

Abstract
Purpose. Complement activation is implicated in the pathogenesis of age-related macular degeneration (AMD). Apolipoprotein E (ApoE) and complement activation products such as membrane attack complex (MAC) are present in eyes of individuals with AMD. Herein, we investigated the effect of complement activation on induction of ApoE accumulation in human retinal pigment epithelial (RPE) cells.
Methods. Cultured human RPE cells were primed with a complement-fixing antibody followed by treatment with C1q-depleted (C1q-Dep) human serum to elicit alternative pathway complement activation. Controls included anti-C5 antibody-treated serum and heat-inactivated C1q-Dep. Total protein was determined on RPE cell extracts, conditioned media, and extracellular matrix (ECM) by Western blot. ApoE and MAC colocalization was assessed on cultured RPE cells and human eyes by immunofluorescent stain. ApoE mRNA expression was evaluated by quantitative PCR (qPCR).
Results. Complement challenge upregulated cell-associated ApoE, but not apolipoprotein A1. ApoE accumulation was blocked by anti-C5 antibody and enhanced by repetitive complement challenge. ApoE mRNA levels were not affected by complement challenge. ApoE was frequently colocalized with MAC in complement-treated cells and drusen from human eyes. ApoE was released into complement-treated conditioned media after a single complement challenge and accumulated on ECM after repetitive complement challenge.
Conclusions. Complement challenge induces time-dependent ApoE accumulation in RPE cells. An understanding of the mechanisms by which complement affects RPE ApoE accumulation may help to better explain drusen composition, and provide insights into potential therapeutic targets.
Keywords: AMD, RPE, complement, ApoE
Complement has been increasingly implicated in age-related macular degeneration (AMD) pathogenesis. In both neovascular and nonneovascular AMD associated with geographic atrophy, complement activation split products are found within the systemic circulation and ocular tissues including drusen within the subretinal pigment epithelial space, Bruch's membrane, and the choroid. Complement membrane attack complex (MAC) accumulates in the choriocapillaris intercapillary pillars adjacent to sites of drusen formation, and is seen on retinal pigment epithelium (RPE) in advanced neovascular and nonneovascular AMD.1–11 Furthermore, there is a strong genetic component that links complement regulatory protein polymorphisms and MAC formation with progression to intermediate AMD.9,11 The genetic and functional linkage of complement to AMD centers on the dysregulation of the alternative pathway that serves to activate complement initially, as well as to amplify the classical and lectin pathways.12,13 Together these findings point to a fundamental role of complement in the development of advanced AMD.1,3,5
Complement proteins found within ocular tissues are derived from local tissues including RPE cells, as well as from extraocular sources such as the liver, which is the primary source of complement proteins present within the circulation.1,14–18 The mechanism(s) by which complement constituents of local and systemic origin interact with RPE cells, and their relative contribution to AMD, both experimentally and clinically, remain to be fully defined.
Complement activation is initiated by stimulation of one or more of the three activation pathways (classical, lectin, and alternative pathways). When complement activation is triggered, there is sequential processing of complement components to generate anaphylatoxins (C3a, C4a, C5a) and corresponding C3 and C5 convertase. The C5 convertase is responsible for the generation of C5b that serves as a scaffold for the sequential assembly of C6, C7, C8, and C9 forming the MAC. The fully formed MAC, when assembled on the cell surface, forms a nonspecific membrane pore leading to cellular stress and if uncontrolled, eventual cell death.19–22
Apolipoprotein E (ApoE) is a 34-kDa multifunctional protein with central roles in lipid metabolism and neurodegenerative diseases. Three common ApoE isoforms (ApoE2, ApoE3, and ApoE4) specific to humans are encoded by a single gene locus on chromosome 19, q13.2. These alleles are differentiated on the basis of cysteine–arginine residue interchanges at sites 112 and 158 in the amino acid sequence. As a result of this polymorphism, six common variants exist: three homozygous (E2E2, E3E3, E4E4) and three heterozygous (E2E3, E2E4, E3E4).
In peripheral tissues, ApoE is primarily produced in the liver and by macrophages. Plasma levels range from 30 to 70 mg/L. In the central nervous system, it is mainly produced by astrocytes.23–27 In the retina, Müller cells and RPE cells are the most prominent ApoE biosynthetic sources.28–33 ApoE is a common component of extracellular plaques and deposits characteristic of many diseases such as atherosclerosis and Alzheimer disease.6,34,35 ApoE is a prominent drusen component and seen in choroid capillary pillars; however, the source of ApoE is not entirely clear.28,30,31,36,37
In the present study, we examined the relationship between complement activation on RPE cells and cell-associated ApoE in the local RPE microenvironment. Previously, in a fetal RPE cell culture model, it was shown that ApoE and MAC accumulate in sub-RPE deposits, and that complement within the deposits was activated by the classical, but not alternative complement pathway.38 However, the effect on RPE cell-associated ApoE accumulation associated with activation of the alternative complement pathway has not previously been examined. Furthermore, the effect of complement on ApoE accumulation in RPE cells from aged human donors has not previously been reported. Herein, we show the effect of alternative complement pathway activation on human RPE cell-associated ApoE. The data suggest a mechanism by which alternative complement pathway activation can induce ApoE accumulation on RPE cells and deposition within the extracellular matrix (ECM).
