

Abstract
The complex life cycle of Mycobacterium tuberculosis requires diverse energy mobilization and utilization strategies facilitated by a battery of lipid metabolism enzymes. Among lipid metabolism enzymes, the Lip family of mycobacterial serine hydrolases is essential to lipid scavenging, metabolic cycles, and reactivation from dormancy. Based on the homologous rescue strategy for mycobacterial drug targets, we have characterized the three-dimensional structure of full length LipW from Mycobacterium marinum, the first structure of a catalytically active Lip family member. LipW contains a deep, expansive substrate-binding pocket with only a narrow, restrictive active site, suggesting tight substrate selectivity for short, unbranched esters. Structural alignment reinforced this strict substrate selectivity of LipW, as the binding pocket of LipW aligned most closely with the bacterial acyl esterase superfamily. Detailed kinetic analysis of two different LipW homologues confirmed this strict substrate selectivity, as each homologue selected for unbranched propionyl ester substrates, irrespective of the alcohol portion of the ester. Using comprehensive substitutional analysis across the binding pocket, the strict substrate selectivity of LipW for propionyl esters was assigned to a narrow funnel in the acyl-binding pocket capped by a key hydrophobic valine residue. The polar, negatively charged alcohol-binding pocket also contributed to substrate orientation and stabilization of rotameric states in the catalytic serine. Together the structural, enzymatic, and substitutional analysis of LipW provide a connection between the structure and metabolic properties of a Lip family hydrolase that refines its biological function in active and dormant tuberculosis infection.
Keywords: Hydrolases, Mycobacterium tuberculosis, Fluorogenic enzyme substrates, Substrate specificity, tuberculosis
Mycobacterium tuberculosis (Mtb) maintains a complex interplay with its host through diverse nutrient cycles.1 In this relationship, Mtb scavenges for host cell fatty acids, cholesterol, amino acids, and other carbon sources.1–3 Mtb can subsist on a single carbon source in the laboratory, but requires multiple nutrient sources during active host infection.1;4;5 During infection, Mtb also shifts nutrient sources to adapt to changing phases of infection and sites of growth.4 This interplay between nutrient cycles and TB infection is exemplified in the lipid filled granulomas that demarcate a TB infection.4;6;7 In these granulomas, Mtb accumulates large congregations of lipid inclusions and foamy macrophages that facilitate the lipid degradation and nutrient scavenging pathways required to sustain a long-term Mtb infection.1;4;6;7
Among enzyme superfamilies identified as upregulated under nutrient limitation and diverse metabolic growth conditions are serine hydrolases.8–10 A diverse enzyme superfamily with significant genetic overrepresentation in the Mtb genome, serine hydrolases catalyze a range of hydrolysis reactions essential to the life cycle of Mtb.11;12 Expression of serine hydrolases also cycle with nutrient starvation and triacylglyerol (TAG) utilization and are colocalized with lipid inclusions and foamy macrophages.7–9;13–15 Although the ability of Mtb to shift carbon sources makes targeting a particular nutrient pathway unlikely to completely inhibit growth, nonselective inhibitors of serine hydrolases downregulate TAG utilization and impede the reactivation of dormant Mtb infections.1;4;16–18
The Lip family of serine hydrolases from Mtb holds special biological importance with validated roles in TAG degradation, immune recognition, and growth and survival in dormant infection.17;19;20 Originally assayed for roles in TAG degradation, only a single Lip family member, LipY, showed significant intracellular and extracellular TAG activity. Instead, a large subsection of the Lip family members falls into the bacterial hormone sensitive lipase (HSL) superfamily with substrate specificity for esters of varying carbon chain lengths and branch patterns.17;21;22 Many Lip family members are, however, likely still connected to metabolism and energy utilization in Mtb, as 11 Lip genes are located adjacent or proximal to triacylglycerol synthetase (tgs) genes in the Mtb genome.7
Across Lip family enzymes, only LipJ has been structurally characterized, but LipJ does not contain a catalytic triad or measurable hydrolase activity.23 LipJ is an unusual lipase homologue, as the function and properties of LipJ revolve around its guanylate cyclase domain instead of its α/β hydrolase protein fold.23 A small number of three-dimensional structures for additional serine hydrolases across mycobacterial species have been reported, but these enzymes from metabolic and cutinase hydrolase families have only limited homology to Lip family members.24;25 Structural coverage of other protein subclasses across Mycobacteria has increased significantly due to intra-genus homologue rescue strategies with 68 structures of mycobacterial drug targets now submitted by a single structural genomics initiative.26
Herein, we report the three-dimensional structure of LipW, a Lip family member, with direct connections to nutrient recovery and dormant TB infection. LipW was structurally aligned to similar members of the bacterial HSL superfamily and to acyl ester hydrolases to assign its broad alcohol and narrow acyl binding pockets. The substrate specificity of two LipW homologues was then determined against diverse libraries of hydrolase substrates. Mutational analysis across the binding pocket and active site was used to identify the structural components responsible for the tight substrate selectivity of LipW. Together the combined structural, biochemical, and enzymatic analysis provided insight into the biological substrates of LipW, its role in tuberculosis infection, and its potential for therapeutic inhibition.
EXPERIMENTAL PROCEDURES
Cloning, expression, and purification
The 313-residue Mycobacterium marinum LipW gene (UniProt accession code B2HLX2; SSGCID target ID MymaA.00277.c) was amplified from genomic DNA and cloned into the pAVA0421 expression vector encoding an N-terminal histidine affinity tag followed by the human rhinovirus 3C protease cleavage sequence (the entire tag sequence is MAHHHHHHMGTLEAQTQGPGS-ORF). The clone (SSGCID construct ID MymaA.00277.c.A1) was transformed into E. coli BL21 (DE3) Rosetta cells, and a starter culture was grown in LB broth with ampicillin (50 μg/ml), carbenicillin (50 μg/ml), and chloramphenicol (34 μg/ml) for ~18 hours at 37 °C. The protein was expressed in 2L of ZYP-5052 auto-induction media in a LEX bioreactor. After 24 hours at 25 °C the temperature was reduced to 15 °C for another 60 hours. The sample was centrifuged at 4000 × g for 20 minutes at 4 °C. Cell paste was flash frozen and stored at −80 °C.
The cells were re-suspended 6:1 v/w in 20 mM HEPES pH 7.4, 300 mM NaCl, 5% v/v glycerol, 0.5% w/v CHAPS, 30 mM imidazole, 10 mM MgCl2, 3 mM β-mercaptoethanol, protease inhibitor cocktail tablets (Roche), and 0.05 mg/mL lysozyme at 4 °C and disrupted on ice for 15 minutes with a Branson Digital 450D Sonifier (70% amplitude, with alternating cycles of five seconds of pulse-on and ten seconds of pulse-off). The cell debris was incubated with 20 μl of Benzonase nuclease at room temperature for 40 minutes. The lysate was clarified by centrifugation with a Sorvall RC5 at 10,000 RPM for 60 min at 4°C in a F14Y Rotor (Thermo Fisher). Clarified solution was syringe filtered through a 0.45 μm cellulose acetate filter (Corning Life Sciences, Lowell, MA). Tagged protein was purified by IMAC using a HisTrap FF 5 ml column (GE Biosciences, Piscataway, NJ) and an ÄKTA Purifier FPLC system (GE Biosciences) equilibrated with binding buffer (20 mM HEPES, pH 7.0, 300 mM NaCl, 5% v/v glycerol, 30 mM imidazole, 1 mM TCEP), and eluted with binding buffer supplemented with 500 mM imidazole. The N-terminal affinity tag was removed with 3C protease. 27,61,35 (61) 61,27 The sample was dialyzed overnight at 4°C against cleavage buffer (20 mM HEPES pH 7.6, 500 mM NaCl, 5% v/v glycerol, 1 mM TCEP) and purified by subtractive nickel affinity chromatography in binding buffer (20 mM HEPES, pH 7.0, 300 mM NaCl, 5% v/v glycerol, 30 mM imidazole, 1 mM TCEP). The protein was concentrated and purified by size exclusion chromatography (SEC) using a Superdex 75 26/60 column (GE Biosciences) equilibrated in SEC buffer (20 mM HEPES, pH 7.0, 300 mM NaCl, 5% v/v glycerol and 1 mM TCEP). After purity analysis by SDS-PAGE, the pooled fractions were concentrated to 45.1 mg/mL and flash-frozen in liquid nitrogen. Samples were stored at −80°C until used in crystallization experiments. LipW from M. smegmatis (MsLipW, SSGCID construct ID MysmA.00277.c.A1) was cloned, expressed, and purified in the same way, and concentrated to 16.4 mg/ml. Both MmLipW (MymaA.00277.c.A1.PS00815) and MsLipW (MysmA.00277.c.A1.PS00565) proteins and plasmids are available from SSGCID.
MmLipW (SSGCID: MymaA.00277.c.A1.PS00815) and MsLipW (SSGCID: MysmA.00277.c.A1.PS00565) used for substrate specificity measurements were diluted to ~ 2 mg/mL in PBS, and their final concentrations confirmed by measuring the absorbance at 280 nm. Absorbance readings were converted to molarity units with an extinction coefficient of ε280 = 26,930 M−1 cm−1 for MmLipW and of ε280 = 33,460 M−1 cm−1 for MsLipW calculated using ExPASy (http://web.expasy.org/protparam).28
Crystallization, data collection, and structure determination
Sparse matrix crystallization screens were set up for both proteins in 96-well sitting drop vapor diffusion format at 16 °C with an equal volume of protein (0.4 μL) and precipitant against reservoir (80 μL). Only poorly diffracting samples were obtained for MsLipW. For MmLipW well diffracting crystals appeared after a month in the PACT screen condition D8 (20% w/v PEG 6000, 0.1 M Tris pH 8.0, 0.2 M ammonium chloride). A crystal was cryo-protected with reservoir supplemented with 25% v/v ethylene glycol and vitrified in liquid nitrogen. A 1.75 Å resolution data set was collected at 100 K using a Rigaku FR-E+ X-ray generator with VariMax optics and a Saturn 944+ CCD detector (Table 1). The resolution limits were in large part defined by the limits of the in house detector system, and scaling to higher resolution resulted in low completeness in the highest resolution shell. The data were reduced with XDS/XSCALE.29
Table 1.
