

Abstract
Ectopic calcifications in intervertebral discs (IVDs) are known characteristics of IVD degeneration that are not commonly reported but may be implicated in structural failure and dysfunctional IVD cell metabolic responses. This study investigated the novel hypothesis that ectopic calcifications in the IVD are associated with advanced glycation end products (AGEs) via hypertrophy and osteogenic differentiation. Histological analyses of human IVDs from several degeneration stages revealed areas of ectopic calcification within the nucleus pulposus and at the cartilage endplate. These ectopic calcifications were associated with cells positive for the AGE methylglyoxal-hydroimidazolone-1 (MG-H1). MG-H1 was also co-localised with Collagen 10 (COL10) and Osteopontin (OPN) suggesting osteogenic differentiation. Bovine nucleus pulposus and cartilaginous endplate cells in cell culture demonstrated that 200 μg/mL AGEs in low-glucose media increased ectopic calcifications after 4 d in culture and significantly increased COL10 and OPN expression. The receptor for AGE (RAGE) was involved in this differentiation process since its inhibition reduced COL10 and OPN expression. We conclude that AGE accumulation is associated with endochondral ossification in IVDs and likely acts via the AGE/RAGE axis to induce hypertrophy and osteogenic differentiation in IVD cells. We postulate that this ectopic calcification may play an important role in accelerated IVD degeneration including the initiation of structural defects. Since orally administered AGE and RAGE inhibitors are available, future investigations on AGE/RAGE and endochondral ossification may be a promising direction for developing non-invasive treatment against progression of IVD degeneration.
Keywords: Intervertebral disc degeneration, ectopic calcification, advanced glycation end products, nucleus pulposus cells
Introduction
Intervertebral disc (IVD) degeneration (IDD) is a progressive condition and presents a major socioeconomic burden for western societies because of its strong association with back pain (Web ref.1; Vos et al., 2012). The causes for IDD are multifactorial and risk factors include heritability, rapid increase in weight, obesity, low physical activity levels and smoking (Samartzis et al., 2011; Wang et al., 2012; Williams et al., 2011). Consequently, environmental factors including diet are likely to play a role in accelerating IDD. A need for alternate and earlier interventions for IDD exists since individuals that underwent spine fusion surgery had a 30–40 % increased risk for developing IDD in adjacent levels (Kumar et al., 2001; Park et al., 2004). Research on regenerative therapies show greatest potential for treating early stages of IDD; however, early IDD can be asymptomatic and difficult to distinguish from aging. Furthermore, injectable treatments at early stages of IDD are not ideal because the resulting needle puncture and injection can accelerate IDD (Carragee et al., 2009; Michalek et al., 2010). Together, the literature suggests a substantial need to develop novel ways to assess or prevent painful IDD.
IDD is most commonly characterised by loss of IVD height, structural defects, loss of water and glycosaminoglycan content, chronic inflammation and catabolic metabolism (Adams et al., 2006; Antoniou et al., 2013; Le Maitre et al., 2007). Although less commonly reported, IDD is also known to be associated with ectopic calcifications in the nucleus pulposus (NP) (Roberts et al., 1998), subchondral endplate sclerosis (Moskowitz et al., 1990) and hypertrophic differentiation, manifested by increased Collagen 10 (COL10) expression and ongoing mineral deposition such as the presence of calcium deposits with progressing IDD (Boos et al., 1997; Hristova et al., 2011; Melrose et al., 2009; Rutges J et al., 2010). Interstitial and pericellular calcification of the NP can lead to cell death, reduced cell turnover and impaired NP viability (Loreto et al., 2011). Ectopic calcifications and other vertebral bone changes were observed with increased IDD in type I & II diabetic rodent models, further supporting the potential association of IDD with dysfunctional calcifications (Illien-Junger et al., 2013; Moskowitz et al., 1990). We recently showed that a high advanced glycation end product (AGE) diet resulted in exogenous AGE accumulation in the mouse spine and was directly associated with ectopic calcification, hypertrophy and IDD (Illien-Junger et al., 2015). If AGE accumulation can induce ectopic calcifications and focal defects in addition to well-known tissue stiffening and brittleness, then there are multiple pathways how AGE accumulation can induce structural disruption and painful IDD (Illien-Junger et al., 2015). Improved knowledge about AGEs and ectopic calcification are therefore a high clinical priority.
AGE formation occurs in tissues as a normal part of aging from non-enzymatic reactions or Maillard reactions between reducing sugars and free amino groups on proteins, lipids and nucleic acids (Ahmed, 2005; Vlassara et al., 2011). Common AGEs including Nε-carboxymethyl-lysine, pentosidine, and glucosepane are associated with protein structural changes, while reactive AGE precursors such as the cytotoxic metabolite methylglyoxal (MG) and its derivatives such as the AGE methylglyoxal-hydroimidazolone-1 (MG-H1) are linked to cellular injury (Cai et al., 2012; Karim et al., 2013; Kim et al., 2010; Monnier et al., 2014; Nemet et al., 2005).
Studies demonstrating mechanisms of AGE induced ectopic calcifications are notably limited. AGEs can induce ectopic calcification in vascular tissues (Pugliese et al., 2015; Wang et al., 2012). Activation of the receptor of AGE (RAGE) (Wei et al., 2013) was recently shown to induce hypertrophy in podocytes (Liebisch et al., 2014) and calcification in smooth muscle cells (Wang et al., 2013b). However, the cellular microenvironment in IVDs is very different from that in more highly vascularised tissues, thereby requiring further investigation.
We hypothesise that AGE accumulation in IVDs is associated with ectopic calcifications and that AGE can directly induce hypertrophy and osteogenic differentiation of IVD cells contributing to IDD. The aim of this study was to evaluate if AGE accumulation is associated with ectopic calcification in human IVDs and to investigate if AGEs are causative for induction of endochondral ossification. Ectopic calcification, hypertrophy and osteogenic differentiation via the AGE/RAGE pathway were assessed to determine extent of endochondral ossification. Investigations of this under-explored pathway could elucidate novel non-invasive treatment options for IDD.
Materials and Methods
Overall study design
Human IVDs from autopsy were evaluated to identify presence of ectopic calcifications and their localisation near structural defects within IVD and endplate tissues. Human IVDs were also histochemically analysed for co-localisation of the AGE precursor MG-H1 with markers of calcifications, hypertrophy and osteogenic differentiation. Bovine NP and cartilage endplate (CEP) cell culture and immunocytochemistry experiments were performed with exogenous AGE stimulation and RAGE induced inhibition to mechanistically test for relationships between AGEs, calcifications and hypertrophic and osteogenic differentiation. AGEs, calcifications and markers identifying hypertrophy and osteogenesis were assessed with immunohistochemistry and western blot. COL10 was chosen as a marker for hypertrophy because it is well known to be expressed in hypertrophic chondrocytes and has been reported to be expressed in degenerated IVDs (Hristova et al., 2011; Rutges JP et al., 2010). Osteopontin (OPN) is known to be associated with ectopic calcification in IVDs and was chosen over alkaline phosphatase, osterix or osteocalcin, because of its dual role in chondrocyte hypertrophy and ectopic calcification, where intact OPN acts as inhibitor (Hunter, 2013) and OPN fragments contribute to ectopic calcification (Shao et al., 2011).
Human IVD preparation
Permission for the use of cadaveric material was obtained prior to specimen procurement. Human lumbar IVDs (Thompson grade 2–5) were isolated from spinal segments from autopsy samples (Table 1). Excess tissue was removed and IVDs were isolated with a band saw (Marmed, Cincinnati, OH, USA) by cutting through the vertebrae adjacent to the IVD retaining a thin layer of the superior and inferior vertebral endplates. Thompson degeneration grading was performed for each IVD segment prior fixation (Thompson et al., 1990). IVDs were fixed in zinc formalin (Z-Fix, Anatech LTD, Battle Creek, MI, USA) for at least 48 h and processed and embedded in methyl-methacrylate (03629-4, Fisher Scientific, Waltham, MA, USA) (Laudier et al., 2007).
Table 1.
