

Abstract
Characterization of proteins that mediate mechanotransduction by hair cells, the sensory cells of the inner ear, is hampered by the scarcity of these cells and their sensory organelle, the hair bundle. Mass spectrometry, with its high sensitivity and identification precision, is the ideal method for determining which proteins are present in bundles and what proteins they interact with. We describe here the isolation of mouse hair bundles, as well as preparation of bundle-protein samples for mass spectrometry. We also describe protocols for data-dependent (shotgun) and parallel-reaction-monitoring (targeted) mass spectrometry that allow us to identify and quantify proteins of the hair bundle. These sensitive methods are particularly useful for comparing proteomes of wild-type and mice with deafness mutations affecting hair-bundle proteins. (120 words; maximum 250)
Keywords: Hair cells, stereocilia, hair bundles, actin, cytoskeleton, espin, Orbitrap, ion trap, shotgun, targeted
1. Introduction
The inner ear, with its auditory and vestibular divisions, measures sound and detects head movements, transmitting this information to the VIIIth cranial nerve and, eventually, the central nervous system. Auditory and vestibular hair cells, the sensory cells, mediate the mechanical-to-electrical transduction at the heart of this process (Fettiplace & Kim, 2014). To carry out mechanotransduction, hair cells use an apical cluster of actin-rich stereocilia, coupled together by various linkages to form a hair bundle (Fig. 1). The stereocilia in the bundle are arranged in staggered rows, giving the structure a beveled appearance. Deflection of bundles by external stimuli, which are transmitted to the hair cells through structures in the outer, middle, and inner ears, leads to open and closing of cation-conducting transduction channels. Special links within the bundle, the tip links, run up the bevel and control the transduction channels; deflection of bundles towards the tallest stereocilia opens channels and depolarizes hair cells. Hair cell depolarization, in turn, triggers neurotransmitter release and the ensuing signal transmission to the central nervous system.
Figure 1. Isolation of utricle hair bundles.
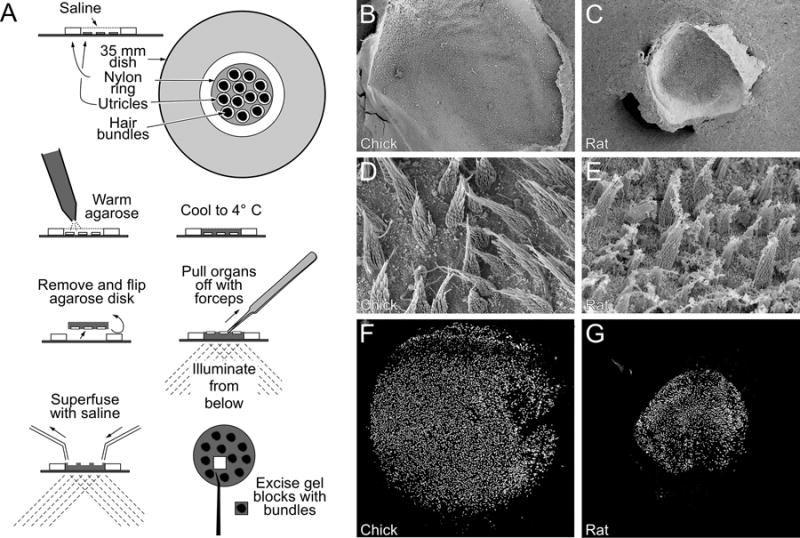
(A) Illustration of twist-off method. Utricles are adhered to the bottoms of plastic culture dishes, and are surrounded by a nylon ring. Warm agarose is applied to the utricles, which is decanted to leave agarose only in the ring. After cooling to gel the agarose, the disk is removed and flipped; the utricles remain in the agarose and their back surfaces are exposed. A quick perpendicular pull on the utricle using forceps leaves the bundles in agarose. Bundles are superfused with saline and cleaned up under a dissecting microscope; small blocks of agarose containing bundles from each ear are excised and frozen. (B–C), Low-power scanning electron micrograph showing chick (B) and rat (D) utricles. (D–E), high-power scanning electron micrograph showing chick (D) and rat (E) hair bundles. (F–G), Hair bundles in agarose, isolated from chick (F) or rat (G) utricles, labeled with phalloidin. Panel full widths: B–C and F–G, 1170 μm; D–E, 41 μm.
Knowledge of the identity, concentrations, and interactions of the proteins that make up hair bundles and control transduction is critical for understanding how these structures operate to carry out transduction with their impressive sensitivity (Barr-Gillespie, 2015). Unfortunately, there are few hair cells in the inner ear; for example, each ear of the mouse has ~10,000 hair cells combined in its cochlea (auditory system) and its five vestibular organs. Moreover, hair bundles account for <1% of the protein in a hair cell, and thus are scarce even within the inner ear. Even more daunting, transduction molecules like the tip link and channel may be present at <1 part in 105 in the bundle. Finally, hair cells are housed in the periotic bone, one of the hardest in the body, and these cells’ sensitivity to minute stimuli renders them extremely delicate, readily damaged in attempts to extricate them. Taken together, these difficulties conspire to slow molecular characterization of this system as compared to others.
Nevertheless, methods have been developed to allow isolation, identification, and quantitation of proteins critical for function of the hair bundle. In particular, the “twist-off” method allows for a clean isolation of bundles from vestibular epithelia, including those of frog, mouse, rat, and chicken. Bundle proteins can be analyzed by shotgun mass spectrometry, which allows determination of the diversity of proteins present and estimation of their concentrations, and by targeted mass spectrometry, which allows accurate measurement of protein abundance. Together these biochemical techniques complement other methods used for characterizing bundle function, including electrophysiology, imaging, and genetics.
2. Isolation of hair bundles
Hair bundles are isolated relatively easily because of the unique structure of their constituent stereocilia (Fig. 1). While stereocilia are 200–500 nm in diameter throughout most of their length, as they approach the apical surface, they taper down to few tens of nanometers wide. Because of this tapering, the stereocilia both flex easily—as part of their physiological role—and are mechanically weak, which means that they can be readily sheared off of the cell. The twist-off method for bundle isolation, originally developed for bullfrog saccule hair bundles (Gillespie & Hudspeth, 1991) and later adapted for hair bundles of mouse (Dumont, Zhao, Holt, Bahler & Gillespie, 2002; Krey, Sherman, Jeffery, Choi & Barr-Gillespie, 2015), rat (Shin, Pagana & Gillespie, 2009; Wilmarth, Krey, Shin, Choi, David & Barr-Gillespie, 2015), and chick (Shin, Streijger, Beynon, Peters, Gadzala, McMillen et al., 2007; Shin, Krey, Hassan, Metlagel, Tauscher, Pagana et al., 2013) utricles, isolates bundles by first trapping them in an agarose matrix, then mechanically displaces them from the utricular epithelium (Fig. 1A).
We describe here isolation of hair bundles from the mouse utricle, a vestibular organ. We have used both shotgun (Francis, Krey, Krystofiak, Cui, Nanda, Xu et al., 2015; Krey et al., 2015; Wilmarth et al., 2015; Ebrahim, Avenarius, Grati, Krey, Windsor, Sousa et al., 2016; Krey, Drummond, Foster, Porsov, Vijayakumar, Choi et al., 2016) and targeted mass spectrometry (Krey et al., 2016; Ebrahim et al., 2016) to characterize mouse utricle proteins; similar methods are used with chick and rat hair bundles (Fig. 1B–G).