Methods
Antibodies and Reagents
Anti-RPE polyclonal antibody denoted S58 (named to indicate the origin from sheep number 58) and monoclonal anti-C5 antibody were obtained from Allergan, Inc. (Irvine, CA, USA). Goat anti-ApoE was purchased from Calbiochem/EMD (1:8000 for Western blot, cat. no. 178479; San Diego, CA, USA). Rabbit anti-ApoE antibody (1:100 for immunostaining, cat. no. PA5-27088; Chicago, IL, USA) and rabbit anti-zonula occludens-1 (ZO-1) were purchased from Thermo Scientific, Inc. (Chicago, IL, USA). Anti-apolipoprotein A1 (ApoA1) antibody was purchased from Cell Signaling Technology (1:1000, cat. no. 3350; Beverly, MA, USA). Monoclonal anti-human C5b-9 clone aE11 was purchased from Dako (Carpinteria, CA, USA). Rabbit anti-fibronectin antibody was purchased from Abcam, Inc. (1:8000, cat. no. ab23750; Cambridge, MA, USA). C1q-depleted human serum (C1q-Dep), C6-depleted human serum (C6-Dep), and purified C6 protein were purchased from Quidel Corp. (San Diego, CA, USA). Recombinant ApoE human protein with N-terminus thioredoxin and polyhistidine (N-TRX.His) tags was purchased from Life Technologies (cat. no. 10817-H30E; Frederick, MD, USA). Protein G–purified normal sheep IgG was purchased from Molecular Innovations, Inc. (Novi, MI, USA). Cycloheximide (CHX), ammonium hydroxide (NH4OH), phalloidin (cat. no. P1951), and collagen type I were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (cat. no. 89900), GelCode blue stain reagent (cat. no. 24590), and In-Gel tryptic digestion kit (cat. no. 89871) were purchased from Thermo Scientific, Inc.
Human RPE Cell Culture and Complement Challenge
Human donor eyes were obtained within 24 hours after death from the North Carolina Organ Donor and Eye Bank, Inc. (Winston-Salem, NC, USA) in accordance with the provisions of the Declaration of Helsinki for research involving human tissue. RPE cells were harvested from eyes obtained from three donors (a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant, a 61-year-old donor with ApoE phenotype E3/E4 and CFHYH402 variant, and a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant) as we have previously described.39,40 ApoE genotype was determined by restriction fragment length polymorphism analysis on polyacrylamide gel as previously described with slight modification.41 Cells were grown in Eagle's minimal essential Medium (MEM; Invitrogen, Chicago, IL, USA) with 10% fetal bovine serum (FBS; Invitrogen) and with 1× penicillin/streptomycin (Pen/Strep; Invitrogen) at 37°C in a humidified environment containing 5% CO2. The identity of RPE cells from three donors was confirmed by cytokeratin-18 and ZO-1 stain (not shown).
Human donor RPE cells from 62-, 51-, and 61-year-old donors were seeded on collagen-coated 24-well plates (Corning-Costar Incorporated, Corning, NY, USA) at 6 × 104, 6 × 104, and 1 × 105 cells/well, respectively. Cells reached 80% to 100% confluence 24 hours after plating, and confluent cells were then grown for an additional 4 or 5 days until growth was density arrested. At this time point, the cells express ZO-1 and phalloidin (actin) (Supplemental Fig. S1). On day 6 after plating, the cells were washed once with serum-free MEM (SF-MEM), primed with a complement-fixing antibody, S58 (0.4 or 0.8 or 1.2 mg/mL), in SF-MEM for 30 minutes at 37°C, washed and incubated with SF-MEM containing either 6% C1q-Dep (eliciting complement activation and cell surface MAC formation) or 6% heat-inactivated C1q-Dep (HiC1q-Dep, 30 minutes at 56°C, as a negative control) at 37°C for various times, as we have described previously.40 In a subset of experiments, unprimed cells treated with C1q-Dep serum were used as negative control. The role of S58 is to provide a complement nucleating source on RPE cells. C1q-Dep was used as a source of complement for all experiments. To block complement activation and MAC formation, anti-C5 antibody was mixed on ice for 10 minutes with C1q-Dep at a concentration of 10 μg/mL to neutralize the complement C5 component. As a control, an aliquot of C1q-Dep without C5 antibody was prepared in an identical manner. A mixture of C1q-Dep+anti-C5 antibody or C1q-Dep alone was then added to cells. To exclude the possibility that C5a solely accounted for ApoE accumulation, S58-primed cells were treated with 6% C6-Dep for 5 hours with or without purified C6 protein, a complement cascade protein downstream of C5a formation. For a cell-suspension model, 2 × 105 and 2 × 105 human RPE cells from 62- and 51-year-old donors, respectively, were added into Eppendorf tubes, primed with either normal sheep IgG (1.2 mg/mL) or S58 (1.2 mg/mL) for 30 minutes, and then treated with 6% C1q-Dep for 4.5 hours at 37°C in an incubator-shaker. To block de novo protein synthesis, cells were preincubated for 1.5 hours with CHX (a protein biosynthesis inhibitor) at various concentrations, and CHX was included continuously during the S58 priming and complement serum incubation period.
