteric modulators in 7TMR endpoint assays. These assays typically provide lower throughput for screening allosteric modulators than kinetic readouts, and end point assays are not as flexible as kinetic assays in terms of assessing multiple properties of a compound in a single assay run (e.g., the Ca2+ mobilization triple-add protocol discussed above that allows for the detection of agonist, PAM, and NAM/antagonist activity in a single well). A significant drawback of endpoint assays is the inability to rapidly adjust agonist concentrations during the assay run in order to achieve optimal agonist concentration for PAM and NAM assessments (i.e., agonist EC20 and EC80, respectively). This can be particularly troublesome if the receptor being studied demonstrates a significant day-to-day variability in agonist potency or if the agonist CRC has a large Hill coefficient. One solution to this problem is to run several concentrations of agonist against test compound CRCs to hedge against missing the optimal EC levels (e.g., running EC10, EC50, and EC90 concentrations of agonist provides two concentrations, EC10 and EC50, for the assessment of PAM activity and two concentrations, EC50 and EC90, for that assessment of NAM activity). It may be advantageous to run a full matrix of agonist and allosteric modulator concentrations, which will yield both modulator potencies and agonist fold-shift data needed to assess the cooperative coefficients of allosteric compounds (see Eq. 1.3). Assays designed in this manner will provide a robust platform for measuring the properties of an allosteric modulator but will also limit throughput.
Novel compounds may exhibit unexpected effects in vivo or lack activity in vivo, thus it is important to study signaling pathways in vitro that are relevant to the in vivo situation to gain a better understanding of allosteric drug action. The high degree of biological relevance that many 7TMR endpoint assays offer makes these assays important components of allosteric drug discovery efforts. The measurement of cAMP and PI hydrolysis are classic endpoint assays used for the detection of 7TMR activity.88,89 These techniques were established several decades ago and were used to tease apart the receptor–G-protein–enzyme (i.e., adenylate cyclase, phospholipase) system.90,91 Measurement of cAMP accumulation and PI hydrolysis continues to be important endpoint assays for 7TMR drug discovery/tool development, but more recently, other endpoint assays have gained ground in this arena, including extracellular regulated kinase (ERK) phosphorylation92–94 and arrestin recruitment.95–97 The assay systems discussed below for the detection of cAMP accumulation, PI hydrolysis, ERK1/2 phosphorylation, arrestin recruitment, and transcriptional regulation will focus particularly on those methods that are (1) nonradioactive, thus requiring fewer safety precautions compared to older methodologies that rely upon radiochemicals, (2) applicable to HTS and robotic automation, (3) significantly less expensive than traditional techniques like radioligand binding, Western blot analysis, and microscopy, particularly when scaled to 384- and 1536-well plates, and (4) detectable on relatively common laboratory plate readers.
7.1. cAMP accumulation
Traditional methods of cAMP quantification like ion-exchange chromatography88 and [3H]cAMP protein kinase A (PKA) competition binding assays98 require the use of radiochemicals, are laborious and low throughput, and come with a prohibitively high cost per data point for the screening of thousands of compounds in typical HTS campaigns or during follow-up chemical optimization and SAR support. Advances in molecular biology and the proliferation of high sensitivity plate readers have allowed for the development of nonradioactive, miniaturized cAMP assays capable of detecting subnanomolar levels of cAMP produced by intact cells, membrane homogenates, and tissue preparations. Two examples of such assay kits are the LANCE® cAMP/LANCE® ultra cAMP assays system (PerkinElmer) and the HitHunter® cAMP assay system (DiscoveRx). Several other assay systems are commercially available (e.g., CatchPoint® cyclic-AMP, Molecular Devices; HTRF® cAMP kits, Cisbio; AlphaScreen® cAMP Assay, PerkinElmer; and various cAMP enzyme-linked immunosorbent assay (ELISA) kits). In this review, the LANCE® cAMP and HitHunter® cAMP assay systems will serve as examples of affordable assay kits whose applications are not limited to proprietary or stably transfected cell lines.
The assay principle of the LANCE® cAMP kits is based on the disruption of a fluorescence resonance energy transfer (FRET) complex formed between a fluorescent europium (Eu) chelator linked to a biotinylated-cAMP molecule though a streptavidin–biotin interaction (or direct covalent linking of the Eu chelator with cAMP in the case of the LANCE® ultra version of this kit) and a fluorescently labeled anti-cAMP antibody. In the absence of unlabeled cAMP, the FRET complex readily forms and maximal FRET is achieved, but in the presence of cAMP, the FRET complex is disrupted by unlabeled cAMP competition with biotinylated-cAMP at the anti-cAMP antibody binding site, and FRET is significantly reduced. Therefore, the concentration of cAMP produced by adenylate cyclase is proportional to FRET output along a sigmoidal cAMP competition curve. The concept of the assay is similar to the classic PKA [3H]cAMP competition binding methodology, in that increased cAMP production yields a reduction in the overall assay signal and the cAMP standard curve conforms to a sigmoidal competition curve. Due to the nonlinear nature of the cAMP standard curve, this assay system requires that FRET values be converted to cAMP levels before data are analyzed or normalized and requires that the investigator take care not to saturate the anti-cAMP antibody by using excessive amounts of cells or membranes, as this would yield inaccurate data. The LANCE® kit’s time-resolved FRET (TR-FRET) readout is resistant to interference by autofluorescence of cells, membranes, test compounds, and the assay plate. This readout is achievable with most filter-based fluorescence plate readers with the appropriate filter sets and xenon flash lamp light source. The LANCE® cAMP kits are not well suited for HTS in cell monolayers; although cAMP levels can be assessed in cell monolayers, for use in HTS applications, adherent cells must be lifted and suspended.
Aside from the assay detection technology, the HitHunter® cAMP system differs from LANCE® cAMP in that increasing cAMP levels yield an increase in the assay signal, though the standard curve is still a sigmoidal one. In this assay, a cAMP tracer is covalently linked to a fragment of the beta-galactosidase enzyme (the enzyme donor, ED); ED-cAMP is captured by an anti-cAMP antibody and is not available to complement with the remaining fraction of beta-galactosidase (the enzyme acceptor, EA); thus, when the beta-galactoside substrate is added, no enzyme activity is present and chemiluminescence is not produced. In the presence of increasing levels of cAMP, the unlabeled cAMP competes with the ED-cAMP at the anti-cAMP antibody, allowing for increased levels of free ED-cAMP capable of complementing the EA fragment and robust chemiluminescence is produced. Like LANCE® cAMP and other competitive assays, the standard curve of this assay is sigmoidal and requires the conversion of raw luminescence units to units of cAMP for the reasons stated above. The potential to saturate the system with unlabeled cAMP is also present; thus, the optimization of cell number or membrane concentration is required. An advantage of the HitHunter® system is that adenylate cyclase activity can be easily measured in cell monolayers. Another advantage of this system is that the luminescent assay readout can be detected on most luminescence-capable plate readers, which are more prevalent and less expensive than the time-resolved fluorescence plate readers required for the LANCE® system. However, the HitHunter® system has lower sensitivity than the LANCE® cAMP kits.
With the LANCE® cAMP assay system, particularly low levels of cAMP can be quantified as the result of its higher affinity anti-cAMP antibody, accommodating the needs of primary cell culture or slow growing cell lines where measurement of subnanomolar levels of cAMP is required.99 Because allosteric modulators may display different properties based on receptor density,100 the ability to use cells from primary culture with endogenous levels of receptor is a significant advantage of these assay systems in allosteric drug development. The sensitivity offered by these assay systems is essential to investigating the effects of allosteric modulators, where the measurement of small changes in agonist potency is often necessary. Both assay systems are capable of measuring cAMP produced by homogenized tissue and membrane preparations, requiring only that an ATP regeneration system is present in the assay medium (e.g., Ref. 101).
7.2. PI hydrolysis
PI hydrolysis, like cAMP production, is a well-studied 7TMR signaling pathway, and, in recent years, several assay systems that are applicable to allosteric modulator HTS efforts have become commercially available to measure PI hydrolysis products. The general principle of this assay is that stimulation of Gq-coupled receptors activates PLC, which in turn hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce inositol 1,4,5-triphosphate (IP3) and diacyl glycerol. IP3 diffuses to the endoplasmic reticulum to activate IP3-sensitive Ca2+ channels, releasing Ca2+ from the endoplasmic reticulum and increasing cytosolic Ca2+ levels. Thus, the measurement of IP3 levels is a highly relevant biological endpoint for studying 7TMR activity; however, the half-life of IP3 is very short compared to its downstream metabolite inositol 1-phosphate (IP1), which accumulates in cells in the presence of lithium (usually LiCl).102 Because of the relative ease of measuring IP1 and the biological significance of IP3, accumulation of both molecules is commonly used to assess Gq-mediated activity.
The only currently available plate-based assay for measuring IP3 is the HitHunter® Inositol 1,4,5 Triphosphate assay system (DiscoveRx), which provides a reasonable degree of assay simplicity fit for automation of HTS campaigns and higher sensitivity than most other IP3 detection methods. In this assay system, an IP3-binding protein is used to slow the rotation of a fluorescent IP3 tracer molecule; in this state, plane-polarized light is minimally disrupted and a high florescence polarization (FP) signal is detected. In the presence of high levels of IP3, the unlabeled IP3 competes for binding at the IP3-binding protein, increasing the amount of unbound fluorescent IP3 tracer molecules free to rotate more rapidly, reducing the FP signal. This assay system is in effect very similar to the LANCE® cAMP readout: increasing IP3 correlates with a reduction in the assay signal along a nonlinear standard curve, thus necessitating the conversion of FP values to units of IP3 amounts before data can be analyzed. The principle advantage of this assay system is its miniaturized format compared to traditional radioactive means of detecting IP3. It should be noted that fluorescent compounds may interfere with FP signals in homogenous assay systems using this method for detection.103
The IP-One HTRF® assay system (Cisbio) is an example of a microtiter plate assay system for the detection of IP1. Because IP1 levels remain relatively stable over extended periods when in the presence of lithium, the measurement of IP1 can be more readily assessed with greater sensitivity than IP3 and lends itself better to HTS applications, particularly for allosteric drug discovery campaigns where assay sensitivity is at a premium. The IP-One HTRF® system provides a miniaturized detection method of IP1 accumulation and is built on TR-FRET technology, which can be detected on any plate reader capable of detecting LANCE® and other HTRF® assays. The principle behind this assay system is again one of competition binding: IP1 labeled with a FRET acceptor competes with cellularly produced IP1 for binding to an anti-IP1 antibody labeled with a FRET donor molecule. In the presence of low cellular IP1, FRET acceptor-labeled IP1 readily interacts with the anti-IP1 antibody, producing a robust FRET signal; high levels of cellular IP1 inhibit the formation of the FRET complex. This, like the LANCE® cAMP assay, produces a TR-FRET signal that inversely correlates with IP1 levels along a nonlinear standard curve, necessitating the conversion of TR-FRET values to units of IP1.
