

Abstract
Bacterial infection caused by intracellular pathogens, such as Mycobacterium, Salmonella, and Brucella, is a burgeoning global health epidemic that necessitates urgent action. However, the therapeutic value of a number of antibiotics, including aminoglycosides, against intracellular pathogenic bacteria is compromised due to their inability to traverse eukaryotic membranes. To address this significant problem, a cleavable conjugate of the antibiotic kanamycin and a non-membrane lytic, broad-spectrum antimicrobial peptide with efficient mammalian cell penetration, P14LRR, was prepared. This approach allows kanamycin to enter mammalian cells as a conjugate linked via a tether that breaks down in the reducing environment within cells. Potent antimicrobial activity of the P14KanS conjugate was demonstrated in vitro, and this reducible conjugate effectively cleared intracellular pathogenic bacteria within macrophage more potently than a conjugate lacking the disulfide moiety. Notably, successful clearance of Mycobacterium tuberculosis within macrophages was observed with the dual antibiotic conjugate, and Salmonella levels were significantly reduced in an in vivo Caenorhabditis elegans model.
Keywords: dual antibiotic, kanamycin, antimicrobial peptide, cell penetrating peptide, intracellular bacteria
Graphical abstract
INTRODUCTION
A significant challenge in the development of effective antibacterial agents arises from bacterial pathogens, including Mycobacterium tuberculosis, Salmonella, Brucella, Listeria, Shigella, and methicillin-resistant S. aureus (MRSA), that have evolved to inhabit mammalian cells, such as phagocytic macrophages.1–4 Moreover, most of the bacterial bioterrorism agents monitored by the U.S. Centers for Disease Control and Prevention are intracellular pathogens, such as multidrug-resistant Mycobacterium (tuberculosis, TB), Yersinia pestis (plague), Salmonella species (salmonellosis), Fancisella tularensis (tularemia), Brucella species (brucellosis), Chlamydia psittaci (psittacosis), Coxiella burnetii (Q fever), Rickettsia prowazekii (typhus fever), Burkholderia mallei (glanders) and Burkholderia pseudomallei (melioidosis). Within these intracellular safe havens the bacteria reproduce and form a repository, often causing chronic infections. Infected patients become life-long carriers of the pathogens,5 and chronically suffer from the infection or die from invasive forms of the pathogen. For example, in 2013, 9 million people around the world became sick with TB disease and there are an estimated 1.5 million TB-related deaths annually. Also, Salmonella infection affects >100 million people worldwide, accounting for 370,000 deaths annually.6–7 In addition, some intracellular pathogens are capable of switching to a dormant stage of growth, thus enabling long-term colonization of the host and relapses, even after prolonged antibiotic therapy.8,9
Whereas these pathogenic bacteria are internalized within macrophages, many of the commonly used classes of antibiotics, such as aminoglycosides, glycopeptides and macrolides, do not effectively accumulate within these cells. This is due, in part, to inefficient membrane penetration and susceptibility to drug efflux transporters.8 As such, the therapeutic value of many antibiotics against intracellular bacteria is severely limited. For instance, it is generally accepted that aminoglycosides have poor internalization within macrophages, with activity developing slowly because of the low rate of uptake. For instance, kanamycin has demonstrated only about 50% reduction in M. tuberculosis levels within THP-1 macrophages after 10 days.10
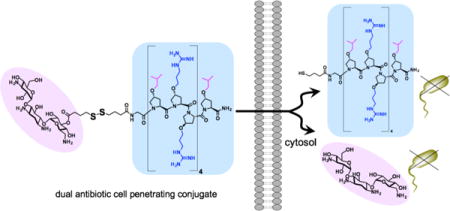
One approach to target intracellular pathogenic bacteria uses delivery vehicles, such as liposomes and micro/nanoparticles, that are loaded with antibiotics.11 While these vehicles have demonstrated cellular delivery of antibiotics and reduction in intracellular bacteria levels, they suffer from instability to biological fluids and difficulties in drug loading.12–13 Recently a non-cleavable conjugate of methotrexate and a short cell penetrating peptide was demonstrated to target intracellular Listeria,14 whereas a cell penetrating peptide with intrinsic antimicrobial activity, Fl-PRPRPL-4 (or P14LRR), was shown to target intracellular Salmonella and Brucella.15 This latter peptide forms a cationic amphiphilic polyproline helix and is a non-membrane lytic, broad spectrum antibiotic. While both of these peptide-based agents display a good reduction in intracellular bacteria, much more potent agents are needed to effectively eradicate pathogenic bacteria from mammalian cells. Herein, we describe a cell-permeable, dual antimicrobial agent that reversibly links the antibacterial P14LRR with the aminoglycoside antibiotic kanamycin (Figure 1) to achieve an agent with synergistic activity against intracellular pathogenic bacteria, including M. tuberculosis.