Crystallographic statistics for LipW
| Overall | Highest shell | |
|---|---|---|
| Wavelength | 1.5418 Å | |
| Space Group | P21212 | |
| Unit Cell |
a = 73.27 Å, b= 86.99 Å, c = 46.19 Å α = β = γ = 90° |
|
| Solvent content | 43.5 % | |
| Vm | 2.18 Å3/Da | |
| Resolution | 50–1.75 Å | 1.79–1.75 Å |
| I/σ | 21.76 | 9.13 |
| Completeness | 98.9% | 95.0% |
| Rmerge | 0.041 | 0.085 |
| CC ½ | 99.8 | 98.5 |
| Multiplicity | 3.2 | 1.9 |
| Reflections | 30,171 | 2,100 |
| Mosaicity | 0.5 | |
| Refinement | ||
| Resolution | 50–1.75 Å | 1.79–1.75 Å |
| Reflections | 30,076 | 1,967 |
| R | 0.147 | 0.165 |
| Rfree | 0.188 | 0.209 |
| Protein Atoms | 2,336 | |
| Waters | 419 | |
| B-factors Mean | 11.4 Å2 | |
| Wilson B-factors | 17.9 Å2 | |
| Validation | ||
| Ramachandran favored | 95.9% | |
| Ramachandran outliers | 0.0% | |
| Rotamer outliers | 0.0% | |
| Clash score | 0.86 | |
| Molprobity score | 1.05 |
The structure of M. marinum LipW was solved by molecular replacement using the Rhodococcus sp. heroin esterase crystal structure 30 trimmed with Chainsaw 31 as a search model in Phaser 32 from the CCP4 suite.33 This search model contains 37% sequence identity over 65% of the length of LipW. The final model was obtained after automated building in ARP/wARP,34 numerous iterative rounds of refinement in REFMAC,35 and manual re-building in COOT.36 The structure was assessed and corrected for geometry and fitness using Molprobity.37 The atomic coordinates and structure factors are available in the Protein Data Bank under accession code 3QH4.
Overexpression and purification of MmLipW and MmLipW variants for kinetic assays
A bacterial expression plasmid (AVA0421; a derivative of pET14b) containing the LipW gene from Mycobacterium marinum strain ATCC BAA-535/M (Genbank: NC_010612; UniProt: B2HLX2; protein name MmLipW) was obtained from BEI Resources (NR-27756). This bacterial plasmid (AVA0421- MmLipW) was transformed into E. coli BL21 (DE3) RIPL cells (Agilent, La Jolla, CA). A saturated overnight culture of E. coli BL21 (DE3) RIPL (AVA0421- MmLipW) in LB media containing ampicillin (200 μg/mL) and chloramphenicol (30 μg/mL) was used to inoculate LB-media (250 mL) containing ampicillin (200 μg/mL) and chloramphenicol (30 μg/mL) and the bacterial culture was grown with constant shaking (225 rpm) at 37 °C. When the OD600 reached 1.0 – 1.2, the temperature of the culture was decreased to 16 °C and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1.0 mM. Protein induction proceeded overnight (~16–20 hours) at 16 °C. Bacterial cultures were collected by centrifugation at 6,000 × g for 10 min at 4 °C. The bacterial cell pellet was resuspended in PBS (3 mL) and stored at −20 °C. To disrupt the bacterial cell wall, lysozyme (50 mg; Sigma-Aldrich) and 10× Bug Buster solution (700 μL; EMD Millipore) were added to the thawed cell pellet; cell lysis proceeded on circular rotator for 2–3 h at 4 °C. To remove insoluble cell material, lysed cells were centrifuged at 16,000 × g for 10 min at 4 °C. Ni-NTA agarose (600 μL; Qiagen, Valencia, CA) was added to the soluble fraction and allowed to incubate at 4 °C for 15 – 20 min. The resin was washed three times with PBS containing increasing concentrations of ice-cold imidazole (30 mL each of PBS containing 10 mM imidazole, 25 mM imidazole, or 50 mM imidazole) and recollected by centrifugation at 2000 × g for 2 min at 4 °C. MmLipW was eluted in PBS containing 250 mM imidazole (1.0 mL) and dialyzed (10K MWCO; Pierce, Rockford, IL) against PBS overnight at 4 °C with constant stirring.
The purity of MmLipW was confirmed by SDS–PAGE on a 4–20% gradient gel (LifeTechnologies) visualized with colloidal Coomassie brilliant blue; the purity was shown to be greater than 95% (Supplemental Figure 1). The concentration of MmLipW was determined by measuring the absorbance at 280 nm and converted to molarity units with an extinction coefficient of ε280 = 26,930 M−1 cm−1 calculated using ExPASy (http://web.expasy.org/protparam).28
Site-directed mutagenesis and purification
Variants of LipW were produced by QuikChange II site-directed mutagenesis of the AVA0421-MmLipW template DNA using the manufacturer’s suggested procedure (Agilent, Santa Clara, CA) and the primers (Integrated DNA Technologies, Coralville, IA) outlined in Supplementary Table 5. Proper mutations in the MmLipW DNA sequence were confirmed by DNA sequencing (Genewiz, South Plainfield, NJ) using T7 and/or T7-terminal sequencing primers. Plasmids coding for MmLipW variants were transformed into E. coli BL21 (DE3) RIPL cells and variants of MmLipW were overexpressed and purified using the same procedure as for wild-type MmLipW.
Kinetic measurements with fluorogenic hydrolase substrates
The enzymatic activity of MmLipW MsLipW and variants of MmLipW was measured using fluorogenic hydrolase substrates (Figure 2) in a 96-well microplate assay.38–40 Fluorogenic substrates were synthesized as previously described 38;41;42. Fluorogenic substrates were prepared as stock solutions in DMSO (10 mM) and were diluted into PBS containing acetylated BSA (Sigma; 0.1 mg/mL) to starting concentrations between 100–1000 μM, depending on the Km value of MmLipW for the substrate. The majority of the substrates (substrates 1–18) had the same starting concentration (100 μM) with fluorine containing substrates 19–21 requiring higher starting concentrations (1000 μM). Eight serial dilutions (1:2; 60 μL into 180 μL total volume) of each substrate were made using PBS–BSA. Fluorogenic substrate dilutions (95 μL) were then transferred to a black 96-well microplate (Corning, Lowell, MA). Enzyme-catalyzed hydrolysis was initiated by addition of MmLipW, MsLipW, or variants of MmLipW (5 μL of 150 μg/mL; final concentration all proteins = 7.5 μg/mL; MmLipW 227 nM final concentration; MmLipW 225 nM final concentration) to diluted fluorogenic substrates in the black 96-well microplate (100 μL final volume) and the fluorescence change (λex = 485 nm, λem = 528 nm) was measured for 7.5 min at 25 °C, collecting data every 50 sec, on a Biotek Synergy H1 multimode plate reader (Biotek Instruments; Winooski, VT). The fluorescence change was converted to molar concentrations using a fluorescein standard curve (300 nM–2.3 nM), whose fluorescence was measured simultaneously. The initial rates of the enzyme-catalyzed reactions were measured in triplicate and plotted versus fluorogenic substrate concentration. The saturation enzyme kinetic traces were fitted to a standard Michaelis–Menten equation using GraphPad Prism 5.0 (Graphpad Software, La Jolla, CA) and values for kcat, KM and kcat/KM calculated.
Figure 2.
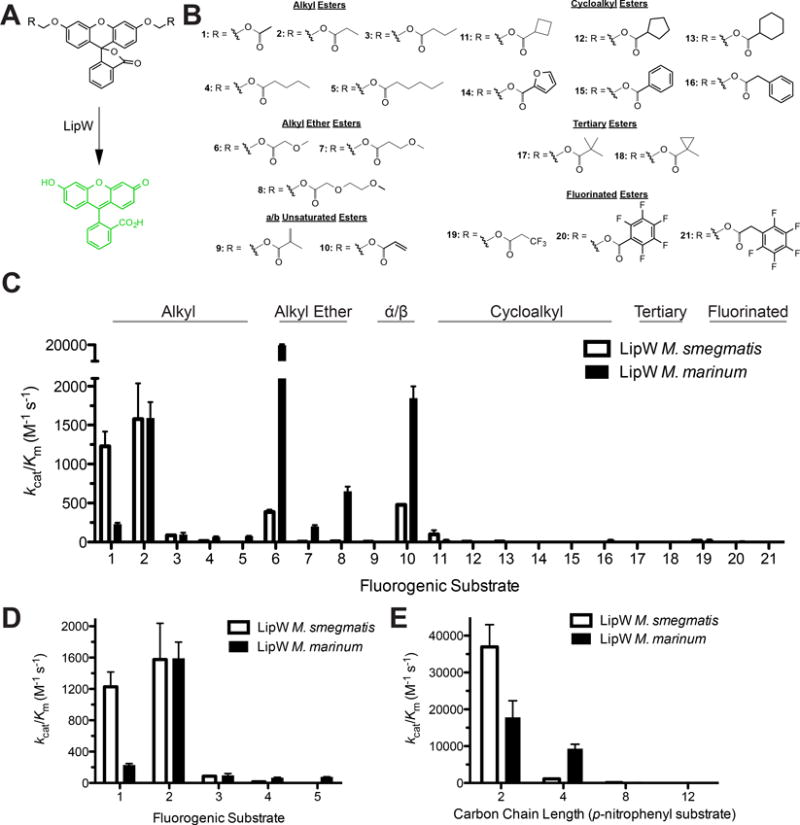
Comparative kinetics of LipW homologues using a fluorogenic substrate library. A) Activation of fluorogenic substrates by LipW. Hydrolysis of the ester bond on the diacyloxymethyl ether fluorescein substrates by LipW converts the fluorescein core from the nonfluorescent lactone form to the highly fluorescent quinoid form. The rate of fluorophore activation is measured at a range of substrate concentrations to determine the kinetic constants for fluorophore activation. B) Each of the substrates is composed of diacyloxymethyl ether fluorescein with varying R-groups. The differing R-groups have been organized into classes based on chemical functionality. All of the substrates were synthesized as described previously.38;42;51 C) Global comparison of the catalytic specificity (kcat/KM) of LipW against each of the 21 substrates (structures and numbering given in Fig. 3B) with ester classes labeled. D) Substrate specificity of LipW against alkyl ester substrates, illustrating the substrate selectivity based on the carbon chain length. E) Catalytic efficiency of LipW against p-nitrophenyl substrates (p-nitrophenyl acetate (2), p-nitrophenyl butyrate (4), p-nitrophenyl octanoate (8), and p-nitrophenyl laurate (12)). Catalytic efficiency values (kcat/Km) are given ± SE. Detailed kinetic results for each substrate are provided in Supplemental Tables 1 and 2 with representative kinetic plots shown in Supplemental Figure 2.