Both the HitHunter® IP3 and IP-One HTRF® assay systems can be applied to native cells and primary cell culture; however, neither assay system is well suited for membrane preparations or tissue homogenates. Considering the greater sensitivity of the IP-One HTRF® assay and the robust nature of TR-FRET readouts, which do not suffer significantly from autofluorescence and fluorescent test compounds,104 the IP-One HTRF® assay system is better suited for allosteric drug HTS efforts, while IP3 detection systems are best used for verifying the activity of small groups of compounds in follow-up assays.
7.3. ERK1/2 phosphorylation
Receptor stimulation of mitogen-activated protein kinase (MAPK) pathways has in recent years become appreciated as major signaling pathways for 7TMRs.105 Various receptors coupled to Gq, Gi/o, and Gs have been found to stimulate the phosphorylation of ERKs (e.g., ERK1/2) either through G-protein-mediated mechanisms or through arrestin signaling.94,95,105,106 With mounting evidence for the biological relevance of 7TMR-mediated ERK1/2 phosphorylation and the availability of plate-based assays, measurement of pERK1/2 is becoming a significant endpoint assay for drug discovery efforts, especially for allosteric modulators, which have been shown to exhibit pERK responses that significantly differ from classical assay systems.15
Traditionally, Western blot analysis has been used to assess levels of ERK1/2 phosphorylation (e.g., Ref. 107); however, this method is time-consuming, expensive, very low throughput, and difficult to accurately quantify. The most widely used microtiter plate-based assay system for measuring pERK accumulation is the AlphaScreen® SureFire® pERK1/2 (Thr202/Tyr204) assay system (PerkinElmer); this method addresses many of the issues that make Western blot analyses unsuitable for detailed characterization of novel compounds. This system was developed from the luminescent oxygen channeling assay (LOCI),108,109 and, much like an ELISA, the principle behind the pERK1/2 AlphaScreen® system is one of immunocapture by an antibody with affinity for one epitope of the ERK molecule and immunocapture by a second phospho-specific antibody. The first antibody is tagged with an AlphaScreen® donor bead that, upon light excitation, produces a short-lived reactive oxygen species (1O2) that diffuse less than 200 nm before settling to a ground state110; if the phospho-specific antibody (tagged with an AlphaScreen® acceptor bead) is nearby, i.e. is bound to the same pERK molecule as the first antibody, the reactive oxygen can interact with the acceptor bead before it returns to a ground state and a chemiluminescent reaction is produced. Low levels of pERK will allow for the formation of very few of these complexes and little luminescence will be detectable; higher levels of pERK will allow for many of these complexes to form and the luminescent signal will increase. However, excessive levels of pERK will cause a reduction in luminescence, as few of the donor bead-conjugated antibodies will interact with the same ERK molecules as the acceptor bead-conjugated antibodies, producing a bimodal standard curve.111 Fortunately, levels of pERK produced in most cells are low enough that this “hook effect” is not commonly observed, though one should still be aware of this caveat. The nature of this assay system and the low levels of pERK produced by most cells allow for a direct, essentially linear correlation between pERK1/2 accumulation and AlphaScreen® signal, thus no conversion of raw Alpha values to units of pERK is necessary. Fetal bovine serum contains a plethora of mitogens that stimulate a robust pERK response and can be used as a positive control for pERK accumulation and a control that experimental values can be normalized to for day-to-day comparisons. Just as with Western blot analysis of pERK levels, various cell types can be used in this assay, including primary cultured cells (e.g., Ref. 112). Being a plate-based assay system, AlphaScreen® pERK1/2 is better suited for the characterization of allosteric modulator potencies; assessing the effects of PAMs or NAMs on agonist EC20 and EC80 concentrations is not easily accommodated by Western blot analysis.
Several caveats accompany this assay system. Care must be taken not to mechanically disrupt cells during the compound treatment phase, as this will typically induce MAPK activity and confound results. Unlike cAMP and PI hydrolysis assays, no convenient inhibitors of the enzymes that dephosphorylate pERK exist, and depending on the cell background, maximal pERK production may occur within as little as 5 min with a precipitous drop-off afterward.111 It is advisable to establish agonist time-courses to identify the optimal assay window for a particular receptor and cell type. pERK assays often cost more to run than other commonly used 7TMR assay systems; however, the higher cost may be warranted since detection of pERK levels relays highly relevant biological data that often sheds light on critical and unique properties of allosteric modulators that would otherwise go unobserved in other assays.
7.4. Arrestin recruitment
Many 7TMRs have the ability to signal through more than just the canonical G-protein-mediated pathways. Arrestins, which were originally identified as molecules that uncouple 7TMRs from their cognate G-proteins and were thus implicated in receptor desensitization,113,114 have now been shown to mediate a variety of signaling cascades upon recruitment to receptors.95,97 Activation of MAPKs has been highlighted as a major arrestin signaling pathway; however, no high-throughput assay systems are available that can clearly isolate arrestin-mediated signaling cascades from G-protein-mediated signaling cascades. Thus, several groups have devised methods to detect arrestin recruitment to various 7TMRs (e.g., Refs. 115–117); these methods, however, tend not to provide the throughput and large signal window required for HTS campaigns. Better suited to HTS needs are the two most common commercially available arrestin recruitment assays: the Tango™ GPCR Assay System (Life Technologies) and the PathHunter® β-Arrestin GPCR Assay system (DiscoveRx). Unfortunately, both systems rely upon proprietary cell lines expressing tagged 7TMRs and tagged arrestin molecules, which limits the flexibility of these assays.
The Tango™ GPCR Assay System is a gene reporter based proximity assay for arrestin recruitment to 7TMRs.118 The receptor of interest is tagged with a viral transcription factor linked to the receptor via a peptide sequence containing a protease cleavage site. This receptor construct is expressed in a cell line co-expressing arrestin molecules tagged with a protease specific for the protease site of the receptor construct. Upon recruitment of arrestin, the protease site is cleaved and the viral transcription factor translocates to the nucleus and promotes the transcription of a beta-lactamase reporter gene. The activity of beta-lactamase is then measured by its ability to cleave a FRET substrate molecule, reducing the FRET signal. This assay, though highly engineered, does provide a robust readout of arrestin recruitment specific to the receptor of interest without cross-reactivity with other endogenously expressed receptors. Though this assay system can be automated and applied to HTS platforms, when planning multiple experiments, investigators should take into account the long incubation times required between compound addition and the detection phase (at least several hours). An advantage of this assay system over others is that it allows for imaging of cellular responses to the FRET substrate118; however, this is typically not a quality required for HTS efforts.
A more streamlined proximity assay for arrestin recruitment is the Path-Hunter® β-Arrestin GPCR Assay system (DiscoveRx). In this system, the receptor of interest is tagged with a portion of the beta-galactosidase enzyme and expressed in cells that also express β-arrestin2 tagged with the remaining portion of the enzyme. Upon recruitment to the tagged 7TMR, the enzyme fragments complement and with the addition of the beta-galactoside substrate, a chemiluminescent reaction is catalyzed that is readily detectable on most luminescence-equipped plate readers. Due to its luminescent readout, the signal-to-noise ratio of this assay is very high and provides Z′ values suitable for most HTS programs.
Neither the PathHunter® nor Tango™ assay systems can be applied to primary cell culture or native (i.e., untagged) receptors, limiting the use of these assays to in vitro screening of compounds. A significant strength of these and other arrestin recruitment assays is that many 7TMRs interact with arrestin,118 and the measurement of arrestin recruitment may be used as a primary measurement of receptor activity when the G-protein coupling of a receptor is unknown or not easily measured.118 Additionally, arrestin recruitment is highly proximal to ligand binding and receptor conformational changes, thus little receptor reserve is typically detected in these assay systems, helping to simplify the interpretation of results. The investigation of allosteric modulators’ abilities to affect arrestin recruitment to 7TMRs is still in its early stages and will undoubtedly yield compelling and crucial data for future allosteric drug development efforts.21
7.5. Transcriptional regulation
Many of the signaling cascades induced by 7TMRs ultimately impact transcription of a wide array of genes. Activation of 7TMRs has been linked to transcription regulated by CREB, STATs, SRF, NFkB, NFAT, Elk-1, c-Jun, and c-Fos. Of these, induction of c-Fos has become an important preclinical marker of antipsychotic efficacy of novel compounds. The exact mechanisms that relate c-Fos induction to antipsychotic action are not fully understood; it is known that c-Fos binds to c-Jun to form the AP-1 transcription factor, which induces transcription of numerous genes involved in cell proliferation, differentiation, cell damage, and apoptosis.119 Drugs known to produce robust antipsychotic activity in humans (e.g., haloperidol, clozapine, olanzapine, risperidone) show a direct correlation of antipsychotic efficacy with their ability to induce c-Fos in striatal and prefrontal cortical regions of the brain.120 In situ hybridization has been frequently used in the past to assess c-Fos expression, and this technique will continue to be vital to the investigation of psychoactive drugs, as the specific brain regions where c-Fos is induced is critical to behavioral outcomes. However, in situ hybridization is not amenable to the characterization of numerous novel compounds; to screen compounds for the ability of induce c-Fos, more efficient cell-based assays have been developed. One of the most commonly used plate-based, nonradioactive assay systems for determining c-Fos activation is the c-Fos EZ-TFA Transcription Factor Assay (Millipore). The principle behind this assay is similar to an ELISA. A double-stranded oligonucleotide, containing the DNA-binding consensus sequence of the TPA response element (5′-TGAG/CTCA-3′), is used as a probe that binds c-Fos/c-Jun dimers present in nuclear extracts prepared from cells treated with drug or control cells. This oligonucleotide probe is biotinylated and, when transferred to a streptavidin-coated micro plate, the oligonucleotide-c-Fos/c-Jun complex adheres to the plate surface via a high-affinity biotin–streptavidin interaction. After a wash step, antibodies specific for c-Fos and c-Jun are added, and with successive washing and addition of a horse radish peroxidase secondary antibody, activated c-Fos levels can be determined. The assay readout is colorimetric and can be detected on a variety of common and inexpensive plate readers. A caveat of this assay system is the relatively laborious preparation of experimental samples, which includes the purification of nuclear extracts. This limits the application of this assay and others like it to follow-up experiments and later in-depth analysis of a novel compound’s properties. These tradeoffs may be warranted if the induction of c-Fos is a critical marker for the development of a new drug. Most published investigations of c-Fos, and the induction of other transcription factors by 7TMRs for that matter, have not employed 7TMRs allosteric modulators. Considering the potential of allosteric compounds to induce these transcription factors, this area of research will be important for understanding the impact of allosteric compounds in vivo and in the clinic.