Human IVD specimen information.
| IVD level | Age | Sex | Degenerative Grade |
|---|---|---|---|
| Lumbar 3/4 | 44 | Male | 2 |
| Lumbar 3/4 | 47 | Female | 4 |
| Lumbar 1/2 | 85 | Male | 5 |
| Lumbar 3/4 | 81 | Female | 3 |
| Lumbar 4/5 | 93 | Male | 4 |
Immunohistochemistry and histology of human IVDs
Human IVDs were assessed histologically for calcifications (von Kossa, Bills et al., 1971) and immunohistochemically (chromogenic and fluorescent for double staining). To assess markers of hypertrophy, osteogenic differentiation and AGEs, 5 μm thick mid-sagittal histological sections were taken from each sample using a rotary microtome (Leica SM2500 base, Wetzlar, Germany). All sections were de-plasticised in a series of toluene (Fisher Chemical, Fail Lawn, NJ, USA) petroleum-ether and ethylene glycol mono ethyl ether (Fisher Chemical, Fail Lawn, NJ, USA) as previously described (Laudier et al., 2007). Immunohistochemistry was performed for Collagen 10 alpha-1 (COL10, concentration: 1:200; ab58632, abcam, Cambridge, MA, USA), a marker for hypertrophy, and osteopontin (OPN, concentration: 1:200; ab33046, abcam, Cambridge, MA, USA), a marker for osteogenic differentiation which is also expressed in atherosclerotic arteries and has been associated with vascular calcification in the IVD (Hristova et al., 2011; Scatena et al., 2007). AGEs were also assessed with immunostaining for methylglyoxal-hydroimidazolone (MG-H1, concentration: 1:250; STA-011, Cell Biolabs, San Diego, CA, USA), an AGE that is associated with atherosclerotic calcifications (Heier et al., 2015) and the receptor for AGE (RAGE, concentration 1:200; ab37647, abcam, Cambridge, MA, USA), a trans-membrane receptor which has been associated with vascular calcification (Cecil et al., 2011). De-plasticised sections were washed in distilled water, incubated for 5 min in Proteinase K (s3020, DAKO, Carpinteria, CA, USA), and followed by another wash. Non-specific binding was blocked with blocking solution (DAKO, Carpinteria, CA, USA) and incubated with the primary antibody for 2 h at room temperature. Sections were then rinsed with distilled water and incubated in secondary antibodies for either chromogenic or fluorescent staining. For chromogenic staining, samples were incubated for 30 min in ImmPRESS detection solution (ImmPRESS reagent kit peroxidase anti rabbit Ig, MP-7401 or ImmPRESS reagent kit peroxidase anti mouse Ig, MP-7402, Vector Laboratories, Burlingame, CA, USA), rinsed again and incubated for 1 min in ImmPACT staining substrate (ImmPACT DAB, peroxidase substrate kit SK-4106; Burlingame, CA, USA). Sections were then washed in distilled water and counterstained with toluidine blue (T-161, Fisher Scientific, Waltham, MA, USA) for 30 s, rinsed in distilled water, ethylene glycol monoethyl ether and toluene and then cover-slipped (Eukitt, Sigma-Aldrich, St. Louis, MO, USA). Negative controls were treated the same but were incubated with a universal negative control (X0902, DAKO, Carpinteria, CA, USA) instead of the primary antibody. We validated immunohistochemistry antibodies by assessing their reactivity in resin-embedded samples (as compared to paraffin-embedded samples described on data sheets) by using negative controls and by having identified specific staining confirmed by our histologist (DML, who was blinded on sample identification).
Co-localisation of MG-H1 staining with calcification was detected with chromogenic stained MG-H1 sections that were subsequently stained for von Kossa. Co-localisation of MG-H1 with COL10 or OPN was detected with double-labelled fluorescent staining where the primary antibodies (mouse anti MG-H1 with rabbit anti COL10 or rabbit anti OPN) were incubated simultaneously in blocking solution (DAKO, Carpinteria, CA, USA) for 2 h at room temperature. Sections were then rinsed with distilled water and incubated for 30 min simultaneously in secondary antibodies (for COL10 or OPN: Cy5 donkey anti rabbit, concentration 1:700; 711-175-152, Jackson ImmunoResearchLab, West Grove, PA USA; for MG-H1: Alexa Fluor 594 goat anti mouse, concentration 1:700; A11032, Molecular Probes, Eugene, OR, USA). Samples were rinsed in distilled water and cover-slipped with ProLong Gold Antifade Reagent with DAPI (Molecular Probes, Eugene, OR, USA). Negative controls were treated the same but were incubated with a universal negative control (DAKO, Carpinteria, CA, USA) instead of the primary antibodies.
RAGE, COL10 and OPN chromogenic samples were imaged using bright field; co-localisation of MG-H1 with von Kossa was determined by using Differential Interference Contrast (DIC) microscopy; co-localisation of MG-H1 with COL10 or OPN was determined by fluorescence by visualising Cy5 in red and Alexa Fluor 594 in green. All imaging was performed using an Axio Imager Z1 (Zeiss, Oberkochen, Germany).
Bovine cell isolation
Bovine tails (n = 6) were obtained from a local abattoir (Green Village Packing, Green Village, NJ, USA) within 24 h of death. Tails were rinsed in 70 % ethanol, the surrounding tissue was removed, 4 IVDs were dissected and NP or CEP tissues were isolated. The NP was isolated with a 8 mm biopsy punch from the centre of the disc and rinsed in 70 % ethanol and minced into 2 mm3 pieces. The CEP was carefully scratched from the bone endplate by using a size 11 scalpel blade and CEP pieces were rinsed in 70 % ethanol. NP and CEP pieces were washed in sterile PBS (Fisher Scientific, Pittsburg, PA, USA) containing 3 % penicillin-streptomycin (Thermo Scientific, Waltham, MA, USA) and 1.5 % Fungizone (Thermo Scientific, Waltham, MA, USA) followed by washing 3× in sterile PBS (centrifuged for 10 min at 0.5 RCF). Tissue pieces were incubated in 25 mL Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Life Technologies, Grand Island, NY, USA) containing 0.05 g pronase from Streptomyces griseus (p5147, Sigma-Aldrich, St. Louis, MO, USA) for 1 h at 37 °C, washed 3× in sterile PBS (centrifuged for 10 min at 0.5 RCF) and incubated for 12 h in 25 mL DMEM containing 0.003 g collagenase type II (#17101015, Life Technologies, Grand Island, NY, USA). Cells were then filtered through a 70 μm cell strainer (#22363548, Fisher Scientific, Waltham, MA, USA) and washed 3× in sterile PBS (centrifuged for 10 min at 0.5 RCF). Supernatant was replaced by standard high glucose DMEM media containing 10 % FBS (900-108, Gemini Bio Products, Broderick, CA, USA) and cell suspensions were transferred to cell culture flasks and expanded in 21 % O2 5 % CO2 at 37 °C in humidified atmosphere.
Bovine cell culture conditions
All cell culture experiments were performed in 5 % O2, 5 % CO2 at 37 °C in humidified atmosphere. Cells were cultured in culture slides (5000 cells/well, 4 wells/slide; Corning, Big Flats, NY, USA) for histology and immunocytochemistry or seeded in 6 well plates (30000 cells/well) for protein analysis. After 24 h pre-culture, cells were stimulated with AGE-BSA media (200 μg/mL; Biovision, Milpitas, CA, USA) in low glucose (1 g/L) DMEM + 1 % ITS (35452, Discovery Labware, Bedford, MA, USA) + 1 % FBS + 0.2 % ascorbic acid (A4544, Sigma, St. Louis, MO, USA) + 0.2 % Primocin (SH300042.01, InvivoGen, San Diego, CA, USA). Control media (BASAL) used BSA (200 μg/mL, Equitech-Bio, Kerrville, TX, USA) instead of AGE-BSA. To assess if AGEs act via the AGE/RAGE pathway, 0.01 mM RAGE Antagonist Peptide (RAP – Calbiochem, EMD Millipore, Billerica, MA, USA) was added to the culture media 1 h before AGE-BSA or BSA supplementation. The experiments were performed for 4 d with media changes on day 1 and day 3.
Immunocytochemistry of bovine IVD cells
Immunocytochemistry was performed with the same antibodies as mentioned above. For COL10 (n = 4), OPN (n = 4) and RAGE (n = 3) immunocytochemistry, the chambers on the culture slides were carefully removed and slides were washed in PBS before cell fixation in 4 % paraformaldehyde (16 % paraformaldehyde, Electron Microscopy Sciences, Hatfield, PA, USA, diluted in PBS). After permeabilisation with 0.1 % Triton-X100 (BP 151-100, Fisher Scientific, Waltham, MA, USA) in PBS, free binding sites were blocked with universal blocking solution (Dakoprot block # x0909; DAKO, Carpinteria, CA, USA) and cells were incubated with the primary antibodies (COL10, concentration 1:200; OPN concentration 1:200; RAGE concentration 1:200) or negative control for 2 h at room temperature. After incubation, the slides were washed in PBS + Tween 20 (PBST, Thermo Scientific, Rockford, IL, USA) and cells were incubated with the secondary antibodies for 30 min at room temperature followed by washing in TBS + Tween 20 (TBST, Bio-Rad Laboratories, Hercules, CA, USA). Slides were cover-slipped with ProLong Gold Antifade Reagent with DAPI (Molecular Probes, Eugene, OR, USA) and stored in the dark at 4 °C. Percent positive cells were quantified using Cellprofiler (http://www.cellprofiler.org/download.shtml). To assess calcification, NP cells (n = 6) or CEP cells (n = 2) were fixed with 4 % paraformaldehyde and mineralisation was visualised by von Kossa staining as described by Puchtler et al., 1978.