Figure 1.
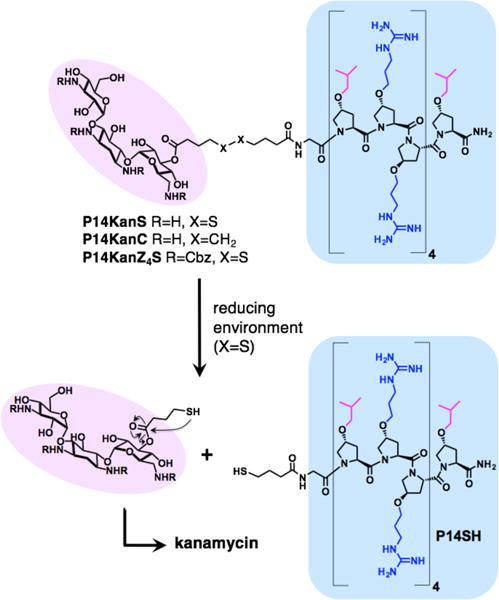
The designed dual agents composed of the antibacterial and cell penetrating peptide P14LRR (blue) and the antibiotic kanamycin (pink). In the reducing environment of mammalian cells, the conjugate containing the disulfide linkage (P14KanS) will breakdown to release kanamycin and P14SH, whereas the conjugate lacking the disulfide (P14KanC) should remain intact. The protected P14KanZ4S was designed to monitor the reductive release of kanamycin by HPLC.
In the design of the dual antibiotic agent, we wished to investigate the importance of reversibility of the conjugation chemistry between P14LRR and kanamycin. Therefore, we chose to link the peptide and kanamycin using a tether either with or without a disulfide moiety to generate P14KanS and P14KanC, respectively (Figure 1). We hypothesized that the disulfide linker within P14KanS would cleave in the reducing environment within the cell to release free kanamycin and the thiol-modified P14SH (Figure 1), whereas P14KanC would remain intact.16–18 The reductive approach with P14KanS would allow dual delivery of both of the individual antibiotics to mammalian cells, thereby increasing the therapeutic value of kanamycin, and potentially more effectively clearing intracellular pathogens than either agent alone.
RESULTS AND DISCUSSION
For the synthesis of the dual agents, the amino groups of kanamycin were first protected using the acid labile tert-butyloxycarbonyl group (Boc) or the carboxybenzyl group (Cbz) to afford Boc4-kanamycin (1a) or Cbz4-kanmaycin (1b), respectively, according to literature procedures.19–20 Treatment of compounds 1a and 1b with either 4,4′-dithiodibutyric acid or sebacic acid and O-(7-azabenzotriazole-1-yl)-N,N,N,N′-tetramethyluronium hexafluorophosphate (HATU) afforded compounds 2a – 2c, either with or without the disulfide in the tethering group (Scheme 1). A mixture of isomers was obtained, and one isomer was easily separated from the others by HPLC. Detailed NMR analysis of this isomer, including COSY, TOCSY and HMBC, was used to confirm the site of tether functionalization at the 3′ hydroxyl group of kanamycin (see Supporting Information). For instance, the 3′ H of compound 2a was found to shift by 1.6 ppm as compared to this proton in 1a (Figures S2 and S3), and a correlation was observed between the 3′ H and the carbonyl carbon of the tether in the HMBC experiment (Scheme 1 and, Figures S5 and S23). Resin-bound and sidechain protected P14LRR on the ChemMatrix Rink amide resin was treated with the modified kanamycins 2a – 2c and HATU to prepare the resin-bound P14LRR-kanamycin conjugates. Treatment of the resins with a trifluoroacetic acid (TFA) cocktail liberated the peptide conjugates from the resin, as well as deprotecting the Boc groups. The dual agents P14KanS, P14KanC and P14KanZ4S (Figure 1) were purified to homogeneity using reverse phase-HPLC and their final structures were confirmed using MALDI mass spectrometry (See Supporting Information).