CREB (cAMP response element-binding) is another well-studied transcription factor that is clearly linked to the activity of 7TMRs. CREB can be activated through several pathways that are linked to 7TMRs. Cellular cAMP, a second messenger of Gs-coupled receptors and inhibited by Gi/o receptors, mediates the activation of CREB by stimulating PKA, which then translocates to the nucleus and phosphorylates CREB. CREB can also be activated via phosphorylation by Ca2+/calmodulin-dependent protein kinases, and can thus be linked to Gq-coupled receptor activity. Additionally, CREB can be activated through G-protein-induced MAPK signaling cascades.121 Once phosphorylated, CREB binds the cAMP response element (CRE), driving the expression of hundreds of genes (e.g., tyrosine hydroxylase, c-Fos, brain-derived neurotrophic factor) and has been most commonly associated with the processes involved in learning and memory122 but has also been implicated in depression, anxiety, and addiction.123 Because of these observations, CREB activation is seen as an important outcome of 7TMR stimulation, and several assay systems have been developed to detect phosphorylated CREB. The AlphaScreen® SureFire® CREB (p-Ser133) Assay (PerkinElmer) is one such kit and is based on the same assay principle at the AlphaScreen® SureFire® pERK kit discussed above and comes with the same benefits and caveats of this assay system. There are also numerous ELISAs available for the detection of phosphorylated CREB, including those that use specific antibodies to capture phosphorylated CREB and those that use oligonucleotides containing the CRE-binding region to capture activated CREB. Both the ELISA methods and AlphaScreen kit present rather expensive approaches for detecting CREB activation in a plate-based format. Alternatively, gene reporter assays present a method for detecting CREB activity that is more amenable to HTS screening. Cells can be transfected with a vector that encodes an enzyme whose activity can be easily measured (e.g., luciferase, GFP) and is under the control of CRE. Compounds that activate CREB would thus result in an increase in the reporter gene’s expression. This method is limited to recombinant cell lines, and as with any gene reporter assay, cells must undergo a prolonged incubation (several hours to overnight) before the activity can be measured.
8. RADIOLIGAND BINDING ASSAYS FOR ALLOSTERIC INTERACTIONS
Due to the reliance on binding assays during HTS campaigns before current functional assays were available, most small molecules discovered were competitive with the orthosteric radioligand or, at least, inhibited its binding to a measureable degree. In the case of allosteric (or putatively allosteric) ligands, binding assays are applied to more thoroughly characterize a compound’s interaction with the receptor to better understand its affinity, site of interaction, and influence on endogenous ligand binding. This information may then be combined with functional data to more fully understand the ligand–target dynamic. The equilibrium dissociation constant of a compound can be derived from the law of mass action and describes the strength of association between the ligand and its compliment target (e.g., a small molecule and a 7TMR). This is expressed as
| [1.4] |
where Kd is the concentration of ligand that occupies half of the target at equilibrium, and koff and kon are dissociation rate (reciprocal time units) and association rate (reciprocal concentration-time units), respectively. It is upon this relation that further equilibrium binding principles are built. This also assumes that the ligand(s) involved in the experiment does not bind at more than one target on the receptor. This basic equation holds true, given certain caveats, regardless of the site where the ligand binds.124,125 However, as discussed below, more complex models are needed when describing systems where more than one site and/or ligand are being considered, as is often the case in studies involving allosteric modulators and orthosteric radioligands.
This section will focus on describing approaches used to determine binding characteristics for an allosteric compound at the target of interest using radiochemical techniques. Practical considerations and applications will be covered with references to other sources for detailed methods including the derivation of quantitative models of allosteric modulation that may be applied to binding data.
8.1. General methodology and initial considerations
8.1.1 Receptor source
Because 7TMRs are transmembrane proteins spanning between the extracellular and cytosolic space, cellular membranes must be isolated by established methods.126 Alternatively, whole cell fractions may be utilized when there is concern of disrupting particular protein–protein interactions that may be necessary for proper receptor conformation; however, this type of preparation may produce higher variability and potentially different results from more purified membrane preparations due to the presence of different effectors (pharmacological and physiochemical).127 As with any assay, consideration should be taken for the system used as the source of the receptor. If possible, choosing a system that is highly enriched in the target receptor, such as in inducible cell lines capable of generating high expression levels or in lines in which the receptor is highly constitutively expressed, will increase both yield and specific binding.
8.1.2 Choice of radioligand
The experiments designed for allosteric binding studies are dictated, to a large degree, by the availability of radioligand(s) for the target of interest. As the orthosteric sites of 7TMRs have been studied for significantly longer than the allosteric sites, radioligands for the former are more readily available. High-affinity radioligands for allosteric sites do exist and these have proven to be invaluable tools for probing PAM and NAM interactions.128–132 The utility of any radioligand is limited by its affinity, such that weakly bound (Kd>10 nM) radioligands will often not provide a strong enough interaction with the binding site to remain bound during postbinding assay steps that involve washing the membranes or separating bound versus free ligand. The development of allosteric radioligands is an active area for drug discovery as more allosteric molecules move through the discovery pipeline.133 This is critical for both in vitro characterization assays and applications of ligands as PET tracers in preclinical and clinical studies. An example of such development is the application of [3H]-methoxy-PEPy to probe one of the allosteric sites of mGlu5. [3H]-methoxy-PEPy is based on the biaryl acetylene scaffold of MPEP, which is a NAM at mGlu5. Several useful tools have been generated from this chemical series, including the neutral allosteric modulator 5-MPEP (Fig. 1.5D) and the PET ligand [18F]-FPEB.134 As explained below, the results from a binding experiment should not be considered as stand-alone evidence of allosteric (or lack of) interaction but should be interpreted in conjunction with other assays.
8.1.3 Experimental conditions
Conditions such as temperature, membrane protein amount needed for optimal signal, radioligand Kd at the receptor being used, and minimum time to reach steady state should be known before beginning experiments with the allosteric modulator of interest. These (with the exception of radioligand Kd) can be determined through straightforward matrix experiments to ensure that the assay design is robust and resources are used wisely throughout the study. When combining binding results with those from functional studies, one should consider aligning the conditions of the two whenever possible, for example, buffer conditions, receptor source, and presence or absence of any component that may affect the compound–target interaction such as the presence of agonist. Note that the presence of the allosteric modulator itself may affect time to reach equilibrium; therefore, it should be considered that this parameter may have been affected when an experiment generates complex binding curves that do not fit to the allosteric ternary complex model (ATCM).135,136
9. COMPETITIVE OR NOT COMPETITIVE
Generally, binding experiments are performed to determine the affinity of a compound at its target. However, since the compound of interest is not standardly the radiolabel, one of the most common questions that binding assays are designed to address is to determine if the compound being studied is competitive with another ligand known to interact at a specific site of the receptor. If a compound appears to completely displace a radiolabel, the next calculation that is performed is to determine the affinity, or Ki value. Competition binding assays to study allosteric compounds are performed in the same way as those in which an orthosteric compound is the compound of interest; some differences exist, however, in the data analysis and interpretation.
Briefly, the experiment is performed under steady-state conditions with a known concentration of radioligand (typically around its Kd but not so low as to result in ligand depletion) and varying concentrations of the competing compound. The radioligand, unlabeled compound being studied, and membrane preparation (at a known, constant protein concentration) are combined and should be solvent-matched if the compound is dissolved in a nonaqueous solvent. A second, unlabeled compound at a concentration known to completely displace the radioligand from the target is included in a condition to determine nonspecific binding. In addition, the binding of the radioligand in the absence of competing compound is included to determine maximal (100%) binding. The experiment is mixed at a constant temperature until the predetermined time to reach steady state. Note that this time to equilibrium may differ in the presence of an allosteric modulator compared to without, and this should be considered during the experimental design phase.136 The contents of the wells are then transferred to a filter plate and rapidly washed to remove unbound radiolabel; this last step is performed quickly using cold buffer to minimize dissociation of the bound radioligand. Once the filter plate(s) is dried (overnight is usually sufficient), scintillant is added and the plate is read using a microplate scintillation counter. The amount of radioligand present in the experiment should be determined empirically for each experiment by taking a count measurement of the radioligand-working solution added to the experiment and converting these counts using standard conversion factors and the instrument efficiency for the isotope used (for an example of this calculation, see http://graphpad.com/curvefit/radioactivity_theory.htm). Scintillation proximity assay beads may also be employed as a homogenous approach to perform a binding experiment, although this is not applied as commonly as the filter plate approach.