Kinetic measurements with p-nitrophenyl substrate
The enzymatic activity of MmLipW and MsLipW were measured against p-nitrophenyl acetate, p-nitrophenyl butyrate, p-nitrophenyl octanoate, and p-nitrophenyl laurate (Sigma – Aldrich) using a 96-well microplate assay (Figure 2).38 All four substrates (p-nitrophenyl acetate (2 M), p-nitrophenyl butyrate (2 M), p-nitrophenyl octanoate (200 mM), and p-nitrophenyl laurate (200 mM)) were prepared as stock solutions in acetonitrile and diluted into PBS containing acetylated BSA (PBS–BSA; 0.1 mg/mL). The starting concentration for p-nitrophenyl acetate and p-nitrophenyl butyrate was 20 mM and for p-nitrophenyl octanoate and p-nitrophenyl laurate was 2 mM. Eight serial dilutions (1:1; 110 μL into 220 μL total volume; 20 mM–156 μM final concentrations for acetate and butyrate and 2 mM–15.6 μM for octanoate and laurate) were made using PBS–BSA containing 1% acetonitrile. Substrate dilutions (95 μL) were transferred to a clear 96-well microplate and MmLipW and MsLipW (5 μL of 150 μg/mL for acetate and butyrate and 5 μL of 300 μg/mL for octanoate and laurate; final concentration MmLipW and MsLipW = 7.50 μg/mL for acetate and butyrate and = 15.0 μg/mL for octanoate and laurate) was added to start the reaction. The absorbance change at 412 nm was measured on a Biotek Synergy H1 multimode plate reader (Biotek Instruments; Winooski, VT) for 4 min at 25 °C. The change in absorbance was converted to molar concentrations using the extinction coefficient of p-nitrophenol (Δε412=1.034 mM−1 cm−1).43 The initial rates of the reactions were measured in triplicate and plotted versus substrate concentration. The saturation enzyme kinetic traces were fitted to a standard Michaelis–Menten equation using GraphPad Prism 5.0 (Graphpad Software, La Jolla, CA) and values for kcat, KM and kcat/KM calculated.
Kinetic measurement with a cephalosporin derivative
The enzymatic activity of MmLipW toward 7-amino cephalosporonic acid (7-ACA; Sigma-Aldrich, St. Louis, MO) was determined using a previously published method.38;44 Briefly, the deacetylation of 7-ACA by MmLipW was determined by measuring the change in pH over time using the pH indicator bromothymol blue (BTB; Sigma-Aldrich, St. Louis, MO). Serial dilutions (1:2) of 7-ACA (300 mM–0.14 mM final concentrations) were made in 5 mM phosphate buffer pH 7.3 containing 0.01% BTB and the serial dilutions transferred to a 96-well clear microplate (95 μL). To start the reaction, wild-type MmLipW (5 μL of a 1 mg/mL solution) was added to a final concentration of 0.05 mg/mL. Plates were preshaken for 30 sec to insure complete mixing and the change in absorbance at 630 nm was measured for 5 min at 25 °C on a Biotek Synergy H1 multimode plate reader. The initial velocity of the reaction was calculated using the following equation:
where CB is the molar concentration of buffer (0.005 M), CIn is the molar concentration of indicator (16 nM), Δε is the difference in the extinction coefficient between protonated and deprotonated BTB (Δε630= 15,700 M−1 cm−1), and l is the pathlength (1 cm). Values for kcat, KM, and kcat/KM were determined by fitting the hyperbolic curve of 7-ACA substrate concentration versus initial velocity to a standard Michaelis–Menten equation using GraphPad Prism 5.0.
Thermal stability measurement
The thermal stability of MmLipW and MmLipW variants was determined using differential scanning fluorimetry.45 MmLipW protein (0.30 mg/mL) was diluted in triplicate in PBS containing a 1:250 dilution of 5000× Sypro Orange dye (Invitrogen, Carlsbad, CA). The samples were heated from 15 °C to 90 °C at 1.0 °C/min in a thermocycler (Bio-rad C1000 Thermocycler with CFX96 Real-time System, Hercules, CA) and the change in Sypro Orange fluorescence followed over time (λex = 450–490 nm, λem = 610–650 nm). The midpoint denaturation temperature (Tm) was determined by plotting the first derivative of fluorescence versus temperature and finding the temperature at the midpoint of the transition.
RESULTS
Structural organization of LipW
LipW displays an overall fold similar to other α/β hydrolases consisting of a central parallel β-sheet surrounded by α-helices (Figure 1A). The catalytic triad common to all α/β hydrolases, consisting of residues Ser162, Asp254, and His284 in LipW, lies at the bottom of a deep cleft in the active site. The catalytic serine nucleophile in LipW is observed in two different rotameric states, as has been observed in other bacterial serine hydrolase structures.46;47 In one rotameric state, the serine nucleophile is pointing toward the acyl-binding pocket and oxyanion hole, is hydrogen bonded to the general base histidine (His284), and is oriented in the direction of the expected hydrolysis reaction. Although the active site cavity is well resolved given the resolution limits, the oxyanion hole is devoid of an ordered water molecule, which would be expected close to Ser162 since this residue only H-bonds to His284. In the second rotameric state, the serine nucleophile shifts 60° toward the alcohol binding pocket and forms hydrogen bonds to the side chains of His284 and Ser161. Similar rotameric diversity in serine hydrolase nucleophiles has been attributed to specific features of the serine hydrolase catalytic mechanism, including facilitating product release in the breakdown of the acyl-enzyme intermediate and downregulating the reverse acylation reaction.46;47 Together hydrogen-bonded residues within the alcohol-binding pocket of LipW may serve dual roles in selecting and orienting substrates for catalysis and in stabilizing this secondary rotameric state for the catalytic serine.
Figure 1.
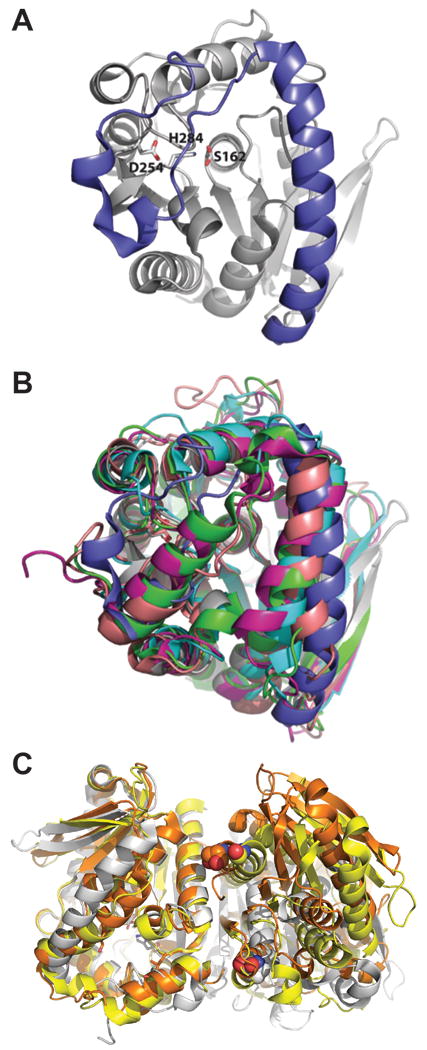
Structure of LipW. A) 1.75 Å resolution crystal structure of the serine hydrolase LipW from Mycobacterium marinum. The core α/β hydrolase domain is shown in gray ribbons with the catalytic triad of Ser162, Asp254, and His284 shown in sticks. Ser162 has a dual rotamer conformation. The cap domain is shown in blue ribbons. B) Overlay of the crystal structure of MmLipW (colored in gray/blue) in comparison with related serine hydrolases Est1 shown in cyan (PDB ID 2C7B),50 Est8 shown in magenta (PDB ID 4YPV), ThaEst2349 shown in green (PDB ID 4V2I),48 and heroin esterase shown in pink (PDB ID 1LZK).30 C) Dimeric interface of LipW. Two protomers of LipW (gray) were aligned with the dimeric structures of two related hydrolases: ThaEst2349 in yellow (PDB ID 4V2I)48 and a thermostable esterase in orange (PDB ID 3ZWQ).49 The final residue in the C-terminal helix at the dimeric interface is shown in spheres for each homologue.