Scheme 1.
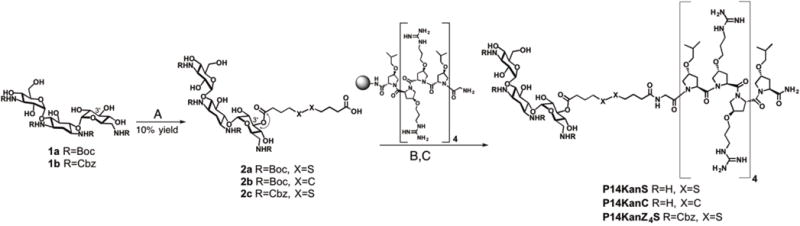
Synthesis of P14KanS, P14KanC and P14KanZ4: A) 2a and 2c: 4,4′-dithiobutyric acid, DIEA, HATU, DMF, 24h; 2b: sebacic acid, DIEA, HATU, DMF, 24h; B) DIEA, HATU, DMF, 24h; C) TFA:H2O:triisoprolylsilane (95:2.5:2.5), 1 h, RT.
The dual antibiotic conjugate P14KanS was designed to enter mammalian cells and release kanamycin and the P14SH peptide within an intracellular reducing environment. To evaluate the half-life of release of these agents under reducing conditions, we employed the P14KanZ4S construct as it will release the UV-active KanZ4 (Figure 1). The conjugate P14KanZ4S (100 μM in a 1:1 mixture of PBS buffer and DMF) was treated with 1 mM dithiothreitol (DTT) and the accumulation of KanZ4 was monitored by HPLC and LC-MS. We observed efficient release from the conjugate with a half-life of 1.5 ± 0.2 h and full release after 4 h (Figure S8). Ideally, the ester bond of P14KanS linking kanamycin to the P14LRR peptide would have sufficient stability to withstand plasma esterases. To investigate whether the ester bond is susceptible to esterase cleavage, P14KanS was incubated in presence of the 100 U of porcine liver esterase. The accumulation of the released peptide was monitored by HPLC for up to 120 h, however only ~15% of the cleavage product was observed. We conclude, therefore, that P14KanS is not susceptible to esterase cleavage presumably due to steric crowding on kanamycin around the ester bond. These data demonstrate that the dual antibiotic conjugate P14KanS is responsive to a reducing environment and produces KanZ4 and P14SH as designed, but should be stable against plasma esterases. The conjugate lacking the disulfide linkage, P14KanC, is not affected by a reducing environment and should remain intact within mammalian cells. This will allow us to evaluate the two different conjugation strategies for antibacterial potency against intracellular pathogens, as kanamycin analogs lacking the 3′ hydroxyl group have been shown to maintain activity against bacteria.21
The addition of kanamycin to P14LRR increases the overall net positive charge of the conjugate from +8 to +12, a change that may modulate mammalian cell accumulation efficiency. Therefore, we prepared a fluorescent conjugate, P14KanC-Fl (See Supporting Information), to monitor cell uptake within J774A.1 cells as compared to P14LRR. This fluorescent analog displayed a potency that was within 2-fold of the activity of P14KanC against a selection of bacteria (Table S1). Both peptides displayed a concentration dependent cell penetration (Figure 2a), and the kanamycin conjugate demonstrated a small increase (~20%) in cell uptake as compared to P14LRR at 10 μM. Although there is a substantial increase in positive charge in the conjugate, the increase in cell accumulation was not large, as has been shown with Arg-based cell penetrating peptides.22
Figure 2.
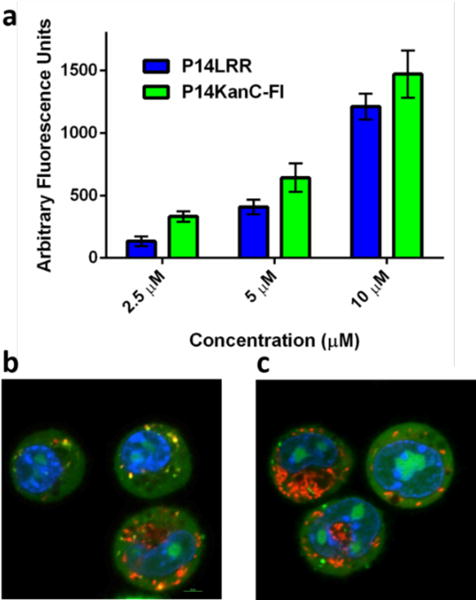
a) Cellular internalization of P14KanC-Fl and P14LRR. J774A.1 cells were incubated with the peptides for 1h and analyzed by flow cytometry. Confocal microscopy images of co-localization studies using P14KanC-Fl (10 μM) and b) LysoTracker or c) MitoTracker stains within J774A.1 cells after 1h.