NP cell lysis and protein concentration determination
After culture (n = 3) cells were harvested by detaching the cells with a cell scraper (Fisher Scientific, Waltham, MA, USA) in ice-cold PBS. Following centrifugation, cell pellets were lysed in lysis buffer (M-Per mammalian protein reaction reagent #78501, Thermo Scientific, Waltham, MA, USA) + proteinase inhibitor (Halt Protease and Phosphatase inhibitor cocktail #78440, Thermo Scientific, Waltham, MA, USA) and sonicated (Sonic dismembrator’ Fisher Scientific, Waltham, MA, USA). To remove cellular debris, the suspension was centrifuged again and the supernatant was transferred into a fresh tube and the protein concentration was determined by DC protein assay (5000111; Bio-Rad Laboratories, Hercules, CA, USA). Samples were stored at −80 °C until use.
Western blot analysis
Proteins were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad Laboratories, Hercules, CA, USA). 20 μg protein samples were loaded on 10 % gels (4 % stacking gel + 10 % resolving gel; all solutions were purchased from Bio-Rad Laboratories, Hercules, CA, USA; protein standards: Precision Plus Protein Dual Color Standard #1610374; Bio-Rad Laboratories, Hercules, CA, USA and SeeBlue Plus2 Pre-stained Protein Standard #LC5925; Invitrogen, Carlsbad, CA, USA). After electrophoresis, proteins were transferred on nitrocellulose blotting membranes (Protran BA Cellulosenitrat; Schleicher & Schuell, Dassel, Germany). Following the protein transfer, membranes were incubated for 30 min in blocking buffer (5 % milk powder in 1 % TBS + Tween 20) and then incubated with the primary antibody over night at 4 °C on an orbital shaker. For COL10 (1:300) and OPN (1:300) the same antibodies as described above were used; β-Actin (1:5000; ab8227; abcam, Cambridge, MA, USA) was used as control because it is ubiquitously expressed in eukaryotic cells. The next day the membranes were washed in TBST and incubated with the secondary antibody for 2 h at room temperature on an orbital shaker. After washing, the membranes were incubated in ECL solution (Pierce ECL Western Blotting Substrate #32106; Thermo Scientific, Waltham, MA, USA) and visualised on X-ray films (Fuji X-Ray film, Cedar Falls, IA, USA). X-ray films were scanned and analysed by ImageJ software (https://imagej.nih.gov/ij/).
Statistics
Paired Student’s t-tests were used to determine significance, a p-value < 0.05 was considered significant.
Results
Ectopic calcifications were present in human IVDs of various degeneration stages and often co-localised with MG-H1
Histological analysis of grade 2, 3, 4 and 5 IVDs revealed areas of ectopic calcification using von Kossa staining (Fig. 1). Ectopic calcifications were identified as ossifications within the normally cartilaginous NP and CEP tissues. Ectopic calcifications were further identified as ossifications within the CEP when the cartilaginous region was disrupted by areas of ossifications. Ossifications were already observed in grade 2 IVDs but increased with degeneration (Fig. 1). At higher magnification, von Kossa stained calcified deposits appeared granulated and were observed in pericellular regions of MG-H1 positive cells which were often located in clusters (representative staining displayed in a grade 3 IVD, Fig. 2 a,b).
Fig. 1.
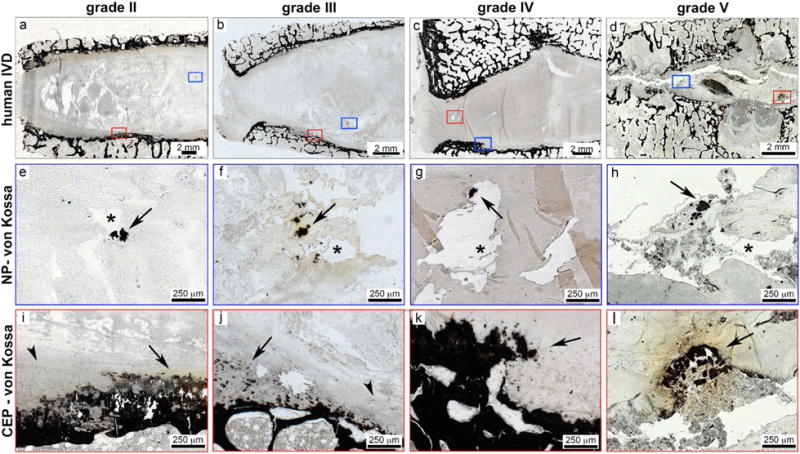
Ectopic calcifications were located at ossification sites and close to fissures in human IVDs of various degeneration stages. Representative overview images of human IVDs of varying degenerative levels including (a) grade II, (b) grade III, (c) grade IV, and (d) grade V. The von Kossa staining identifies ossifications as dark black/brown staining and boxes mark region of interest in NP (blue box) and CEP (red box) regions. In NP regions (e-h), calcified deposits were located close to fissures. Arrows denote calcified deposits and asterisks denote fissures in NP tissues. In CEP regions (i-l), irregular ossified structures were also identified in all degenerative grades. Arrows denote calcified deposits and arrow heads denote intact CEP.
Fig. 2.
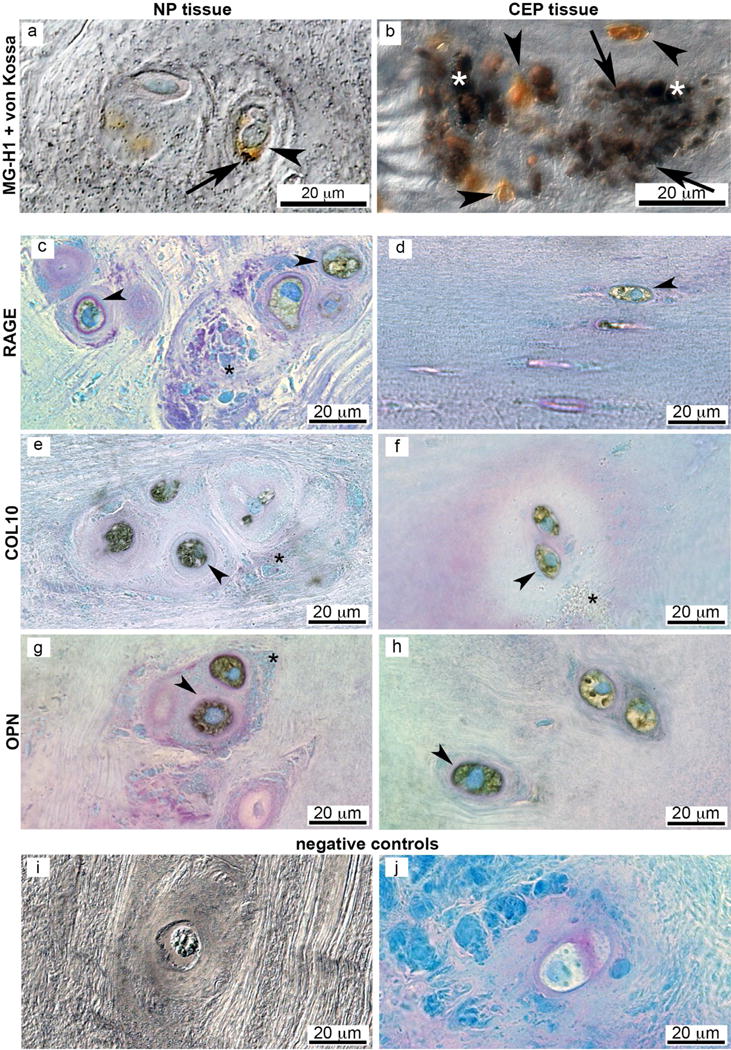
Large COL10, OPN, MG-H1 and RAGE positive cells are often located in clusters and surrounded by granulated matrix. Human grade 3 IVD: Differential interference contrast image of (a) NP and (b) CEP tissue. Calcified deposits (von Kossa) are located pericellular to brown MG-H1 positive cells. Bright field images of (c,d) RAGE, (e,f) COL10 and (g,h) OPN positive NP cells (c,e,g) and CEP cells (d,f,h) co-stained with toluidine blue to visualise cell morphology (blue). (i,j) Negative controls (NP tissue). Arrow heads denote positive cells (brown chromogenic staining); arrows denote calcified particles (dark von Kossa stain); asterisks denote granulated structures.
Markers for endochondral ossification and RAGE were associated with MG-H1 expression
In human IVDs, the NP regions contained granulated structures and clusters of large cells that were positive for RAGE, COL10 and OPN (Fig. 2 c–h). The co-localisation of MG-H1 with COL10 and OPN was identified in NP and CEP regions in IVDs of multiple degenerative grades and they were often associated with structural defects. Little positive staining was observed in grade 2 IVDs, while moderate presence in grade 3 and 4 IVDs and severe presence in the grade 5 IVD was observed. The co-localisation of immunopositive stains was further validated with immunofluorescent staining that demonstrated dual staining of IVD cells for MG-H1 with COL10 and OPN in clustered cells (grade 3 and 5 IVDs shown in Fig. 3).
Fig. 3.