2.1 Mouse utricle dissection and twist-off bundle isolation
Originally named for the twisting motion applied to the agarose relative to the fixed utricular epithelia, in the twist-off procedure, the epithelia are adhered to the bottom of a chamber, formed with a small nylon washer, and the buffer solution surrounding them is displaced with molten low-melting-point agarose. After the agarose infiltrates the bundles, it is cooled to allow it to gel firmly. The epithelia are then tugged off, parallel to the agarose, and leaving the trapped bundles in the agarose. With the right illumination angle, bundles (and cellular debris) can be visualized on a dissecting microscope, allowing cleaning of the embedded bundles prior to excision.
2.1.1 Dissecting mouse utricles
Prepare dissection solution by adding 1 M HEPES at pH 7.4 to Leibovitz’s L-15 Medium (without phenol red) to a final concentration of 5 mM. Hank’s Balanced Salt Solution (HBSS) can also be used. Oxygenate for 5–15 min.
Euthanize mice using CO2 asphyxiation; use decapitation for animals younger than postnatal day 8 (P8). Utricles from 20–25 mice can be processed at one time.
Decapitate mice, cut the skull sagittally along the midline, remove brain, and cut out periotic bone containing ear. Place temporal bones in dissection solution in petri dish on ice. Repeat for all mice.
Transfer each periotic bone to new petri dish with cold dissection solution. For mice older than P8, use dissection scissors to cut the bones encasing the semicircular canals, then use forceps to chip open the inner-ear capsule and expose the utricle. This procedure can be done entirely with forceps for younger animals. Carefully remove the utricle, using forceps to cut the nerve and semicircular canals. Transfer utricles to new dish; a cut 200 μl plastic pipette tip that has been passivated with a 10% BSA solution works well as a transfer pipette for mouse utricles.
Use forceps to complete removal of the nerve from back of utricle and remove the endolymphatic sac. Carefully remove the otolithic membrane using a fine eyelash. Once all utricles have been dissected, transfer them to new petri dish with fresh dissection solution. Wash 1–2 times with dissection solution.
2.1.2 Embedding utricles in agarose
Prepare a 4.5% agarose solution by adding 10 ml dissection medium to 0.45 g low-melting point agarose, swirling to mix. Microwave until agarose is dissolved (~30 sec), stopping to swirl the tube every 5 sec. Filter through a 5 μm syringe filter into a new 50 ml conical tube and allow to cool to 40–42°C in a water bath.
While cooling, fill a new uncoated 35 mm bacteriological petri dish (BD Falcon; cat #351008) with dissection solution. Carefully transfer utricles to the dish with the transfer pipette. Under a dissection microscope, use forceps to stick down the organs (nerve side down) to the bottom of the dish. Use leftover tissue flaps from the endolymphatic sac to help flatten the utricles as much as possible without touching the sensory epithelium. To give a guide to utricles placement, trace the inside of the washer on the bottom side of the dish with a marker prior to adding dissection solution.
Once all utricles are adhered to the dish, carefully place a nylon washer ring into the dish (angling it into the surface of the liquid) to surround the utricles. Using a disposable plastic transfer pipette, slowly and carefully pipette the agarose solution onto the utricles and fill the inside of the washer ring (the agarose should be level with or higher than the top of the washer ring). It is helpful to do this under a dissection microscope to ensure even dispersal of the agarose.
Leave the dish untouched at room temperature (RT) until the agarose has turned opaque, then remove remaining saline. Place the covered dish at 4°C for at least 15 min to fully solidify the agarose.
2.1.3 Twist-off
Set up the isolation apparatus in a laminar flow hood. The apparatus includes a dissecting microscope and stage (30 cm wide × 13 cm deep × 7.5 cm tall) that has a 7.5 × 3.8 cm rectangular opening that allows placement of a glass slide, on which a nylon ring is permanently attached. A fiber-optic light is placed underneath for visualization of the bundles within the agarose. Superfusion and suction tubes are attached to stage and a peristaltic pump is used to pump cold dissection solution across the agarose during the procedure.
Use a clean scalpel blade to cut away excess agarose from around the washer. Turn washer on its side and slice away any excess agarose from the top so that only an even disk of agarose inside of the washer remains. The organs should remain embedded in the agarose on one side. Remove the agarose disk from the washer and place it organ side up inside the washer affixed to the glass slide on the stage. Illuminate from below with the fiber optic light and start superfusion with cold dissection media.
Using forceps, grab the peripheral edge of each organ and quickly pull from the agarose, parallel to the surface of the agarose disk. Discard the organs. Adjust the position of the light to allow clear visualization of the bundles; they act as light pipes, so oblique illumination increases their contrast with the surrounding agarose. Use a fine scalpel blade or tungsten needle to cut away any agarose surrounding the bundles that contains cellular debris. It is easiest to carve around the regions containing bundles, leaving them on an agarose pedestal. Use a fine tungsten needle to slice underneath the bundles and then pierce the chunk of agarose containing the bundles and transfer it to a low-protein-binding microfuge tube. Store bundles at −80°C until processing.
Purified hair bundles always contain some contaminants from the whole epithelium. Purity of bundles can be assessed in several ways. First, bundles and contaminants can be visualized by fluorescence microscopy directly in the agarose; stereocilia actin can be stained with fluorescent phalloidin, nuclei with DAPI or any other nuclear dye, and contaminants with selective antibodies (Shin et al., 2007; Shin et al., 2013; Krey et al., 2015). While not quantitative, fluorescence microscopy can reveal whether certain contaminants (e.g., cuticular plates) are present along with bundles. Mass spectrometry can also be used to assess purity (see below).
2.2 Biochemical manipulation of hair bundles in agarose
Hair bundles embedded in agarose can be subjected to biochemical manipulation, including selective extraction to determine properties of individual proteins (Gillespie & Hudspeth, 1991; Walker, Hudspeth & Gillespie, 1993) or manipulation of nucleotide state (Gillespie, Wagner & Hudspeth, 1993; Yamoah & Gillespie, 1996). Although purified bundles are quite dilute—bundles from one ear, ~5 ng of total protein, can be recovered in about 1 μl of agarose—the agarose prevents the bundles from being lost to tube surfaces and allows easy sedimentation. Extraction of bundle proteins therefore requires addition of a carrier, such as a purified protein, that prevents nonspecific binding but does not interfere with analysis; in different experiments, we have successfully used lysozyme, myoglobin, hemoglobin, bovine serum albumin, and maltose-binding protein (Gillespie & Hudspeth, 1991; Gillespie et al., 1993; Walker et al., 1993). We do not add carrier proteins, however, when carrying out mass spectrometry analyses.
2.3 Sample preparation for mass spectrometry using in-gel digestion
Prior to mass spectrometry, hair-bundle proteins must be reduced, alkylated, and digested by trypsin to peptides. Because of the small amounts of protein, efficiency of these steps is of utmost importance. In-gel digestion of proteins (Shevchenko, Tomas, Havlis, Olsen & Mann, 2006) relies on SDS-PAGE to eliminate contaminants that otherwise might interfere with mass spectrometry; moreover, little handling is required, and proteins remain in high concentrations of SDS, minimizing protein loss. In-gel digestion has limitations, however. Some proteins aggregate strongly and never enter the gel matrix, preventing their analysis; in addition, small proteins can diffuse out of the gel matrix and are lost during initial steps. Finally, unless handled with extreme precautions, gels attract keratins during processing and thus usually have higher levels of contamination than in-solution digests. Recovery from gels can be improved by addition of a degradable surfactant (ProteaseMAX or RapiGest) to digestion solution. In-gel digestion also allows for fractionation of the sample by cutting the gel lane into multiple pieces corresponding to different protein masses. While fractionation is associated with some additional sample loss, the multiple shotgun runs performed on each sample can increase proteome coverage.