9.1. Allosteric radioligand competition
The availability of an allosteric radioligand typically depends on the site being well characterized such that there has been time for a high-affinity compound to be developed for that site. The choice of test compounds for these experiments will come from those that exhibit features of allosterism in upstream assays or are of a similar structural class as other known allosteric ligands for that target. In these cases, relatively straightforward analyses of the data using the law of mass action and standard curve fitting of the percent radioligand bound versus log[ligand] may be applied in which the log(IC50) of the ligand is determined using nonlinear regression of a three-parameter equation as shown below. These experiments are treated the same as orthosteric radioligand–orthosteric compound competition experiments. The more complex situation where the radioligand and compound are not expected to bind to the same site is addressed in Section 9.3.
| [1.5] |
In the above equation, “log[ligand]” values are the log of the molar concentration of the competing ligand (allosteric modulator) added to the experiment and the “% Bound” values are the corresponding values calculated from the experiment and are usually expressed as % Specific Binding using the total and nonspecific binding conditions. By knowing the concentration of the radioligand in the experiment and the Kd of the radioligand under the same experimental conditions, if behavior consistent with a competitive mode of action (full displacement) is observed, one may calculate the Ki (which is the more physiologically relevant parameter compared to the IC50) of the compound of interest at the target site, which may be determined by fitting experimental data in a similar manner as above using the following equation
| [1.6] |
where [radioligand] is its molar concentration and Kd is the radioligand’s equilibrium binding constant at the target. However, caution should be applied when concluding the competitive nature of a compound for a radioligand at a specific site. Confirmation of full competition (i.e., that the compound being studied and the radioligand have fully overlapping sites of interaction on the target) should be determined using an orthogonal assay that also measures the competition of one ligand versus the other, for example, saturation isotherms of radioligand at different competing ligand concentrations (with no change in Bmax indicating a competitive interaction), lack of effect on rate of radioligand dissociation (see Section 9.3), or a functional assay using a neutral ligand that is known to bind to the same site as the radioligand (described above in Section 3.3 and Fig. 1.5D). The challenge with performing a functional assay for this kind of confirmation is assuring that it is being performed under steady-state conditions. For experimental results that do not show full displacement of the radioligand, the interpretation of this type of binding data may be qualitative in nature although there are approaches that use models specifically describing allosteric interactions such as ATCM, which will determine the affinity of the allosteric modulator independent of site of interaction of the radioligand as discussed in Section 9.2. Lastly, studies utilizing site-directed mutagenesis of the receptor are a common approach for shedding light on the location of allosteric modulator binding as well the effect on signaling.15,35,137–143 It should be emphasized that knowing the nature of the interaction of the compound (in these examples, allosteric modulator) with the radioligand is not a purely academic pursuit. For drug discovery purposes, this knowledge is directly applied to the development of PET tracers that are used for confirming target engagement and determining receptor occupancy needed for therapeutic effect during preclinical and clinical studies. Although translation across species cannot be guaranteed, the in vitro characterization of a ligand is an important first step before moving into resource-intensive studies in whole animal and humans.
9.2. Orthosteric radioligand competition
Many orthosteric 7TMR radioligands are available as tools for competition binding studies. In these studies, increasing concentrations of allosteric modulator are incubated with a single concentration of radioligand to give qualitative confirmation that the modulator is indeed interacting at a site topographically separate from the orthosteric site due to its lack of full displacement of the radioligand (except in the case of a NAM with high cooperativity, which may displace 100%). If there is a directional effect (enhancement or decrement) on radiolabel binding, the mode of action of the allosteric modulator for the ligand being used as the radiolabel may be ascertained; an increase above “100% specific binding” denotes a PAM that acts through enhancing the affinity of the radioligand at the orthosteric site, while a decrease indicates that the modulator is a NAM (Fig. 1.8). A constraint of this interpretation is that this determined mode of action cannot be assumed to be the same across radioligands (an example of probe dependence discussed earlier). In addition, if the modulator has no effect on radioligand affinity, then there will be no change observed for the binding, lending no information to its role as a PAM or NAM. For quantitative analysis of allosteric modulator effect on orthosteric radioligand binding, the ATCM, which accounts for both allosteric and orthosteric binding and the effect of the former on the latter, may be applied.144,145 Fitting the data to this model will allow determination of the affinity of the allosteric modulator and its cooperativity factor, α, for the radioligand, which is a measure of the degree that an allosteric modulator affects the binding of the orthosteric ligand (0<α<1 for NAMs; α>1 for PAMs).4 In the case where the modulator does not alter the affinity (α=1), no change in binding will be observed. Care should be taken not to interpret this as there being no allosteric modulation (especially if evidence has been found in previous functional assays); but, instead, that there is no allosteric modulation on the binding of the orthosteric compound being used as the probe in the experiment. The equation derived from the ATCM that may be used for fitting the experimental data can be found in graphing and fitting software such as GraphPad Prism 5 and is shown in a modified form below:
| [1.7] |
where
| [1.8] |
In this equation, % Bound0 is the amount of radioligand bound with no ligand (allosteric modulator) present; note that this is not termed “Top” as before since there may be some enhancement of binding under PAM application. Kapp is the apparent KD and is made up of the KD of the radioligand modified by a factor consisting of modulator concentration, [ligand], the affinity cooperativity factor, α, and the dissociation constant of the modulator, KB. Figure 1.8 shows examples of the effect of PAMs and NAMs with different α values on orthosteric radioligand binding using a simulation of the above equation. Note the effect not only on the change in amount bound but also on the left shift in the curve that occurs upon larger absolute values of log α.
Figure 1.8.
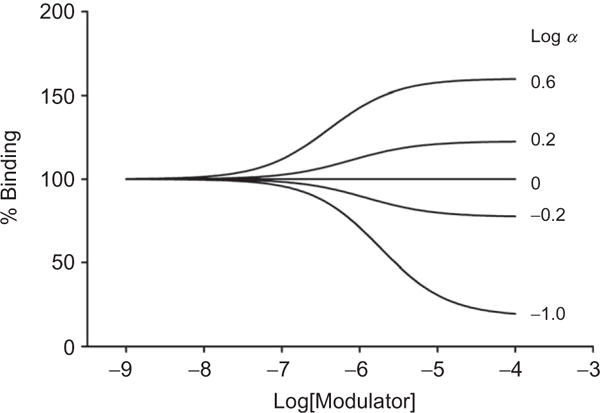
Simulation of the effect of different values of the allosteric modulator affinity parameter, α, on the binding of orthosteric radioligand using the ATCM. For the simulation, radioligand concentration=radioligand KD=1 nM and modulator affinity (KB) = 10−6 M. Curves were generated with GraphPad Prism 5 using the preloaded allosteric modulator titration equation.
One may also determine the effect of the allosteric modulator on an unlabeled orthosteric ligand by performing a standard competition binding assay using a single radiolabel concentration and titrating with cold orthosteric ligand of interest in the presence of increasing concentrations of allosteric modulator. This will show what, if any, effect the modulator has on the affinity of the unlabeled orthosteric ligand based on the shift imparted on the curve. This is useful when one is interested in the effect on an orthosteric ligand that is not labeled or is not suitable as a radioligand.146 This effect may be expressed as a fold-increase on ligand affinity by the allosteric modulator for the purposes of confirmation of effect and relative comparisons across orthosteric and allosteric ligands. Similar to the application of the ATCM above, the model may be extended to account for more than one orthosteric ligand and the data sets fit globally to determine the parameters of allosteric modulator affinity and α values (modulator for cold ligand and modulator for radioligand) as detailed in Langmead (2011).145
9.3. Dissociation kinetic assays
Another primary means of using binding as a way to detect or confirm allosterism at a receptor is to observe the effect on dissociation kinetics of an orthosteric radioligand. When an allosteric compound binds to its target, the effect on the receptor can be detected by a change in the rate of association or dissociation of a bound orthosteric ligand if the conformational change impacts the orthosteric ligand’s affinity. However, because a competitive compound would also have an effect on association due solely to its presence at the orthosteric binding site, this design should not be used as the first pass in a kinetic assay. Rather, a change in the koff of a radioligand once it has reached equilibrium may be assigned unambiguously to engagement of the test compound at an allosteric site. The design and caveats of this assay are described elsewhere in detail.136 Briefly, to test the presence and/or degree of allosterism of a test compound in relation to the site of the radioligand, a single concentration of radioligand is allowed to come to equilibrium at the receptor of interest (using a predetermined time). The test compound, along with a cold orthosteric compound at a high enough concentration to block radioligand reassociation, is added at different time points (including a zero-concentration compound condition) before experiment termination (i.e., harvesting). The remaining bound radioligand is measured and plotted as a function of time. Alternatively, one may use the “infinite dilution” design that may not be practical in some cases due to the volumes involved, but avoids any effect that the modulator may have on the unlabeled ligand used to prevent reassociation of the radioligand.147 Equations describing monoexponential decay kinetics and the effect of one or more allosteric modulator concentrations on radioligand off rate may be combined (by substituting the right side of the latter for koffobs in the monoexponential decay equation) and the data for one or more modulator concentrations fit simultaneously to solve for koff, KoffB, and Kb/α (note that Kb and α cannot be resolved from each other using this approach).
| [1.9] |
| [1.10] |
In the above equations, Boundt is an expression of the amount of radioligand bound at a specific time, Boundt=0 is the amount bound at equilibrium (no allosteric ligand), koff, koffB, and koffobs are the radioligand dissociation rate constant at the receptor in the absence of the modulator, for the receptor–modulator complex, and observed experimental rate constant, respectively. KB is the modulator equilibrium dissociation constant, [B] is the modulator concentration, and α is the affinity cooperativity factor discussed earlier. As [B] approaches zero, koffobs=koff as represented in the control curve that is generated. Likewise, an increase in the modulator concentration, affinity (i.e., lower Kb), and/or α (i.e., greater cooperativity) values brings the observed rate closer to the dissociation rate from the modulator-occupied receptor.