To evaluate the serum contribution of ApoE to RPE cell ApoE accumulation, recombinant ApoE human protein with N-TRX.His tags was added to serum at levels equivalent to that in normal serum level (4 μg/mL; 30–70 μg/mL in 100% serum equivalent to 4 μg/mL in 6% serum).25,27,42,43
Mass Spectrometry (LC-MS/MS) Analysis
Cells were incubated for 6 hours with C1q-Dep/HiC1q-Dep, and total protein was extracted in RIPA buffer after two washes with cold PBS to reduce the amount of nonspecifically associated proteins. Equal amounts of proteins were separated on SDS-PAGE gels, which were then were washed twice with distilled water and fixed in a mixture of 50% methanol and 7% acetic acid for 20 minutes followed by two rinses. Gels were stained for 30 minutes with GelCode blue stain reagent (Thermo Scientific, Inc.) and destained overnight with distilled water. For each sample, 15 gel bands were excised and subjected to in-gel tryptic digestion using an in-gel tryptic digestion kit (Thermo Scientific, Inc.). Peptide mixes obtained from in-gel trypsin digest were analyzed using a nanoAcquity UPLC system coupled to a Synapt G2 HDMS mass spectrometer (Waters Corp., Milford, MA, USA). Peptides were separated on a 75-μm × 100-mm column with 1.7-μm C18 BEH particles (Waters Corp.) using a 90-minute gradient of 6% to 32% acetonitrile with 0.1% formic acid at a flow rate of 0.3 μL/min and 45°C column temperature. For each sample, a data-dependent analysis (DDA) was performed with a 0.8-second mass spectrometry (MS) scan followed by MS/MS acquisition (fragmentation) on the top four ions with charge greater than 1. MS/MS scans for each ion used an isolation window of ∼3 Da, a maximum of 3 seconds per precursor, and dynamic exclusion for 120 seconds within 1.2 Da. DDA data were converted to searchable files using a ProteinLynx Global Server 2.5 (Waters Corp.) and searched against the human IPI database v.3.79 using Mascot server 2.2 (Matrix Science, Boston, MA, USA) with the following parameters: maximum one missed cleavage site, carbamidomethylation at Cys residues as fixed modification and Met oxidation, N-terminal acetylation, Asn, Gln deamidation as variable modifications. Precursor ion mass tolerance was set to 20 ppm, and fragment mass tolerance was set to 0.2 Da. Mascot data were imported into Scaffold 4.0 (Proteome Software, Inc., Portland, OR, USA) to merge all the data for a sample represented by multiple gel bands, to identify a false discovery rate for protein identification, to group proteins, and to perform spectral counting-based protein quantification. Acceptance criteria for protein identification required identification of at least two peptides for each protein with a confidence interval percentage, CI% over 99.5%, corresponding to a false discovery rate of 0.5%.
Three independent protein quantifications were conducted by three spectra counting methods: the weighted spectra, the normalized spectral abundance factor (NSAF),44 and the exponentially modified protein abundance index (emPAI).45 The assays were replicated in three independent experiments on cells from a 62-year-old donor.
Western Blot and Immunofluorescent Staining
A cell-associated ApoE accumulation assay was performed as follows: Total protein extracts were prepared at various time points after C1q-Dep/HiC1q-Dep treatment. Western blot analysis was performed as we have previously described.46 For the ApoE release assay, cells were treated for 6 hours with C1q-Dep/HiC1q-Dep, washed twice, and incubated with SF-MEM for various times. Both conditioned media and cell lysates were collected after 1 hour (as a baseline) and 24, 48, and 72 hours of SF-MEM incubation. The conditioned media were concentrated with a Savant SpeedVac concentrator (SC110; Savant instruments, Inc., Holbrook, NY, USA).
To determine whether anti-C5 antibody attenuated MAC deposition, cells were incubated for 30 minutes with C1q-Dep/HiC1q-Dep in the presence or absence of anti-C5 antibody (10 μg/mL), then fixed for 15 minutes in 4% paraformaldehyde (PFA). Immunofluorescent staining was performed as we have previously described using anti-human C5b-9 (aE11) antibody (1:25 diluted in 1% BSA/PBS).40 The assays were replicated in two independent experiments on RPE cells from 62- and 61-year-old donors. Duplicate wells were run for each experiment. To examine colocalization of ApoE and MAC in culture, RPE cells were incubated for 5 hours with serum. Cells were stained for ApoE (rabbit anti-human ApoE 1:100 diluted in 1% BSA/PBS) and MAC as described above. To examine codistribution of ApoE and MAC in situ, human eyes from 72- and 94-year-old individuals were fixed, embedded, and sectioned as previously described.47 Sections were postfixed in cold acetone for 5 minutes at room temperature and stained for MAC (C5b-9 antibody diluted 1:50 in 1% BSA/PBS) and ApoE (goat anti-human ApoE antibody diluted 1:2000 in 1% BSA/PBS) as described above. To evaluate the effect of repetitive complement challenge on cytoskeletal and cell junction proteins, the cells were fixed for 12 minutes in 4% PFA, permeabilized with 0.5% Triton X-100 for 12 minutes, and costained for phalloidin (1:500) and ZO-1 (1:100 diluted in 5% normal goat serum/0.3% Triton X-100).
Real-Time RT-PCR Analysis
Total RNA was isolated using RNeasy Plus Mini Kit (Qiagen, Inc., Valencia, CA, USA) according to the manufacturer's specifications, and real-time quantitative reverse transcription–polymerase chain reaction (qPCR) was performed as we have described previously.46 Primer pairs for ApoE, interleukin 6 (IL-6), and ribosomal protein, large, P0 (RPLP0) were as follows (5′ to 3′): ApoE, forward: GTT GCT GGT CAC ATT CCT GG; reverse: GCA GGT AAT CCC AAA AGC GAC; IL-6, forward: AGT GAG GAA CAA GCC AGA GCT GTG; reverse: TTT GTG GTT GGG TCA GGG GTG; RPLP0, forward: GGA CAT GTT GCT GGC CAA TAA; reverse: GGG CCC GAG ACC AGT GTT.
Repetitive Complement Challenge Model
Repetitive challenge was performed by challenging cells every other day for 1 week (total of three treatments). For repetitive challenge, cells were plated and primed with S58 as described above followed by treatment with either 6% C1q-Dep or 6% HiC1q-Dep and returned to incubator until the next challenge. For the second and third complement challenge, cells were washed once, reprimed with S58 followed by incubation with C1q-Dep or HiC1q-Dep. To determine ApoE deposition on ECM, at 48 hours after a third complement challenge, cells were removed by incubation with 0.02 N ammonium hydroxide for 15 minutes with gentle shaking at room temperature and then washed twice with cold PBS. The culture wells were examined with a microscope to ensure that RPE cells were removed thoroughly. Proteins were extracted with an equal amount of lysis buffer in each well. Protein concentration was measured by Pierce 660 nm Protein Assay-NanoDrop (Thermo Scientific, Inc.) with bovine serum albumin used to generate the curve, according to the manufacturer's instructions.
