

Abstract
Purpose
To determine the ocular consequences of a dominant-negative mutation in the p85α subunit of phosphatidylinositol 3-kinase (PIK3R1) using a knock-in mouse model of SHORT syndrome, a syndrome associated with short stature, lipodystrophy, diabetes, and Rieger anomaly in humans.
Methods
We investigated knock-in mice heterozygous for the SHORT syndrome mutation changing arginine 649 to tryptophan in p85α (PIK3R1) using physical examination, optical coherence tomography (OCT), tonometry, and histopathologic sections from paraffin-embedded eyes, and compared the findings to similar investigations in two human subjects with SHORT syndrome heterozygous for the same mutation.
Results
While overall eye development was normal with clear cornea and lens, normal anterior chamber volume, normal intraocular pressure, and no changes in the retinal structure, OCT images of the knock-in mouse eyes revealed a significant decrease in thickness and width of the iris resulting in increased pupil area and irregularity of shape. Both human subjects had Rieger anomaly with similar defects including thin irides and irregular pupils, as well as a prominent ring of Schwalbe, goniosynechiae, early cataract formation, and glaucoma. Although the two subjects had had diabetes for more than 30 years, there were no signs of diabetic retinopathy.
Conclusions
A dominant-negative mutation in the p85α regulatory subunit of PI3K affects development of the iris, and contributes to changes consistent with anterior segment dysgenesis in both humans and mice.
Keywords: iris, anterior segment dysgenesis, optical coherence tomography
SHORT syndrome (Online Mendelian Inheritance in Man [OMIM] 269880) is a rare, autosomal dominantly inherited disorder. Originally, the syndrome was defined by its acronym, representing Short stature, Hyperextensibility of joints and/or inguinal hernia, Ocular depression, Rieger anomaly, and Teething delay.1 More recently defined clinical manifestations include intrauterine growth retardation and adult short stature, facial dysmorphism, partial lipodystropy, insulin resistance, and in most cases diabetes.2–6
The ophthalmic anomalies of SHORT syndrome are characterized by anterior segment dysgenesis (ASD), that is, the failure of normal development of the tissues of the anterior eye segment. In SHORT syndrome, ASD has mainly been described as Axenfeld-Rieger anomaly.7–10 The Axenfeld anomaly is an ocular defect characterized by anteriorly displaced Schwalbe's line and iris bands extending to the cornea,11 while the Rieger anomaly, in addition, includes iris and pupil anomalies such as prominent strands and marked atrophy of the iris stroma, with hole or pseudo-hole formation in the iris and corectopia.12
Recently, we and others have shown that SHORT syndrome is caused by autosomal dominant mutations in the PIK3R1 gene that encodes the p85α regulatory subunit of phosphatidylinositol-3 kinase (PI3K). PI3K, acting through AKT and mTOR signaling, is a central component of insulin and growth factor signaling.7–10 The most prevalent mutation in SHORT syndrome is an Arg649Trp missense mutation in the C-terminal SH2 domain of p85α. In a recent review, 23 of 32 cases of SHORT syndrome (representing 16 of 24 families) were found to be heterozygous for the Arg649Trp mutation.6 We have shown that this mutation markedly reduces the ability of p85α to bind to tyrosine phosphorylated proteins like insulin receptor substrate-1 (IRS-1), leading to reduced insulin-dependent activation of the PI3K pathway in vitro.7 Recently, we have demonstrated that a knock-in mouse with the same p85α Arg649Trp mutation develops many of the metabolic features of SHORT syndrome including a reduction in body length, systemic insulin resistance, and reduced fat mass.13
Here, we have characterized the ophthalmologic phenotype of these mice in order to investigate the role of PI3K in eye development and evaluate this mouse model as a potential tool for further studies on ASD. We have further compared the findings in this mouse to two subjects with SHORT syndrome (a mother and son), with the same missense Arg649Trp p85α mutation.
Methods
Animals
Knock-in mice heterozygous for the phosphatidylinositol 3-kinase (PIK3R1) Arg649Trp mutation were kept on a C57Bl/6 background and housed on a 12-hour light/12-hour dark cycle with ad libitum access to water and food (mouse diet F9; PMI Nutrition International, Richmond, IN, USA).13 All animal experiments were approved by and conducted in accordance with guidelines established by the Institutional Animal Care and Use Committee at the Joslin Diabetes Center and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Mouse Optical Coherence Tomography Measurements
Anterior chamber phenotypes were assessed by spectral-domain optical coherence tomography (SD-OCT) (840SDOCT; Bioptigen, Durham, NC, USA) using a 25-mm cornea lens attached to the optical imaging head. For all examinations mice were anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (13.5 mg/kg). Rectangular SD-OCT volumes of the anterior chamber were obtained from undilated mouse eyes at a setting of 1000 A-line by 100 B-line scans. For each eye, a 2.5- by 2.5-mm area was scanned of the overall anterior chamber. Corresponding en face images provided pupil dimensions, while B-scan bisections provided anterior chamber dimensions. Measurements were made with calipers using the InVivoVue Clinic v1.3 software (Durham, NC, USA). For iris width and thickness measurements, six measurements distributed across the iris at approximately equal distance were averaged into one single value. A 1.5- by 1.5-mm area was scanned of the iris and anterior chamber angles at the temporal and nasal locations. For retinal scans, a standard mouse lens was attached to the optical head. Pupils were dilated with 1% tropicamide, and a rectangular SD-OCT volume at 1.5 by 1.5 mm of the retina centered on the optic nerve head was obtained.