Structural alignment of LipW
Structurally, LipW aligns well against a number of other α/β hydrolases (E.C. 3.1.1.-) such as the metagenome-derived esterase Est8 (rmsd 1.4 Å, PDB ID 4YPV, no primary citation), esterase ThaEst2349 from marine bacterium Thalassospira sp. GB04J01 (rmsd 1.5 Å, PDB ID 4V2I),48 esterase PestE (rmsd 1.6 Å, PDB ID 3ZWQ),49 and a bacterial heroin esterase (rmsd 1.70 Å, PDB ID 1LZK).30 All of these enzymes belong to the bacterial acyl esterase superfamily with specificity for short chain esters. Although the core α/β hydrolase domains remain the same among these enzymes, the largest differences are observed in the N-terminal “cap” domain which covers the active site cavity (Figure 1B). The first 25 amino acids of the LipW cap are slightly unstructured, but likely form two small helices. However, a third, longer helix spanning residues 26–53 appears in a similar, but slightly more open conformation than the equivalent helix in other, structurally related α/β hydrolases (Figure 1B). The distance between the Val18 and Arg40 Cα atoms is 13.1 Å in LipW, but the distances between equivalent Cα atoms in the other structures is 10.2 Å (Leu10 and Ile37 of Est8), 10.1 Å (Phe11 and Ala38 of ThaEst2349), 11.2 (Ile10 and Ser36 of PestE), and 12.0 Å (Ala14 and Leu36 of heroin esterase).
In addition to changes in the cap domain, LipW also appears as a monomer in the crystal structure, which differs from the dimeric architecture of many homologues.48;50 Alignment of LipW to these dimeric homologues (PDB IDs 3ZWQ and 4V2I) shows their common dimer interface with strong interactions between the C-terminal α-helices stabilizing dimer formation (Figure 1C). When LipW with a second protomer is overlayed with these dimeric homologues, the overall dimer interface of LipW is shifted, as evidenced by the relative positioning of the last residue in the C-terminal helix (Figure 1C). Yet, size exclusion chromatography of LipW indicated that it has some weak self-assembly, which may not be conserved with the dimeric stabilization observed in homologous α/β hydrolases (Supplemental Figure 3).
Substrate specificity of LipW from M. marinum
The structure of the LipW binding pocket and the biological functions of its structural homologues placed LipW into the acyl ester hydrolase superfamily with proposed selectivity for short chain esters. To refine this substrate specificity profile, the global substrate specificity of LipW from M. marinum (MmLipW) was determined against a diverse library of fluorogenic hydrolase substrates (Figure 2). These fluorogenic substrates utilize the well-established fluorescent to nonfluorescent equilibrium in fluorescein to provide a highly sensitive, low background, and modular substrate library for interrogating the substrate specificity of serine hydrolases (Figure 2A).41;42;51 Additionally, the fluorogenic library encompasses acyl substituents covering the major classes of serine hydrolase substrates and provides single bond resolution of substrate specificity (Figure 2B).38;39;42;52 Measuring the Michaelis-Menten kinetics of MmLipW against the full range of fluorogenic substrates provided a detailed description of the substrate specificity profile for LipW (Figure 2C–2E and Supplemental Table 1).
Based on comparisons of overall enzymatic efficiency values (kcat/KM), MmLipW was highly selective for small ester substituents (Figure 2). This pattern is exemplified in its comparative activity toward substrates 1–5 with only single carbon bond changes in the alkyl chain length leading to large shifts in the enzymatic efficiency (Figure 2D). Among substrates 1–5, MmLipW had highest activity against a propionyl ester substrate (2) with 7-fold selectivity over a one carbon subtraction (acetyl ester; 1) and 16-fold selectivity over a one carbon addition (butyl ester; 3). The majority of the measured decrease in the enzymatic efficiency (kcat/KM) of MmLipW for substrates 1 and 3 versus substrate 2 was driven by a decrease in the kcat value (Supplemental Table 1). The large shifts in the catalytic efficiency (kcat) with small changes in carbon chain length showed the strict selectivity of LipW based on positioning of the catalytic triad and likely reflected the shallow, acyl-binding pocket of LipW. Reinforcing this narrow substrate selectivity pattern, MmLipW also had nearly identical activity (kcat/KM = 1800 M−1 s−1) against substrate 10, which contains a terminal alkene, but like substrate 2 has two carbons past the ester carbonyl (Figure 2C). Thus, LipW selects primarily for absolute substrate length irrespective of carbon bond saturation. MmLipW does however strongly select against branched alkyl chains, as the addition of a single methyl branch (substrate 9 versus 10) decreased the catalytic activity by 100-fold and further branching (compounds 17 and 18) resulted in near complete loss of activity.
In addition to two-carbon alkyl esters, MmLipW had high activity against substrate 6, a four-atom alkyl ether ester (three-carbons and one oxygen), which had >10-fold higher activity in comparison with substrate 2 or >1000-fold higher activity in comparison with substrate 3 of the same length. Substrate 6 has been the most active substrate across a range of serine hydrolases and thus the high reactivity of MmLipW toward this substrate is likely not indicative of its natural reactivity.42;52 The high reactivity of substrate 6 is instead likely due to electronic effects of the β-oxygen increasing the electrophilicity of the carbonyl. This effect is reinforced by the increased reactivity of substrate 8, with its β-oxygen, over substrate 7, with its further spaced γ-oxygen (Figure 2A).
MmLipW, also, displayed high activity against the classic, and highly reactive, p-nitrophenyl ester substrates (Figure 2E). Reinforcing the patterns from the fluorogenic substrates, MmLipW had highest activity against the substrates with the shortest alkyl chains with highest activity against the methyl (C2) and propyl (C4) esters and >100-fold lower activity against longer carbon chains (C8, C12) (Supplemental Table 1). Unlike with the fluorogenic substrates, MmLipW had less selectivity based on the addition of a single carbon atom, as the C4 substrate had only a 2-fold decrease in catalytic activity. This decreased selectivity may reflect the smaller alcohol component of the p-nitrophenyl substrates or the increased reactivity of these p-nitrophenyl substrates. Supporting the selectivity of LipW based on alcohol substituent, MmLipW displayed no measurable activity against 7-aminocephalosporonic acid (7-ACA; Supplemental Table 1). Structurally homologous acyl esterases to MmLipW have previously catalyzed the efficient deacetylation of cephalosporin and simplified derivatives like 7-ACA, classifying LipW away from the acetyl esterase subfamily of α/β hydrolases.38;47;53;54
Comparative substrate specificity of homologous LipW enzymes from M. marinum and M. smegmatis
To determine whether this strict substrate selectivity was conserved across LipW homologues from other mycobacterial species, the substrate specificity of MmLipW was compared to the substrate specificity of LipW from Mycobacterium smegmatis (MsLipW) across fluorogenic and p-nitrophenyl ester substrates (Figures 2 and 3). MsLipW shares 58% sequence identity to MmLipW (Supplemental Table 4). MsLipW also strongly preferred propionyl ester substrates with highest activity toward substrate 2 and substantial activity against substrates 10 (Figures 3C and 3D). Like MmLipW, this substrate selectivity for propionyl esters in MsLipW was also driven by large decreases in the kcat values (>100-fold) with small increases in alkyl chain length or substrate branching (Supplemental Table 2). These strict substrate selectivity patterns for MsLipW also carried over to p-nitrophenyl substrates, as MsLipW had 10-fold higher catalytic efficiency to the acetyl ester (2C) over the butyl ester (4C) (Figure 2E). Together this global comparison of the substrate specificity between two mycobacterial LipW homologues reinforced the strict substrate selectivity of this enzyme superfamily for propionyl ester containing substrates.
Figure 3.
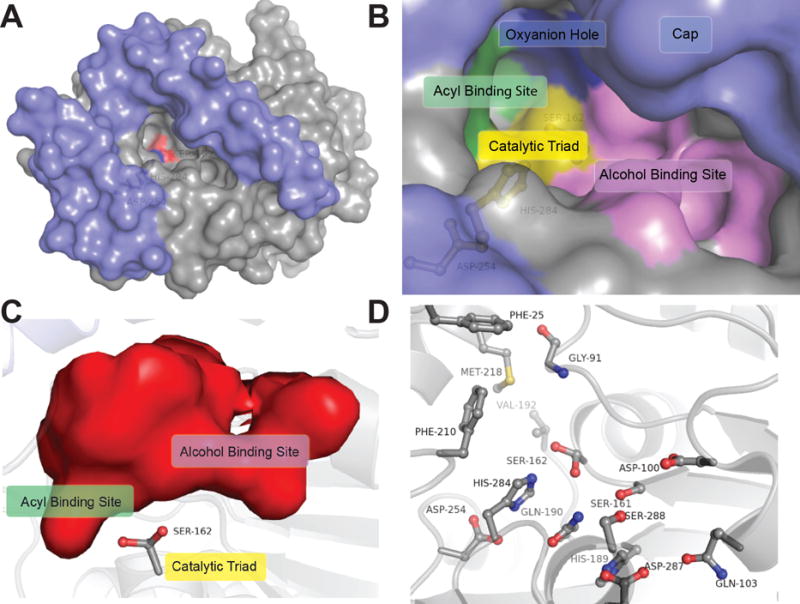
Binding pocket architecture of LipW. A) Cap domain (blue) and binding pocket surface of LipW (grey) with the nucleophilic serine (Ser162) in red and catalytic histidine (His284) in blue. B) Surface representation of the binding pocket and active site of LipW. Individual subsections within the LipW binding pocket are labeled. C) Interior surface of the LipW binding pocket with Ser162 labeled. Binding pocket rotated 90° clockwise from part B and oriented from the interior of the protein. Subsections labeled as in part B. D) Binding pocket and catalytic residues substituted with alanine in LipW. Each of the residues shown in ball and stick was individually substituted with alanine and the relative contribution of each side-chain to the catalytic activity and thermal stability determined (Figure 4).
MsLipW did, however, show some subtle differences in substrate selectivity, which might reflect minor structural shifts. MsLipW had significantly lower (>50-fold) catalytic activity toward the larger alkyl ether substrates (6 and 8), but 5-fold higher activity toward the methyl ester substrate (1). This shift for MsLipW toward preferring smaller substrates was confirmed in the p-nitrophenyl substrates with a significantly larger decrease in activity between C2 and C4 substrates for MsLipW as compared to MmLipW (Supplemental Tables 1 and 2). Overall, this comparison supported the structural and enzymatic similarity between mycobacterial LipW homologues, indicating that the MmLipW structure is likely indicative of the structural organization of LipW homologues across the genus Mycobacterium.