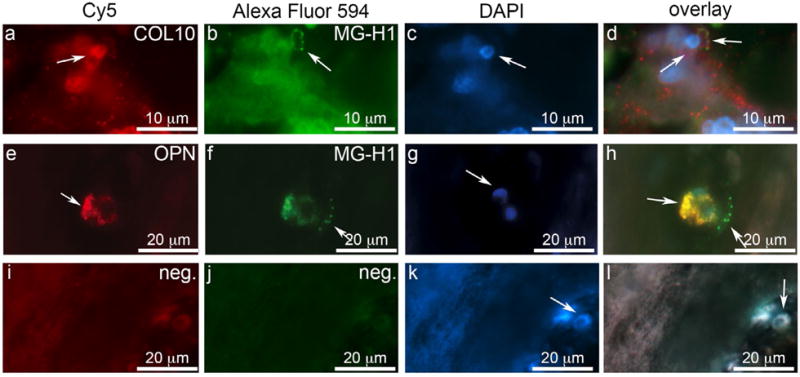
Co-localisation of COL10 and OPN with MG-H1 suggest a role of AGEs in endochondral ossification. Representative images of NP tissue of Grade 5 (COL10) and grade 3 (OPN) expression; (a) COL10 and (e) OPN co-localise with (b,f) MG-H1 in human NP tissue; (i-l) negative control. Arrows indicate positive staining and DAPI nuclei staining.
Markers for endochondral ossifications, especially in CEPs, increased with more severe IDD, implicating that the AGE/RAGE pathway is involved with ectopic calcifications in human IVDs and is likely contributing to IDD.
AGE stimulation increased von Kossa, COL10 and OPN staining in bovine NP and CEP cells
von Kossa staining of bovine NP and CEP cells revealed increased mineralised deposits in intracellular and pericellular regions following 4 d in culture with AGE media (Fig. 4). Immunocytochemical analysis of bovine NP cells cultured with AGE media showed significantly increased expression of the hypertrophy marker COL10 (BASAL: 7.2 ± 3.0 %; AGE: 35.4 ± 11.7 %, n = 4; p = 0.01) and the osteogenic marker OPN (BASAL: 9.5 ± 12.3 %; AGE: 57.8 ± 24.4 %, n = 4; p = 0.046). COL10 immunostaining was mainly present within the elongated processes of interconnecting cells and OPN was detected intracellularly. RAGE staining was faintly expressed intracellularly in both conditions (BASAL: 43.2 ± 14.9 %; AGE: 63.6 ± 2.3 %; n = 3; Fig. 4).
Fig. 4.

Mineralised deposits and expression of COL10 and OPN after 4 d AGE exposure indicate a direct relationship of AGEs on endochondral ossification in bovine cell culture. (a) Mineralised deposits were visible after 4 d AGE exposure in NP (top) and CEP (bottom). Inserts show higher magnification of representative regions. Arrows mark ectopic calcifications. (b) Immunocytochemistry with RAGE (top), COL10 (middle) and OPN (bottom) after 4 d culture. White arrows mark positive stained cells. (c) Quantification of RAGE, COL10 and OPN positive cells after 4 d culture; * p < 0.05; n = 4, COL10 and OPN; n = 4, RAGE.
AGE-mediated COL10 and OPN expression was downregulated after RAGE inhibition
Western blot analysis confirmed that AGE exposure increased COL10 (~58 kDa) and OPN protein expression in bovine NP cells (Fig. 5). OPN was detected as full-length protein (66 kDa) and the MMP cleaved fragment (32 kDa), which is known to contribute to ectopic calcification (Shao et al., 2011). Addition of RAGE inhibitor to the culture media decreased COL10 and OPN expression, further suggesting that AGEs induce endochondral ossification via the AGE/RAGE axis.
Fig. 5.
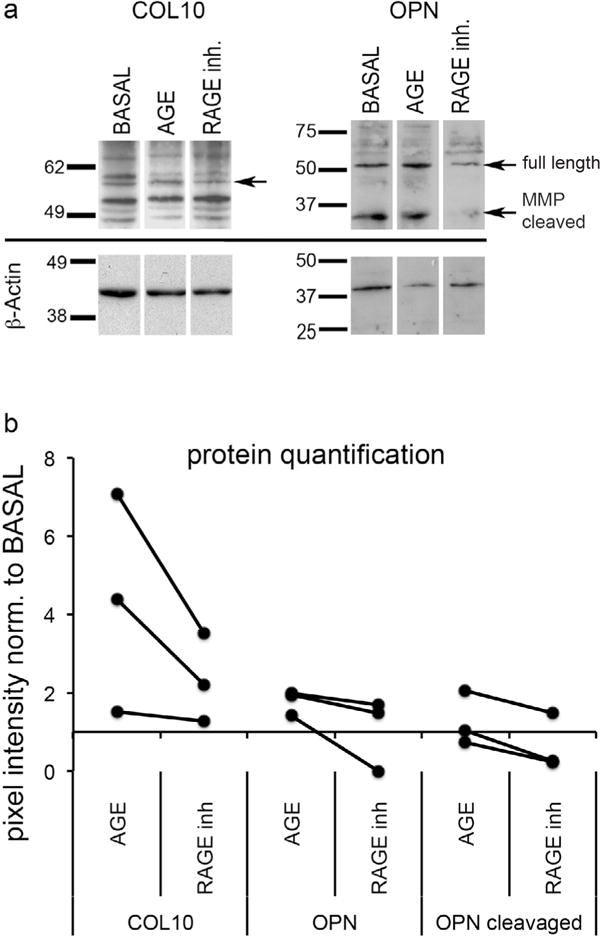
Inhibition of RAGE reduces effect of AGEs on NP cell differentiation, indicating potential pathway of endochondral ossification via the AGE/RAGE axis. (a) Representative images of protein expression determined by western blot of (left) COL10 (58 kDa) and (right) OPN (full length: 66 kDa & MMP cleaved: 32 kDa); (b) quantitative analysis of protein expression normalised to BASAL control (n = 3).
Discussion
Understanding initiators for IDD is essential for developing treatments to prevent the onset and progression of IDD. We hypothesise that AGE accumulation in IVDs is associated with IVD calcifications and that AGEs can directly induce hypertrophy and osteogenic differentiation of IVD cells. This hypothesis is based on the knowledge that AGE accumulation is associated with calcifications in vascular tissues (Hangai et al., 2016; Heier et al., 2015; Wang et al., 2012) but has not been shown in the IVD or collagenous tissues. Our results indicated that the AGE MG-H1 was associated with ectopic calcifications in human IVDs from various degeneration stages and that MG-H1 was also associated with markers of endochondral ossification, hypertrophy and the involvement of RAGE. These human IVD tissue associations were further borne out with mechanistic studies on bovine IVD cells. AGE stimulation of bovine NP and CEP cells in culture induced ectopic calcifications, resulted in slightly elevated RAGE expression in NP cells and significantly increased expression of COL10 and OPN, indicating a direct role of AGEs in endochondral ossification. Furthermore, inhibition of COL10 and OPN by exposure to a RAGE inhibitor peptide demonstrated a role for AGEs in ectopic calcifications and suggested that AGEs act through the AGE/RAGE pathway on IVD cells, providing a potential target for future preventive IDD treatments. With this study we added to the postulation that AGEs can lead to focal defects and structural disruptions that result in IVD degeneration, as previously described (Illien-Junger et al., 2015). Structural disruptions distinguish aging from IVD degeneration and can result in pain (Adams and Roughley, 2006).
Ectopic calcifications and fissures are known markers for IDD (Rutges et al., 2010). We demonstrated here that ectopic calcifications were located close to fissures within the NP of IVDs from various degeneration stages. It is known that hard inclusions in soft materials create stress concentrations (Iatridis et al., 2004) and the proximity of the observed calcifications in the relatively soft cartilaginous IVD tissues might have been nucleation sites that initiated cracks and fissures. Although CEP fissures are less frequently observed in IDD, excessive ectopic calcifications might play a role in more generalised vertebral endplate sclerosis commonly associated with reduced IVD nutritional pathways and degeneration (Benneker et al., 2005; Roberts et al., 1996). Similar to our findings, microscopic calcifications have been reported to be present in degenerative and non-degenerative IVDs and were observed within and around cells (Rutges et al., 2010), further suggesting that IVD cells can undergo osteogenic differentiation and produce ectopic calcifications.
Few studies have investigated endochondral ossification as a potential pathway for IDD (Table 2). COL10 is synthesised by hypertrophic chondrocytes during endochondral ossification in the growth plate of IVDs (Mwale et al., 2000; Tchetina et al., 2003) and is expressed in aged and degenerated IVDs (Boos et al., 1997). OPN is a highly phosphorylated and glycosylated protein that is associated with mineralisation in osseous tissues where it acts as mineralisation inhibitor by regulating osteoclast activity (Chellaiah et al., 2003). In bone fragments of type 2 diabetic patients, OPN together with other key factors of osteoblast metabolism such as runt-related transcription factor 2 (Runx2) and integrin-binding sialoprotein (IBSP) were increased by more than 50 % and thought to potentially build the biomolecular basis for higher bone mass and at the same time a higher risk of bone fracture of type 2 diabetic patients (Haug et al., 2014). Melrose et al. (2009) reported that calcifications in aged ovine IVDs may precede or predispose to IDD in agreement with our findings that calcifications were present in the NP and CEP and increased with severity of degeneration. In contrast, Melrose et al. (2009) reported that neither COL10 nor OPN were present in calcified deposits – suggesting that ectopic calcification did not occur by endochondral ossification in the ovine model, but concluded that ossification mechanism in the ovine model are likely to be distinct from human IVDs and suggested that a direct comparison between sheep and human IVDs would be inappropriate. While species differences may account for different ossification mechanism it remains possible that calcifications in human IVDs occur by endochondral and non-endochondral mechanisms.