Mouse Intraocular Pressure Measurements
Intraocular pressure (IOP) was measured using the noninvasive Icare TONOLAB tonometer (Icare LAB; Icare Finland Oy, Espoo, Finland) calibrated for mice according to the manufacturer's protocol. In brief, mice were anesthetized using a mixture of ketamine (75 mg/kg) and xylazine (13.5 mg/kg) and secured. The tonometer probe tip was held at a 1- to 4-mm distance from the cornea, and for measurements the tip hit the central cornea. Six measurements were made consecutively, and the results averaged. The pressure was recorded 10 to 15 minutes after anesthesia, when it has been shown to be most reproducible.14
Histologic Examination of Mouse Eyes
Mice were killed using CO2 inhalation. Eyes were enucleated and fixed in 4% paraformaldehyde overnight at 4°C. The eyes were subsequently rinsed in PBS and stored in 70% ethanol before being embedded in paraffin. Sections of 6 μm were stained with hematoxylin and eosin and examined by light microscopy.
Human Subjects
The human subjects, a male proband and his mother, both with SHORT syndrome, were first described in 1983 and reexamined in 2013.4,7 In the original 1983 description of the family, the proband was examined through childhood, starting at 3 years, and was examined along with his mother. In 2013, the family (family 1, male proband as subject III-3 and mother as subject II-2) was found to carry the same mutation in PIK3R1 as another family with strikingly similar features.7 The son also had a third examination in 2015, due to the present study. A signed informed letter of consent was obtained. The study was approved by the Regional Committee for Medical and Health Research Ethics, Western Norway (IRB no. 00001872), reference no. 2079/2009, and performed according to the latest version of the Declaration of Helsinki. The ophthalmologic examination included visual acuity measurements (Snellen chart), slit-lamp examination, gonioscopy, Goldmann and Icare tonometry, corneal topography (Pentacam; Oculus, Wetzlar, Germany), anterior and posterior segment OCT (Spectralis; Heidelberg Engineering, Heidelberg, Germany), axial length measurements (IOL Master; Carl Zeiss Meditec, Oberkochen, Germany), and visual field examination (Humphrey Visual Field Analyzer HFA-2, SITA Fast strategy 30-2; Carl Zeiss Meditec). Complete hospital records were obtained for both individuals.
Results
PIK3R1 Mutation Causes Iris Morphology Consistent With Anterior Segment Dysgenesis (ASD) in Mice
To assess the impact of PI3K mutations of SHORT syndrome in eye development we evaluated the ophthalmologic anomalies of knock-in mice carrying the Arg649Trp missense mutation found in most humans with SHORT syndrome. We have previously shown that this acts as a dominant-negative mutation to inhibit PI3K activity (Fig. 1). We performed OCT imaging of the anterior chamber of 1-year-old wild-type (WT) control mice and heterozygous Arg649Trp knock-in mice. We observed that knock-in animals had thinner irides and wider pupillary diameters than the control littermates (Fig. 2A). These observations were confirmed by measurements averaged to derive a value of thickness or length as described in Methods. Consistent with the images, there was a significant thinning of iris (71.2 ± 2.1 vs. 57.9 ± 0.9 μm, P < 0.0001) (Fig. 2B) and a decrease in iris width (1010 ± 60.5 vs. 736.8 ± 42.3 μm, P < 0.002) (Fig. 2C). In addition, in more than half of the mice, we observed ectropiation of the iris at the pupillary margin, a feature that can be observed in iridogoniodysgenesis and Rieger anomaly. Furthermore, the anchoring of the iris appeared to be displaced more anteriorly in the mutant mice compared to control mice, resembling a plateau iris with a narrow chamber angle (Fig. 2A). Despite the observed thinning and decreased width of the iris using OCT imaging, the morphology of the iris, trabecular meshwork, and ciliary body in mutant mice was indistinguishable from that observed in normal mice (Figs. 2D, 2E).
Figure 1.