2.3.1 Sample preparation
To avoid keratin contamination, all steps are performed a in laminar flow hood, with lab coat and nitrile gloves, taking care to rinse gloves often. All solutions are made fresh on the day of the procedure. Sample preparation, including SDS-PAGE and reduction and alkylation, has been described in detail elsewhere (Shin, Longo-Guess, Gagnon, Saylor, Dumont, Spinelli et al., 2010; Shin et al., 2013; Wilmarth et al., 2015; Krey et al., 2015). Isolated hair-bundle samples are thawed on ice, then run on a 4–12% Bis-Tris 1.5 mm 10-well gradient gel (Invitrogen) at 150 V for 10 min until top of dye front is ~1 cm from bottom of wells. The gel is washed with water, stained for 5 hr with Imperial Stain (ThermoFisher) at RT, and washed with water overnight. Keep gel covered at all times. Each lane of the gel is then cut into six 1–2 mm wide slices, each of which is cut into four pieces.
Reduction is carried out with 25 mM DTT in 50 mM ammonium bicarbonate pH 8 for 20 min at 56°C, then the sample is alkylated with 55 mM iodoacetamide in 50 mM ammonium bicarbonate pH 8 in the dark for 20 min at RT (Shin et al., 2010; Shin et al., 2013; Wilmarth et al., 2015; Krey et al., 2015). The gel slices are then washed, stepped into 100% acetonitrile, and are dried by vacuum concentration or air-drying for 5 min in a laminar flow hood.
2.3.3 In-gel digestion
We have had the greatest success using the procedure outlined in the ProteaseMAX manual (Promega) for low abundance protein samples. Gel pieces are rehydrated with 30 μl of 6 ng/μl trypsin, 0.01% ProteaseMAX, 50 mM ammonium bicarbonate pH 8 for 30 min at 4°C. The samples are overlaid with 20 μl of 0.01% ProteaseMAX and digested for 3 hr at 37°C. The digest solution is removed after centrifugation, then 30 μl of 2.5% trifluoroacetic acid (in HPLC-grade H2O) is added to gel pieces and vortexed for 15 min. The combined supernatants are transferred to 0.45 μm filter tubes (Millipore Ultrafree centrifugal filters, #UFC0HV00), and are spun 5 min at 4000 rpm. The filtrated is dried in a vacuum concentrator until all solution is evaporated; the tubes with dried peptides are stored at −80°C until analysis.
2.4 Sample preparation for mass spectrometry using eFASP
In-solution digestion is the least labor-intensive and allows for optimal peptide recovery as samples are not transferred from tube to tube. We have achieved the best results with bundles using the eFASP (enhanced filter-assisted sample preparation) method (Erde, Loo & Loo, 2014). The eFASP method carries out each of the preparation steps in an ultrafiltration unit, which allows solution exchange and prevents losses from tube to tube transfers. Since solution exchange occurs, the eFASP method is compatible with samples solubilized in SDS, and subsequently uses the surfactant deoxycholate to enhance trypsinization. The methodology also removes traces of agarose that interfere with mass spectrometry and is compatible with multiple-dimension chromatography separations.
To carry out eFASP-mediated reduction, alkylation, and trypsin digestion, we follow the “eFASP with Passivated Ultrafiltration Unit” procedure described by Erde et al. (2014). Proteins are reduced with TCEP, alkylated with iodoacetamide, and digested with 200 ng sequencing-grade modified trypsin (Promega) in the filter unit; a total volume of 100 μl digestion buffer is used and the reaction is carried out at 37°C for 12–16 hr. After isolating peptides by centrifugation, they are extracted with ethyl acetate to remove remaining deoxycholic acid.
3. Shotgun mass spectrometry
Shotgun mass spectrometry, which uses the mass spectrometer’s data-dependent acquisition (DDA) modality, is the method of choice when the proteins present in a sample are unknown (Figs. 2–3). Peptides eluting from the LC system are first subjected to mass analysis (the MS1 spectrum), and the instrument then chooses the most abundant 5–10 precursor ions for fragmentation. While biased for more abundant peptides, some breadth of coverage is obtained by the use of a dynamic exclusion list where recently fragmented ions are ignored for a set period of time (e.g., 30 sec), increasing the chance that lower-abundance ions can be analyzed. Ions chosen are fragmented by collision-induced or higher-energy-induced dissociation, and the m/z patterns of fragment ions are acquired. All of the m/z patterns (the MS2 spectra) are searched against theoretical MS2 spectra, derived from a protein database. Despite some biases against small proteins, proteins with unusual amino acid compositions, and membrane proteins, as long as a protein is present in the database, theoretically it can be detected. Given the poorly characterized proteome of the hair bundle, shotgun mass spectrometry has been the logical first step in defining which proteins are present.
Figure 2. Flow chart illustrating sample preparation and analysis by shotgun and targeted mass spectrometry.
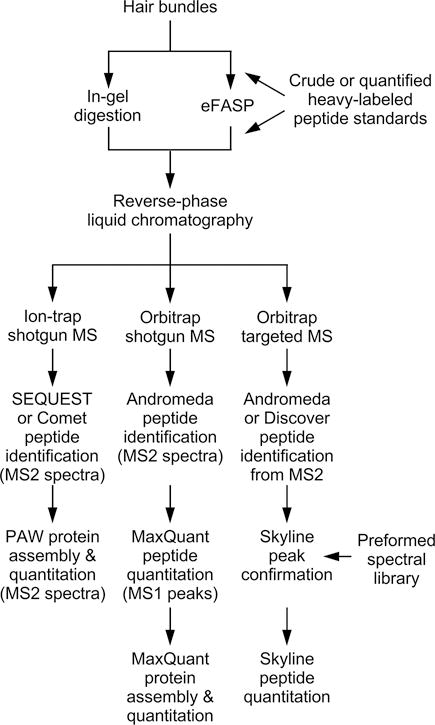
Hair bundle proteins can be prepared for mass spectrometry using either in-gel or eFASP digestion; if used, standards (e.g., heavy-isotope labeled peptides) are added after digestion. Peptides are separated by reverse-phase liquid chromatography, introduced into the mass spectrometer by electrospray ionization, and are subjected to tandem mass spectrometry. If data are acquired on an ion-trap mass spectrometer, MS2 spectra are analyzed by Comet peptide searching and PAW protein quantitation. Shotgun data acquired on an Orbitrap (high resolution) mass spectrometer are analyzed by Andromeda peptide searching of MS2 data and MaxQuant protein quantitation using MS1 peak intensities, while targeted data are analyzed by Skyline after Andromeda or Protein Discoverer peptide searching.
Figure 3. Shotgun and targeted mass spectrometry.