WST-1 Assay
Cell plating and treatments were performed as described above. Cell viability was measured using a cell proliferation reagent, WST-1, as we have previously described.40
Statistical Analysis
Data are expressed as the mean ± SD. One-way ANOVA and Student's t-test were used to determine whether there were statistically significant differences between treatment groups as determined by Western blot and qPCR.
Results
Complement Challenge Increases Cell-Associated ApoE Protein but Not ApoA1, as Determined by LC-MS/MS Analysis
To better understand the mechanisms and pathways evoked by complement-mediated RPE cell injury, we challenged RPE cells with complement-active serum and heat-inactive serum under conditions that elicited sublytic cell surface MAC formation40 followed by LC-MS/MS analyses on total protein extracts. Over 1000 proteins were identified and quantified. The complement proteins C3, C5, and C9 were among those that differentially associated with the cell surface. Their identification was consistent with complement activation (C3) and MAC formation (C5 and C9), although the LC-MS/MS analysis does not discriminate between intact and activated forms of C3 and C5. Interestingly, we did not detect cell-associated Factor B (FB), which is approximately six and two times more abundant in serum than either C5 or C9, respectively (FB 2.2 μM, C5 0.370 μM, and C9 0.83 μM). These data imply that two washes removed the majority of serum components. In addition to complement proteins, apolipoprotein ApoE, but not ApoA1, was differentially associated with RPE cells in response to sublytic complement challenge (Table). Apolipoprotein O (ApoO), ApoO-like protein, apolipoprotein C (ApoC)-I, ApoC-III, apolipoprotein B (ApoB)-100, and apolipoprotein L (ApoL)-2 were also detected. We did not find reproducible changes in the levels of these proteins in response to complement challenge, further demonstrating the specificity of ApoE accumulation. We did not detect drusen components clusterin and apolipoprotein J (ApoJ) in our system.
Table.
Complement Challenge Upregulated Cell-Associated ApoE Protein but Not ApoA1, as Determined by LC-MS/MS Analysis
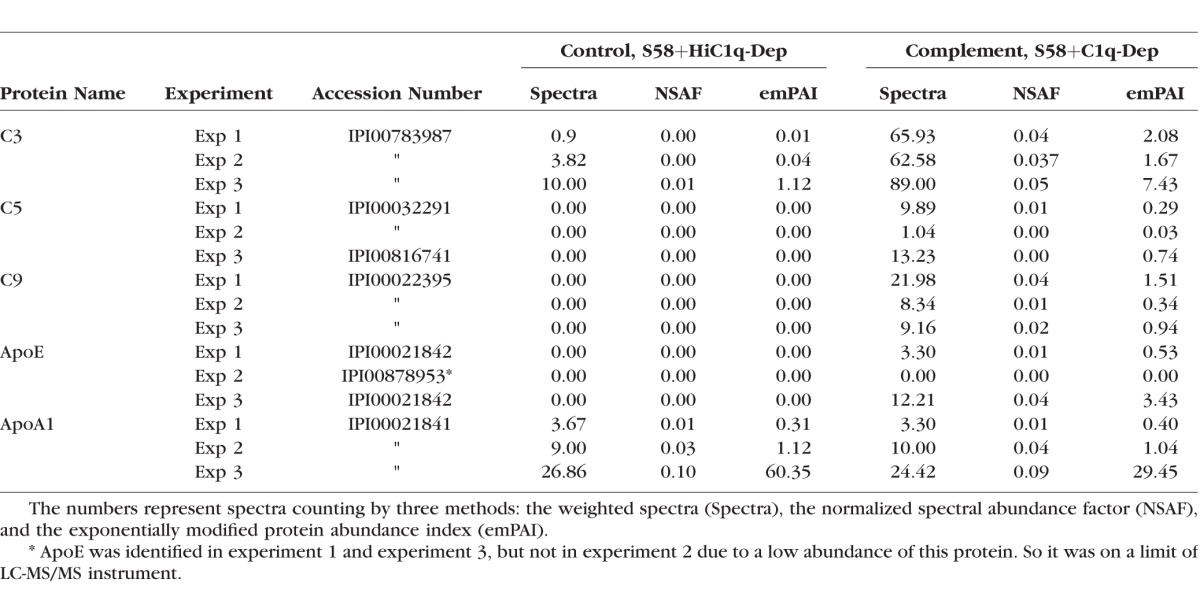
To confirm the LC-MS/MS results, Western blot analysis was conducted on the same lysates used for LC-MS/MS analysis (experiments 1 and 2 in the Table). In agreement with the findings of the LC-MS/MS analysis, ApoE protein levels were increased in the presence of complement challenge and not from cells treated with heat-inactive serum (Fig. 1). We found that basal levels of ApoE protein were present at variable levels that were much lower than the levels seen in complement-challenged cells, when assessed under our experimental conditions.
Figure 1.

Western blot confirmed that complement activation increases cell-associated ApoE protein determined by LC-MS/MS analysis. RPE cells from a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant were primed with S58 (1.2 mg/mL) for 30 minutes and then treated with either 6% C1q-Dep or HiC1q-Dep for 6 hours. Total proteins (30 μg) were separated by SDS-PAGE. Lanes 1 and 3 used the same total protein samples as those in experiment 1 in the Table. Lanes 2 and 4 used the same total protein samples as those in experiment 2 in the Table.