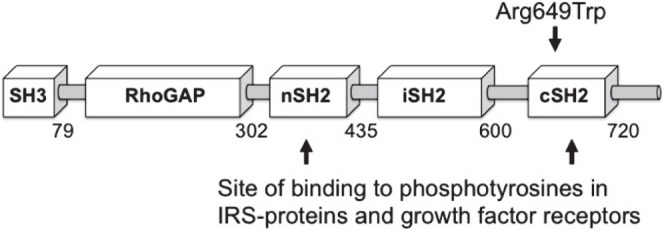
A mutation in PIK3R1 causes SHORT syndrome and anterior segment dysgenesis. A schematic model of the p85α protein encoded by PIK3R1 showing the domain structure and the location of the mutation. The p85α protein contains the following domains: SH3, Src-homology region 3; RhoGap, Rho GTPase-activating protein domain; nSH2, N-terminal Src homology region 2; iSH2, interterminal Src homology region 2; cSH2, N-terminal Src homology region 2. The nSH2 and cSH2 domains bind to phosphotyrosines in IRS proteins and growth factor receptors, to initiate activation of PI3-kinase.
Figure 2.
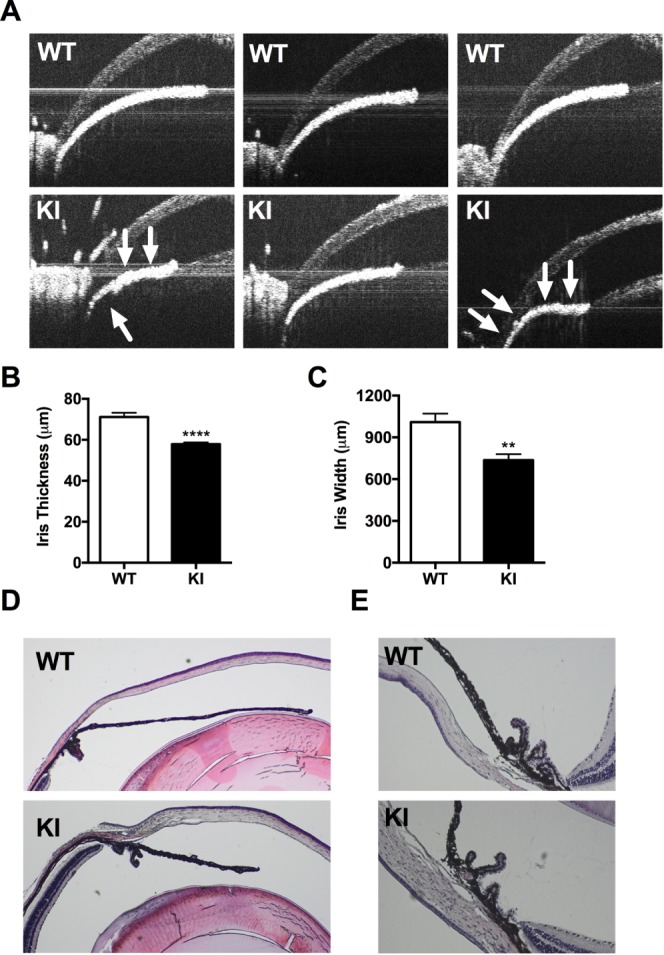
An Arg649Trp PIK3R1 mutation in mice replicates human iris morphology seen in affected individuals with SHORT syndrome. (A) A panel of OCT images showing the irides of WT control mice (top) and PIK3R1 knock-in mice (bottom). Note the thinner and shorter irides in the knock-in animals. Iris plateau formation and narrow chamber angle is marked with arrows. (B) Iris thickness and (C) iris width were quantified from the OCT images using the average of six independent measurements. (D) Hematoxylin- and eosin-stained sections of anterior eye in WT control mice (top) and mutant mice (bottom). (E) Close-up of iridocorneal angle shows similar morphology between the two groups. Results for width and thickness are shown as mean ± SEM (n = 7–13). **P < 0.01; ****P < 0.0005, unpaired Student's t-test.
Anterior Chamber Volume and Intraocular Pressure Remain Normal
Anterior chamber dysgenesis and changes in iris morphology can impair aqueous humor drainage. Using OCT imaging we observed no changes in anterior chamber volume (Fig. 3A). Using a central pupil-to-cornea measurement, however, we observed a significant decrease in depth (402 ± 5.3 vs. 382.3 ± 4.5 μm, P < 0.0112) (Fig. 3B). IOP of mutant and WT control mice was measured every 4 weeks from the age of 12 to 24 weeks and was unchanged between the two groups (Fig. 3C).
Figure 3.