Analysis of the binding pocket and active site of LipW
To identify the residues controlling the strict substrate selectivity of LipW, the binding pocket and active site of LipW were structurally and phylogenetically analyzed for residues contributing to substrate binding and catalysis. Residues selected for substitutional analysis encircled the active site and included the catalytic triad amino acids Ser162, His284, and Asp254 (Figure 3). Residues also spanned the alcohol-binding pocket composed of a congregation of charged and polar residues (Asp100, Gln103, Ser161, His189, Gln190, Asp287, Ser288; Figure 3A and 3D). Selected binding pocket residues had a wide range of evolutionary conservation from the completely conserved catalytic triad residues and highly conserved oxyanion hole residue (Gly91) to highly variable charged/polar residues in the alcohol binding pocket of LipW (Figure 4C and Supplemental Table 4). Hydrophobic residues (Phe25, Val192, Phe210, and Met 218) surrounding the shallow acyl binding pocket showed significant evolutionary variation, but across diverse homologues maintained the highly hydrophobic nature of this section of the binding pocket (Figure 3B and 3D). To characterize the role of these binding pocket residues in controlling the strict substrate selectivity of LipW, each of these fifteen residues surrounding the binding pocket and active site were individually substituted to alanine (Supplemental Table 5). The resulting protein variants were purified to homogeneity (Supplemental Figure 1) and the relative change in enzymatic activity and protein stability was characterized (Figure 4 and Supplemental Table 4).
Figure 4.
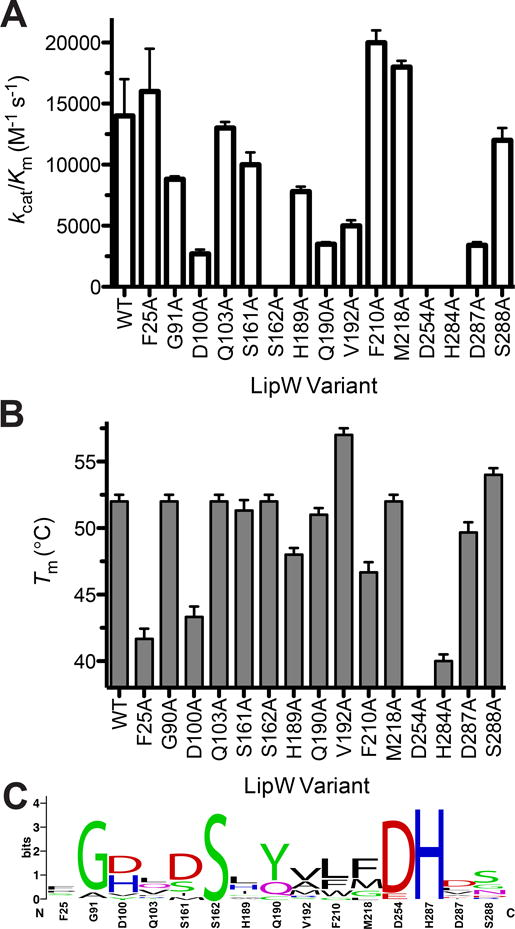
Importance of the binding pocket and active site amino acids to the catalytic activity and thermal stability of LipW. A) Relative catalytic activity of LipW binding pocket variants (Figure 3). The catalytic activity of each of the LipW variants was determined against substrate 2 (Figure 2A). B) Thermal stability of LipW variants. The thermal stability of each LipW variant was determined by measuring the increase in Sypro Orange fluorescence in response to increasing temperature. Results are shown with their standard error values. Detailed kinetic and thermal stability analysis for binding pocket variants are given in Supplemental Table 3. C) Conservation of binding pocket residues across bacterial homologues of LipW. Sequences aligned using Clustal Omega and relative weightings performed using Weblogo 75;76. Detailed sequence analysis along with percent identity between homologues given in Supplemental Table 4.
Confirming the biological significance of the mutational analysis, substitution of each of the catalytic amino acids with alanine completely ablated the catalytic activity of LipW against its preferred propionyl ester substrate (2; Figure 4A). Substitution of residues Asp100, His189, Gln190, and Asp287 in the alcohol-binding pocket also decreased the catalytic efficiency of LipW (>3-fold), mostly driven by decreased kcat values (>3-fold; Supplemental Table 3). Importantly, substitution of closely spaced polar residues (Gln103, Ser161, and Ser288) maintained wild-type catalytic efficiency (Figure 4A). In contradiction to the substitution of these polar amino acids, the majority of the nonpolar amino acid substitutions (Phe25, Phe210, and Met218) slightly increased the catalytic efficiency with small, but significant decreases in their KM values (Supplemental Table 3). The outlier to this pattern was Val192 positioned at the bottom of the shallow acyl-binding cavity (Figure 3D), which decreased the kcat value by 4-fold (Supplemental Table 3).
In comparison to the diverse changes in the catalytic efficiency of LipW upon substitution, these binding pocket residues made only minor contributions to the folded stability of LipW, as the majority of the alanine variants maintained wild-type thermal stability (Figure 4B). These small changes in thermal stability upon substitution showed that the measurable changes in catalytic activity upon substitution were not due to indirect effects of the substitutions on the folded structure of LipW, but rather direct effects on the catalytic mechanism or substrate binding. Similar to other serine hydrolases, the side-chains of the buried catalytic triad residues Asp254 and His284 significantly contributed to the thermal stability of LipW, as individual substitution of each residue to alanine decreased the TM value by >10 °C (Figure 4B).42;52 Outside of these catalytic triad residues, only one additional buried hydrophobic (Phe25) and one polar (Asp100) residue contributed greater than 5 °C to the thermal stability of LipW. Interestingly, the V192A variant was again an outlier with a significant increase (5 °C) in the thermal stability from wild-type LipW, which combined with the decreased activity may indicate that this mutant enzyme gets trapped in a stable conformational state which is less catalytically active than wild-type (Figure 4B). Overall, the binding pocket of LipW was highly tolerant of substitution with diverse contributions across the acyl and alcohol binding pocket to the strict substrate selectivity and catalytic power of LipW, but only minor contributions to its folded stability.
DISCUSSION
Mycobacterial serine hydrolases and especially Lip family hydrolases control important processes in the mycobacterial life cycle, including immune recognition, energy mobilization, and dormancy.8;20;55 Genomically, the LipW gene (Rv0217c) is located in close proximity to multiple fatty acid- and lipid-responsive genes, including TAG synthetases, strongly implicating LipW as being involved in lipid metabolism in Mycobacteria.7 Similar genomic organizations of lipolytic enzyme genes in close proximity or adjacent to TAG synthetase genes have encoded modules of lipid metabolism enzymes for the break down and transport of host cell fatty acids.7 The importance of this genomic module and potentially LipW to the survival and virulence of Mycobacterium is further supported by the loss of pathogenicity of a Mycobacterium avium strain upon deletion of this genomic fragment.56 Identification of lipase genes overexpressed in host cells with high TAG, however, did not identify LipW as significantly upregulated in high TAG, potentially implicating LipW as involved in active TB processes like cell wall synthesis.15
By activity-based protein profiling (ABPP), the enzymatic activity of LipW has been quantitated in M. bovis and M. tuberculosis, confirming general expression and activation of LipW across Mycobacteria.8;9 Matching with the gene expression profile, LipW enzymatic activity peaked in logarithmic growth conditions, confirming the highest expression of LipW under active mycobacterial growth.9 Together, these genomic and proteomic studies of mycobacterial hydrolases start to build a picture of LipW, as intimately involved in active mycobacterial metabolism with significant enzymatic activity and expression in diverse mycobacterial species.
LipW as a bacterial HSL
By sequence and structural alignment, LipW fits firmly into the bacterial hormone sensitive lipase (HSL) superfamily.57 Within the Jaegger classification system, bacterial HSLs are grouped into Family IV, which contains two distinct subfamilies, the larger GDSAG motif subfamily and the newly characterized GTSAG subfamily.57 Each of these subfamilies is defined based on the sequence motif surrounding the catalytic serine. Although MmLipW has a GSSAG motif, which overlaps with the most highly represented motif in the small, newly defined GTSAG family, MmLipW was firmly placed into the larger GDSAG based on complete phylogenetic analysis (Figure 4C).57 Twelve additional bacterial HSL family members are present in Mycobacteria and are jointly termed the “Lip-HSL” family.17 Among the “Lip-HSL” family, LipW from M. tuberculosis showed high activity against p-nitrophenyl butyrate, but no measurable activity against vinyl esters or TAG, unlike other members of the “Lip-HSL” family.17
The narrow substrate specificity of LipW was reinforced in our experiments with two LipW homologues from two different mycobacterial species (Figure 2). With two distinct classes of ester-based substrates, both LipW homologues strongly selected for short, alkyl, unbranched esters (Figure 2 and Supplemental Tables 1 and 2). Using the molecular detail present in the fluorogenic hydrolase substrate library, the substrate preference of LipW was refined to the very narrow subclass of propionyl esters, but with residual catalytic activity toward acetyl and butyl esters (Figure 2). Although the LipW homologues from M. marinum and M. smegmatis varied slightly in their substrate specificity across the library of fluorogenic substrates, the main substrate specificity pattern of strict selectivity for propionyl esters was reproduced across both homologues (Figures 2C and 2D). The p-nitrophenyl ester substrates did not provide this same level of molecular detail about the substrate specificity, but did reaffirm the high activity of LipW toward short, unbranched substrates (Figure 2E).