Complement Activation Increases Cell-Associated ApoE Protein in a Time-Dependent Manner
The time dependency of cell-associated ApoE accumulation was determined at various times after serum treatment. As shown in Figures 2A and 2B, cellular ApoE levels were increased as early as 1 hour and remained elevated out to 24 hours post serum treatment. ApoE accumulation occurred in both anti-RPE antibody-primed and unprimed RPE cells. However, in four independent experiments, ApoE levels were 2.1- to 2.5-fold greater in primed cells relative to unprimed cells and 3.1- to 10.4-fold increased over primed cells treated with heat-inactivated serum. The effect was specific to ApoE, as there was little change in ApoA1, an apolipoprotein with similar molecular weight and higher plasma concentration (1200–1600 mg/L)25,27 than that of ApoE.
Figure 2.

Time-dependent ApoE accumulation in complement-treated cells. RPE cells from a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant were primed with or without S58 (0.8 or 1.2 mg/mL) for 30 minutes and then treated with either 6% C1q-Dep or HiC1q-Dep (control) for 1 to 6 hours (A) and 6 to 24 hours (B) Total proteins (15 μg) were separated by SDS-PAGE. The quantity of ApoE protein and ApoA1 protein is shown below each lane.
Previously, we used a combination of calcium signaling and vital dyes to establish conditions under which MAC formation caused pore formation and induced minimal cell death.40 In that previous publication, minimal induction of a calcium influx (an indicator of pore formation) was observed at an antibody priming concentration of 0.8 mg/mL in the presence of 6% serum. At this induction level, minimal cell death was observed.40 In the current study, we observed that similar ApoE levels accumulated on cells primed with S58 at 0.8 mg/mL, when compared to the cells primed with S58 at 1.2 mg/mL (Figs. 2A, 2B) and, accordingly, we conducted most subsequent experiments with an S58 priming concentration of 1.2 mg/mL. As shown in Supplemental Figure S2, cell viability was not diminished after cells were exposed to complement for 6 hours relative to that at 2.5 hours at a S58 priming concentration of 1.2 mg/mL.
The presence of ApoE within the RPE monolayer suggests an association with the RPE cell surface. However, this observation may also reflect ApoE associated with complement activation and deposition on the tissue culture plate plastic surface.48 To exclude the influence of nonbiological complement activation on ApoE accumulation, RPE cells were challenged while in suspension. As shown in Figure 3, cell-associated ApoE accumulation occurred on suspended cells, in a manner similar to that observed in cells when challenged as a monolayer. In contrast, ApoE did not accumulate on control cells in which a nonspecific sheep IgG was substituted for the anti-RPE complement priming antibody. These data indicate cellular ApoE association in response to sublytic complement activation.
Figure 3.

Cell suspension model confirmed cell-associated ApoE accumulation. (A) Suspended RPE cells from a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant were incubated with either normal sheep IgG (1.2 mg/mL) or S58 (1.2 mg/mL) for 30 minutes and then treated with 6% C1q-Dep for 4.5 hours at 37°C in a shaking incubator followed by cell lysis and SDS-PAGE analysis of total proteins (30 μg). (B) Quantitation of cell-associated ApoE relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) shown in (A) was determined by densitometry. *P = 0.02 versus sheep IgG+C1q-Dep. Data are representative of two separate experiments in two donors with similar results.
Cell-Associated ApoE Accumulation Is Dependent on MAC Formation
Previous studies have shown that ApoE and complement split products are localized in drusen, and ApoE has been colocalized with MAC in the ECM deposited by RPE cells.38,49 To determine if ApoE accumulation is dependent on MAC formation, we assayed ApoE levels in the presence and absence of anti-C5 monoclonal antibody to block C5b formation and MAC deposition. As shown in Figure 4A, MAC was deposited on primed RPE cells in response to complement challenge but not on primed RPE cells in which serum was treated with the anti-C5 monoclonal antibody (Fig. 4A). Corresponding to the inhibition of MAC deposition, the anti-C5 antibody significantly reduced accumulation of cell-associated ApoE protein in RPE cells (Figs. 4B, 4C). To show a MAC-dependent effect on ApoE accumulation, and not one specifically related to C5a generation, which is upstream of MAC, anti-RPE antibody-primed and unprimed cells were treated with C6-Dep with or without purified C6 protein. As shown in Figures 4D and 4E, ApoE accumulation was significantly increased when C6-Dep was reconstituted with C6 but not in C6-Dep alone. Similar levels of MAC formation were observed by immunostaining of anti-RPE antibody-primed cells when C6-Dep was reconstituted with each of three C6 concentrations (3.65, 7.3, and 65 μg/mL) (not shown).
Figure 4.

Cell-associated ApoE accumulation was dependent on MAC deposition. RPE cells were primed with or without S58 (1.2 mg/mL) for 30 minutes and then treated with 6% serum for 30 minutes (A), 6 hours (B, C), or 5 hours (D, E). (A) Induction of MAC accumulation on RPE cells from a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant. After serum treatments in the presence or absence of anti-C5 antibody (10 μg/mL), cells were fixed in 4% PFA for 15 minutes and stained with mouse anti-human C5b-9 (aE11) antibody. Data are representative of two separate experiments in two donors with similar results. Red stain indicates MAC deposition. Blue stain corresponds to 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei. Scale bar: 100 μm. (B) Accumulation of ApoE was prevented when C5 was blocked by anti-C5 antibody. Total proteins (15 μg) obtained from RPE cells of a 61-year-old donor with ApoE phenotype E3/E4 and CFHYH402 variant were separated by SDS-PAGE 6 hours post complement challenge in the presence or absence of anti-C5 antibody (10 μg/mL). (C) The quantity of ApoE relative to GAPDH shown in (B) was determined by densitometry. *P = 0.001 vs. C1q-Dep and C5 Ab+C1q-Dep. **P = 0.002 vs. S58+C1q-Dep. Data are representative of three separate experiments in three donors with similar results. (D) Accumulation of ApoE was blocked by absence of C6. Total proteins (30 μg) obtained from RPE cells of a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant were separated by SDS-PAGE after treatment with C6-Dep in the presence or absence of C6 protein at 7.3 or 65 μg/mL. (E) The quantity of ApoE relative to GAPDH shown in (D) was determined by densitometry. *P < 0.05 vs. C6q-Dep and S58+C6-Dep. Data are representative of two separate experiments in two donors with similar results.