For a better understanding of the behavior that may be observed in this assay, consider the following scenarios. In one case, an allosteric modulator may not affect affinity of the orthosteric ligand, in which case koffB=koff=koffobs, whereby no effect is seen and therefore a negative result is not proof of the ligand being orthosteric. This is once more an example of the necessity to perform more than one type of assay for determining allosterism. In a second example, consider two allosteric compounds that impart an identical koffB to the orthosteric compound but have differing Kb/α ratios (Fig. 1.9). The compound with the lower Kb/α (greater affinity and/or greater affinity cooperativity) will have a larger effect on koffobs at the same concentration. Although the use of association kinetics is not the best approach as an initial investigation of a putative allosteric compound, once the allosteric nature is corroborated by two or more assays, one may measure the effect on the orthosteric rate of association as a further characterization of the compound.139
Figure 1.9.
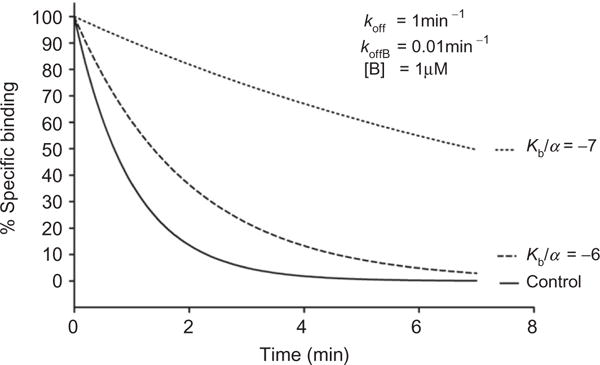
Simulation of the effect of the Kb/α parameter of an allosteric modulator on the observed dissociation rate, koffobs, of an orthosteric radioligand. At a submaximally effective concentration and same koffB, the modulator that has the greater absolute affinity:cooperativity ratio will impart a larger effect on koffobs.
This same principle can be applied using other means to detect the dissociating ligand. For example, the agonist ABA-X-BY630 was used as a fluorescent probe at human A1 and A3 adenosine receptors and was shown to be allosteric as observed by an increase in the dissociation kinetics upon addition of orthosteric agonists and antagonists.148 The experimental design was different as described for binding, but, the same effect was observed to determine the allosteric nature of the compound of interest. Another unconventional study using dissociation kinetics to confirm allosterism was performed by Gomes et al.149 to support the interaction between sites on the δ and μ opioid subunits of the corresponding heteromer.
10. CONCLUSIONS AND FUTURE DIRECTIONS
As the allosteric modulator field has emerged, in vitro pharmacological approaches to detect and quantify their activity have rapidly developed. These techniques are now aiding in the translation of allosteric modulator pharmacology into clinical development. One area in which this is beginning to emerge for allosteric ligands of 7TMRs is in the development of PET ligands to detect target engagement.134,150–158 The availability of a PET ligand that is truly competitive with a clinical candidate offers confidence that the relevant receptor site has been occupied; additionally, these findings can help relate occupancy to efficacy and aid in setting appropriate doses in the clinic. It should be noted that careful molecular pharmacological profiling of a test compound with a radioligand/PET ligand should be undertaken to ensure that the relationship between the two compounds is truly competitive. To this end, PET ligands unique to a given allosteric scaffold may be required for true competition, often necessitating new PET tracer development as different scaffolds progress. Additionally, because of the cooperativity, it may be much easier and straightforward to develop a NAM as a PET tracer when compared to a PAM. For NAMs, the affinity and potency of a compound are often closely aligned, whereas PAMs can show high degrees of positive cooperativity. Molecular pharmacology techniques (e.g., progressive fold-shift experiments) can aid in the determination of affinity estimates; compounds that saturate in their fold-shift values at low concentrations would be predicted to have higher affinities than those that continue to shift. This method can be used as one technique to aid in prioritization of compounds for labeling as tracers. Additional experiments with tools such as SAMs can further aid in understanding if an interaction between two compounds is truly competitive in nature.
Which assays are the best to use to find and characterize allosteric modulators? This question, of course, depends on the needs of the individual investigator. Label-free techniques certainly have the potential to cast a wide net and are being embraced for their ability to capture multiple, potentially unknown, signaling events. However, the cost and need for extensive secondary assays to eliminate nonspecific compounds and convolute signals captured in the kinetic trace information may not make them an ideal choice for some investigators if testing large numbers of compounds is required. Other notes from our own experience come from the ability of many allosteric modulators to “mode switch” during a chemical optimization program. To support a chemistry effort, the utility to screen multiple modes of pharmacology in a first pass experiment, such as that shown in Fig. 1.3 with multiple compound and agonist additions, is quite helpful. Additionally, experiments designed to definitively categorize a compound as allosteric, for example, by determining that a compound cannot compete for binding of an orthosteric radioligand, can be quite important when coupled with functional assays in terms of understanding compound profiles.
Functionally selective effects bring with them their own set of requirements. Certainly, label-free technology can act as an initial window into compound profiles, highlighting compounds that may engage distinct signaling events. The ability to measure multiple, discrete signaling events can be quite useful in “binning” compounds and then attempting to correlate in vitro profiles with in vivo efficacy. The emergence of the importance of G-protein independent events, such as β-arrestin signaling, suggests that assays to monitor these activities should be examined for a receptor of interest and to further profile allosteric modulator pharmacology.
A common question posed in small molecule allosteric modulators programs is: which parameter of an allosteric modulator is most important for in vivo efficacy? Unfortunately, the answer is not clear and may vary by 7TMR, allosteric binding site, allosteric ligand chemotype and therapeutic indication. In a chemical optimization program, potency is generally an important optimization parameter, but it should be noted again that efficacy (e.g., leftward fold shift of an agonist CRC) is balanced with affinity. A compound that shifts a curve 20-fold at 1 μM and then 70-fold at 10 μM would not be predicted to have high affinity; additionally, depending on pharmacokinetic parameters, it is not clear what levels of such a drug could even be achieved (i.e., is a fold shift of 70 relevant if the highest concentration of drug available is 1 μM?). A compound that shifts 10-fold at 1 and 10 μM has saturated in its effect, suggesting that it has completely occupied its binding site at 1 μM. This compound could be a better choice if higher affinity is required.
In conclusion, the number and variety of assays designed to detect and characterize allosteric modulators continue to develop at a rapid pace. The potential ability to correlate in vitro pharmacological and functionally selective effects with relevant in vivo efficacy or side effect profiles can be described as a new frontier of critical importance as basic science findings are translated into new tools and drug candidates. It is anticipated that new techniques, as well as evolution of existing methods and practices, will continue to be developed and refined by pharmacological community.
Acknowledgments
The authors would like to acknowledge funding from the NIH (NS078262, NS31373 MH84659, MH093366), The Molecular Libraries Probe Centers Network, Seaside Therapeutics, Janssen Pharmaceuticals, Inc., and the International Rett Syndrome Foundation.