The diverse substrate specificity patterns observed across the bacterial HSL superfamily have been correlated with the size and organization of the bacterial HSL cap domain, which takes the place of the N-terminal regulatory domain from human HSL.58–60 MmLipW contains a fairly small cap domain with two unstructured helices connected by a small loop that frames the binding pocket (Figure 3A). Minimal cap domains like those seen in MmLipW are correlated with greater substrate binding promiscuity, especially for diverse alcohol-containing substrates.59;61 MmLipW showed some substrate promiscuity based on alcohol substituents with high activity to the large fluorogenic substrates and small p-nitrophenyl substrates, but failed to catalyze the deacetylation of the cephalosporin derivative, 7-ACA (Figure 2 and Supplemental Table 1). Similar to two homologous bacterial HSL enzymes from Rhizomucor miehei, the impingement of the cap domain on the acyl binding pocket of LipW may serve to limit the size of acyl substrates and to create a more funnel-shaped binding pocket, limiting the substrate promiscuity (Figure 3).61
The cap domain also serves a second role in maintaining the stability of bacterial HSLs, where the tight packing between the cap domain and the α/β hydrolase domain increases their stability.60 MmLipW showed above average thermal stability (Tm = 52 °C) for a mesophilic bacterial HSL with high tolerance for substitution (Figure 4B). Only four amino acid substitutions out of 15 total binding pocket substitutions decreased the thermal stability by more than 5 °C with one alanine substitution (V192A) interestingly increasing the thermal stability by 5 °C (Figure 4B). The combined stability and catalytic control imparted by the cap domain have made bacterial HSLs common targets for biocatalyst design.59;60;62
Structural features controlling the strict substrate specificity of LipW
The binding pocket for LipW can be subdivided into distinct subsites for substrate recognition and catalysis (Figure 3B).61 These different subsites influence catalysis directly through the conserved catalytic triad and indirectly through transition state stabilization in the oxyanion hole (Figure 3B and 3C). The subsites also bind and recognize distinct components of the ester substrates, including the alcohol and acylated portion of the substrate.61 Unlike the ability of LipW to accept varying substrates within its alcohol-binding site, the acyl-binding site provides strict substrate selectivity based on the length and branching of the acyl substituents (Figures 2 and 3). Within bacterial HSLs, these two substrate recognition pockets, however, function semi-independently and provide distinct substrate recognition profiles, making de novo predictions of their roles difficult.59;61 By using substitutional analysis of 15 amino acids surrounding the acyl and alcohol binding pockets, oxyanion hole, and catalytic triad, we were able to dissect the relative contributions of these different sections of the binding pocket to the catalytic activity and substrate selectivity of LipW (Figures 4A and 5).
Figure 5.
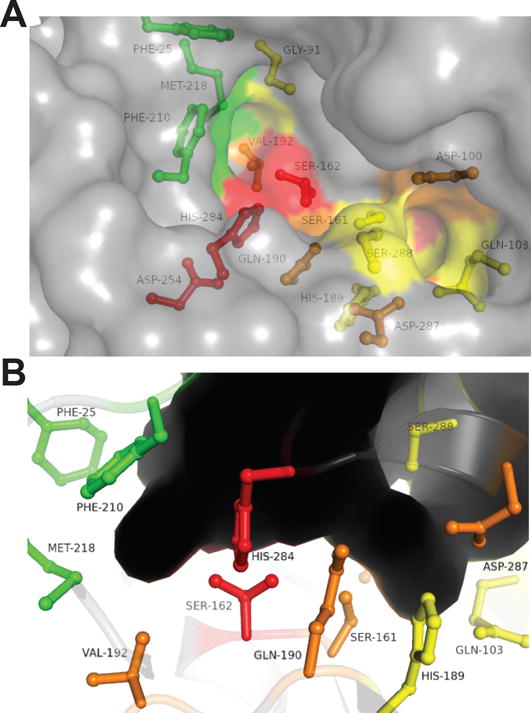
Relative contributions of binding pocket and active site residues to the catalytic activity of LipW. Exterior surface and stick (5A) and interior surface representations (5B) of the binding pocket of LipW color coded for their relative contributions to the catalytic activity of LipW against substrate 2. Colors represent relative changes to the catalytic activity (kcat/KM) upon substitution to alanine: red (>10-fold decrease); orange (>3-fold decrease); yellow (<3-fold decrease); green (>1-fold increase).
The alcohol-binding pocket of LipW is highly polar, partially negatively charged, and mostly open with partial occlusion by the cap domain (Figure 3B). Based on the substitutional analysis, the alcohol-binding pocket plays a fairly large role in controlling the overall enzymatic activity of LipW (Figure 5). Converting each of the seven residues in the alcohol-binding pocket to alanine lowered the catalytic activity of MmLipW by between 3-fold to 5-fold. The residue Asp100 exemplifies this effect, as it is distantly positioned from the catalytic triad, but its conversion to alanine decreased the catalytic activity by 5-fold (Figure 4A). Another part of this effect on the catalytic activity could be attributed to regulating the two rotameric states of the catalytic serine (Figures 1 and 3E).47 Reinforcing the importance of these two rotameric states in catalysis by LipW, substitution of Ser161 and surrounding stabilization residues (His189 and Gln190) all significantly decreased the catalytic activity of LipW (Figures 4 and 5).
In comparison to the generalized role of the alcohol-binding pocket in controlling the catalytic activity of MmLipW, substitution of residues in the acyl-binding pocket did not significantly affect the catalytic activity of MmLipW with the exception of a modest decrease for Val192 (3-fold) (Figures 4A and 5). This lack of change in catalytic activity with acyl pocket substitution may reflect the use of the optimal acyl substrate (2) for these comparative kinetics measurements (Figure 2). This short substrate may not directly interact with the hydrophobic walls of the acyl-binding pocket (Phe25, Phe210, Met218; Figure 3B and 3D). In comparison to the walls of the acyl binding pocket, the floor of the acyl binding funnel does play an important role in controlling substrate orientation for catalysis, as shown by the decreased overall catalytic efficiency (kcat/KM) and catalytic turnover (kcat) of the V192A variant (Supplemental Table 3). The importance of this valine residue in catalysis and in the strict substrate selectivity of LipW was reinforced by the increased thermal stability of the V192A variant, suggesting that the valine residue was selected for its catalytic role against its negative effect on stability (Figure 4). This valine residue with its competing roles in substrate selectivity and folded stability also represents a good starting point for biocatalytic design, as substitution of similarly positioned residues in bacterial HSLs greatly expanded their substrate specificity, introduced greater promiscuity, and created greater enantioselectivity.62–64
Potential biological substrates
The strict selectivity of LipW for propionyl esters complicates the prediction of its biological function (Figure 2). Hydrolases, such as propionyl-CoA and malonyl-CoA hydrolases, have been identified that prefer three carbon substrates.65;66 LipW is, however, unlikely to be a coenzyme A dependent thioester hydrolase, as the cap domain and substrate binding architecture of LipW are not arranged to accommodate the extended structure of the coenzyme A cofactor.65;67 Coenzyme A dependent hydrolases commonly contain a flexible cap domain that undergoes a conformational change to recognize the coenzyme A portion of the substrate.65;67 Instead of serving as a separate flexible domain, the LipW cap encircles the binding pocket and is unlikely to undergo a significant conformational change upon substrate binding (Figure 3).
Acetyl esterases with strict selectivity for acetyl esters from lipid, carbohydrate, and protein-based substrates are also present across bacterial species, but these enzymes select against propionyl and butyl esters.38;68 The binding pocket architecture of LipW does resemble the open alcohol and narrow acyl binding sites of bacterial acetyl esterases and acetyl xylan esterases (Figure 5).47;53;54 The inactivity of LipW toward the cephalosporin derivative, 7-ACA, however, differentiates it from these bacterial acetyl esterases and indicates a unique substrate recognition pattern for LipW.38;68 Additionally, although M. tuberculosis can subsist on a variety of carbon sources and contains cellulases and other enzymes necessary for the breakdown of cellulose, no xylan degradative enzymes or modules have been found in M. tuberculosis, making LipW unlikely to be an acetyl xylan esterase.69–71
For LipW, the genomic architecture, gene and protein expression patterns, and strict substrate selectivity profile for propionyl esters suggest a mycobacterial specific biological substrate, including mycobacterial membrane and storage lipids.7;9;15 Mycobacterial membrane and storage lipids are complex, composing the unique mycolic acid layer in the mycobacterial cell wall.72;73 These membrane lipids are constantly broken down and recycled under normal growth conditions, matching with the genomic and proteomic expression patterns of LipW under normal growth conditions.15 Thus, based on the structure and substrate specificity profile, LipW presents an opportunity for identifying a potentially novel mycobacterial specific hydrolase substrate.
CONCLUSIONS
Mycobacterial serine hydrolases are exciting therapeutic targets with confirmed biological roles in dormancy, metabolic regulation, and immune recognition. Among mycobacterial serine hydrolases, LipW has proposed roles in lipid metabolism across Mycobacteria and potentially in the virulence and survival of M. avium.7;56 LipW is a member of the Lip-HSL subfamily with features of the larger bacterial HSL superfamily, including a conserved GSSAG motif, and some substrate promiscuity with regards to alcohol substituent. The structure of LipW showcased its limited cap domain, its distinct alcohol- and acyl-binding pockets, and its narrow, funnel-shaped, acyl-binding pocket. This funnel-shaped, acyl-binding site strictly selects for short, unbranched, alkyl substrates with highest selectivity for propionyl esters. This unusual substrate specificity profile of LipW combined with its binding pocket structure and its genetic and proteomic expression profiles strongly indicate that LipW catalyzes metabolic reactions with a mycobacterial specific ester substrate. Inhibitor-bound structures have been reported for homologues of LipW, including bacterial heroin esterase30 and a HSL-like carboxylesterase from Sulfolobus tokodaii,74 and the structure of LipW combined with its substrate specificity profile suggest that it may be possible to target LipW with similar small molecule inhibitors.
Supplementary Material
Acknowledgments
We thank CH463 students from Butler University for assistance with preliminary enzyme purification and enzymatic analysis, Ariel Abramov for protein production, SSGCID PI Peter Myler, and the rest of the SSGCID team.
Funding: R.J.J. and M.G.M. were supported by a grant from the National Science Foundation (DUE-1140526) and National Institutes of Health (NIH 1 R15 GM110641-01A1). J.A. and T.E.E. were supported with federal funds from the National Institute of Allergy and Infectious Diseases National Institutes of Health, Department of Health and Human Services under Contract Nos. HHSN272201200025C and HHSN272200700057C.