ApoE Is Colocalized With MAC on Complement-Activated RPE Cells and Drusen
To examine whether ApoE is colocalized with MAC on cultured RPE cells, we costained cells with anti-ApoE and anti-MAC antibodies. In MAC-positive regions, ApoE was frequently colocalized with ApoE in clusters on the cell surface. In some areas, ApoE did not colocalize with MAC. However, in these regions, ApoE stained diffusely on cells, and not in clusters (Fig. 5A). No MAC staining was observed in control cells that included nonspecific sheep IgG-primed cells treated with C1q-Dep (not shown). To assess codistribution of MAC and ApoE in situ, human eyes were costained with MAC and ApoE. As shown in Figures 5B and 5C, colocalization of MAC and ApoE (shown in yellow) was observed in drusen.
Figure 5.

ApoE was colocalized with MAC on complement-activated RPE cell culture and drusen. (A) RPE cells from a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant were primed with S58 (1.2 mg/mL) for 30 minutes and then treated with 6% C1q-Dep for 5 hours. Cells were fixed in 4% PFA for 15 minutes and costained with rabbit anti-human ApoE antibody (green) and mouse anti-human C5b-9 (aE11) antibody (red). Blue stain corresponds to DAPI-stained nuclei. MAC and ApoE were colocalized frequently in the cells. Data are representative of three separate experiments in three donors with similar results. (B) Confocal microscopic colocalization (yellow) of MAC (green) and ApoE (red) in drusen from a 72-year-old donor. Similar results were observed in 94-year-old donor eye (not shown). (C) Primary antibodies were omitted in section of a 72-year-old donor. Scale bar: 50 μm.
Recruitment of ApoE From Serum Onto RPE Cells
Accumulation of ApoE in response to MAC deposition may represent recruitment of ApoE derived from the serum or through de novo protein synthesis. To determine if ApoE is recruited from the media by complement activation, recombinant human ApoE protein with N-TRX.His tags was added to the serum during complement challenge (there is no specific role of TRX in the experiment). Cleaved recombinant ApoE bands (indicative of the exogenously added recombinant ApoE) were detected in cells under complement challenge (Fig. 6). ApoE accumulation was also observed in control cells treated with N-TRX.His tagged human ApoE protein, although the levels were less than that observed in RPE challenged by activated complement. These data indicate that RPE cells alone are able to bind recombinant human ApoE protein when added to the media, irrespective of complement activation. However, cell-associated levels of both tagged ApoE and untagged ApoE were enhanced in the presence of complement challenge, which implies that serum is a likely source of cell-associated ApoE.
Figure 6.

Detection of cleaved recombinant ApoE human protein in serum-treated cells. RPE cells from a 61-year-old donor with ApoE phenotype E3/E4 and CFHYH402 variant were primed with or without S58 (1.2 mg/mL) for 30 minutes and then treated with 6% either C1q-Dep or HiC1q-Dep for 5 hours with or without recombinant ApoE human protein with N-TRX.His tags (4 μg/mL). Total proteins (15 μg) were separated by SDS-PAGE. Data are representative of four separate experiments in two donors with similar results. *S58 IgG heavy chain.
To address the ApoE contribution from endogenous synthesis, the effect of complement challenge on ApoE mRNA was first determined by qPCR. ApoE steady-state mRNA levels were not increased in response to complement challenge when compared to controls (Fig. 7A). In contrast, increased IL-6 mRNA expression was observed in response to C1q-Dep treatment, which was further enhanced by complement challenge; maximal induction was seen at 3 hours post stimulation (Fig. 7B). Thus while the conditions were suitable to detect alterations in IL-6 mRNA expression, no changes in steady-state ApoE mRNA expression were observed up to 5 hours post stimulation (Fig. 7). CHX, a protein synthesis inhibitor,50,51 was used to determine if increased ApoE levels could be explained, at least in part, from increased protein translation. Treatment of RPE cells with CHX (20 μg/mL) reduced ApoE levels in response to C1q-Dep treatment and complement challenge, although the degree of ApoE inhibition was variable (Figs. 8A, 8B). Similar responses were observed with 5 to 100 μg/mL CHX (not shown).
Figure 7.

Steady-state ApoE mRNA levels were not affected by complement challenge. RPE cells were primed with or without S58 (1.2 mg/mL) for 30 minutes and then treated with either 6% C1q-Dep or HiC1q-Dep for various times. RNA in triplicate samples was extracted, reverse transcribed to cDNA, amplified with ApoE- (A) or IL-6- (B) specific primer pairs, and quantified by qPCR. Data are representative of four separate experiments in a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant with similar results. *P < 0.05 vs. S58+HiC1q-Dep.
Figure 8.

CHX partially inhibited complement-mediated ApoE accumulation. RPE cells from a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant (A) and a 61-year-old donor with ApoE phenotype E3/E4 and CFHYH402 variant (B) were preincubated with CHX at 20 μg/mL for 1.5 hours, primed with or without S58 (1.2 mg/mL) for 30 minutes, and then treated with either 6% C1q-Dep or HiC1q-Dep for 5 hours. CHX was continuously present when cells were primed with S58 and incubated with complement serum. Total proteins (15 μg) were separated by SDS-PAGE for Western blot. The quantity of ApoE relative to GAPDH shown in (A, B) was determined by densitometry (labeling 1–5 represents five treatment groups). *P < 0.05 vs. C1q-Dep. **P < 0.05 vs. S58+C1q-Dep. Data are representative of six separate experiments in three donors with similar results.