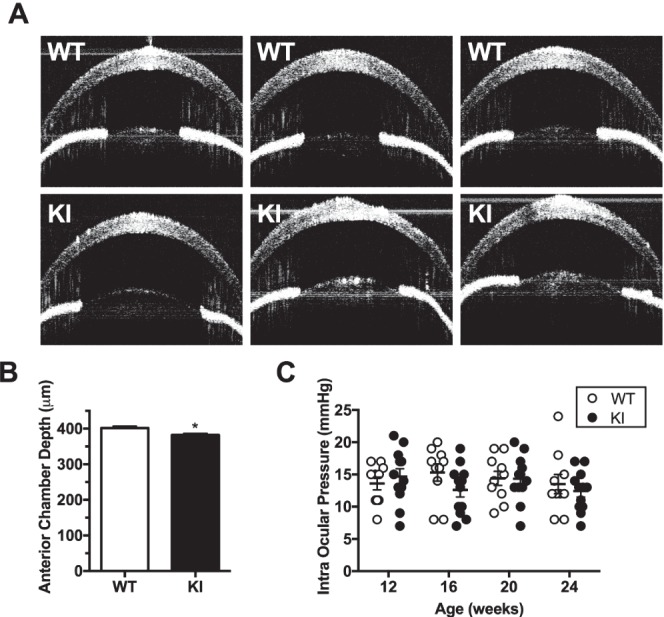
Arg649Trp knock-in mice show no changes in anterior chamber volume or intraocular pressure. (A) A panel of OCT images showing the anterior chamber of WT control (top) and knock-in mice (bottom). There is no difference in volume between the two groups. (B) Quantified depth of anterior chamber at 1 year of age showing a slight decrease in chamber depth. Results are shown as mean ± SEM (n = 8–11). (C) Intraocular pressure measurements of WT and mutant animals from 12 to 24 weeks of age (n = 10–12) reveal that the pressure is normal in the mutant animals compared to WT littermates.
Larger and Misshapen Pupils in Eyes From Knock-In Mice
ASD and iris hypoplasia can be associated with an open pupil. The pupil may also be irregular, and corectopia may be present. OCT images of WT control and knock-in animals revealed a trend of larger pupil area in the knock-in animals, but did not reach statistical significance when quantified (Figs. 4A, 4B). The observed larger pupil area, however, correlated with the significantly shorter iris. Additionally, we observed a tendency of the mutant animals to have misshapen pupils; more than 50% of the animals had irregular and at times a pupil pulled to one side (corectopia), consistent with the syndrome phenotype.
Figure 4.
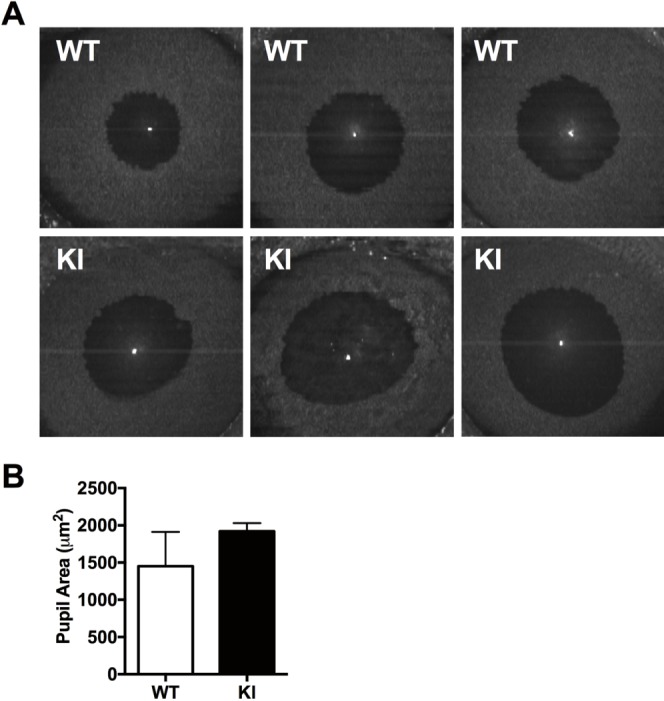
Arg649Trp p85α heterozygous knock-in animals have wide and irregular pupils. (A) Panel of OCT images showing pupil area of WT control mice (top) and knock-in animals (bottom). (B) Quantification of pupil area indicating a trend of larger pupil area of mutant animals compared to WT. Results are shown as mean ± SEM (n = 3–6).
No Changes in Retinal Morphology
Retinal thickness was measured by SD-OCT. We observed no differences in retinal layer thickness between retinal B-scans of WT control animals and knock-in animals (Fig. 5A). Histologic analysis also confirmed no differences in retinal layers (Fig. 5B).
Figure 5.

Arg649Trp knock-in animals have normal posterior segment. (A) Quantification of retinal layers from retinal B-scans and (B) histologic images of retinal layers do not reveal any differences in retinal layers between mutant mice and WT littermates.