Footnotes
ASSOCIATED CONTENT: The Supporting Information is available free of charge on the ACS Publications website at DOI: . Further structural analysis of LipW, detailed kinetic data for LipW and its variants, example kinetic traces, sequence alignment for binding pocket residues in LipW, PCR primers used for mutagenesis, and gel analysis of LipW and its variants.
References
- 1.Lovewell RR, Sassetti CM, VanderVen BC. Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Current opinion in microbiology. 2016;29:30–36. doi: 10.1016/j.mib.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Baek SH, Li AH, Sassetti CM. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011;9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baer CE, Rubin EJ, Sassetti CM. New insights into TB physiology suggest untapped therapeutic opportunities. Immunological reviews. 2015;264:327–343. doi: 10.1111/imr.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gouzy A, Poquet Y, Neyrolles O. Nitrogen metabolism in Mycobacterium tuberculosis physiology and virulence. Nat Rev Microbiol. 2014;12:729–737. doi: 10.1038/nrmicro3349. [DOI] [PubMed] [Google Scholar]
- 6.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–366. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 7.Stehr M, Elamin AA, Singh M. Lipid inclusions in mycobacterial infections. INTECH Open Access Publisher; 2013. [Google Scholar]
- 8.Ortega C, Anderson LN, Frando A, Sadler NC, Brown RW, Smith RD, Wright AT, Grundner C. Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell chemical biology. 2016;23:290–298. doi: 10.1016/j.chembiol.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravindran MS, Rao SP, Cheng X, Shukla A, Cazenave-Gassiot A, Yao SQ, Wenk MR. Targeting lipid esterases in mycobacteria grown under different physiological conditions using activity-based profiling with tetrahydrolipstatin (THL) Mol Cell Proteomics. 2014;13:435–448. doi: 10.1074/mcp.M113.029942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dedieu L, Serveau-Avesque C, Kremer L, Canaan S. Mycobacterial lipolytic enzymes: a gold mine for tuberculosis research. Biochimie. 2013;95:66–73. doi: 10.1016/j.biochi.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 13.Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, Peters PJ, Luirink J, Manganelli R, Bitter W. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem. 2011;286:19024–19034. doi: 10.1074/jbc.M110.204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rastogi S, Agarwal P, Krishnan MY. Use of an adipocyte model to study the transcriptional adaptation of Mycobacterium tuberculosis to store and degrade host fat. Int J Mycobacteriology. 2015 doi: 10.1016/j.ijmyco.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 16.West NP, Cergol KM, Xue M, Randall EJ, Britton WJ, Payne RJ. Inhibitors of an essential mycobacterial cell wall lipase (Rv3802c) as tuberculosis drug leads. Chem Commun (Camb) 2011;47:5166–5168. doi: 10.1039/c0cc05635a. [DOI] [PubMed] [Google Scholar]
- 17.Delorme V, Diomande SV, Dedieu L, Cavalier JF, Carriere F, Kremer L, Leclaire J, Fotiadu F, Canaan S. MmPPOX inhibits Mycobacterium tuberculosis lipolytic enzymes belonging to the hormone-sensitive lipase family and alters mycobacterial growth. PLoS One. 2012;7:e46493. doi: 10.1371/journal.pone.0046493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxena AK, Roy KK, Singh S, Vishnoi SP, Kumar A, Kashyap VK, Kremer L, Srivastava R, Srivastava BS. Identification and characterisation of small-molecule inhibitors of Rv3097c-encoded lipase (LipY) of Mycobacterium tuberculosis that selectively inhibit growth of bacilli in hypoxia. Int J Antimicrob Ag. 2013;42:27–35. doi: 10.1016/j.ijantimicag.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem. 2006;281:3866–3875. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen G, Singh K, Chandra D, Serveau-Avesque C, Maurin D, Canaan S, Singla R, Behera D, Laal S. LipC (Rv0220) is an immunogenic cell surface esterase of Mycobacterium tuberculosis. Infect Immun. 2012;80:243–253. doi: 10.1128/IAI.05541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh G, Jadeja D, Kaur J. Lipid hydrolizing enzymes in virulence: Mycobacterium tuberculosis as a model system. Crit Rev Microbiol. 2010;36:259–269. doi: 10.3109/1040841X.2010.482923. [DOI] [PubMed] [Google Scholar]
- 22.Jadeja D, Dogra N, Arya S, Singh G, Singh G, Kaur J. Characterization of LipN (Rv2970c) of Mycobacterium Tuberculosis H37Rv and its Probable Role in Xenobiotic Degradation. Journal of cellular biochemistry. 2016;117:390–401. doi: 10.1002/jcb.25285. [DOI] [PubMed] [Google Scholar]
- 23.Sinha SC, Wetterer M, Sprang SR, Schultz JE, Linder JU. Origin of asymmetry in adenylyl cyclases: structures of Mycobacterium tuberculosis Rv1900c. The EMBO journal. 2005;24:663–673. doi: 10.1038/sj.emboj.7600573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng X, Guo J, Xu L, Li H, Zhang D, Zhang K, Sun F, Wen T, Liu S, Pang H. Crystal structure of a novel esterase Rv0045c from Mycobacterium tuberculosis. PLoS One. 2011;6:e20506. doi: 10.1371/journal.pone.0020506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crellin PK, Vivian JP, Scoble J, Chow FM, West NP, Brammananth R, Proellocks NI, Shahine A, Le Nours J, Wilce MC, Britton WJ, Coppel RL, Rossjohn J, Beddoe T. Tetrahydrolipstatin inhibition, functional analyses, and three-dimensional structure of a lipase essential for mycobacterial viability. J Biol Chem. 2010;285:30050–30060. doi: 10.1074/jbc.M110.150094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baugh L, Phan I, Begley DW, Clifton MC, Armour B, Dranow DM, Taylor BM, Muruthi MM, Abendroth J, Fairman JW, Fox D, 3rd, Dieterich SH, Staker BL, Gardberg AS, Choi R, Hewitt SN, Napuli AJ, Myers J, Barrett LK, Zhang Y, Ferrell M, Mundt E, Thompkins K, Tran N, Lyons-Abbott S, Abramov A, Sekar A, Serbzhinskiy D, Lorimer D, Buchko GW, Stacy R, Stewart LJ, Edwards TE, Van Voorhis WC, Myler PJ. Increasing the structural coverage of tuberculosis drug targets. Tuberculosis. 2015;95:142–148. doi: 10.1016/j.tube.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexandrov A, Dutta K, Pascal SM. MBP fusion protein with a viral protease cleavage site: one-step cleavage/purification of insoluble proteins. Biotechniques. 2001;30:1194–1198. doi: 10.2144/01306bm01. [DOI] [PubMed] [Google Scholar]
- 28.Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. The proteomics protocols handbook. Springer; 2005. Protein identification and analysis tools on the ExPASy server; pp. 571–607. [Google Scholar]
- 29.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Larsen NA, Basran A, Bruce NC, Wilson IA. Observation of an arsenic adduct in an acetyl esterase crystal structure. J Biol Chem. 2003;278:2008–2014. doi: 10.1074/jbc.M210103200. [DOI] [PubMed] [Google Scholar]
- 31.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr. 2008;41:641–643. [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hedge MK, Gehring AM, Adkins CT, Weston LA, Lavis LD, Johnson RJ. The structural basis for the narrow substrate specificity of an acetyl esterase from Thermotoga maritima. Biochim Biophys Acta. 2012;1824:1024–1030. doi: 10.1016/j.bbapap.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Filippova EV, Weston LA, Kuhn ML, Geissler B, Gehring AM, Armoush N, Adkins CT, Minasov G, Dubrovska I, Shuvalova L, Winsor JR, Lavis LD, Satchell KJ, Becker DP, Anderson WF, Johnson RJ. Large scale structural rearrangement of a serine hydrolase from Francisella tularensis facilitates catalysis. J Biol Chem. 2013;288:10522–10535. doi: 10.1074/jbc.M112.446625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson RJ, Hoops GC, Savas CJ, Kartje Z, Lavis LD. A Sensitive and Robust Enzyme Kinetic Experiment Using Microplates and Fluorogenic Ester Substrates. Journal of chemical education. 2014;92:385–388. [Google Scholar]
- 41.Lavis LD, Chao T-Y, Raines RT. Synthesis and utility of fluorogenic acetoxymethyl ethers. Chem Sci. 2011;2:521–530. doi: 10.1039/C0SC00466A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis EE, Adkins CT, Galovska NM, Lavis LD, Johnson RJ. Decoupled roles for the atypical, bifurcated binding pocket of the ybfF hydrolase. ChemBioChem. 2013;14:1134–1144. doi: 10.1002/cbic.201300085. [DOI] [PubMed] [Google Scholar]
- 43.Kim S, Lee SB. Thermostable esterase from a thermoacidophilic archaeon: purification and characterization for enzymatic resolution of a chiral compound. Biosci Biotechnol Biochem. 2004;68:2289–2298. doi: 10.1271/bbb.68.2289. [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Martinez I, Montoro-Garcia S, Lozada-Ramirez JD, Sanchez-Ferrer A, Garcia-Carmona F. A colorimetric assay for the determination of acetyl xylan esterase or cephalosporin C acetyl esterase activities using 7-amino cephalosporanic acid, cephalosporin C, or acetylated xylan as substrate. Anal Biochem. 2007;369:210–217. doi: 10.1016/j.ab.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 45.Johnson RJ, Savas CJ, Kartje Z, Hoops GC. Rapid and Adaptable Measurement of Protein Thermal Stability by Differential Scanning Fluorimetry: Updating a Common Biochemical Laboratory Experiment. Journal of chemical education. 2014;91:1077–1080. [Google Scholar]