ApoE Release Into Conditioned Media Post Complement Challenge
Complement challenge and MAC formation induced RPE cellular ApoE accumulation. However, after cellular accumulation ApoE may be again released. To evaluate ApoE release into the media, serum-containing medium was exchanged with serum-free medium 6 hours after complement challenge, and levels of released ApoE were determined at various time points. As shown in Figure 9, decreased cell-associated ApoE was accompanied by increased release of ApoE into conditioned media at days 1, 2, and 3 post complement challenge, when compared to controls. Interestingly, cell-associated anti-RPE priming antibody was also decreased from RPE cells, similar to that observed with ApoE protein (Fig. 9) in both control and complement-challenged cells. We used goat anti-human ApoE antiserum that had been defibrinated, delipidized, and dialyzed against a Tris-HCl buffer. We observed multiple bands that were not affected by the treatments. These bands could be nonspecific binding or ApoE aggregates (Supplementary Fig. S3).
Figure 9.

ApoE protein in conditioned media was elevated in response to complement challenge. RPE cells were primed with or without S58 (1.2 mg/mL) for 30 minutes and then treated with either 6% C1q-Dep or 6% HiC1q-Dep for 6 hours. The medium was then exchanged with serum-free medium. After media exchange, cultures were incubated for either 1, 24, 48, or 72 hours and levels of soluble ApoE in the media were determined. Total cellular lysate proteins (15 μg) and concentrated conditioned media (30 μL) were separated by SDS-PAGE. The data are representative of two separate experiments in a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant with similar results.
Repetitive Complement Challenge Increased ApoE Accumulation and Caused ECM Localization
Complement activation products Ba and C3d are significantly increased in the systemic circulation of patients with AMD,17 suggesting sustained chronic complement activation. To assess whether cell-associated ApoE accumulation is influenced by persistent complement activation, RPE cells were exposed to complement challenge every other day over a 1-week period. As shown in Figure 10, three repeated complement challenges dramatically enhanced ApoE accumulation in RPE cells.
Figure 10.

Repetitive complement challenge enhanced cell-associated ApoE accumulation. RPE cells from a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant were primed with or without S58 (1.2 mg/mL) for 30 minutes and then treated with either 6% C1q-Dep or HiC1q-Dep for 24 or 48 hours. For the second and third complement challenges, cells were washed once, reprimed with or without S58, and then incubated with 6% C1q-Dep or HiC1q-Dep for 48 hours. Repetitive challenge was performed every other day for 1 week (three challenges). Total proteins (15 μg) were separated by SDS-PAGE. The relative quantity of ApoE protein normalized to GAPDH is shown below each lane. Blot images are representative of seven separate experiments in three donors with similar results. The bar graph depicts pooled data from two separate experiments in which cells from a single donor were treated under identical conditions.
To determine whether ApoE is deposited in RPE ECM in response to repeated sublytic complement challenge, matrix-associated ApoE levels were determined. As shown in Figure 11, repetitive complement challenge caused ApoE ECM deposition and did not induce an apparent change in cell viability, morphology, or phalloidin–ZO-1 staining, suggesting that repetitive challenge at sublytic levels did not induce significant cell death or cell morphology alteration as determined by light microscopy (Figs. 11A–E). Fibronectin was detected on RPE matrices treated with repetitive complement challenge (Fig. 11F).
Figure 11.

Repetitive sublytic complement challenge caused ApoE deposition in ECM. RPE cells from a 51-year-old donor with ApoE phenotype E3/E3 and CFHHH402 variant were primed with either normal sheep IgG (0.4 mg/mL) or S58 (0.4 mg/mL) for 30 minutes and then treated with 6% C1q-Dep or HiC1q-Dep for 48 hours. Repetitive sublytic challenge was produced as described in the Methods section. RPE cells were removed after the third complement challenge by incubating cells with 0.02 N ammonium hydroxide (NH4OH). (A) Similar cell morphology was observed across treatment groups prior to adding NH4OH and complete cell removal after adding NH4OH. (B) Equal lysate volumes were separated by SDS-PAGE and probed with ApoE. (C) The quantity of ApoE shown in (B) was determined by densitometry and normalized to protein concentration. *P < 0.05 vs. S58+HiC1q-Dep and IgG+C1q-Dep. Data are representative of four separate experiments in two donors with similar results. (D) Following three repetitive complement challenges, as described in the Methods section, RPE cells from a 62-year-old donor with ApoE phenotype E3/E3 and CFHYY402 variant were fixed in 4% PFA for 12 minutes and costained with rabbit anti-ZO-1 (green) and phalloidin (red). Blue stain corresponds to DAPI-stained nuclei. Twelve-micrometer Z-stacks were captured. Confocal horizontal (X–Y) sections are shown above and vertical (X–Z) sections are shown below. Data are representative of two separate experiments in two donors that showed similar results. Scale bar: 20 μm. (E) Following three repetitive challenges as described in the Methods section, cell viability was determined by WST-1 assay. (F) RPE cells from two donors were repetitively primed with S58 (0.4 mg/mL) and treated with 6% C1q-Dep and removed as described in (A, C). Equal lysate volumes were separated by SDS-PAGE and probed with fibronectin.
Discussion
In the current study, we have demonstrated that complement challenge on cultured human RPE cells induced cell-associated ApoE protein but not ApoA1 in a time-dependent manner. The cell-associated ApoE accumulation was dependent on C5 cleavage and MAC formation, but was not dependent on C5a generation. The source of ApoE was likely derived from both serum and cellular generation. ApoE was frequently colocalized with MAC on complement-challenged cells. In eyes of elderly individuals, MAC and ApoE colocalized to drusen. Post cellular accumulation, ApoE was released into tissue culture media and upon rechallenge ApoE was deposited into the RPE ECM.