We also investigated the subcellular localization of P14KanC-Fl within J774A.1 cells. Whereas P14LRR has been shown to localize to endosomes and mitochondria,15 P14KanC-Fl was found throughout the cell, including the cytosol, nucleus and endosomes (Figure 2b with Lysotracker stain), but did not localize with the mitochondria (Figure 2c with Mitotracker stain).
With the knowledge of mammalian cell penetration, we evaluated the antimicrobial activity of the P14KanS and P14KanC conjugates with a series of Gram positive and negative bacteria, including a range of intracellular pathogens (Table 1). This activity was compared to that obtained for P14LRR, P14SH, kanamycin and a 1:1 mixture of kanamycin and P14LRR. Across the series of bacteria P14KanS is highly active with MIC values ranging from 0.12 to 2 μM, including M. tuberculosis. With four strains of bacteria P14KanS is 2- to 32-times more active, respectively, than the non-covalent mixture of P14LRR and kanamycin. With the exception of E. coli and M. tuberculosis, this conjugate is also 2- to 16-fold more potent than the non-reducible P14KanC conjugate. We also investigated the activity of P14KanS on clinical isolates of S. aureus and S. epidermis that form biofilms, and the conjugate was 8- to 16-fold more potent than the P14LRR peptide, respectively. The antibacterial activity of P14LRR has previously been shown to occur in a non-membrane lytic fashion.15 We also investigated if the P14KanS and P14KanC conjugates disrupted bacteria membranes by monitoring β-galactosidase release from E. coli upon addition of the conjugates. At five times their MIC values, no significant release of β-galactosidase was observed (Figure S9), whereas melittin at 5 times its MIC displayed a substantial level of release as has previously been shown.15 These data demonstrate that the kanamycin-peptide conjugates do not lyse membranes as the mechanism for their antibacterial activity, as was observed for P14LRR.
Table 1.
MIC [a] values [μM] for the antibacterial activity of the dual conjugates compared to individual components
| E. coli | S. aureus | S. enteritidis | B. abortus | S. flexneri | M. smegmatis | M. tuberculosis | S. aureus[c] | S. epidermis[c] | |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| P14LRR | 4 | 16 | 32 | 16 | 8 | 8 | 16 | 16 | 8 |
| P14SH | 4 | 16 | 4 | 2 | 4 | 4 | 8 | – | – |
| kanamycin | 2 | 2 | 16 | 4 | 2 | 8 | 2 | 4 | >128 |
| P14LRR:kanamycin [b] | 2 | 2 | 8 | 4 | 2 | 4 | 1 | – | – |
| P14KanS | 2 | 2 | 2 | 0.12 | 1 | 1 | 1 | 1 | 1 |
| P14KanC | 2 | 16 | 8 | 2 | 2 | 2 | 1 | – | – |
The minimum inhibitory concentration,
1:1 mixture of P14LRR and kanamycin
clinical isolate that forms biofilms
Since the conjugates effectively enter mammalian cells and display antibacterial activity, we investigated the ability of P14KanS and P14KanC and their components to clear intracellular pathogenic bacteria from J774A.1 macrophage cells, including S. enteritidis, B. abortus, M. smegmatis, M. tuberculosis and S. flexneri with an in cyto bacterial assay (Figure 3).23 We have shown that the conjugates and the components display limited cytotoxicity to J774A.1 cells across a range of concentrations, including the conditions of these