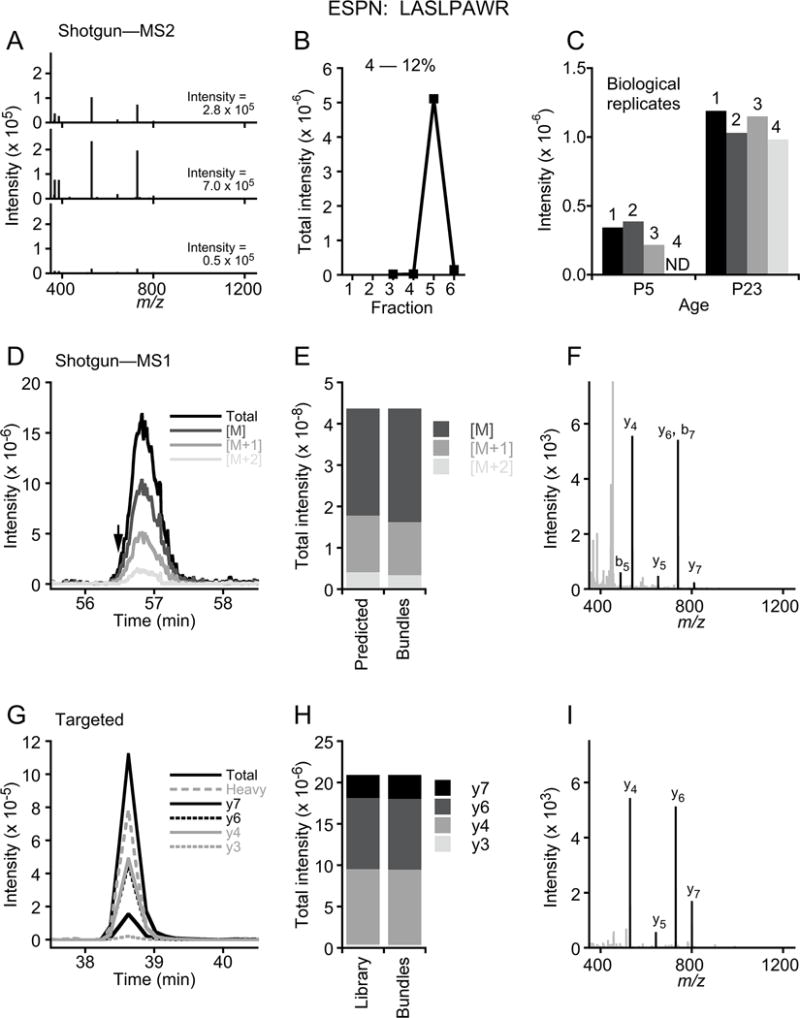
Comparison of ESPN peptide LASLPAWR using shotgun MS2 (A–C), shotgun MS1 (D–F), and targeted (G–I) analyses. Data for A–C is from ProteomeXchange PXD002167, while data for G–I is from Ebrahim et al. (2016). (A) Examples of three matched LASLPAWR spectra, all from a P23 mouse hair-bundle replicate, and the summed intensities of the matched ions. (B) All matched intensities for four P23 bundle samples, separated by gel slice number (high to low mass). Note that almost all LASLPAWR intensity is in the fifth fraction, appropriate for the small ESPN isoform molecular masses of 28–34 kD. (C) Reproducibility of LASLPAWR detection among mouse bundle biological replicates. ND, not detected. (D) Chromatographic profile for LASLPAWR peptide; the single MS2 identification is indicated with arrow. Isotopic distribution is shown; [M] corresponds to the monoisotopic peak (parent), while [M+1] and [M+2] are the ions with 1 or 2 additional mass units due to natural heavy isotopes. Together the isotopic peaks have an intensity of 4.4 × 108 units; to quantify ESPN, iBAQ values for all peptides matched to ESPN are summed and divided by the number of peptides theoretically observable (i.e., between 6 and 30 amino acids). (E) Isotopic peak distribution predicted based on peptide sequence and measured from bundle ESPN peptide. (F) MS2 spectrum identified from peptide peak region. Matched y- and b-ions are labeled and are black; unmatched peaks are gray. (G) Chromatographic profile for LASLPAWR peptide; there were 12 MS2 spectra acquired across the peak (not shown). A heavy-atom peptide standard was co-injected, and it co-elutes with the quantified bundle peptide peak. The four monitored daughter ions are indicated. To quantify ESPN, the areas for each daughter ion were summed, then the amount was determined by comparison to the heavy peptide area and its starting concentration. Note that LASLPAWR elutes earlier in G because of the shorter gradient than that in D. (H)) Intensities for each daughter ion, as compared to those in the spectral library. The dot product of 0.99 indicates high correspondence. (I) Example MS2 spectrum identified from peptide peak region.
3.1 Instrumentation
We have used several instrument configurations for shotgun mass spectrometry of hair-bundle samples. We used two Orbitrap (high resolution) instruments: (1) a Thermo Electron Orbitrap Velos mass spectrometer and a Protana nanospray ion source, interfaced to a reversed-phase capillary column of 8 cm length × 75 μm internal diameter, self-packed with Phenomenex Jupiter C18 particles of 10 μm size; and (2) a Thermo Orbitrap Fusion Tribrid mass spectrometer coupled to a Thermo/Dionex Ultimate 3000 Rapid Separation UPLC system and EasySpray source, using an Acclaim PepMap C18 trap and a 25 cm × 75 μm EasySpray PepMap RSLC C18 column, with 2 μm particle size.
Ion-trap configurations were: (3) a Thermo LTQ linear ion trap mass spectrometer and a Thermo Ion Max source, with a Michrom trap cartridge and an Agilent 25 cm × 500 μm Zorbax SB-C18 column with 5 μm particles controlled with an Agilent 1100 series capillary LC system; (4) a Thermo LTQ Velos linear ion trap mass spectrometer and a Thermo Ion Max source, also with Michrom trap and Zorbax column controlled by an Agilent 1100 series LC; (5) a Thermo LTQ Velos linear ion trap mass spectrometer and a Microm CaptiveSpray source, with a Symmetry C18 trap cartridge and a Waters 25 cm × 75 μm nanoACQUITY BEH130C18 column with 1.7 μm particles controlled with a Waters nanoACQUITY LC system.
Prior to mass spectrometry, peptide samples are dissolved in 5% formic acid and transferred to autosampler vials for injection. Details of sample loading, chromatography, electrospray ionization, and mass spectrometry are described elsewhere (Shin et al., 2013; Krey, Wilmarth, Shin, Klimek, Sherman, Jeffery et al., 2014; Francis et al., 2015; Wilmarth et al., 2015; Krey et al., 2015; Krey et al., 2016; Ebrahim et al., 2016). Data from these ThermoFisher instruments are saved as RAW files, which are then analyzed using the PAW and MaxQuant pathways described below.
3.2 Data analysis using the PAW pipeline
We use two different methods for analysis of shotgun mass spectrometry data. For all ion-trap data, and for some data collected on Orbitrap instruments, we used a pipeline that includes peptide identification with SEQUEST or Comet, followed by peptide validation and protein assembly using the PAW pipeline developed at OHSU (Wilmarth, Riviere & David, 2009). The PAW pipeline employs a “best practices” philosophy for proteomics data analysis. Linear-discriminant score transformations are used to increase sensitivity (Keller, Nesvizhskii, Kolker & Aebersold, 2002), target/decoy database strategies are used to control identification error rates (Elias & Gygi, 2007), and false-discovery rates are controlled independently on different subclasses of peptides (Ma, Dasari, Chambers, Litton, Sobecki, Zimmerman et al., 2009). Support for accurate mass on high-resolution instruments has been added; narrow tolerances on mass differences between measured and calculated peptide masses are used to generate conditional score distributions, which are used in conjunction with wider tolerance searches to maximize sensitivity. This approach to Orbitrap data was developed several years ago (Hsieh, Hoopmann, MacLean & MacCoss, 2010) but has been largely ignored in proteomics data analysis.