References
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–6. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–41. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 4.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–74. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 5.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 2007;190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 7.Kolakowski LF., Jr GCRDb: a G-protein-coupled receptor database. Receptors Channels. 1994;2:1–7. [PubMed] [Google Scholar]
- 8.Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-12-reviews3013. REVIEWS3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, et al. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012;55:1445–64. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 13.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–9. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, et al. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–69. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valant C, Felder CC, Sexton PM, Christopoulos A. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol. 2012;81:41–52. doi: 10.1124/mol.111.074872. [DOI] [PubMed] [Google Scholar]
- 16.Suratman S, Leach K, Sexton P, Felder C, Loiacono R, Christopoulos A. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162:1659–70. doi: 10.1111/j.1476-5381.2010.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–65. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wootten D, Simms J, Koole C, Woodman OL, Summers RJ, Christopoulos A, et al. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J Pharmacol Exp Ther. 2011;336:540–50. doi: 10.1124/jpet.110.176362. [DOI] [PubMed] [Google Scholar]
- 19.Mathiesen JM, Christopoulos A, Ulven T, Royer JF, Campillo M, Heinemann A, et al. On the mechanism of interaction of potent surmountable and insurmountable antagonists with the prostaglandin D2 receptor CRTH2. Mol Pharmacol. 2006;69:1441–53. doi: 10.1124/mol.105.017681. [DOI] [PubMed] [Google Scholar]
- 20.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 21.Digby GJ, Conn PJ, Lindsley CW. Orthosteric- and allosteric-induced ligand-directed trafficking at GPCRs. Curr Opin Drug Discov Dev. 2010;13:587–94. [PMC free article] [PubMed] [Google Scholar]
- 22.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–22. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–69. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenakin T. A holistic view of GPCR signaling. Nat Biotechnol. 2010;28:928–9. doi: 10.1038/nbt0910-928. [DOI] [PubMed] [Google Scholar]
- 25.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, et al. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol. 2009;75:577–88. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenakin T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res. 2010;30:313–21. doi: 10.3109/10799893.2010.503964. [DOI] [PubMed] [Google Scholar]
- 28.Niswender CM, Johnson KA, Miller NR, Ayala JE, Luo Q, Williams R, et al. Context-dependent pharmacology exhibited by negative allosteric modulators of metabotropic glutamate receptor 7. Mol Pharmacol. 2010;77:459–68. doi: 10.1124/mol.109.058768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol. 2012;81:120–33. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weaver CD. Triple-addition assay protocols for detecting and characterizing modulators of seven-transmembrane receptors. Current Protocols in Chemical Biology. 2011;3:119–40. doi: 10.1002/9780470559277.ch110060. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J-H, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, et al. A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol. 2003;64:731–40. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 33.Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg Med Chem Lett. 2008;18:4098–101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol. 2005;68:1793–802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- 35.Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, et al. Discovery of a novel chemical class of mGlu(5) allosteric ligands with distinct modes of pharmacology. ACS Chem Neurosci. 2010;1:702–16. doi: 10.1021/cn100051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coward P, Chan SDH, Wada HG, Humphries GM, Conklin BR. Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal Biochem. 1999;270:242–8. doi: 10.1006/abio.1999.4061. [DOI] [PubMed] [Google Scholar]
- 37.Kostenis E, Waelbroeck M, Milligan G. Techniques: promiscuous Ga proteins in basic research and drug discovery. Trends Pharmacol Sci. 2005;26:595–602. doi: 10.1016/j.tips.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Yokoyama T, Kato N, Yamada N. Development of a high-throughput bioassay to screen melatonin receptor agonists using human melatonin receptor expressing CHO cells. Neurosci Lett. 2003;344:45–8. doi: 10.1016/s0304-3940(03)00419-1. [DOI] [PubMed] [Google Scholar]
- 39.Blattermann S, Peters L, Ottersbach PA, Bock A, Konya V, Weaver CD, et al. A biased ligand for OXE-R uncouples Galpha and Gbetagamma signaling within a heterotrimer. Nat Chem Biol. 2012;8:631–8. doi: 10.1038/nchembio.962. [DOI] [PubMed] [Google Scholar]
- 40.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen. 2004;9:671–7. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 41.Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, et al. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol. 2011;79:42–50. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol. 2009;76:1094–103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, et al. A novel assay of Gi/o-linked G protein coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol. 2008;73:1213–24. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- 44.Gentles RG, Grant-Young K, Hu S, Huang Y, Poss MA, Andres C, et al. Initial SAR studies on apamin-displacing 2-aminothiazole blockers of calcium-activated small conductance potassium channels. Bioorg Med Chem Lett. 2008;18:5316–9. doi: 10.1016/j.bmcl.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 45.Gentles RG, Hu S, Huang Y, Grant-Young K, Poss MA, Andres C, et al. Preliminary SAR studies on non-apamin-displacing 4-(aminomethylaryl)pyrrazolopyrimidine K(Ca) channel blockers. Bioorg Med Chem Lett. 2008;18:5694–7. doi: 10.1016/j.bmcl.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 46.Huang XP, Mangano T, Hufeisen S, Setola V, Roth BL. Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev Technol. 2010;8:727–42. doi: 10.1089/adt.2010.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q, Rottlander M, Xu M, Christoffersen CT, Frederiksen K, Wang MW, et al. Identification of novel KCNQ4 openers by a high-throughput fluorescence-based thallium flux assay. Anal Biochem. 2011;418:66–72. doi: 10.1016/j.ab.2011.06.040. [DOI] [PubMed] [Google Scholar]
- 48.Schmalhofer WA, Swensen AM, Thomas BS, Felix JP, Haedo RJ, Solly K, et al. A pharmacologically validated, high-capacity, functional thallium flux assay for the human Ether-a-go-go related gene potassium channel. Assay Drug Dev Technol. 2010;8:714–26. doi: 10.1089/adt.2010.0351. [DOI] [PubMed] [Google Scholar]
- 49.Titus SA, Beacham D, Shahane SA, Southall N, Xia M, Huang R, et al. A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel. Anal Biochem. 2009;394:30–8. doi: 10.1016/j.ab.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wojtovich AP, Williams DM, Karcz MK, Lopes CM, Gray DA, Nehrke KW, et al. A novel mitochondrial K(ATP) channel assay. Circ Res. 2010;106:1190–6. doi: 10.1161/CIRCRESAHA.109.215400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou B, Yu H, Babcock JJ, Chanda P, Bader JS, McManus OB, et al. Profiling diverse compounds by flux- and electrophysiology-based primary screens for inhibition of human ether-a-go-go related gene potassium channels. Assay Drug Dev Technol. 2010;8:743–54. doi: 10.1089/adt.2010.0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emkey R, Rankl NB. Screening G protein-coupled receptors: measurement of intracellular calcium using the fluorometric imaging plate reader. Methods Mol Biol. 2009;565:145–58. doi: 10.1007/978-1-60327-258-2_7. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, et al. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol. 2010;78:1105–23. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead CJ, Fry VA, Forbes IT, Branch CL, Christopoulos A, Wood MD, et al. Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M(1) receptor: direct pharmacological evidence that AC-42 is an allosteric agonist. Mol Pharmacol. 2006;69:236–46. doi: 10.1124/mol.105.017814. [DOI] [PubMed] [Google Scholar]
- 55.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 56.Stables J, Green A, Marshall F, Fraser N, Knight E, Sautel M, et al. A bioluminescent assay for agonist activity at potentially any G-protein-coupled receptor. Anal Biochem. 1997;252:115–26. doi: 10.1006/abio.1997.2308. [DOI] [PubMed] [Google Scholar]
- 57.Poul EL, Hisada S, Mizuguchi Y, Dupriez VJ, Burgeon E, Detheux M. Adaptation of aequorin functional assay to high throughput screening. J Biomol Screen. 2002;7:57–65. doi: 10.1177/108705710200700108. [DOI] [PubMed] [Google Scholar]
- 58.Gilchrist MA, Cacace A, Harden DG. Characterization of the 5-HT2b receptor in evaluation of aequorin detection of calcium mobilization for miniaturized GPCR high-throughput screening. J Biomol Screen. 2008;13:486–93. doi: 10.1177/1087057108319212. [DOI] [PubMed] [Google Scholar]
- 59.Jones B, Holskin B, Meyer S, Ung T, Dupriez V, Flores SY, et al. Aequorin functional assay for characterization of G-protein-coupled receptors: implementation with cryopreserved transiently transfected cells. Anal Biochem. 2010;400:184–9. doi: 10.1016/j.ab.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 60.Silvano E, Millan MJ, la Cour CM, Han Y, Duan L, Griffin SA, et al. The tetrahydroisoquinoline derivative SB269,652 is an allosteric antagonist at dopamine D3 and D2 receptors. Mol Pharmacol. 2010;78:925–34. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters MF, Knappenberger KS, Wilkins D, Sygowski LA, Lazor LA, Liu J, et al. Evaluation of cellular dielectric spectroscopy, a whole-cell, label-free technology for drug discovery on Gi-coupled GPCRs. J Biomol Screen. 2007;12:312–9. doi: 10.1177/1087057106298637. [DOI] [PubMed] [Google Scholar]
- 62.Peters MF, Vaillancourt F, Heroux M, Valiquette M, Scott CW. Comparing label-free biosensors for pharmacological screening with cell-based functional assays. Assay Drug Dev Technol. 2010;8:219–27. doi: 10.1089/adt.2009.0232. [DOI] [PubMed] [Google Scholar]
- 63.McGuinness RP, Proctor JM, Gallant DL, van Staden CJ, Ly JT, Tang FL, et al. Enhanced selectivity screening of GPCR ligands using a label-free cell based assay technology. Comb Chem High Throughput Screen. 2009;12:812–23. doi: 10.2174/138620709789104861. [DOI] [PubMed] [Google Scholar]