Table 2.
Summary of studies investigating associations of hypertrophy (COL10) and osteogenic differentiation (OPN) with ectopic calcifications and endochondral ossification in IDD.
| Reference | Species | Summary |
|---|---|---|
| (Yeh et al., 2016) | Human AF cells (surgery) | Endochondral ossification in IDD was induced by resident progenitor cells in the annulus fibrosus. Osteogenic differentiation was induced in normal and degenerated AF cells with osteogenic medium. Mineralisation, increased gene expression of Runx2, alkaline phosphatase and osteocalcin was more prominent in degenerated AF cells. Overexpression of microRNA-221 decreased the osteogenic potential of degenerated AF cells, indicating that endochondral ossification can be regulated by microRNA-221. |
| (Marfia et al., 2015) | Human IVD-MSC (surgery) | OPN gene expression was significantly increased in MSCs from degenerated IVDs compared to MSCs from herniated IVDs. OPN protein was only detected in MSCs from degenerated IVDs, suggesting that OPN plays an important role in IDD. |
| (Hristova et al., 2011) | Human IVD (scoliosis surgery) (autopsy) | COL10 positive cells, ectopic calcifications and endochondral ossifications were increased in degenerated and scoliotic IVDs, suggesting that IDD is associated with ongoing mineral deposition and might be related to degenerative processes. |
| (Bian et al., 2011) | Rat IVD (in vivo) | Prolonged upright posture induced cartilage end-plate calcification and hypertrophy in rat lumbar spine as detected by μCT, histology, immunohistochemistry and gene expression for COL10, VEGF and TGFβ3. |
| (Rutges et al., 2010) | Human IVD (autopsy) | Degenerated NP tissue was more positive for COL10, OPG and Runx2. Osteoprotegerin levels correlated significantly with degeneration grade and number of microscopic calcifications measured on μCT and von Kossa staining. Results indicated that hypertrophic differentiation occurs during IDD. |
| (Melrose et al., 2009) | Ovine IVD | Ectopic calcification in aged ovine IVDs likely precedes or predisposes IVD degeneration and occurred independently from COL10 and OPN expression. |
| (Roberts et al., 1998) | Human IVD (fusion surgery) | COL10 positive enlarged hypertrophic chondrocyte-like cells were most commonly found in NP tissue of degenerated IVDs, in areas of disorganised architecture, such as clefts, and in areas of CEP that calcified ectopically, suggestive of IDD. |
| (Boos et al., 1997) | Human IVD (autopsy) | COL10 was localised in degenerated IVDs in the pericellular region of the ECM and increased with aging and degeneration. |
| (Moskowitz et al., 1990) | Rat IVD (motion segment) | Aging diabetic rats exhibited IDD including IVD ossification, ligamentous calcifications, subchondral endplate sclerosis as well as IVD-space narrowing, chondrocyte replication and IVD necrosis. |
It is widely accepted that OPN is strongly upregulated in pathological ectopic calcifications such as vascular calcifications thought to be associated with arterial stiffness in coronary artery diseases (Berezin et al., 2013), or renal crystal formation, gallstone formation and pathologic IVD mineralisation (Giachelli et al., 1993; Hirose et al., 2012; Imano et al., 2010; Kahles et al., 2014; Marfia et al., 2015). The observed expression of COL10 and OPN positive cells within granulated structures in IVDs of all degeneration stages indicates possible focal endochondral ossifications in IVDs.
In atherosclerosis, ectopic calcifications can be caused by AGE accumulation (Wang et al., 2012). Morphological assessment of human IVDs demonstrated that hypertrophy, osteogenic differentiation and ectopic calcifications were associated with MG-H1 and RAGE, suggesting a potential correlation between AGE accumulation and ectopic calcification in IVDs. Further, AGE stimulation of bovine NP and CEP cells led to increased presence of calcified particles, supporting a direct effect of AGEs on ectopic calcification independent from glucose. Yet it is possible that AGEs increased oxidative stress or induced alternate pathophysiological conditions with ectopic calcifications as a downstream effect.
Detrimental effects of AGEs on NP cells were also described by Tsai et al. (2004) who demonstrated upregulation of MMP2 and RAGE after AGE exposure to diabetic NP cells from rat and human. They concluded that hyperglycaemia contributed to AGE formation due to the increased sugar concentrations leading to activation of MMP2 and matrix degradation. In our cell culture study exposure to AGEs resulted in increased OPN cleavage, which has been shown to act as a pro-inflammatory cytokine and a pro-angiogenic factor facilitating vascular mineralisation (Scatena et al., 2007) and osteogenic differentiation (Shao et al., 2011). Interestingly, we observed a baseline RAGE expression after culture in BASAL medium, which did not increase significantly after AGE supplementation. RAGE is known to be present in low levels on several cell types (Kierdorf et al., 2013) and that once AGE accumulation reaches a critical threshold it could induce RAGE activation (Lander et al., 1997; Ramasamy et al., 2012). RAGE involvement in the observed AGE-induced endochondral ossification is likely, especially, since RAGE inhibition resulted in decreased COL10 and OPN expression. Therefore, we believe that AGE stimulation may have increased RAGE activation, rather than increased RAGE expression. However, RAGE staining on our bovine samples was faint and sample size relatively low so that future studies are required to better clarify if increased RAGE activation or expression is responsible for the observed effects.
Taken together with the literature, the current results suggest that AGEs may play a role in ectopic calcifications in multiple cell types and the observed co-localisation of MG-H1 with COL10 and OPN suggests that AGE accumulation is associated with endochondral ossifications. The RAGE inhibitor reduced COL10 and OPN in bovine NP cells, suggesting that AGE-induced endochondral ossification in IDD could be mediated via the AGE/RAGE pathway and suggests that RAGE could be a potential target candidate for drug treatment. The transmembrane receptor RAGE is upregulated in the presence of AGEs (Tanikawa et al., 2009), has been associated with AGE accumulation and vascular calcification (Cecil and Terkeltaub, 2011) and is correlated to the pathogenesis of accelerated atherosclerosis by multiple ligands (Suga et al., 2011; Tanikawa et al., 2009; Wang et al., 2013a).
Only few studies have investigated the AGE/RAGE pathway in relation to IDD (Fields et al., 2015; Tsai et al., 2014; Yokosuka et al., 2006; Yoshida et al., 2009). Yokosuka et al., 2006 demonstrated that AGEs and RAGE co-localised in human IVDs and showed that exposure to AGEs down-regulated aggrecan expression. The same group demonstrated later in bovine NP cells that when NP cells were exposed to AGE and Interleukin-1β, aggrecan expression decreased, which was reversed after inhibiting RAGE, suggesting a role of RAGE in IDD (Yoshida et al., 2009). Fields et al. (2015) demonstrated in a type-2 diabetes rat model that diabetes induced endplate sclerosis, increased oxidative stress and AGE and RAGE presence in the NP, suggesting that AGE/RAGE-mediated interactions could be important factors for IVD pathology.
AGEs are well known to be elevated during hyperglycaemia and to be risk factors for diabetic complications (Ahmed, 2005; Nemet et al., 2005; Vlassara and Striker, 2011). Besides hyperglycaemia, factors that are independent of the diabetic status, such as consumption of a diet high in foods processed at elevated temperatures can also result in AGE accumulation (Illien-Junger et al., 2015; Vlassara and Striker, 2011). We do not have records about the diabetic status of the IVD autopsy donors and have limited details on their medical histories; therefore, it is possible that other factors caused the observed endochondral ossifications independent from AGE accumulation. Nevertheless, our previous study on diabetic mice demonstrated that oral delivery of anti-AGE and anti-inflammatory drugs improved IVD health in diabetic mice (Illien-Junger et al., 2013), suggesting that targeting AGEs could be a promising treatment strategy to prevent the progression of IVD degeneration including endochondral ossification.
There is a need for non-invasive therapies to prevent or reverse early degenerative IVD changes. Currently there is a phase 3 clinical trial using the orally bioavailable RAGE inhibitor Azeliragon (TTP488; trial for Mild Alzheimer’s disease), suggesting additional anti-AGE drugs are available. A clinical study further reported that restriction of oral AGE intake reduced systemic AGE levels and improved insulin resistance in humans with type 2 diabetes (Uribarri et al., 2011), suggesting that effects of AGEs might be reversible. Importantly, we observed indications for endochondral ossifications in human IVDs already in grade II IVDs, a stage at which preventative treatment could still inhibit further degeneration. In conclusion, accumulation of the AGE MG-H1 was associated with endochondral ossifications, hypertrophy and osteogenic differentiation in human IVDs and mechanistic investigations on IVD cells showed a direct relationship involving RAGE, suggesting that AGE/RAGE could be a potential therapeutic target. Further investigations in animal experiments are warranted to assess whether targeting AGEs via the AGE/RAGE axis can potentially provide a non-invasive treatment option for preventing progression of IDD.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R01 AR069315 and R01AR064157 and a grant from the North American Spine Society. The authors gratefully acknowledge Alexander Real and Dr Andrew Hecht, Dr Thomas Naidich, Dr Helen Vlassara and Dr Gary Striker for helpful discussions and technical contributions.