Typical analyses start with conversions of the instrument RAW files into human-readable MS2-formatted files (Tabb, McDonald & Yates, 2002) using MSConvert from the Proteowizard toolkit (Chambers, Maclean, Burke, Amodei, Ruderman, Neumann et al., 2012). Protein sequence databases are prepared using utilities available at www.ProteomicAnalysisWorkbench.com. Database searches are performed using Comet (Eng, Jahan & Hoopmann, 2013), an open source implementation of the original SEQUEST algorithm (Eng, McCormack & Yates, 1994). Some Comet parameter settings depend on whether the instrument is a low-resolution ion trap or an Orbitrap; other parameters are independent of instrument. Common independent search parameters are protein database, tryptic enzyme specificity, missed enzymatic cleavages (maximum of two), static modification on cysteine residues (alkylation +57.0215 Da), variable oxidation of methionine modification (+15.9949 Da), and monoisotopic fragment ion mass tolerance of 1.0005. For low-resolution instruments, the parent ion mass tolerance is an average mass window of 2.5 Da. For Orbitraps, we use a monoisotopic parent ion mass tolerance of 1.25 Da. The delta mass values in Da (difference between measured peptide mass and assigned sequence calculated mass) for all identifications are histogrammed in small bins separately for target identifications and for decoy identifications. Narrow windows are set around the 0 Da peak, and the 0.984 Da/1.003 Da doublet (deamidation and first isotopic peaks, respectively) to create sets of score histograms conditioned on accurate delta mass. Score histograms for all delta masses not located inside of these two narrow regions are also generated. Orbitrap instruments frequently cannot determine accurate monoisotopic masses or charge states for low abundance peptide signals, such as those from hair-bundle samples. Many correct identifications do not have precisely measured parent ion masses, and these identifications can be lost in searches with narrow parent ion mass tolerances.
Post-processing of search results to filter peptides to desired false discovery rates, as well as protein inference using standard and extended parsimony principles, are done with a suite of Python programs. Spectral counting (Liu, Sadygov & Yates, 2004) and MS2-intensity weighted spectral counting (Spinelli, Klimek, Wilmarth, Shin, Choi, David et al., 2012; Krey et al., 2014) are supported.
When using PAW to analyze ion-trap mass spectrometry data from purified hair bundles (Shin et al., 2013; Wilmarth et al., 2015; Krey et al., 2015), we exclusively use MS2 intensities for quantitation (Fig. 3A–B). All intensities from matched MS2 spectra for a given peptide, for example the ESPN (espin) peptide LASLPAWR (Fig. 3A,C), are summed for a given biological replicate. Technical replicates are not used for purified-bundle experiments. Because a single sample is fractionated into six gel slices, each of which is processed and run separately, the peptide’s identification can occur in different fractions (Fig. 3B). Detection of a single peptide is reasonably reproducible when it is present at moderate to high levels, but becomes more stochastic as its levels drop (Fig. 3C). To generate the intensity for a single protein in a single replicate, intensities from all identified peptides for that protein are summed. Further processing for quantitation is described in section 3.5.
3.3 Data analysis using MaxQuant
We also use MaxQuant (Cox & Mann, 2008) to analyze data collected on Orbitrap mass spectrometers. MaxQuant identifies peptides using the search engine Andromeda (Cox, Neuhauser, Michalski, Scheltema, Olsen & Mann, 2011) and quantifies them by measuring MS1 peak intensity (Fig. 3D–F); the program carries out intensity-based absolute quantitation (iBAQ), which we use for quantitation (Shin et al., 2013; Krey et al., 2014).
MaxQuant (available at http://www.coxdocs.org/doku.php?id=maxquant:start) is installed as described. With the exception of iBAQ quantitation and “Match Between Runs” (occasionally used), all settings are the default ones. The principal output (“proteinGroups.txt”) is processed further using a custom Mathematica program and using Excel spreadsheet manipulation. The Mathematica program (a) rearranges the table to reduce the number of columns; (b) replaces the description and symbol for the “best protein”; (c) removes entries flagged by MaxQuant as contaminants (reversed entries are maintained to allow calculation of the false-discovery rate); (d) finds symbols for all matched identifiers; (e) finds all the MaxQuant groups that share a defined fraction of their peptides, which is defined by the user (we usually use ≥20%); (f) consolidates those groups with shared peptides together into a “super-group”, making a list with all contributing protein symbols; (g) sums the number of unique peptides and the iBAQ values for the super-group; and writes an output file. Further processing for quantitation is described in section 3.5.
3.4 Protein databases
We generally use Ensembl databases for searching shotgun data, acquired using the BioMart tool (http://uswest.ensembl.org/biomart/martview/). Because they include predicted and verified splice products, Ensembl databases are relatively large and have greater tryptic peptide redundancy. This increases processing times and increases peptide-level quantification ambiguity (which we address through protein grouping). Advantages of Ensembl databases are completeness of gene products and rich gene-level annotation.
3.5 Quantitation of proteins in bundles
For samples analyzed by Comet and PAW, the summed intensities (i) for each protein or protein group in each separate replicate are divided by the protein’s molecular mass; each i/MW value is then divided by the sum of all non-reversed, non-contaminant i/MW values for that run, giving the relative molar intensity for each protein (im). im values are averaged across all replicate runs, giving an average im. Stochastic sampling of MS2 spectra during shotgun analysis leads to considerable variability, and at least four biological replicates are favored for each condition. Analysis using known standards diluted in a complex protein background showed that im and riBAQ methods for protein quantitation are on average accurate (Krey et al., 2014), with somewhat better performance by riBAQ. While this correspondence does hold on average, it is not necessarily true for an individual protein. Thus the estimates of the amounts of each protein present in a proteome—as determined with shotgun mass spectrometry—must be used with caution.
For samples analyzed by MaxQuant, the Mathematica output file is opened in Excel and iBAQ values for all non-reversed, non-contaminant entries are summed for each replicate; the iBAQ for a given protein from that replicate is then divided by the summed iBAQ, yielding riBAQ. For each protein, riBAQ values from each replicate are averaged together to give the mean riBAQ for that protein under that condition.
Bundle-to-epithelium enrichment, which helps identify which proteins are concentrated in hair bundles and which are present as contaminants, is calculated by dividing each protein’s mean im or riBAQ value for bundles by the mean im or riBAQ value for the epithelium. In addition, we calculate molecules per stereocilium for each bundle protein; the im or riBAQ value for each protein is divided by the value for actin, then multiplied by 400,000 actin monomers per stereocilium. While this value for actin varies from species to species and certainly within a bundle (stereocilia in a single chick or rodent utricle bundle range from 2–15 μm in length), it is useful to use a single value to allow comparison between species.