Clinical Characteristics of Anterior Segment Dysgenesis in Human PIK3R1 Mutation Carriers
We compared the findings in these mice to two affected individuals, a son and his mother, in a Norwegian family with SHORT syndrome carrying the same missense Arg649Trp mutation in p85α as the mice.
The son was seen by an ophthalmologist at the age of 3 after being diagnosed with several features of SHORT syndrome. He presented with an accommodative esotropia; a prominent ring of Schwalbe was observed in both eyes; and the irides were thin without discernible crypts. Both pupils were irregular and responded poorly to light. At 8 years of age, he was noted to have deeply situated eyes. IOP was 18 mm Hg in both eyes, and visual field examination was normal. Gonioscopy revealed poorly pigmented angles without anterior synechiae, and corneal diameter was 11.5 mm in the right eye and 11 mm in the left eye. At the age of 12 years, IOP was 19 mm Hg in both eyes, and visual field examination revealed a possible Bjerrum scotoma. One year later, IOP was 23 mm Hg in the right eye and 22 mm Hg in the left eye, and pilocarpine was started in both eyes. Three months later, IOP was 17 mm Hg in both eyes. At 20 years of age, opacities were observed in both lenses. He had cataract surgery with phacoemulsification and intraocular lens implantation in the right eye at the age of 22 years and in the left eye at the age of 24 years. At 23 years of age, increased excavation of the right optic disc was observed, and he had a trabeculectomy in his right eye. At 36 years of age, laser trabeculoplasty was performed twice in his right eye followed at 37 years of age by another trabeculectomy. At 38 years of age, a trabeculectomy was performed in his left eye.
By age 42, best-corrected visual acuity (BCVA) was 0.63+, and IOP was 16 mm Hg in both eyes. He had normal motility and no diplopia, but cover test revealed an esophoria of approximately 12 prism diopters (PD). He had loss of volume of the superior sulcus as a consequence of his lipodystrophy. Proptosis measured by a Hertel exophthalmometer was 15 mm on both sides (15-102-15). Slit-lamp examination (Fig. 6A) revealed embryotoxon posterior in both eyes. The corneas were otherwise clear with central corneal thickness (CCT) of 698 μm in the right eye and 673 μm in the left eye. The irides appeared darker and thinner than normal, which was confirmed by anterior segment OCT (Fig. 6B). The pupil was irregular and fibrotic, and responded poorly to mydriatics. The iridectomies were open and filtration blebs were present. Gonioscopy revealed open angles with poorly pigmented trabecular meshwork. Small goniosynechiae were seen, more in the right than in the left eye. Visual field defects were observed in both eyes (Supplementary Fig. S1). The patient was also diagnosed with diabetes mellitus at age 22 and started on insulin. At age 35 there were no signs of diabetic retinopathy. OCT revealed thinning of the nerve fiber layer, and the pigment epithelium appeared poorly pigmented (Fig. 6C).
Figure 6.
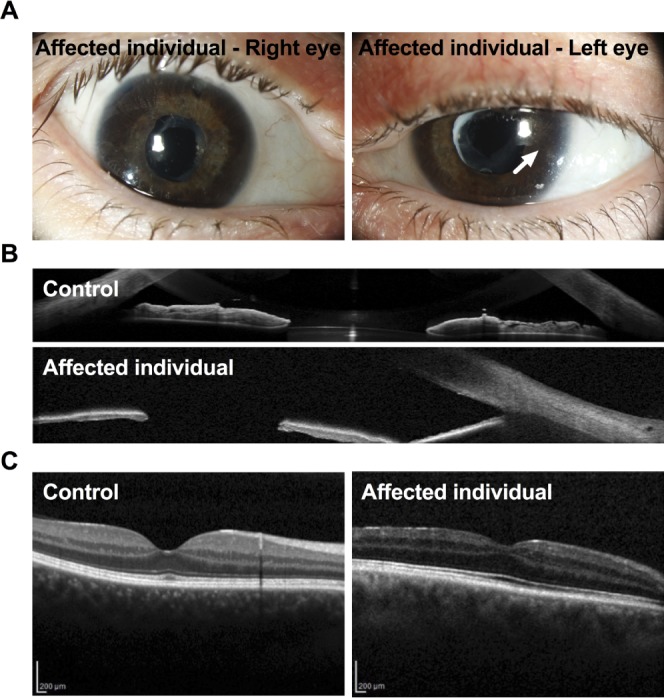
(A) Slit-lamp images of the eyes of an affected human individual with SHORT syndrome revealing embryotoxon posterior (arrow) and iris atrophy. In addition, two iridotomies after previous trabeculectomies can be seen in the upper part of the iris in the right eye, and anterior capsule opacification after cataract surgery in both eyes. (B) A representative OCT image showing the iris of an unaffected individual (top) and an affected individual (the son) with SHORT syndrome (bottom), showing thinner irides in the affected compared to the unaffected individual. (C) A representative OCT image showing the retina of an unaffected individual (left) and the retina of the same affected individual as above (the son) with SHORT syndrome, showing thinning of the nerve fiber layer. The pigment epithelium appeared poorly pigmented.