Footnotes
Discussion with Reviewers
Sibylle Grad: This work focuses on accumulation of ectopic calcifications in intervertebral discs that is likely mediated by AGEs. Besides, other effects of AGEs that are RAGE dependent and independent are expected. Which additional effects may be triggered by AGEs that could impair IVD function?
Authors: Our recently published mouse in vivo study indicated that high amounts of dietary AGEs were associated with ectopic calcifications in IVDs (Illien-Junger et al., 2015). The current study shows an association between AGEs and ectopic calcifications in human IVD tissues and indicated that AGEs act directly on IVD cells to induce ectopic calcifications. Hyperglycaemic conditions increase endogenous AGE accumulation and contribute to IDD by causing IVD matrix degradation (Fields et al., 2015; Tsai et al., 2013; Yokosuka et al., 2006; Tsuru et al., 2002) because of increased oxidative stress and pro-inflammatory responses (Fields et al., 2015; Yoshida et al., 2009; Illien-Junger et al., 2013). AGEs also directly crosslink collagenous tissues to increase tissue stiffness (Fields et al., 2015; Wagner et al., 2006) and reduce IVD water content (Jazini et al., 2012).
Fackson Mwale: When RAGE binds AGEs, NF-KB is activated in many diseases leading to inflammation. Is a similar situation happening in the disc?
Authors: Since receptors for both AGEs and NF-KB are present in IVDs and it has been shown that AGEs can induce inflammation in IVDs, it is expected that RAGE binding of AGEs can induce pro-inflammatory responses via NF-KB in IVDs.
Fackson Mwale: Any suggestions on how to prevent breakdown crosslinks or manage them in the disc?
Authors: Several therapeutic interventions exist that may be useful to manage the effects of AGEs. First, diets low in AGE were protective of age-accelerated IVD degeneration in mice (Illien-Junger et al., 2015). Second, oral treatment with the AGE inhibitor pyridoxamine and the anti-inflammatory drug pentosan polysulphate were able to reduce diabetes-induced IVD degeneration in mice (Illien-Junger et al., 2013). Furthermore, the AGE crosslink breaker Alagebrium reversed AGE crosslinking in atherosclerosis after oral administration in diabetic mice (Watson et al., 2011) and may offer another way to manage AGE effects on IVD tissues. As a result, we believe there are several ways that may be helpful to manage the effects of AGEs on IVD degeneration.
Editor’s note: The Scientific Editor responsible for this paper was Brian Johnstone.
References
- Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine (Phila Pa 1976) 2006;31:2151–2161. doi: 10.1097/01.brs.0000231761.73859.2c. [DOI] [PubMed] [Google Scholar]
- Ahmed N. Advanced glycation endproducts – role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Antoniou J, Epure LM, Michalek AJ, Grant MP, Iatridis JC, Mwale F. Analysis of quantitative magnetic resonance imaging and biomechanical parameters on human discs with different grades of degeneration. J Magn Reson Imaging. 2013;38:1402–1414. doi: 10.1002/jmri.24120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneker LM, Heini PF, Alini M, Anderson SE, Ito K. 2004 Young Investigator Award Winner: vertebral endplate marrow contact channel occlusions and intervertebral disc degeneration. Spine (Phila Pa 1976) 2005;30:167–173. doi: 10.1097/01.brs.0000150833.93248.09. [DOI] [PubMed] [Google Scholar]
- Berezin AE, Kremzer AA. Circulating osteopontin as a marker of early coronary vascular calcification in type two diabetes mellitus patients with known asymptomatic coronary artery disease. Atherosclerosis. 2013;229:475–481. doi: 10.1016/j.atherosclerosis.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Bills CE, Eisenberg H, Pallante SL. Complexes of organic acids with calcium phosphate: the von Kossa stain as a clue to the composition of bone mineral. Johns Hopkins Med J. 1971;128:194–207. [PubMed] [Google Scholar]
- Boos N, Nerlich AG, Wiest I, von der Mark K, Aebi M. Immunolocalisation of type X collagen in human lumbar intervertebral discs during ageing and degeneration. Histochem Cell Biol. 1997;108:471–480. doi: 10.1007/s004180050187. [DOI] [PubMed] [Google Scholar]
- Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA. 2012;109:15888–15893. doi: 10.1073/pnas.1205847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragee EJ, Don AS, Hurwitz EL, Cuellar JM, Carrino JA, Herzog R. 2009 ISSLS Prise Winner: Does discography cause accelerated progression of degeneration changes in the lumbar disc: a ten-year matched cohort study. Spine (Phila Pa 1976) 2009;34:2338–2345. doi: 10.1097/BRS.0b013e3181ab5432. [DOI] [PubMed] [Google Scholar]
- Cecil DL, Terkeltaub RA. Arterial calcification is driven by RAGE in Enpp1−/− mice. J Vasc Res. 2011;48:227–235. doi: 10.1159/000318805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellaiah MA, Kiser N, Biswas R, Alvarez U, Strauss-Schoenberger J, Rifas L, Rittling SR, Denhardt DT, Hruska KA. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol Biol Cell. 2003;14:173–189. doi: 10.1091/mbc.E02-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AJ, Berg-Johansen B, Metz LN, Miller S, La B, Liebenberg EC, Coughlin DG, Graham JL, Stanhope KL, Havel PJ, Lotz JC. Alterations in intervertebral disc composition, matrix homeostasis and biomechanical behavior in the UCD-T2DM rat model of type 2 diabetes. J Orthop Res. 2015;33:738–746. doi: 10.1002/jor.22807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Investigat. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangai M, Takebe N, Honma H, Sasaki A, Chida A, Nakano R, Togashi H, Nakagawa R, Oda T, Matsui M, Yashiro S, Nagasawa K, Kajiwara T, Takahashi K, Takahashi Y, Satoh J, Ishigaki Y. Association of advanced glycation end products with coronary artery calcification injapanese subjects with type 2 diabetes as assessed by skin autofluorescence. J Atheroscler Thromb. 2016;10:1178–1187. doi: 10.5551/jat.30155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug AT, Braun KF, Ehnert S, Mayer L, Stockle U, Nussler AK, Pscherer S, Freude T. Gene expression changes in cancellous bone of type 2 diabetics: a biomolecular basis for diabetic bone disease. Langenbeck’s Arch of Surg. 2014;399:639–647. doi: 10.1007/s00423-014-1188-4. [DOI] [PubMed] [Google Scholar]
- Heier M, Margeirsdottir HD, Torjesen PA, Seljeflot I, Stensaeth KH, Gaarder M, Brunborg C, Hanssen KF, Dahl-Jorgensen K. The advanced glycation end product methylglyoxal-derived hydroimidazolone-1 and early signs of atherosclerosis in childhood diabetes. Diab Vasc Dis Res. 2015;12:139–145. doi: 10.1177/1479164114560910. [DOI] [PubMed] [Google Scholar]
- Hirose M, Tozawa K, Okada A, Hamamoto S, Higashibata Y, Gao B, Hayashi Y, Shimizu H, Kubota Y, Yasui T, Kohri K. Role of osteopontin in early phase of renal crystal formation: immunohistochemical and microstructural comparisons with osteopontin knock-out mice. Urol Res. 2012;40:121–129. doi: 10.1007/s00240-011-0400-z. [DOI] [PubMed] [Google Scholar]
- Hristova GI, Jarzem P, Ouellet JA, Roughley PJ, Epure LM, Antoniou J, Mwale F. Calcification in human intervertebral disc degeneration and scoliosis. J Orthop Res. 2011;12:1888–1895. doi: 10.1002/jor.21456. [DOI] [PubMed] [Google Scholar]
- Hunter GK. Role of osteopontin in modulation of hydroxyapatite formation. Calcif Tissue Int. 2013;93:348–354. doi: 10.1007/s00223-013-9698-6. [DOI] [PubMed] [Google Scholar]
- Iatridis JC, ap Gwynn I. Mechanisms for mechanical damage in the intervertebral disc annulus fibrosus. J Biomech. 2004;37:1165–1175. doi: 10.1016/j.jbiomech.2003.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illien-Junger S, Grosjean F, Laudier DM, Vlassara H, Striker GE, Iatridis JC. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLoS One. 2013;8:e64302. doi: 10.1371/journal.pone.0064302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illien-Junger S, Lu Y, Qureshi SA, Hecht AC, Cai W, Vlassara H, Striker GE, Iatridis JC. Chronic ingestion of advanced glycation end products induces degenerative spinal changes and hypertrophy in aging pre-diabetic mice. PLoS One. 2015;10:e0116625. doi: 10.1371/journal.pone.0116625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imano M, Satou T, Itoh T, Takeyama Y, Yasuda A, Peng YF, Shinkai M, Haji S, Yasuda C, Nakai T, Yasuda T, Imamoto H, Okuno K, Shiozaki H, Ohyanagi H. An immunohistochemical study of osteopontin in pigment gallstone formation. Am Surg. 2010;76:91–95. [PubMed] [Google Scholar]