- 46.Zhu X, Larsen NA, Basran A, Bruce NC, Wilson IA. Observation of an arsenic adduct in an acetyl esterase crystal structure. Journal of Biological Chemistry. 2003;278:2008–2014. doi: 10.1074/jbc.M210103200. [DOI] [PubMed] [Google Scholar]
- 47.Levisson M, Han GW, Deller MC, Xu Q, Biely P, Hendriks S, Ten Eyck LF, Flensburg C, Roversi P, Miller MD, McMullan D, von Delft F, Kreusch A, Deacon AM, van der Oost J, Lesley SA, Elsliger MA, Kengen SW, Wilson IA. Functional and structural characterization of a thermostable acetyl esterase from Thermotoga maritima. Proteins. 2012 doi: 10.1002/prot.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Santi C, Leiros HK, Di Scala A, de Pascale D, Altermark B, Willassen NP. Biochemical characterization and structural analysis of a new cold-active and salt-tolerant esterase from the marine bacterium Thalassospira sp. Extremophiles. 2016;20:323–336. doi: 10.1007/s00792-016-0824-z. [DOI] [PubMed] [Google Scholar]
- 49.Palm GJ, Fernandez-Alvaro E, Bogdanovic X, Bartsch S, Sczodrok J, Singh RK, Bottcher D, Atomi H, Bornscheuer UT, Hinrichs W. The crystal structure of an esterase from the hyperthermophilic microorganism Pyrobaculum calidifontis VA1 explains its enantioselectivity. Appl Microbiol Biotechnol. 2011;91:1061–1072. doi: 10.1007/s00253-011-3337-9. [DOI] [PubMed] [Google Scholar]
- 50.Byun JS, Rhee JK, Kim ND, Yoon J, Kim DU, Koh E, Oh JW, Cho HS. Crystal structure of hyperthermophilic esterase EstE1 and the relationship between its dimerization and thermostability properties. BMC Struct Biol. 2007;7:47. doi: 10.1186/1472-6807-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian L, Yang Y, Wysocki LM, Arnold AC, Hu A, Ravichandran B, Sternson SM, Looger LL, Lavis LD. Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc Natl Acad Sci USA. 2012;109:4756–4761. doi: 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukowski JK, Savas CP, Gehring AM, McKary MG, Adkins CT, Lavis LD, Hoops GC, Johnson RJ. Distinct substrate selectivity of a metabolic hydrolase from Mycobacterium tuberculosis. Biochemistry. 2014;53:7386–7395. doi: 10.1021/bi501108u. [DOI] [PubMed] [Google Scholar]
- 53.Vincent F, Charnock SJ, Verschueren KH, Turkenburg JP, Scott DJ, Offen WA, Roberts S, Pell G, Gilbert HJ, Davies GJ, Brannigan JA. Multifunctional xylooligosaccharide/cephalosporin C deacetylase revealed by the hexameric structure of the Bacillus subtilis enzyme at 1.9A resolution. J Mol Biol. 2003;330:593–606. doi: 10.1016/s0022-2836(03)00632-6. [DOI] [PubMed] [Google Scholar]
- 54.Montoro-Garcia S, Gil-Ortiz F, Garcia-Carmona F, Polo LM, Rubio V, Sanchez-Ferrer A. The crystal structure of the cephalosporin deacetylating enzyme acetyl xylan esterase bound to paraoxon explains the low sensitivity of this serine hydrolase to organophosphate inactivation. Biochem J. 2011;436:321–330. doi: 10.1042/BJ20101859. [DOI] [PubMed] [Google Scholar]
- 55.Deb C. A Novel Lipase Belonging to the Hormone-sensitive Lipase Family Induced under Starvation to Utilize Stored Triacylglycerol in Mycobacterium tuberculosis. Journal of Biological Chemistry. 2006;281:3866–3875. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marsh IB, Bannantine JP, Paustian ML, Tizard ML, Kapur V, Whittington RJ. Genomic comparison of Mycobacterium avium subsp. paratuberculosis sheep and cattle strains by microarray hybridization. Journal of Bacteriology. 2006;188:2290–2293. doi: 10.1128/JB.188.6.2290-2293.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li PY, Ji P, Li CY, Zhang Y, Wang GL, Zhang XY, Xie BB, Qin QL, Chen XL, Zhou BC, Zhang YZ. Structural basis for dimerization and catalysis of a novel esterase from the GTSAG motif subfamily of the bacterial hormone-sensitive lipase family. J Biol Chem. 2014;289:19031–19041. doi: 10.1074/jbc.M114.574913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ngo TD, Ryu BH, Ju H, Jang E, Park K, Kim KK, Kim TD. Structural and functional analyses of a bacterial homologue of hormone-sensitive lipase from a metagenomic library. Acta Crystallographica Section D: Biological Crystallography. 2013;69:1726–1737. doi: 10.1107/S0907444913013425. [DOI] [PubMed] [Google Scholar]
- 59.Sayer C, Szabo Z, Isupov MN, Ingham C, Littlechild JA. The Structure of a Novel Thermophilic Esterase from the Planctomycetes Species, Thermogutta terrifontis Reveals an Open Active Site Due to a Minimal ‘Cap’ Domain. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li PY, Chen XL, Ji P, Li CY, Wang P, Zhang Y, Xie BB, Qin QL, Su HN, Zhou BC. Interdomain Hydrophobic Interactions Modulate the Thermostability of Microbial Esterases from the Hormone-Sensitive Lipase Family. Journal of Biological Chemistry. 2015;290:11188–11198. doi: 10.1074/jbc.M115.646182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang S, Qin Z, Duan X, Yan Q, Jiang Z. Structural insights into the substrate specificity of two esterases from the thermophilic Rhizomucor miehei. Journal of lipid research. 2015;56:1616–1624. doi: 10.1194/jlr.M060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dou S, Kong XD, Ma BD, Chen Q, Zhang J, Zhou J, Xu JH. Crystal structures of Pseudomonas putida esterase reveal the functional role of residues 187 and 287 in substrate binding and chiral recognition. Biochemical and Biophysical Research Communications. 2014;446:1145–1150. doi: 10.1016/j.bbrc.2014.03.072. [DOI] [PubMed] [Google Scholar]
- 63.Ma BD, Yu HL, Pan J, Liu JY, Ju X, Xu JH. A thermostable and organic-solvent tolerant esterase from Pseudomonas putida ECU1011: catalytic properties and performance in kinetic resolution of alpha-hydroxy acids. Bioresour Technol. 2013;133:354–360. doi: 10.1016/j.biortech.2013.01.089. [DOI] [PubMed] [Google Scholar]
- 64.Ma BD, Kong XD, Yu HL, Zhang ZJ, Dou S, Xu YP, Ni Y, Xu JH. Increased Catalyst Productivity in alpha-Hydroxy Acids Resolution by Esterase Mutation and Substrate Modification. ACS Catalysis. 2014;4:1026–1031. [Google Scholar]
- 65.Park SY, Lee SH, Lee J, Nishi K, Kim YS, Jung CH, Kim JS. High-resolution structure of ybfF from Escherichia coli K12: a unique substrate-binding crevice generated by domain arrangement. J Mol Biol. 2008;376:1426–1437. doi: 10.1016/j.jmb.2007.12.062. [DOI] [PubMed] [Google Scholar]
- 66.Garras A, Asiedu DK, Berge RK. Subcellular localisation and induction of NADH-sensitive acetyl-CoA hydrolase and propionyl-CoA hydrolase activities in rat liver under lipogenic conditions after treatment with sulfur-substituted fatty acids. Biochim Biophys Acta. 1995;1255:154–160. doi: 10.1016/0005-2760(94)00236-r. [DOI] [PubMed] [Google Scholar]
- 67.Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Human fatty acid synthase: structure and substrate selectivity of the thioesterase domain. Proc Natl Acad Sci U S A. 2004;101:15567–15572. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tian Q, Song P, Jiang L, Li S, Huang H. A novel cephalosporin deacetylating acetyl xylan esterase from Bacillus subtilis with high activity toward cephalosporin C and 7-aminocephalosporanic acid. Applied Microbiology and Biotechnology. 2014;98:2081–2089. doi: 10.1007/s00253-013-5056-x. [DOI] [PubMed] [Google Scholar]
- 69.Medie FM, Salah IB, Drancourt M, Henrissat B. Paradoxical conservation of a set of three cellulose-targeting genes in Mycobacterium tuberculosis complex organisms. Microbiology. 2010;156:1468–1475. doi: 10.1099/mic.0.037812-0. [DOI] [PubMed] [Google Scholar]
- 70.Varrot A, Leydier S, Pell G, Macdonald JM, Stick RV, Henrissat B, Gilbert HJ, Davies GJ. Mycobacterium tuberculosis strains possess functional cellulases. Journal of Biological Chemistry. 2005;280:20181–20184. doi: 10.1074/jbc.C500142200. [DOI] [PubMed] [Google Scholar]
- 71.Medie FM, Davies GJ, Drancourt M, Henrissat B. Genome analyses highlight the different biological roles of cellulases. Nature Reviews Microbiology. 2012;10:227–234. doi: 10.1038/nrmicro2729. [DOI] [PubMed] [Google Scholar]
- 72.Layre E, Sweet L, Hong S, Madigan CA, Desjardins D, Young DC, Cheng TY, Annand JW, Kim K, Shamputa IC, McConnell MJ, Debono CA, Behar SM, Minnaard AJ, Murray M, Barry CE, 3rd, Matsunaga I, Moody DB. A comparative lipidomics platform for chemotaxonomic analysis of Mycobacterium tuberculosis. Chem Biol. 2011;18:1537–1549. doi: 10.1016/j.chembiol.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sartain MJ, Dick DL, Rithner CD, Crick DC, Belisle JT. Lipidomic analyses of Mycobacterium tuberculosis based on accurate mass measurements and the novel “Mtb LipidDB”. J Lipid Res. 2011;52:861–872. doi: 10.1194/jlr.M010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Angkawidjaja C, Koga Y, Takano K, Kanaya S. Structure and stability of a thermostable carboxylesterase from the thermoacidophilic archaeon Sulfolobus tokodaii. FEBS J. 2012;279:3071–3084. doi: 10.1111/j.1742-4658.2012.08687.x. [DOI] [PubMed] [Google Scholar]
- 75.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.