We assess the degree of contamination by the epithelium in a hair-bundle sample by quantifying its histone content. Relative molar abundance for each contaminant is calculated as discussed below; as no histones should be present in the bundle samples, the bundle-to-epithelium ratio for each histone directly indicates the degree of contamination. Histone analysis indicates that chick or mouse bundle preparations have 2–20% contamination, depending on the particular experiment. Other contaminants may be present at higher levels, however, if they are more readily captured by the agarose than is whole epithelium debris. For example, small amounts of apical microvilli, cuticular plates, and mitochondria all can be visualized in the bundle preparation using transmission electron microscopy (Gillespie & Hudspeth, 1991). If proteins making up these components are known, their enrichment can be calculated separately from the shotgun proteomics data; their enrichment can be compared to histone enrichment to determine if they are selectively isolated by the twist-off method.
4. Targeted mass spectrometry
While shotgun mass spectrometry allows determination of the spectrum of proteins present in a given sample, accuracy and precision of protein abundance in these experiments suffers from protein-to-protein variability inherent with data-dependent acquisition. In addition, shotgun proteomics tends to be biased towards detection of higher abundance peptides, often failing to detect very low abundance proteins. Targeted proteomics offers a different strategy for protein detection and quantitation, and is well suited for comparison of proteins to standards and thus absolute quantitation (Liebler & Zimmerman, 2013). In targeted assays, specific peptides from each protein of interest are isolated and fragmented within the mass spectrometer, and the intensity of specific fragment ions from each peptide are monitored over the entire chromatographic elution (Fig. 3G–I). Targeted proteomics can be performed using selected reaction monitoring (SRM) or parallel reaction monitoring (PRM), depending on the instrument. We use PRM (Gallien & Domon, 2015), which takes advantage of a high-resolution hybrid mass spectrometer (quadrupole-Orbitrap), the same instrument we now also use for shotgun analysis of bundle samples. While both SRM and PRM have comparable sensitivity, reproducibility, and precision, PRM requires less effort in building assay methods. PRM acquisition produces high-resolution fragmentation (MS2) spectra across a wide m/z range that can also be used to confirm the sequence of each peptide, and chromatographic peaks from each fragment ion that can be quantified to accurately measure the amount of the peptide in the sample. The intensities of multiple peptides are then used to infer the abundance of the corresponding protein. Targeted assays have been shown to quantify proteins in the attomole range and thus are well suited towards detection of scarce proteins in low protein amount samples like purified hair bundles.
4.1 Designing a targeted mass spectrometry method
Targeted proteomic assays for hair-bundle proteins require optimal proteotypic peptides for each protein. Proteotypic peptides are unique to the protein of interest and, when analyzed by mass spectrometry, have favorable fragmentation characteristics and high intensity per mole (Mallick, Schirle, Chen, Flory, Lee, Martin et al., 2007). Response characteristics of peptides derived from any protein vary enormously due to differences in sequence-specific physicochemical properties; for this reason, choosing the highest-responding peptides for each protein can greatly boost sensitivity. Proteotypic peptides can be identified from shotgun experiments, retrieved from public spectral databases (www.peptideatlas.org, www.thegpm.org), or predicted using online tools (e.g., Prego, CONSeQuence, ESP). Proteotypic peptides must be unique to the protein of interest and typically are 7–26 amino acids in length, have doubly or triply charged m/z values within the mass range of the instrument, are efficiently produced during enzymatic digestion, and have no known modification sites or amino acids prone to variable modification during sample processing (e.g., methionine oxidation).
We have successfully used peptides that were routinely identified in shotgun runs with relatively high MS1 peak intensities. The shotgun runs also provide useful instrument-specific information for these peptides, including retention time, precursor charge state, fragmentation pattern, and relative fragment ion intensities. When targeting very low-abundance proteins with minimal shotgun data, databases or prediction algorithms can be used, although peptides predicted by these approaches are not always detected. Digestion methods used and the modification state of the endogenous protein both can influence which theoretical tryptic peptides actually get cleaved or detected. Empirical data acquired by analyzing all possible tryptic peptides using a recombinant protein is perhaps the ideal approach for identifying the highest responding peptides for a protein, accounting for variables associated with chosen digestion and LC-MS/MS methods (Bollinger, Stergachis, Johnson, Egertson & MacCoss, 2016).
Once optimal peptides have been identified for each protein, multiplexed targeted experiments are designed to measure the best 3 to 5 peptides for each of the group of proteins to be assayed simultaneously. With PRM, because a full MS2 spectrum is collected for each peptide, only the precursor m/z (and charge state) of the targeted peptide is entered into the isolation list. Precursor isolation lists for all peptides are exported from Skyline (http://proteome.gs.washington.edu/software/skyline/), a freely available software package (MacLean, Tomazela, Shulman, Chambers, Finney, Frewen et al., 2010), into Orbitrap control software.
4.2 Internal standards for targeted quantitation methods
Quantitation with targeted proteomics can use either label-free or label-based methods. In label-based quantitation methods, heavy isotope-labeled peptides or proteins are spiked into the sample during processing and are used to calibrate signals of the endogenous peptides. If the concentration of the heavy peptide or protein is known, “absolute” quantitation of the endogenous peptide can be carried out. If heavy peptides are not used, label-free quantitation methods can be used to measure the relative amounts of each peptide in different samples. While heavy peptides or proteins can both be used as internal standards, proteins offer some advantages as they are spiked into the sample at the beginning of processing, minimizing variability introduced during sample processing. Indeed, the use of heavy peptides assumes complete efficiency of trypsin digestion for the endogenous peptide, which is not always the case. Full-length heavy protein standards are very expensive and labor-intensive to make, however, so the use of heavy peptides is advantageous when seeking to quantify multiple proteins within a sample. We will focus on the use of heavy peptides as internal standards, the approach we have taken with targeted quantitation of hair-bundle proteins.
4.2.1 Label-based quantitation
The most commonly used internal standards for label-based quantitation are heavy isotope-labeled peptides with 13C and 15N atoms in the C-terminal arginine or lysine residue. These peptides are ordered to be identical in sequence (with any modifications) to chosen endogenous proteotypic peptides. While the precursor mass for the heavy peptide will be higher (and thus distinguishable from the endogenous peptide in the mass spectrometer), properties such as the retention time and fragmentation pattern will be identical to the endogenous peptide. The peptides are spiked into the samples following trypsin digestion (or along with trypsin if eFASP processing or SpikeTides peptides are used, see below). By including them in the sample processing, one can detect and correct for any technical variability between runs and thereby achieve more reproducible quantitation. Both heavy and light peptide precursor masses are included in the isolation list used during the LC-MS/MS run, and the heavy/light ratio of the co-eluted fragment ion peak areas is calculated to determine the endogenous peptide amount (Fig. 2D). For absolute quantitation, the heavy peptide must be highly purified (with high isotopic purity) so that accurate quantitation of the synthesized peptide can be performed. Purified heavy peptides can be very expensive but we have had success with the relatively inexpensive SpikeTides_TQL from JPT Technologies (www.jpt.com). To avoid artifacts, the spiked-in heavy peptide should produce a peak area that is similar to that of the endogenous peak. A calibration curve for each peptide should be produced to ensure that it performs linearly around the measurement range and that the measurements are not being performed outside the linear range of quantitation. Heavy labeled peptides can also be used in relative quantitation experiments (e.g., bundles collected at different developmental time points), spiking the same amount into each sample. In this case, relatively inexpensive crude heavy peptides, which are supplied with an imprecise concentration, can be used. In this case, the ratio between heavy and light peptide peaks is calculated and compared between samples to determine the relative concentration of each protein.