- Kahles F, Findeisen HM, Bruemmer D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol Metab. 2014;3:384–393. doi: 10.1016/j.molmet.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim L, Tang SY, Sroga GE, Vashishth D. Differences in non-enzymatic glycation and collagen cross-links between human cortical and cancellous bone. Osteoporos Int. 2013;24:2441–2447. doi: 10.1007/s00198-013-2319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94:55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim NH, Sohn E, Kim CS, Kim JS. Methylglyoxal induces cellular damage by increasing argpyrimidine accumulation and oxidative DNA damage in human lens epithelial cells. Biochem Biophys Res Commun. 2010;391:346–351. doi: 10.1016/j.bbrc.2009.11.061. [DOI] [PubMed] [Google Scholar]
- Kumar MN, Baklanov A, Chopin D. Correlation between sagittal plane changes and adjacent segment degeneration following lumbar spine fusion. Eur Spine J. 2001;10:314–319. doi: 10.1007/s005860000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- Laudier D, Schaffler MB, Flatow EL, Wang VM. Novel procedure for high-fidelity tendon histology. J Orthop Res. 2007;25:390–395. doi: 10.1002/jor.20304. [DOI] [PubMed] [Google Scholar]
- Le Maitre CL, Hoyland JA, Freemont AJ. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res Ther. 2007;9:R77. doi: 10.1186/ar2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebisch M, Bondeva T, Franke S, Daniel C, Amann K, Wolf G. Activation of the receptor for advanced glycation end products induces nuclear inhibitor of protein phosphatase-1 suppression. Kidney Int. 2014;86:103–117. doi: 10.1038/ki.2014.3. [DOI] [PubMed] [Google Scholar]
- Loreto C, Musumeci G, Castorina A, Loreto C, Martinez G. Degenerative disc disease of herniated intervertebral discs is associated with extracellular matrix remodeling, vimentin-positive cells and cell death. Ann Anat. 2011;193:156–162. doi: 10.1016/j.aanat.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Marfia G, Navone SE, Di Vito C, Tabano S, Giammattei L, Di Cristofori A, Gualtierotti R, Tremolada C, Zavanone M, Caroli M, Torchia F, Miozzo M, Rampini P, Riboni L, Campanella R. Gene expression profile analysis of human mesenchymal stem cells from herniated and degenerated intervertebral discs reveals different expression of osteopontin. Stem Cells Dev. 2015;24:320–328. doi: 10.1089/scd.2014.0282. [DOI] [PubMed] [Google Scholar]
- Melrose J, Burkhardt D, Taylor TK, Dillon CT, Read R, Cake M, Little CB. Calcification in the ovine intervertebral disc: a model of hydroxyapatite deposition disease. Eur Spine J. 2009;18:479–489. doi: 10.1007/s00586-008-0871-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek AJ, Buckley MR, Bonassar LJ, Cohen I, Iatridis JC. The effects of needle puncture injury on microscale shear strain in the intervertebral disc annulus fibrosus. Spine J. 2010;10:1098–1105. doi: 10.1016/j.spinee.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier VM, Sun W, Sell DR, Fan X, Nemet I, Genuth S. Glucosepane: a poorly understood advanced glycation end product of growing importance for diabetes and its complications. Clin Chem Lab Med. 2014;52:21–32. doi: 10.1515/cclm-2013-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz RW, Ziv I, Denko CW, Boja B, Jones PK, Adler JH. Spondylosis in sand rats: a model of intervertebral disc degeneration and hyperostosis. J Orthop Res. 1990;8:401–411. doi: 10.1002/jor.1100080312. [DOI] [PubMed] [Google Scholar]
- Mwale F, Billinghurst C, Wu W, Alini M, Webber C, Reiner A, Ionescu M, Poole J, Poole AR. Selective assembly and remodelling of collagens II and IX associated with expression of the chondrocyte hypertrophic phenotype. Dev Dyn. 2000;218:648–662. doi: 10.1002/1097-0177(200008)218:4<648::AID-DVDY1022>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Nemet I, Turk Z, Duvnjak L, Car N, Varga-Defterdarovic L. Humoral methylglyoxal level reflects glycemic fluctuation. Clin Biochem. 2005;38:379–383. doi: 10.1016/j.clinbiochem.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Park P, Garton HJ, Gala VC, Hoff JT, McGillicuddy JE. Adjacent segment disease after lumbar or lumbosacral fusion: review of the literature. Spine (Phila Pa 1976) 2004;29:1938–1944. doi: 10.1097/01.brs.0000137069.88904.03. [DOI] [PubMed] [Google Scholar]
- Puchtler H, Meloan SN. Demonstration of phosphates in calcium deposits: a modification of von Kossa’s reaction. Histochemistry. 1978;56:177–185. doi: 10.1007/BF00495978. [DOI] [PubMed] [Google Scholar]
- Pugliese G, Iacobini C, Blasetti Fantauzzi C, Menini S. The dark and bright side of atherosclerotic calcification. Atherosclerosis. 2015;238:220–230. doi: 10.1016/j.atherosclerosis.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Yan SF, Schmidt AM. The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascular Pharmacol. 2012;57:160–167. doi: 10.1016/j.vph.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S, Bains MA, Kwan A, Menage J, Eisenstein SM. Type X collagen in the human invertebral disc: an indication of repair or remodelling? Histochem J. 1998;30:89–95. doi: 10.1023/a:1003278915981. [DOI] [PubMed] [Google Scholar]
- Roberts S, Urban JP, Evans H, Eisenstein SM. Transport properties of the human cartilage endplate in relation to its composition and calcification. Spine (Phila Pa 1976) 1996;21:415–420. doi: 10.1097/00007632-199602150-00003. [DOI] [PubMed] [Google Scholar]
- Rutges J, Creemers LB, Dhert W, Milz S, Sakai D, Mochida J, Alini M, Grad S. Variations in gene and protein expression in human nucleus pulposus in comparison with annulus fibrosus and cartilage cells: potential associations with aging and degeneration. Osteoarthritis Cartilage. 2010;18:416–423. doi: 10.1016/j.joca.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Rutges JP, Duit RA, Kummer JA, Oner FC, van Rijen MH, Verbout AJ, Castelein RM, Dhert WJ, Creemers LB. Hypertrophic differentiation and calcification during intervertebral disc degeneration. Osteoarthritis Cartilage. 2010;18:1487–1495. doi: 10.1016/j.joca.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Samartzis D, Karppinen J, Mok F, Fong DY, Luk KD, Cheung KM. A population-based study of juvenile disc degeneration and its association with overweight and obesity, low back pain, and diminished functional status. J Bone Joint Surg Am. 2011;93:662–670. doi: 10.2106/JBJS.I.01568. [DOI] [PubMed] [Google Scholar]
- Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- Shao JS, Sierra OL, Cohen R, Mecham RP, Kovacs A, Wang J, Distelhorst K, Behrmann A, Halstead LR, Towler DA. Vascular calcification and aortic fibrosis: a bifunctional role for osteopontin in diabetic arteriosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:1821–1833. doi: 10.1161/ATVBAHA.111.230011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga T, Iso T, Shimizu T, Tanaka T, Yamagishi S, Takeuchi M, Imaizumi T, Kurabayashi M. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb. 2011;18:670–683. doi: 10.5551/jat.7120. [DOI] [PubMed] [Google Scholar]
- Tanikawa T, Okada Y, Tanikawa R, Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vasc Res. 2009;46:572–580. doi: 10.1159/000226225. [DOI] [PubMed] [Google Scholar]
- Tchetina E, Mwale F, Poole AR. Distinct phases of coordinated early and late gene expression in growth plate chondrocytes in relationship to cell proliferation, matrix assembly, remodeling, and cell differentiation. J Bone Miner Res. 2003;18:844–851. doi: 10.1359/jbmr.2003.18.5.844. [DOI] [PubMed] [Google Scholar]