- 64.Verdonk E, Johnson K, McGuinness R, Leung G, Chen YW, Tang HR, et al. Cellular dielectric spectroscopy: a label-free comprehensive platform for functional evaluation of endogenous receptors. Assay Drug Dev Technol. 2006;4:609–19. doi: 10.1089/adt.2006.4.609. [DOI] [PubMed] [Google Scholar]
- 65.Shiau AK, Massari ME, Ozbal CC. Back to basics: label-free technologies for small molecule screening. Comb Chem High Throughput Screen. 2008;11:231–7. doi: 10.2174/138620708783877807. [DOI] [PubMed] [Google Scholar]
- 66.Coffman F, Hamid R, Cohen MC, Garippa R, Cohen S. Biophysical profiling of tumor cell lines. Anal Cell Pathol (Amst) 2011;34:225–34. doi: 10.3233/ACP-2011-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dodgson K, Gedge L, Murray DC, Coldwell M. A 100K well screen for a muscarinic receptor using the Epic label-free system—a reflection on the benefits of the label-free approach to screening seven-transmembrane receptors. J Recept Signal Transduct Res. 2009;29:163–72. doi: 10.1080/10799890903079844. [DOI] [PubMed] [Google Scholar]
- 68.Li G, Lai F, Fang Y. Modulating cell-cell communication with a high-throughput label-free cell assay. J Lab Autom. 2012;17:6–15. doi: 10.1177/2211068211424548. [DOI] [PubMed] [Google Scholar]
- 69.Schmidt J, Liebscher K, Merten N, Grundmann M, Mielenz M, Sauerwein H, et al. Conjugated linoleic acids mediate insulin release through islet G protein-coupled receptor FFA1/GPR40. J Biol Chem. 2011;286:11890–4. doi: 10.1074/jbc.C110.200477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee PH, Gao A, van Staden C, Ly J, Salon J, Xu A, et al. Evaluation of dynamic mass redistribution technology for pharmacological studies of recombinant and endogenously expressed g protein-coupled receptors. Drug Dev Technol. 2008;6:83–94. doi: 10.1089/adt.2007.126. [DOI] [PubMed] [Google Scholar]
- 71.Fang Y, Ferrie A, Fontaine N, Ki Yuen P. Optical biosensors for monitoring dynamic mass redistribution in living cells mediated by epidermal growth factor receptor activation. Conf Proc IEEE Eng Med Biol Soc. 2005;1:666–9. doi: 10.1109/IEMBS.2005.1616501. [DOI] [PubMed] [Google Scholar]
- 72.Fang Y, Ferrie AM. Optical biosensor differentiates signaling of endogenous PAR1 and PAR2 in A431 cells. BMC Cell Biol. 2007;8:24. doi: 10.1186/1471-2121-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang Y, Ferrie AM. Label-free optical biosensor for ligand-directed functional selectivity acting on beta(2) adrenoceptor in living cells. FEBS Lett. 2008;582:558–64. doi: 10.1016/j.febslet.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 74.Fang Y, Ferrie AM, Fontaine NH, Mauro J, Balakrishnan J. Resonant waveguide grating biosensor for living cell sensing. Biophys J. 2006;91:1925–40. doi: 10.1529/biophysj.105.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fang Y, Ferrie AM, Tran E. Resonant waveguide grating biosensor for whole-cell GPCR assays. Methods Mol Biol. 2009;552:239–52. doi: 10.1007/978-1-60327-317-6_17. [DOI] [PubMed] [Google Scholar]
- 76.Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007;55:314–22. doi: 10.1016/j.vascn.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 77.Verrier F, An S, Ferrie AM, Sun H, Kyoung M, Deng H, et al. GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat Chem Biol. 2011;7:909–15. doi: 10.1038/nchembio.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng H, Hu H, He M, Hu J, Niu W, Ferrie AM, et al. Discovery of 2-(4-methylfuran-2(5H)-ylidene)malononitrile and thieno[3,2-b]thiophene-2-carboxylic acid derivatives as G protein-coupled receptor 35 (GPR35) agonists. J Med Chem. 2011;54:7385–96. doi: 10.1021/jm200999f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goral V, Jin Y, Sun H, Ferrie AM, Wu Q, Fang Y. Agonist-directed desensitization of the beta2-adrenergic receptor. PLoS One. 2011;6:e19282. doi: 10.1371/journal.pone.0019282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferrie AM, Sun H, Fang Y. Label-free integrative pharmacology on-target of drugs at the beta(2)-adrenergic receptor. Sci Rep. 2011;1:33. doi: 10.1038/srep00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferrie AM, Wu Q, Fang Y. Resonant waveguide grating imager for live cell sensing. Appl Phys Lett. 2010;97:223704. doi: 10.1063/1.3522894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Codd EE, Mabus JR, Murray BS, Zhang SP, Flores CM. Dynamic mass redistribution as a means to measure and differentiate signaling via opioid and cannabinoid receptors. Assay Drug Dev Technol. 2011;9:362–72. doi: 10.1089/adt.2010.0347. [DOI] [PubMed] [Google Scholar]
- 83.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, et al. (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 84.Marino MJ, Williams DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci USA. 2003;100:13668–73. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008;74:1345–58. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012;33:372–84. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henriksen U, Fog J, Loechel F, Praestegaard M. Profiling of multiple signal pathway activities by multiplexing antibody and GFP-based translocation assays. Comb Chem High Throughput Screen. 2008;11:537–44. doi: 10.2174/138620708785204081. [DOI] [PubMed] [Google Scholar]
- 88.Salomon Y, Londos C, Rodbell M. A highly sensitive adenylate cyclase assay. Anal Biochem. 1974;58:541–8. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- 89.Agranoff BW, Murthy P, Seguin EB. Thrombin-induced phosphodiesteratic cleavage of phosphatidylinositol bisphosphate in human platelets. J Biol Chem. 1983;258:2076–8. [PubMed] [Google Scholar]
- 90.Birnbaumer L, Pohl SL, Michiel H, Krans MJ, Rodbell M. The actions of hormones on the adenyl cyclase system. Adv Biochem Psychopharmacol. 1970;3:185–208. [PubMed] [Google Scholar]
- 91.Gilman AG. Guanine nucleotide-binding regulatory proteins and dual control of adenylate cyclase. J Clin Invest. 1984;73:1–4. doi: 10.1172/JCI111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, et al. Betaarrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–7. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 93.Osmond RI, Crouch MF, Dupriez VJ. An emerging role for kinase screening in GPCR drug discovery. Curr Opin Mol Ther. 2010;12:305–15. [PubMed] [Google Scholar]
- 94.Werry TD, Christopoulos A, Sexton PM. Mechanisms of ERK1/2 regulation by seven-transmembrane-domain receptors. Curr Pharm Des. 2006;12:1683–702. doi: 10.2174/138161206776873725. [DOI] [PubMed] [Google Scholar]
- 95.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 96.Lefkowitz RJ, Inglese J, Koch WJ, Pitcher J, Attramadal H, Caron MG. G-protein-coupled receptors: regulatory role of receptor kinases and arrestin proteins. Cold Spring Harb Symp Quant Biol. 1992;57:127–33. doi: 10.1101/sqb.1992.057.01.016. [DOI] [PubMed] [Google Scholar]
- 97.Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–33. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- 98.Gilman AG. A protein binding assay for adenosine 3′:5′-cyclic monophosphate. Proc Natl Acad Sci USA. 1970;67:305–12. doi: 10.1073/pnas.67.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith C, Toohey N, Knight JA, Klein MT, Teitler M. Risperidone-induced inactivation and clozapine-induced reactivation of rat cortical astrocyte 5-hydroxytryptamine(7) receptors: evidence for in situ G protein-coupled receptor homodimer protomer cross-talk. Mol Pharmacol. 2011;79:318–25. doi: 10.1124/mol.110.069278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32:8532–44. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Klein M, Teitler M. Distribution of 5-ht(1E) receptors in the mammalian brain and cerebral vasculature: an immunohistochemical and pharmacological study. Br J Pharmacol. 2012;166:1290–302. doi: 10.1111/j.1476-5381.2012.01868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Naccarato WF, Ray RE, Wells WW. Biosynthesis of myo-inositol in rat mammary gland. Isolation and properties of the enzymes. Arch Biochem Biophys. 1974;164:194–201. doi: 10.1016/0003-9861(74)90022-8. [DOI] [PubMed] [Google Scholar]
- 103.Turek-Etienne TC, Small EC, Soh SC, Xin TA, Gaitonde PV, Barrabee EB, et al. Evaluation of fluorescent compound interference in 4 fluorescence polarization assays: 2 kinases, 1 protease, and 1 phosphatase. J Biomol Screen. 2003;8:176–84. doi: 10.1177/1087057103252304. [DOI] [PubMed] [Google Scholar]
- 104.Hemmilä I, Webb S. Time-resolved fluorometry: an overview of the labels and core technologies for drug screening applications. Drug Discov Today. 1997;2:373–81. [Google Scholar]
- 105.Hawes BE, van Biesen T, Koch WJ, Luttrell LM, Lefkowitz RJ. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J Biol Chem. 1995;270:17148–53. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- 106.Fukuhara S, Marinissen MJ, Chiariello M, Gutkind JS. Signaling from G protein-coupled receptors to ERK5/Big MAPK 1 involves Galpha q and Galpha 12/13 families of heterotrimeric G proteins. Evidence for the existence of a novel Ras AND Rho-independent pathway. J Biol Chem. 2000;275:21730–6. doi: 10.1074/jbc.M002410200. [DOI] [PubMed] [Google Scholar]
- 107.Kramer RM, Roberts EF, Hyslop PA, Utterback BG, Hui KY, Jakubowski JA. Differential activation of cytosolic phospholipase A2 (cPLA2) by thrombin and thrombin receptor agonist peptide in human platelets. Evidence for activation of cPLA2 independent of the mitogen-activated protein kinases ERK1/2. J Biol Chem. 1995;270:14816–23. doi: 10.1074/jbc.270.24.14816. [DOI] [PubMed] [Google Scholar]
- 108.Ullman EF, Kirakossian H, Switchenko AC, Ishkanian J, Ericson M, Wartchow CA, et al. Luminescent oxygen channeling assay (LOCI): sensitive, broadly applicable homogeneous immunoassay method. Clin Chem. 1996;42:1518–26. [PubMed] [Google Scholar]
- 109.Ullman EF, Kirakossian H, Singh S, Wu ZP, Irvin BR, Pease JS, et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc Natl Acad Sci USA. 1994;91:5426–30. doi: 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peppard J, Glickman F, He Y, Hu SI, Doughty J, Goldberg R. Development of a high-throughput screening assay for inhibitors of aggrecan cleavage using luminescent oxygen channeling (AlphaScreen) J Biomol Screen. 2003;8:149–56. doi: 10.1177/1087057103252308. [DOI] [PubMed] [Google Scholar]
- 111.Osmond RI, Sheehan A, Borowicz R, Barnett E, Harvey G, Turner C, et al. GPCR screening via ERK 1/2: a novel platform for screening G protein-coupled receptors. J Biomol Screen. 2005;10:730–7. doi: 10.1177/1087057105277968. [DOI] [PubMed] [Google Scholar]