4.2.2 Label-free quantitation
Label-free quantitation, a much simpler and more cost-effective approach, is ideal for experiments in which only semi-quantitative measurements need to be collected or in which large changes in protein abundance are expected between samples (e.g., when comparing hair bundles collected from wild-type and knockout mice). In a label-free approach, equal amounts of bundle samples are digested and run without including any heavy peptides. Fragment-ion peak areas are normalized to the peak areas for actin peptides, which are also spiked into each sample to control for differences in bundle recovery and concentration within each sample. While truly label-free targeted proteomics can be carried out, use of crude heavy labeled peptides (e.g., SpikeTides_L from JPT) is highly recommended to demonstrate that the monitored peptide is correctly identified by having the same elution time, parent-ion isotopic distribution, and fragmentation pattern as the heavy peptide.
4.3 Peptide analysis using PRM
We perform our targeted assays using an Orbitrap Tribrid Fusion quadrupole-Orbitrap hybrid mass spectrometer coupled to a Thermo/Dionex Ultimate 3000 Rapid Separation UPLC system and EasySpray nanosource. Samples are loaded onto an Acclaim PepMap C18, 5 μm particle, 100 μm × 2 cm trap using a 5 μl/min flow rate and then separated on a EasySpray PepMap RSLC, C18, 2 μm particle, 75 μm × 25 cm column at a 300 nl/min flow rate. Solvent A is water and solvent B is acetonitrile, each containing 0.1% (v/v) formic acid. After loading at 2% B for 5 min, peptides are separated using a 55-min gradient from 7.5–30% B, 10-min gradient from 30–90% B, 6-min at 90% B, followed by a 19 min re-equilibration at 2% B. This chromatographic gradient produces sharp peaks with an average width of ~30 sec. Increasing the length of the gradient is not advisable for targeted runs as it will decrease the peak sharpness. In the mass spectrometer, precursor ions for each peptide are isolated within the quadrupole, then full high-resolution MS2 scans are performed on each precursor with the Orbitrap detector at 30,000 resolution following higher-energy collisional dissociation. Targeted MS2 parameters for our bundle experiments typically include an isolation width of 2 m/z for each precursor of interest, collision energy of 30%, AGC (automatic gain control) target of 5 × 104, maximum ion injection time of 100 ms, spray voltage of 2400 V, and ion transfer temperature of 275°C. These parameters should be optimized, however, for the particular sample, instrument, and chromatographic gradient used. For reliable quantification, at least 6–8 scans are acquired across the chromatographic peak for each peptide. The cycle time, or time it takes the instrument to cycle through the entire list of peptides, should be adjusted based on the average peak width to ensure adequate sampling across the peak. The cycle time is determined by the number of peptides in the isolation list, the injection time, and the resolution used to collect the MS2 scan. With our instrument, we have found empirically that no more than 75 precursors can be targeted in each run. Because hair-bundle samples have very small amounts of protein, we use a relatively long injection time, which allows collection of as many ions as possible and increases the sensitivity for low-abundance proteins. To shorten cycle time and increase peak coverage without changing injection times, the number of peptides analyzed in each run can be reduced, the resolution can be decreased, or peptide acquisition can be scheduled for a specific time during the gradient. Higher abundance peptides are more likely to reach the AGC target before the maximum injection time is reached and thus cycle time will be shorter for these.
4.4 Data analysis using Skyline
Skyline software is optimized for the analysis of targeted MS/MS data (MacLean et al., 2010). Tutorials, webinars, and other resources are available at the Skyline website and are extremely useful for training of new users. Chromatographic and spectral data from the RAW files are loaded into Skyline and manually analyzed to confirm fragment ion peaks corresponding to each peptide, most often using spectral libraries generated from shotgun results. RAW files are also processed using Proteome Discoverer (ThermoFisher) software in order to match MS2 spectra to an Ensembl protein database using Sequest HT. Once the data have been imported, the different precursor masses targeted for each peptide can be scanned and the extracted-ion chromatograms of all detected fragment ions, retention times, and quality of match to MS2 spectra in a spectral library can be visualized. The mass error (difference between the expected and measured mass) of the fragment ions can also be examined, helping identify background peaks within the chromatogram.
Skyline automatically identifies the boundaries of each chosen peak, but manual inspection and correction of the boundaries should be performed. The peak boundaries of the light and heavy peptides should be identical. Fragment-ion peaks for each peptide are chosen according to the following criteria: 1) three or more co-eluting fragment ions contribute to the peak signal, 2) six or more data points collected across the peak, and 3) one or more MS2 spectra within the peak match to the correct peptide sequence within the spectral database. Any fragment-ion peaks that have co-eluting, interfering background ions can be removed. When MS2 spectra are not directly identified by Sequest, peaks are integrated provided a) the retention time of the chosen peak is within 2 min of the retention time of an identified peak for that peptide in another sample, and b) the type of daughter ions contributing to the peak match the identified peptide peak from another sample. If no peaks matching these criteria are found in a particular sample, the peak area is counted as zero. Chromatographic peak areas from all detected fragment ions for each peptide are integrated and summed to give a final peptide peak area. The peptide peak areas for each protein of interest are averaged for each sample. For calibration curve samples, a linear regression of the heavy peptide peak area in each of four calibration samples is performed and tested for linearity in the measurement range. Peptides that do not perform linearly (R2 > 0.98) are excluded from analysis.
4.5 Quantitation using targeted mass spectrometry
For samples including spiked-in peptides, chromatographic peak areas from all detected fragment ions for the light and heavy version of each peptide are integrated and summed, and then the peak-area ratio between the light and heavy peptides is calculated. This ratio is multiplied by the amount of spiked heavy peptide to give the molar amount of each light (endogenous) peptide in the sample. With purified hair bundles, because the amount of bundle protein varies from replicate to replicate, we reference each peptide’s signal to that of actin in that same replicate. As with shotgun data, we then use a constant for actin (400,000 molecules per typical stereocilium) to calculate the protein’s abundance. The normalized amounts for different peptides from a protein are averaged for each replicate, then the protein amounts are averaged for (typically) four biological replicates of each condition to give an average abundance for each protein.
In the absence of calibrated heavy standards, relative protein abundance can be estimated by comparing two or more conditions, for example hair bundles from wild-type and knockout mice (Krey et al., 2016). Again, results are improved by normalizing the signal in each sample to actin or another protein that is not expected to change between the samples.
5. Summary
Mass spectrometry has had a profound impact on the biochemical characterization of hair bundles and bundle function. While antibody-based methods can be sufficiently sensitive to characterize the negligible amounts of protein present in purified bundles, the performance of antibodies varies dramatically, necessarily focusing attention only on those proteins for which antibodies work. By contrast, proteins are much more democratically detected with mass spectrometry, with little bias between larger and reasonably abundant proteins where many distinct peptides can be detected. While shotgun mass spectrometry is outstanding for detecting the breadth of proteins present in a sample such as purified bundles, it is less favorable for quantitation. Instead, targeted proteomics, particularly parallel reaction monitoring, offers much more precise and accurate quantitation of bundle proteins.