assays (Figure S10). This selective toxicity towards bacteria but not mammalian cells is a crucial feature for application of the conjugate against intracellular bacteria. Whereas the 1:1 mixture of P14LRR and kanamycin demonstrated modest reductions in Salmonella, Brucella and Shigella levels in the infected cells over the components alone, the P14KanS conjugate displayed a marked increase in efficacy with significant reduction in these intracellular bacteria (Figure 3a – 95, 85 and 70%, respectively). Interestingly, the non-reducible conjugate P14KanC was closer in activity to the 1:1 mixture of components with Salmonella, Brucella and Shigella than P14KanS, indicating the importance of the reducible linker on intracellular potency. With Mycobacteria, the 1:1 mixture of P14LRR and kanamycin demonstrated very modest reductions of bacteria levels as compared to the individual components. P14KanS, however, very effectively lowered levels of these bacteria within macrophages by 95 and 93% for M. smegmatis and M. tuberculosis, respectively – a significant increase in efficacy over the individual antibiotics or their mixture at two concentrations (Figure 3b). These data illustrate that neither kanamycin nor P14LRR is capable of effectively clearing intracellular bacteria on their own, even when co-administered. Only the P14KanS conjugate potently clears intracellular pathogens by taking advantage of the non-membrane lytic, mammalian cell penetrating activity of P14LRR to co-deliver kanamycin. When comparing the antimicrobial activity of P14KanS against Salmonella and Brucella, this agent is 16-fold more potent against Brucella in vitro (Table 1), but the activity trends switches in cyto with a more effective clearance of Salmonella intracellularly (Figure 3a). Both of these pathogens enter macrophages via vacuoles, but ultimately reside in sub-cellular locations such as endosomes and the endoplasmic reticulum, respectively.24–25 Since P14KanS localizes to endosomes, it is reasonable that this agent is more effective against Salmonella within mammalian cells. The notable difference in potency between P14KanS andP14KanC demonstrates that it is crucial for the two antibiotics to separate and go their own way within macrophages for optimal performance.
Figure 3.
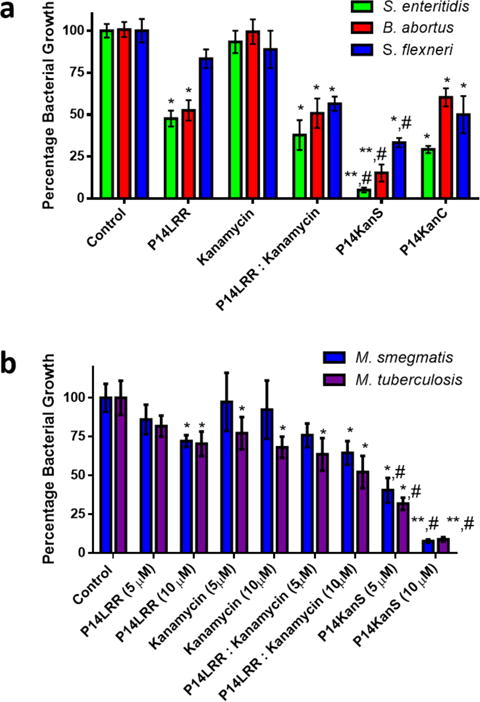
Intracellular antibacterial activity of a) P14LRR (10 μM), kanamycin (10 μM), a 1:1 mix of P14LRR and kanamycin (10 μM), P14KanS (10 μM) and P14KanC(10 μM) in J774A.1 cells infected with Salmonella enteritidis, Brucella abortus and Shigella flexneri, and b) P14LRR, kanamycin, a 1:1 mix of P14LRR and kanamycin, and P14KanS (each at 5 and 10 μM) in J774A.1 cells infected with Mycobacterium smegmatis and Mycobacterium tuberculosis after 9 h of treatment. Statistical analysis was calculated using one-way ANOVA, with post hoc Tukey’s multiple comparisons test. P values of < 0.05 were considered significant. One asterisk (*) and two asterisks (**) indicates significance from the control P < 0.05 and P < 0.01, respectively. Symbol (#) indicates significance from kanamycin, P14LRR, and P14LRR:kanamycin (P < 0.05). Results are expressed as means from three biological replicates ± standard deviation.