The mother was diagnosed with several of the SHORT syndrome features at age 40. From childhood she had dark irides, and at age 46 years, she was diagnosed with an anterior segment anomaly classified as a variant of Rieger anomaly. In both eyes, posterior embryotoxon was present, and there were anterior synechiae extending to the ring of Schwalbe. The iris was thin without crypts, and the outline of the lens could be seen through the iris. The pupillary border was atrophic, and there was slight corectopia with temporal displacement of the pupil. The pupil was round in the right eye and irregular in the left. Both pupils responded poorly to light. Gonioscopy revealed poorly pigmented trabecular meshwork with multiple goniosynechiae. IOP was 20 mm Hg in both eyes but increased by the age of 51 years, leading to treatment with timolol and pilocarpine, and a bilateral laser trabeculoplasty. She had cataract surgery in the left eye at the age of 60 years and in the right eye at the age of 62 years. Postoperative BCVA was 0.8 in both eyes. Corneal pachymetry was 623 μm in the right eye and 603 μm in the left eye. Her optic discs remained normal, and she also had normal visual fields until 72 years of age (Supplementary Fig. S1). At this point in time, IOP was 19 mm Hg in the right eye and 16 mm Hg in the left eye. Pressure then started to rise mainly in her right eye, and a trabeculectomy was performed at the age of 75 years. Preoperatively, IOP was 34 mm Hg in the right eye and 16 mm Hg in the left eye. In the same period her right visual field deteriorated, but the left visual field remained normal (Supplementary Fig. S1). Also the right optic disc became excavated while the left remained normal. At present, IOP is 11 mm Hg in the right eye without any medication, and 10 mm Hg in the left eye with a combination of bimatoprost, timolol, dorzolamide, and pilocarpine. In addition, she was diagnosed with type 2 diabetes mellitus at the age of 40 years and started insulin treatment at the age of 56 years. She is now 80 years old, but free of diabetic retinopathy.
Discussion
We have previously reported that a mouse knock-in model of SHORT syndrome with an Arg649Trp mutation in the C-terminal SH2 domain of the p85α gene recapitulates multiple aspects of the human disease. This phenotype includes reduced linear growth, a reduction in subcutaneous adipose tissue, and development of insulin resistance and type 2 diabetes.13 In this study, we further investigated the pathophysiology of the disease and the role of the PI3K pathway by characterizing the ocular abnormalities associated with SHORT syndrome. Our study evaluated the ocular phenotype in this knock-in mouse, comparing it to two human subjects carrying the same mutation associated with the disease.
The ocular phenotype of SHORT syndrome is mostly associated with abnormalities of the anterior segment, and is illustrated by the two subjects evaluated here. Both had Rieger anomaly with thinner irides, irregular pupils, and a prominent ring of Schwalbe. Likewise, the knock-in animals, like the human subjects, showed thinner and narrower irides than their WT controls on OCT imaging. In some mice a degree of ectropiation of the iris was present, commonly seen in iridogoniodysgenesis and Rieger anomaly. In addition, we observed a plateau-like morphology of the iris causing narrowing of the chamber angle. While the human subjects showed early cataract formation and glaucoma, mice had no increase in IOP and no evidence of either glaucoma or cataract. Impaired drainage of aqueous humor due to malformations in the anterior chamber angle, trabecular meshwork, and canal of Schlemm can lead to glaucoma in individuals with ASD.15 The severity of the malformations does not, however, strictly correlate with IOP, and many ASD patients do not develop increased IOP until late in childhood or in adulthood. Other factors are therefore likely to be involved in glaucoma development in ASD patients.
Despite the widespread nature of PI3K in cells, regulating diverse biological processes, including cell growth, differentiation, survival, proliferation, migration, and metabolism, the malformations in the mouse were strictly located to the anterior segment of the eye, with no detection of changes in the posterior part of the eye including the retina. The extent to which these iris abnormalities might impair vision is unclear; however, the iris plays an important role in visual function by regulating the amount of light that enters the eye, as well as focal adjustment of closer objects.
Our previous studies of the Arg649Trp mutation show that the mutation causes resistance to several growth factors including insulin, insulin-like growth factor 1 (IGF-1), epidermal growth factor (EGF), and platelet-derived growth factor (PDGF), indicating a general growth factor resistance.13 Resistance to other growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) has not yet been studied but is very likely, and this would have an important impact on events such as embryonic development, cell migration, and angiogenesis. Thus, it is not surprising that homozygous Arg649Trp mutant animals are embryonic lethal, while the heterozygous knock-in animals show features of SHORT syndrome such as lipodystrophy and insulin resistance and reduced overall body weight and growth, as well as iris malformations.