In addition to characterizing the identities and concentrations of components of the hair-bundle proteome, both mass spectrometry methods are also valuable for other biochemical characterization experiments with bundles. For example, we are using both shotgun and targeted proteomics to analyze experiments immunoprecipitating key bundle proteins; quantitation with either method, especially when comparing a protein’s concentration in the starting material with that in the immunoprecipitate, allows us to identify proteins that specifically co-precipitate with the protein of interest. Using these experiments, we will be able to build on our quantitation information for bundle proteins, characterizing their interactions as well. This information will be invaluable when describing how hair bundles operate at a biochemical level.
Figure 4. Comparison of shotgun and targeted quantitation.
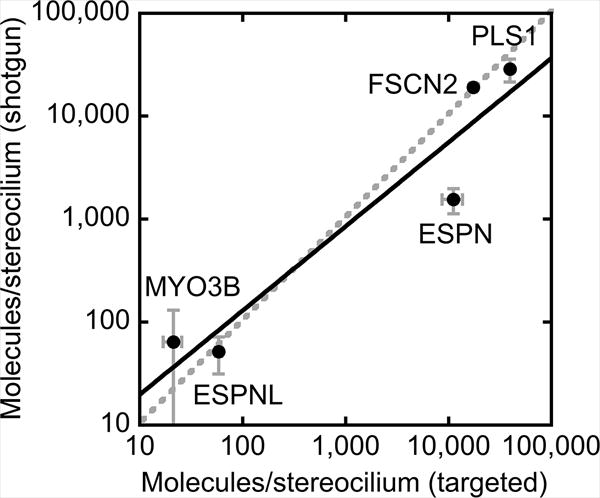
Estimated molecules per stereocilum (± SEM) for five proteins with both shotgun and targeted mass spectrometry data. Linear regression (log-log plot) gives y = 3.01*×0.82, with R2 = 0.87. Dashed line, unity line. Note that ESPN is an outlier; its small size (several isoforms are ~30 kD) allow it to diffuse out of SDS gels used in in-gel sample preparation for shotgun mass spectrometry; by contrast, ESPN is not lost in the eFASP procedure, used for targeted mass spectrometry.
Acknowledgments
PGBG was supported by NIH grants R01DC002368, R01DC011034, and P30DC005983; JFK by R03DC014544; and PAW and LLD by R01EY007755 and P30EY010572. We obtained mass spectrometry support from the University of Virginia W.M. Keck Biomedical Mass Spectrometry Lab and from the OHSU Proteomics Shared Resource (partial support from NIH core grants P30 EY010572 & P30 CA069533; Orbitrap Fusion S10 OD012246).
References
- Barr-Gillespie PG. Assembly of hair bundles, an amazing problem for cell biology. Mol Biol Cell. 2015;26:2727–2732. doi: 10.1091/mbc.E14-04-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger JG, Stergachis AB, Johnson RS, Egertson JD, MacCoss MJ. Selecting Optimal Peptides for Targeted Proteomic Experiments in Human Plasma Using In Vitro Synthesized Proteins as Analytical Standards. Methods Mol Biol. 2016;1410:207–221. doi: 10.1007/978-1-4939-3524-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Dumont RA, Zhao YD, Holt JR, Bahler M, Gillespie PG. Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J Assoc Res Otolaryngol. 2002;3:375–389. doi: 10.1007/s101620020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim S, et al. Stereocilia-staircase spacing is influenced by myosin III motors and their cargos espin-1 and espin-like. Nat Commun. 2016;7:10833. doi: 10.1038/ncomms10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Erde J, Loo RR, Loo JA. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. Journal of Proteome Research. 2014;13:1885–1895. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace R, Kim KX. The physiology of mechanoelectrical transduction channels in hearing. Physiol Rev. 2014;94:951–986. doi: 10.1152/physrev.00038.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SP, et al. A short splice form of Xin-actin binding repeat containing 2 (XIRP2) lacking the Xin repeats is required for maintenance of stereocilia morphology and hearing function. Journal of Neuroscience. 2015;35:1999–2014. doi: 10.1523/JNEUROSCI.3449-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Domon B. Advances in high-resolution quantitative proteomics: implications for clinical applications. Expert Rev Proteomics. 2015;12:1–10. doi: 10.1586/14789450.2015.1069188. [DOI] [PubMed] [Google Scholar]
- Gillespie PG, Hudspeth AJ. High-purity isolation of bullfrog hair bundles and subcellular and topological localization of constituent proteins. Journal of Cell Biology. 1991;112:625–640. doi: 10.1083/jcb.112.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Wagner MC, Hudspeth AJ. Identification of a 120 kd hair-bundle myosin located near stereociliary tips. Neuron. 1993;11:581–594. doi: 10.1016/0896-6273(93)90071-x. [DOI] [PubMed] [Google Scholar]
- Hsieh EJ, Hoopmann MR, MacLean B, MacCoss MJ. Comparison of database search strategies for high precursor mass accuracy MS/MS data. Journal of Proteome Research. 2010;9:1138–1143. doi: 10.1021/pr900816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Krey JF, et al. Annexin A5 is the most abundant membrane-associated protein in stereocilia but is dispensable for hair-bundle development and function. Sci Rep. 2016;6:27221. doi: 10.1038/srep27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Sherman NE, Jeffery ED, Choi D, Barr-Gillespie PG. The proteome of mouse vestibular hair bundles over development. Sci Data. 2015;2:150047. doi: 10.1038/sdata.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, et al. Accurate label-free protein quantitation with high- and low-resolution mass spectrometers. Journal of Proteome Research. 2014;13:1034–1044. doi: 10.1021/pr401017h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical Chemistry. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- Ma ZQ, et al. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. Journal of Proteome Research. 2009;8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick P, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nature Biotechnology. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Shin JB, et al. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, et al. Molecular architecture of the chick vestibular hair bundle. Nature Neuroscience. 2013;16:365–374. doi: 10.1038/nn.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, et al. The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. Journal of Neuroscience. 2010;30:9683–9694. doi: 10.1523/JNEUROSCI.1541-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, Pagana J, Gillespie PG. Twist-off purification of hair bundles. Methods Mol Biol. 2009;493:241–255. doi: 10.1007/978-1-59745-523-7_14. [DOI] [PubMed] [Google Scholar]
- Spinelli KJ, et al. Distinct energy metabolism of auditory and vestibular sensory epithelia revealed by quantitative mass spectrometry using MS2 intensity. Proceedings of the National Academy of Science, USA. 2012;109:E268–77. doi: 10.1073/pnas.1115866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, Yates JR. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. Journal of Proteome Research. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RG, Hudspeth AJ, Gillespie PG. Calmodulin and calmodulin-binding proteins in hair bundles. Proc Natl Acad Sci USA. 1993;90:2807–2811. doi: 10.1073/pnas.90.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmarth PA, Krey JF, Shin JB, Choi D, David LL, Barr-Gillespie PG. Hair-bundle proteomes of avian and mammalian inner-ear utricles. Sci Data. 2015;2:150074. doi: 10.1038/sdata.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmarth PA, Riviere MA, David LL. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. Journal of Ocular Biology, Diseases, and Informatics. 2009;2:223–234. doi: 10.1007/s12177-009-9042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamoah EN, Gillespie PG. Phosphate analogs block adaptation in hair cells by inhibiting adaptation-motor force production. Neuron. 1996;17:523–533. doi: 10.1016/s0896-6273(00)80184-1. [DOI] [PubMed] [Google Scholar]