- 112.Sava A, Formaggio E, Carignani C, Andreetta F, Bettini E, Griffante C. NMDA-induced ERK signalling is mediated by NR2B subunit in rat cortical neurons and switches from positive to negative depending on stage of development. Neuropharmacology. 2012;62:925–32. doi: 10.1016/j.neuropharm.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 113.Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–8. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Natl Acad Sci USA. 1987;84:8879–82. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oakley RH, Hudson CC, Cruickshank RD, Meyers DM, Payne RE, Jr, Rhem SM, et al. The cellular distribution of fluorescently labeled arrestins provides a robust, sensitive, and universal assay for screening G protein-coupled receptors. Assay Drug Dev Technol. 2002;1:21–30. doi: 10.1089/154065802761001275. [DOI] [PubMed] [Google Scholar]
- 116.Namkung Y, Dipace C, Javitch JA, Sibley DR. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J Biol Chem. 2009;284:15038–51. doi: 10.1074/jbc.M900388200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993;268:16879–82. [PubMed] [Google Scholar]
- 118.Hanson BJ, Wetter J, Bercher MR, Kopp L, Fuerstenau-Sharp M, Vedvik KL, et al. A homogeneous fluorescent live-cell assay for measuring 7-transmembrane receptor activity and agonist functional selectivity through beta-arrestin recruitment. J Biomol Screen. 2009;14:798–810. doi: 10.1177/1087057109335260. [DOI] [PubMed] [Google Scholar]
- 119.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–73. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 120.Butini S, Gemma S, Campiani G, Franceschini S, Trotta F, Borriello M, et al. Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: design, synthesis, and effects on behavior. J Med Chem. 2009;52:151–69. doi: 10.1021/jm800689g. [DOI] [PubMed] [Google Scholar]
- 121.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–51. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saura CA, Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev Neurosci. 2011;22:153–69. doi: 10.1515/RNS.2011.018. [DOI] [PubMed] [Google Scholar]
- 123.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–45. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 124.Kenakin TP. Pharmacologic analysis of drug-receptor interaction. 3rd. Philadelphia: Lippincott-Raven Publishers; 1997. [Google Scholar]
- 125.Limbird LE. Cell surface receptors: a short course on theory & methods. 3rd. New York: Springer Science+ Business Media; 2005. [Google Scholar]
- 126.McKinney M, Raddatz R. Practical aspects of radioligand binding. Current Protocols in Pharmacol. 2006;33:1.3.1–1.3.42. doi: 10.1002/0471141755.ph0103s33. [DOI] [PubMed] [Google Scholar]
- 127.Kenakin T. The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol Rev. 1996;48:413–63. [PubMed] [Google Scholar]
- 128.Bernat V, Heinrich MR, Baumeister P, Buschauer A, Tschammer N. Synthesis and application of the first radioligand targeting the allosteric binding pocket of chemokine receptor CXCR3. ChemMedChem. 2012;7:1481–9. doi: 10.1002/cmdc.201200184. [DOI] [PubMed] [Google Scholar]
- 129.Cosford NDP, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, et al. [H-3]-methoxymethyl-MTEP and [H-3]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg Med Chem Lett. 2003;13:351–4. doi: 10.1016/s0960-894x(02)00997-6. [DOI] [PubMed] [Google Scholar]
- 130.Heitman LH, Oosterom J, Bonger KM, Timmers CM, Wiegerinck PHG, IJzerman AP. [3H]Org 43553, the first low-molecular-weight agonistic and allosteric radioligand for the human luteinizing hormone receptor. Mol Pharmacol. 2008;73:518–24. doi: 10.1124/mol.107.039875. [DOI] [PubMed] [Google Scholar]
- 131.Shin N, Solomon K, Zhou N, Wang KH, Garlapati V, Thomas B, et al. Identification and characterization of INCB9471, an allosteric noncompetitive small-molecule antagonist of C-C chemokine receptor 5 with potent inhibitory activity against monocyte migration and HIV-1 infection. J Pharmacol Exp Ther. 2011;338:228–39. doi: 10.1124/jpet.111.179531. [DOI] [PubMed] [Google Scholar]
- 132.Tränkle C, Mies-Klomfass E, Cid MHB, Holzgrabe U, Mohr K. Identification of a [3H]ligand for the common allosteric site of muscarinic acetylcholine M2 receptors. Mol Pharmacol. 1998;54:139–45. doi: 10.1124/mol.54.1.139. [DOI] [PubMed] [Google Scholar]
- 133.Jeffrey Conn P, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang JQ, Tueckmantel W, Zhu A, Pellegrino D, Brownell AL. Synthesis and preliminary biological evaluation of 3-[(18)F]fluoro-5-(2-pyridinylethynyl)benzonitrile as a PET radiotracer for imaging metabotropic glutamate receptor subtype 5. Synapse. 2007;61:951–61. doi: 10.1002/syn.20445. [DOI] [PubMed] [Google Scholar]
- 135.Avlani V, May LT, Sexton PM, Christopoulos A. Application of a kinetic model to the apparently complex behavior of negative and positive allosteric modulators of muscarinic acetylcholine receptors. J Pharmacol Exp Ther. 2004;308:1062–72. doi: 10.1124/jpet.103.059840. [DOI] [PubMed] [Google Scholar]
- 136.Leach K, Sexton PM, Christopoulos A. Quantification of allosteric interactions at G protein-coupled receptors using radioligand binding assays. Curr Protoc Pharmacol. 2011 doi: 10.1002/0471141755.ph0122s52. Chapter 1 Unit 1.22. [DOI] [PubMed] [Google Scholar]
- 137.de Kruijf P, Lim HD, Roumen L, Renjaän VA, Zhao J, Webb ML, et al. Identification of a novel allosteric binding site in the CXCR2 chemokine receptor. Mol Pharmacol. 2011;80:1108–18. doi: 10.1124/mol.111.073825. [DOI] [PubMed] [Google Scholar]
- 138.Gregory KJ, Dong EN, Meiler J, Conn PJ. Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology. 2011;60:66–81. doi: 10.1016/j.neuropharm.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, et al. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem. 2008;283:29312–21. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhong H, Hansen KB, Boyle NJ, Han K, Muske G, Huang X, et al. An allosteric binding site at the human serotonin transporter mediates the inhibition of escitalopram by R-citalopram: kinetic binding studies with the ALI/VFL–SI/TT mutant. Neurosci Lett. 2009;462:207–12. doi: 10.1016/j.neulet.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 141.Leach K, Simms J, Sexton PM, Christopoulos A. Structure-function studies of muscarinic acetylcholine receptors. Handb Exp Pharmacol. 2012;208:29–48. doi: 10.1007/978-3-642-23274-9_2. [DOI] [PubMed] [Google Scholar]
- 142.Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–74. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koole C, Wootten D, Simms J, Savage EE, Miller LJ, Christopoulos A, et al. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) differentially regulates orthosteric but not allosteric agonist binding and function. J Biol Chem. 2012;287:3659–73. doi: 10.1074/jbc.M111.309369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gregory KJ, Sexton PM, Christopoulos A. Overview of receptor allosterism. Curr Protoc Pharmacol. 2010 doi: 10.1002/0471141755.ph0121s51. Chapter 1, Unit 1.21. [DOI] [PubMed] [Google Scholar]
- 145.Langmead CJ. Determining allosteric modulator mechanism of action: integration of radioligand binding and functional assay data. Methods Mol Biol. 2011;746:195–209. doi: 10.1007/978-1-61779-126-0_10. [DOI] [PubMed] [Google Scholar]
- 146.Nawaratne V, Leach K, Felder CC, Sexton PM, Christopoulos A. Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: identification of ligand-specific and global activation mechanisms. J Biol Chem. 2010;285:19012–21. doi: 10.1074/jbc.M110.125096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Christopoulos A, Lanzafame A, Ziegler A, Mitchelson F. Kinetic studies of co-operativity at atrial muscarinic M2 receptors with an “infinite dilution” procedure. Biochem Pharmacol. 1997;53:795–800. doi: 10.1016/s0006-2952(96)00814-3. [DOI] [PubMed] [Google Scholar]
- 148.May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine-A3 receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J. 2011;25:3465–76. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gomes I, IJzerman AP, Ye K, Maillet EL, Devi LA. G protein-coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol Pharmacol. 2011;79:1044–52. doi: 10.1124/mol.110.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sephton SM, Dennler P, Leutwiler DS, Mu L, Schibli R, Kramer SD, et al. Development of [(18)F]-PSS223 as a PET tracer for imaging of metabotropic glutamate receptor subtype 5 (mGluR5) Chimia (Aarau) 2012;66:201–4. doi: 10.2533/chimia.2012.201. [DOI] [PubMed] [Google Scholar]
- 151.Wanger-Baumann CA, Mu L, Honer M, Belli S, Alf MF, Schubiger PA, et al. In vitro and in vivo evaluation of [18F]-FDEGPECO as a PET tracer for imaging the metabotropic glutamate receptor subtype 5 (mGluR5) Neuroimage. 2011;56:984–91. doi: 10.1016/j.neuroimage.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 152.Baumann CA, Mu L, Wertli N, Kramer SD, Honer M, Schubiger PA, et al. Syntheses and pharmacological characterization of novel thiazole derivatives as potential mGluR5 PET ligands. Bioorg Med Chem. 2010;18:6044–54. doi: 10.1016/j.bmc.2010.06.070. [DOI] [PubMed] [Google Scholar]
- 153.Baumann CA, Mu L, Johannsen S, Honer M, Schubiger PA, Ametamey SM. Structure-activity relationships of fluorinated (E)-3-((6-methylpyridin-2-yl)ethynyl) cyclohex-2-enone-O-methyloxime (ABP688) derivatives and the discovery of a high affinity analogue as a potential candidate for imaging metabotropic glutamate recepors subtype 5 (mGluR5) with positron emission tomography (PET) J Med Chem. 2010;53:4009–17. doi: 10.1021/jm901850k. [DOI] [PubMed] [Google Scholar]
- 154.Ametamey SM, Treyer V, Streffer J, Wyss MT, Schmidt M, Blagoev M, et al. Human PET studies of metabotropic glutamate receptor subtype 5 with 11C-ABP688. J Nucl Med. 2007;48:247–52. [PubMed] [Google Scholar]
- 155.Treyer V, Streffer J, Ametamey SM, Bettio A, Blauenstein P, Schmidt M, et al. Radiation dosimetry and biodistribution of 11C-ABP688 measured in healthy volunteers. Eur J Nucl Med Mol Imaging. 2008;35:766–70. doi: 10.1007/s00259-007-0638-4. [DOI] [PubMed] [Google Scholar]
- 156.Treyer V, Streffer J, Wyss MT, Bettio A, Ametamey SM, Fischer U, et al. Evaluation of the metabotropic glutamate receptor subtype 5 using PET and 11C-ABP688: assessment of methods. J Nucl Med. 2007;48:1207–15. doi: 10.2967/jnumed.107.039578. [DOI] [PubMed] [Google Scholar]
- 157.Nordquist RE, Steckler T, Wettstein JG, Mackie C, Spooren W. Metabotropic glutamate receptor modulation, translational methods, and biomarkers: relationships with anxiety. Psychopharmacology (Berl) 2008;199:389–402. doi: 10.1007/s00213-008-1096-9. [DOI] [PubMed] [Google Scholar]
- 158.Hostetler ED, Eng W, Joshi AD, Sanabria-Bohorquez S, Kawamoto H, Ito S, et al. Synthesis, characterization, and monkey PET studies of [(1)(8)F]MK-1312, a PET tracer for quantification of mGluR1 receptor occupancy by MK-5435. Synapse. 2011;65:125–35. doi: 10.1002/syn.20826. [DOI] [PubMed] [Google Scholar]