Hypoplasia of the iris is a rather frequent feature of genetic disorders that involve failure of normal cell migration, such as Axenfeld-Rieger syndrome (ARS), which in the eye closely resembles the clinical features seen in SHORT syndrome. In ARS, the ocular features are proposed to be caused by an arrest in the development of neural crest derived–tissues in the anterior segment. This leads to retention of primordial endothelial tissue on the iris and across the anterior chamber angle, which produces the iris changes. PI3K has been demonstrated as a key factor in stimulating proliferation and migration of neural crest and other cell types. It is also important for cell migration both through direct actions and through cross-talk with other pathways such as Rho GTPase signaling.16,17 It is not unlikely that migration of neural crest and other cell types involved in iris development is impaired to some extent in our mutant mice, resulting in the iris phenotype.
Two other signaling pathways that participate in the development of the irides are the bone morphogenic protein (BMP) and Wnt signaling pathways.18,19 Wnt signaling pathway regulates developmental processes such as proliferation, cell migration, axon guidance, and cell-fate determination, and altered Wnt signaling has been implicated in numerous diseases, including type 2 diabetes and the metabolic syndrome. Wnt and insulin signaling pathways cross-talk at multiple levels where Wnt, dependent on insulin/IGF-1 receptors, induces phosphorylation of AKT, ERK1/2, and GSK3β.20 The BMP growth factors, initially discovered for their ability to induce bone formation, have also been shown to be essential for cell differentiation and morphogenesis during embryonic development, and are known to modulate the phosphatidylinositol-3-kinase/AKT pathway.21,22 In fact, BMP4 deletions have previously been detected in subjects with anterior segment anomalies; one of these had SHORT syndrome.23
The phenotype of the anterior segment abnormalities in SHORT syndrome varies between families and between individuals within the same family. In a recent report of 32 individuals with SHORT syndrome and mutations in PIK3R1, Rieger abnormality was present in only 13 of 30 cases. However, in cases without Rieger abnormality, some patients (5/16 cases) presented with other abnormalities of the anterior chamber of the eye.6 This kind of phenotypic heterogeneity is not unexpected and has also been observed for mutations in other genes that cause ASD such as PITX2, FOXC1, and COL4A1.24,25
While not all SHORT syndrome patients have diabetes, both subjects that we studied are diabetic. Interestingly, neither has evidence of diabetic retinopathy despite at least 30 years of exposure to hyperglycemia. In types 1 and 2 diabetes, retinopathy affects up to 75% of patients after 15 years of the disease, and up 7.5% when diagnosis is made prior to 30 years of age. Inhibition of the PI3K/AKT/mTOR pathway has been proposed as a potential therapeutic target for diabetic retinopathy because of the role of PI3K in vascular proliferation and endothelial integrity.26 Indeed, the SHORT syndrome mutation, which affects many growth factors working through PI3K, including VEGF, might be responsible for slowing progression of vasculopathies as in diabetic retinopathy, since enhanced migration of endothelial cells is a requirement for neovascularization.
In summary, we show that an Arg649Trp heterozygous mutation in PIK3R1, the most common mutation in SHORT syndrome, affects normal development of the iris and anterior chamber of the eye, causing changes consistent with ASD. While the phenotype in mice is somewhat milder than observed in the human subjects, the knock-in mouse clearly reflects the human disease. In both mice and humans the eye phenotype is likely due to a general growth factor resistance and impaired neural crest cell migration. Our studies show that PI3K plays an important role in anterior segment eye development and that impaired PI3K signaling can lead to ASD.
Supplementary Material
Acknowledgments
The authors thank the family for participating in this study. They thank Lloyd Paul Aiello for helpful discussion, Christie Penniman for technical assistance during IOP measurements, and Unni Larsen for her assistance with mouse tissue histology.
Supported by National Institutes of Health R01 Grant DK055545 (CRK) and the Mary K. Iacocca Professorship. MHS was supported in part by funds from the University of Bergen, the KG Jebsen Foundation, the Norwegian Society of Endocrinology, Eckbo's Foundation, Tom Wilhelmsen Foundation, and the Norwegian Diabetes Association. PRN was supported by the European Research Council (293574), the Research Council of Norway, the University of Bergen, Stiftelsen Kristian Gerhard Jebsen, and the Western Regional Health Authorities. ER was supported by the Western Regional Health Authorities (911746). AM was supported by Novo Nordisk Foundation.