Encouraged by the potent antibacterial activity of the peptide conjugate P14KanS in vitro and in the in cyto bacterial clearing assay, we investigated the ability of the conjugate to clear S. enteritidis using an in vivo C. elegans model (Figure 4).26 First we evaluated the toxicity of P14KanS, P14LRR, kanamycin and melittin at 50 μM against C. elegans. After 3 days of treatment with P14KanS, P14LRR or kanamycin the C. elegans were highly viable (>90%), whereas no live worms were observed with melittin after this time (Figure S11). These data serve to highlight the non-toxic nature of the kanamycin-peptide conjugate. To monitor antibacterial activity, S. enteritidis infected C. elegans were treated with a range of concentrations of P14KanS, P14LRR, kanamycin, and a 1: 1 mixture of P14LRR and kanamycin for 24 hr, and the bacteria levels were monitored. P14KanS demonstrated significantly reduced levels of Salmonella in the C. elegans host as compared to the 1:1 mixture of antibiotics across all concentrations, and at the highest dose approximately 90% of the bacteria had been cleared in vivo. The C. elegans infection model confirms that P14KanS does exhibit potent in vivo antimicrobial activity and has significant promise to be used as a novel treatment of intracellular pathogens.
Figure 4.
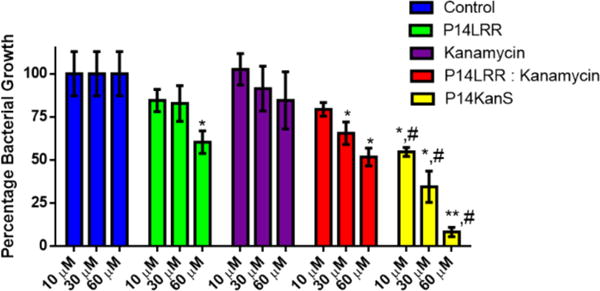
Treatment of Caenorhabditis elegans infected with Salmonella enteritidis with P14LRR, kanamycin, a 1:1 mix of P14LRR and kanamycin, and P14KanS for 24 h. Statistical analysis was calculated using one-way ANOVA, with post hoc Tukey’s multiple comparisons test. P values of < 0.05 were considered significant. One asterisk (*) and two asterisks (**) indicates significance from the control P < 0.05 and P < 0.01, respectively. Symbol (#) indicates significance from kanamycin, P14LRR, and P14LRR:kanamycin (P < 0.05). Results are expressed as means from three biological replicates ± standard deviation.
CONCULSION
A number of pathogenic bacteria invade and then reside within mammalian host cells. At the same time, many of the most commonly used antibiotics, such as β-lactams and aminoglycosides, are unable to achieve therapeutic concentrations within these same cells. Therefore, there is a great need to develop antibiotics with the ability to enter mammalian cells and target intracellular pathogens.27 In this work we have found that very effective targeting of intracellular bacteria, including Salmonella, Brucella, Shigella and Mycobacterium, is possible using a dual antibiotic approach, where one of the antibiotics also has mammalian cell penetrating properties. The reducible nature of the linker within these dual antibiotic conjugates was found to play a significant role in the intracellular activity. P14KanC lacks the reducible disulfide moiety, and, as such the kanamycin travels within macrophages to the preferred intracellular sites of the antibiotic peptide. Synergistic antibacterial activity is realized when the dual components of the conjugate are released within the reducing environment of the cell. Herein, we have demonstrated that the P14KanS conjugate clears notoriously difficult to treat intracellular pathogens such as S. enteritidis, B. abortus, and M. tuberculosis within J774A.1 cells, but also displays a significant reduction of S. enteritidis in an in vivo C. elegans model. Combining the cell penetrating ability and non-membrane lytic mechanism of antimicrobial action of P14LRR with the kanamycin antibiotic has proven to be very effective tool in combating intracellular pathogenic bacteria.
Supplementary Material
Acknowledgments
This manuscript is dedicated to Professor Ronald Breslow on the occasion of his 85th birthday. The National Science Foundation (1012316-CHE) and the Purdue Research Foundation are acknowledged for financial support.
Footnotes
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org. Experimental Procedures, Spectra, Figures and Supplementary References.