- Thompson JP, Pearce RH, Schechter MT, Adams ME, Tsang IK, Bishop PB. Preliminary evaluation of a scheme for grading the gross morphology of the human intervertebral disc. Spine (Phila Pa 1976) 1990;15:411–415. doi: 10.1097/00007632-199005000-00012. [DOI] [PubMed] [Google Scholar]
- Tsai TT, Ho NY, Lin YT, Lai PL, Fu TS, Niu CC, Chen LH, Chen WJ, Pang JH. Advanced glycation end products in degenerative nucleus pulposus with diabetes. J Orthop Res. 2014;32:238–244. doi: 10.1002/jor.22508. [DOI] [PubMed] [Google Scholar]
- Uribarri J, Cai W, Ramdas M, Goodman S, Pysik R, Chen X, Zhu L, Striker GE, Vlassara H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34:1610–1616. doi: 10.2337/dc11-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassara H, Striker GE. AGE restriction in diabetes mellitus: a paradigm shift. Nat Rev Endocrinol. 2011;7:526–539. doi: 10.1038/nrendo.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Bahalim AN, Barker-Collo S, Barrero LH, Bartels DH, Basanez MG, Baxter A, Bell ML, Benjamin EJ, Bennett D, Bernabe E, Bhalla K, Bhandari B, Bikbov B, Bin Abdulhak A, Birbeck G, Black JA, Blencowe H, Blore JD, Blyth F, Bolliger I, Bonaventure A, Boufous S, Bourne R, Boussinesq M, Braithwaite T, Brayne C, Bridgett L, Brooker S, Brooks P, Brugha TS, Bryan-Hancock C, Bucello C, Buchbinder R, Buckle G, Budke CM, Burch M, Burney P, Burstein R, Calabria B, Campbell B, Canter CE, Carabin H, Carapetis J, Carmona L, Cella C, Charlson F, Chen H, Cheng AT, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahiya M, Dahodwala N, Damsere-Derry J, Danaei G, Davis A, De Leo D, Degenhardt L, Dellavalle R, Delossantos A, Denenberg J, Derrett S, Des Jarlais DC, Dharmaratne SD, Dherani M, Diaz-Torne C, Dolk H, Dorsey ER, Driscoll T, Duber H, Ebel B, Edmond K, Elbaz A, Ali SE, Erskine H, Erwin PJ, Espindola P, Ewoigbokhan SE, Farzadfar F, Feigin V, Felson DT, Ferrari A, Ferri CP, Fevre EM, Finucane MM, Flaxman S, Flood L, Foreman K, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabbe BJ, Gabriel SE, Gakidou E, Ganatra HA, Garcia B, Gaspari F, Gillum RF, Gmel G, Gosselin R, Grainger R, Groeger J, Guillemin F, Gunnell D, Gupta R, Haagsma J, Hagan H, Halasa YA, Hall W, Haring D, Haro JM, Harrison JE, Havmoeller R, Hay RJ, Higashi H, Hill C, Hoen B, Hoffman H, Hotez PJ, Hoy D, Huang JJ, Ibeanusi SE, Jacobsen KH, James SL, Jarvis D, Jasrasaria R, Jayaraman S, Johns N, Jonas JB, Karthikeyan G, Kassebaum N, Kawakami N, Keren A, Khoo JP, King CH, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lalloo R, Laslett LL, Lathlean T, Leasher JL, Lee YY, Leigh J, Lim SS, Limb E, Lin JK, Lipnick M, Lipshultz SE, Liu W, Loane M, Ohno SL, Lyons R, Ma J, Mabweijano J, MacIntyre MF, Malekzadeh R, Mallinger L, Manivannan S, Marcenes W, March L, Margolis DJ, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGill N, McGrath J, Medina-Mora ME, Meltzer M, Mensah GA, Merriman TR, Meyer AC, Miglioli V, Miller M, Miller TR, Mitchell PB, Mocumbi AO, Moffitt TE, Mokdad AA, Monasta L, Montico M, Moradi-Lakeh M, Moran A, Morawska L, Mori R, Murdoch ME, Mwaniki MK, Naidoo K, Nair MN, Naldi L, Narayan KM, Nelson PK, Nelson RG, Nevitt MC, Newton CR, Nolte S, Norman P, Norman R, O’Donnell M, O’Hanlon S, Olives C, Omer SB, Ortblad K, Osborne R, Ozgediz D, Page A, Pahari B, Pandian JD, Rivero AP, Patten SB, Pearce N, Padilla RP, Perez-Ruiz F, Perico N, Pesudovs K, Phillips D, Phillips MR, Pierce K, Pion S, Polanczyk GV, Polinder S, Pope CA, 3rd, Popova S, Porrini E, Pourmalek F, Prince M, Pullan RL, Ramaiah KD, Ranganathan D, Razavi H, Regan M, Rehm JT, Rein DB, Remuzzi G, Richardson K, Rivara FP, Roberts T, Robinson C, De Leon FR, Ronfani L, Room R, Rosenfeld LC, Rushton L, Sacco RL, Saha S, Sampson U, Sanchez-Riera L, Sanman E, Schwebel DC, Scott JG, Segui-Gomez M, Shahraz S, Shepard DS, Shin H, Shivakoti R, Singh D, Singh GM, Singh JA, Singleton J, Sleet DA, Sliwa K, Smith E, Smith JL, Stapelberg NJ, Steer A, Steiner T, Stolk WA, Stovner LJ, Sudfeld C, Syed S, Tamburlini G, Tavakkoli M, Taylor HR, Taylor JA, Taylor WJ, Thomas B, Thomson WM, Thurston GD, Tleyjeh IM, Tonelli M, Towbin JA, Truelsen T, Tsilimbaris MK, Ubeda C, Undurraga EA, van der Werf MJ, van Os J, Vavilala MS, Venketasubramanian N, Wang M, Wang W, Watt K, Weatherall DJ, Weinstock MA, Weintraub R, Weisskopf MG, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams SR, Witt E, Wolfe F, Woolf AD, Wulf S, Yeh PH, Zaidi AK, Zheng ZJ, Zonies D, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shan J, Yang W, Zheng H, Xue S. High mobility group box 1 (HMGB1) mediates high-glucose-induced calcification in vascular smooth muscle cells of saphenous veins. Inflammation. 2013a;36:1592–1604. doi: 10.1007/s10753-013-9704-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Videman T, Battie MC. ISSLS Prise Winner: Lumbar vertebral endplate lesions: associations withdisc degeneration and back pain history. Spine (Phila Pa 1976) 2012;37:1490–1496. doi: 10.1097/BRS.0b013e3182608ac4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang ZY, Chen XQ, Wang X, Cao H, Liu SW. Advanced glycation end products promote human aortic smooth muscle cell calcification in vitro via activating NF-kappaB and down-regulating IGF1R expression. Acta Pharmacol Sin. 2013b;34:480–486. doi: 10.1038/aps.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, An Y, Chen D. Advanced glycation end-product nepsilon-carboxymethyl-Lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis. 2012;221:387–396. doi: 10.1016/j.atherosclerosis.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Wei Q, Ren X, Jiang Y, Jin H, Liu N, Li J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc Disord. 2013;13:13. doi: 10.1186/1471-2261-13-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams FM, Popham M, Sambrook PN, Jones AF, Spector TD, MacGregor AJ. Progression of lumbar disc degeneration over a decade: a heritability study. Ann Rheum Dis. 2011;70:1203–1207. doi: 10.1136/ard.2010.146001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka K, Park JS, Jimbo K, Yamada K, Sato K, Tsuru M, Takeuchi M, Yamagishi S, Nagata K. Advanced glycation end-products downregulating intervertebral disc cell production of proteoglycans in vitro. J Neurosurg Spine. 2006;5:324–329. doi: 10.3171/spi.2006.5.4.324. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Park JS, Yokosuka K, Jimbo K, Yamada K, Sato K, Takeuchi M, Yamagishi S, Nagata K. Up-regulation in receptor for advanced glycation end-products in inflammatory circumstances in bovine coccygeal intervertebral disc specimens in vitro. Spine (Phila Pa 1976) 2009;34:1544–1548. doi: 10.1097/BRS.0b013e3181a98390. [DOI] [PubMed] [Google Scholar]
Web references
- Andersson G. United States bone and joint initiative: The Burden of Musculoskeletal Diseases in the United States (BMUS) Amer Acad Orthop Surg. 2014 http://www.boneandjointburden.org/2014-report/iia0/definition.
Additional References
- Jazini E, Sharan AD, Morse LJ, Dyke JP, Aronowitz EB, Chen LKH, Tang SY. Alterations in T2 relaxation magnetic resonance imaging of the ovine intervertebral disc due to nonenzymatic glycation. Spine (Phil Pa 1976) 2012;37:209–215. doi: 10.1097/BRS.0b013e31822ce81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuru M, Nagata K, Jimi A, Irie K, Yamada A, Nagai R, Horiuchi S, Sata M. Effect of AGEs on human disc herniation: intervertebral disc hernia is also effected by AGEs. Kurume Med J. 2002;49:7–13. doi: 10.2739/kurumemedj.49.7. [DOI] [PubMed] [Google Scholar]
- Wagner DR, Reiser KM, Lotz JC. Glycation increases human annulus fibrosus stiffness in both experimental measurements and theoretical predictions. J Biomech. 2006;39:1021–1029. doi: 10.1016/j.jbiomech.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Watson AM, Soro-Paavonen A, Sheehy K, Li J, Calkin AC, Koitka A, Rajan SN, Brasacchio D, Allen TJ, Cooper ME, Thomas MC, Jandeleit-Dahm KJ. Delayed intervention with AGE inhibitors attenuates the progression of diabetes-accelerated atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia. 2011;54:681–689. doi: 10.1007/s00125-010-2000-9. [DOI] [PubMed] [Google Scholar]