Disclosure: M.H. Solheim, None; A.C. Clermont, None; J.N. Winnay, None; E. Hallstensen, None; A. Molven, None; P.R. Njølstad, None; E. Rødahl, None; C.R. Kahn, None
References
- 1. Gorlin RJ,, Cervenka J,, Moller K,, Horrobin M,, Witkop CJ., Jr. Malformation syndromes. A selected miscellany. Birth Defects Orig Artic Ser. 1975; 11: 39–50. [PubMed] [Google Scholar]
- 2. Graettinger WF,, Lipson JL,, Klein RC,, Cheung DG,, Weber MA. Comparison of antihypertensive therapies by noninvasive techniques. Chest. 1989; 96: 74–79. [DOI] [PubMed] [Google Scholar]
- 3. Koenig R,, Brendel L,, Fuchs S. SHORT syndrome. Clin Dysmorphol. 2003; 12: 45–49. [DOI] [PubMed] [Google Scholar]
- 4. Aarskog D,, Ose L,, Pande H,, Eide N. Autosomal dominant partial lipodystrophy associated with Rieger anomaly, short stature, and insulinopenic diabetes. Am J Med Genet. 1983; 15: 29–38. [DOI] [PubMed] [Google Scholar]
- 5. Schwingshandl J,, Mache CJ,, Rath K,, Borkenstein MH. SHORT syndrome and insulin resistance. Am J Med Genet. 1993; 47: 907–909. [DOI] [PubMed] [Google Scholar]
- 6. Avila M,, Dyment DA,, Sagen JV,, et al. Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: towards recommendation for molecular testing and management. Clin Genet. 2015; 89: 501–506. [DOI] [PubMed] [Google Scholar]
- 7. Chudasama KK,, Winnay J,, Johansson S,, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013; 93: 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dyment DA,, Smith AC,, Alcantara D,, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013; 93: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schroeder C,, Riess A,, Bonin M,, et al. PIK3R1 mutations in SHORT syndrome. Clin Genet. 2014; 86: 292–294. [DOI] [PubMed] [Google Scholar]
- 10. Thauvin-Robinet C,, Auclair M,, Duplomb L,, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013; 93: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Axenfeld T. Embryotoxon cornea posterius. Klin Monatsbl Augenheilkd. 1920; 65: 381–382. [Google Scholar]
- 12. Rieger H. Beitraege zur Kenntnis seltener Missbildungen der Iris: ueber Hypoplasie des Irisvorderblattes mit Verlagerung und Entrundung der Pupille. Albrecht von Graefes Arch Klin Exp Ophthal. 1935; 133: 602–635. [Google Scholar]
- 13. Winnay JN,, Solheim MH,, Dirice E,, et al. PI3-kinase mutation linked to insulin and growth factor resistance in vivo. J Clin Invest. 2016; 126: 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu Y,, Yang H,, Lei B. Effects of three commonly used anesthetics on intraocular pressure in mouse. Curr Eye Res. 2014; 39: 365–369. [DOI] [PubMed] [Google Scholar]
- 15. Gould DB,, John SW. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet. 2002; 11: 1185–1193. [DOI] [PubMed] [Google Scholar]
- 16. Focke PJ,, Swetlik AR,, Schilz JL,, Epstein ML. GDNF. and insulin cooperate to enhance the proliferation and differentiation of enteric crest–derived cells. J Neurobiol. 2003; 55: 151–164. [DOI] [PubMed] [Google Scholar]
- 17. Eberle H,, Forrest N,, Hrynyszyn J,, Van Knapp J. Regulation of DNA synthesis and capacity for initiation in DNA temperature sensitive mutants of Escherichia coli I. Reinitiation and chain elongation. Mol Gen Genet. 1982; 186: 57–65. [DOI] [PubMed] [Google Scholar]
- 18. Zhao GQ. Consequences of knocking out BMP signaling in the mouse. Genesis. 2003; 35: 43–56. [DOI] [PubMed] [Google Scholar]
- 19. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127: 469–480. [DOI] [PubMed] [Google Scholar]
- 20. Palsgaard J,, Emanuelli B,, Winnay JN,, Sumara G,, Karsenty G,, Kahn CR. Cross-talk between insulin and Wnt signaling in preadipocytes: role of Wnt co-receptor low density lipoprotein receptor-related protein-5 (LRP5). J Biol Chem. 2012; 287: 12016–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang RN,, Green J,, Wang Z,, et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014; 1: 87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009; 19: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reis LM,, Tyler RC,, Schilter KF,, et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet. 2011; 130: 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reis LM,, Tyler RC,, Volkmann Kloss BA,, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012; 20: 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodahl E,, Knappskog PM,, Majewski J,, et al. Variants of anterior segment dysgenesis and cerebral involvement in a large family with a novel COL4A1 mutation. Am J Ophthalmol. 2013; 155: 946–953. [DOI] [PubMed] [Google Scholar]
- 26. Jacot JL,, Sherris D. Potential therapeutic roles for inhibition of the PI3K/Akt/mTOR pathway in the pathophysiology of diabetic retinopathy. J Ophthalmol. 2011; 2011: 589813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.