References
- 1.Ray K, Marteyn B, Sansonetti PJ, Tang CM. Nat Rev Microbiol. 2009;7(5):333. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 2.Flannagan RS, Cosío G, Grinstein S. Nat Rev Microbiol. 2009;7(5):355. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 3.Diacovich L, Gorvel JP. Nat Rev Microbiol. 2010;8(2):117. doi: 10.1038/nrmicro2295. [DOI] [PubMed] [Google Scholar]
- 4.LaRock DL, Chaudhary A, Miller SI. Nat Rev Microbiol. 2015;13(4):191. doi: 10.1038/nrmicro3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monack DM, Mueller A, Falkow S. Nat Rev Microbiol. 2004;2(9):747. doi: 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- 6.Crump JA, Luby SP, Mintz ED. Bull World Health Organ. 2004;82(5):346. [PMC free article] [PubMed] [Google Scholar]
- 7.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O’Brien SJ, Jones TF, Fazil A, Hoekstra RM, International Collaboration on Enteric Disease “Burden of Illness” Studies Clin Infect Dis Off Publ Infect Dis Soc Am. 2010;50(6):882. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 8.Carryn S, Chanteux H, Seral C, Mingeot-Leclercq MP, Van Bambeke F, Tulkens PM. Infect Dis Clin North Am. 2003;17(3):615. doi: 10.1016/s0891-5520(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 9.Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. Science. 2014;343(6167):204. doi: 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giovagnoli S, Marenzoni ML, Nocchetti M, Santi C, Blasi P, Schoubben A, Ricci M. J Pharm Pharmacol. 2014;66(1):106. doi: 10.1111/jphp.12162. [DOI] [PubMed] [Google Scholar]
- 11.Briones E, Colino CI, Lanao JM. J Control Release Off J Control Release Soc. 2008;125(3):210. doi: 10.1016/j.jconrel.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 12.Prior S, Gander B, Blarer N, Merkle HP, Subirá ML, Irache JM, Gamazo C. Eur J Pharm Sci. 2002;15(2):197. doi: 10.1016/s0928-0987(01)00218-4. [DOI] [PubMed] [Google Scholar]
- 13.Perrett S, Golding M, Williams WP. J Pharm Pharmacol. 1991;43(3):154. doi: 10.1111/j.2042-7158.1991.tb06657.x. [DOI] [PubMed] [Google Scholar]
- 14.Lei EK, Pereira MP, Kelley SO. Angew Chem Int Ed. 2013;52(37):9660. doi: 10.1002/anie.201302265. [DOI] [PubMed] [Google Scholar]
- 15.Kuriakose J, Hernandez-Gordillo V, Nepal M, Brezden A, Pozzi V, Seleem MN, Chmielewski J. Angew Chem Int Ed. 2013;52(37):9664. doi: 10.1002/anie.201302693. [DOI] [PubMed] [Google Scholar]
- 16.Jones LR, Goun EA, Shinde R, Rothbard JB, Contag CH, Wender PA. J Am Chem Soc. 2006;128(20):6526. doi: 10.1021/ja0586283. [DOI] [PubMed] [Google Scholar]
- 17.Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Bioorg Med Chem Lett. 2006;16(20):5350. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 18.Namanja HA, Emmert D, Davis DA, Campos C, Miller DS, Hrycyna CA, Chmielewski J. J Am Chem Soc. 2012;134(6):2976. doi: 10.1021/ja206867t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Disney MD, Barrett OJ. Biochemistry (Mosc) 2007;46(40):11223. doi: 10.1021/bi701071h. [DOI] [PubMed] [Google Scholar]
- 20.Chen G, Pan P, Yao Y, Chen Y, Meng X, Li Z. Tetrahedron. 2008;64(38):9078. [Google Scholar]
- 21.Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, Böttger EC. Antimicrob Agents Chemother. 2012;56(12):6104. doi: 10.1128/AAC.01326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. Adv Drug Delivery Rev. 2008;60(4–5):452. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seleem MN, Jain N, Pothayee N, Ranjan A, Riffle JS, Sriranganathan N. FEMS Microbiol Lett. 2009;294(1):24. doi: 10.1111/j.1574-6968.2009.01530.x. [DOI] [PubMed] [Google Scholar]
- 24.LaRock DL, Chaudhary A, Miller SI. Nat Rev Microbiol. 2015;13:191. doi: 10.1038/nrmicro3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celli J, Tsolis RM. Nat Rev Microbiol. 2015;13:71. doi: 10.1038/nrmicro3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uccelletti D, Zanni E, Marcellini L, Palleschi C, Barra D, Mangoni ML. Antimicrob Agents Chemother. 2010;54(9):3853. doi: 10.1128/AAC.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereira MP, Shi J, Kelley SO. ACS Infect Dis. 2015;1:586. doi: 10.1021/acsinfecdis.5b00099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.