The phenomenon of complement challenge–induced ApoE accumulation was observed by LC-MS/MS analysis. Existing methods for differential proteomics fall into two categories: spectral counting methods and peptide chromatographic peak intensity methods. Spectral counting methods are straightforward to employ and correctly detect known differences between samples. Accordingly, they are widely used to quantify protein levels in mass spectrometry–based experiments. We used three protein quantification techniques: weighted spectra, the normalized spectral abundance factor (NSAF), and protein abundance index (PAI), which are based on spectral counting of the identified peptides in which the number of spectra that map to a given protein across multiple experiments is determined. Weighted spectra uses the sum of all weighted spectra associated with a specific protein and within a sample, where the weight is a measure of how much a spectrum is shared by other proteins (Scaffold version 4.0, user's manual; Proteome Software, Inc.). The NSAF quantitative method is used when comparing the abundance of individual proteins in multiple independent samples and is typically applied to quantify the expression changes in various complexes.44 The PAI is defined as the number of observed peptides divided by the number of all possible tryptic peptides from a particular protein, that are within the mass range of the employed mass spectrometer. The exponentially modified PAI (emPAI) is equal to 10PAI minus 1, which is proportional to protein content in a protein mixture for the determination of absolute abundance of proteins.45 Spectral counting methods are less accurate than peptide chromatographic peak intensity methods, but provide broader protein coverage in complex biological mixtures. There is always a compromise between more accurate quantification for less protein or less accurate quantification for more proteins. We have chosen the latter option for this study for a primary screen of protein changes. A sizable variability in our study came from the sample preparation, the mass spectrometry measurements, and the quantification method. A high variability is typical for mass spectrometry–based protein quantification.52
Our data suggest that RPE ApoE accumulates from both serum and cellular sources. However, RPE cell–derived ApoE was not likely related to transcriptional regulation, as complement challenge did not alter ApoE steady-state mRNA levels under conditions that upregulated IL-6 expression.20 In contrast, CHX inhibited complement-mediated ApoE accumulation, suggesting that cell-derived ApoE depended on protein synthesis. Whether this inhibition is a direct effect on ApoE protein synthesis or due to production of proteins required for ApoE synthesis and/or export remains to be determined. The studies to demonstrate RPE cell accumulation of recombinant ApoE when added to the media, at physiological ApoE concentrations (30–70 mg/L),25,27,42,43 support the notion that complement challenge can increase serum-derived cell-associated ApoE accumulation on RPE cells. We speculate that in AMD, plasma ApoE transits Bruch's membrane, and in the context of complement dysregulation, leads to RPE cell ApoE accumulation in association with MAC and subsequent release of the complex into sub-RPE deposits. The permeability of Bruch's membrane to serum proteins greater than 100 kDa,53 which could allow passage of serum-derived ApoE across Bruch's membrane, is consistent with this hypothesis.
Our data indicate that ApoE does not accumulate because of a general property of lipoproteins or through entry into the cell via nonspecific pores formed by MAC. ApoA1, an apolipoprotein, is similar in molecular weight (∼28 kDa) to ApoE and is present at higher serum concentration than ApoE (1200–1600 mg/L).25,27 However, ApoA1 levels were not significantly altered by complement challenge, unlike ApoE, which was markedly increased after complement challenge.
The mechanism by which MAC-mediated RPE cellular ApoE accumulates is not entirely clear. Our data support a hypothesis that interactions between serum ApoE present in media and MAC contribute to increased cell-associated ApoE. ApoE is known to bind to low-density lipoprotein receptor (LDLR) family members24,26 through an extracellular ligand-binding domain consisting of cysteine-rich repeats of approximately 40 amino acids, also called class A domains.54–58 Interestingly, similar cysteine-rich repeats are present in functionally unrelated proteins including the complement proteins C6, C7, C8, and C9 present within the MAC. Class A domains, which are thought to bind ligands, are, therefore, referred to as complement type repeats.59–64 The LDLR class A domains of these complement proteins may play a role in ApoE association with MAC on the cell surface. Further studies will be required to determine whether LDLR class A domains present in MAC-associated proteins bind directly to ApoE. It has been reported that MAC deposition activates intracellular calcium stores and that ApoE secretion in macrophages is calcium dependent.65 A key finding of the current study was that complement induced cellular accumulation of ApoE. At later time points, we observed ApoE release into the media. At these later times, we were not inducing complement activation and, thus, a complement-mediated calcium flux was not present. Furthermore, we have previously shown that calcium chelation within our system, to prevent a rise in intracellular calcium, results in a significant increase in cell death.40 Therefore, we are unable to directly determine whether complement induces calcium-dependent ApoE release independent of the effect on cell viability.
In our model, in which RPE cells from aged donors were used to simulate RPE cells in older individuals with AMD, we observed that ApoE was readily detected after complement challenge, but only minimal levels were observed under basal conditions. Although beyond the scope of the current report, it would be of interest to determine whether complement induces polarized secretion of ApoE in these aged RPE cells. However, the current model has limitations in this regard, as we and others have found that it is technically challenging to develop a polarized monolayer with tight junctions that generate a high transepithelial resistance in these RPE cells from older individuals.66
RPE cell release of ApoE mirrored that of S58 IgG. It is possible that release of ApoE and S58 resulted from MAC-induced membrane integrity compromise. However, once cells are exposed to complement challenge, C5b-9 formed on the cell surface modulates C5b-9 membrane levels either by directly budding off from the cell surface or through its initial internalization and then exocytosis. Thus, it is possible also that the shedding of ApoE and IgG is in response to this activity.
The complement system is continuously active at a low level in the normal eye, and chronic complement activation products C5a, Ba, and C3d are significantly elevated in serum from patients with AMD compared to those without retinal pathology.17,67 We show here that repetitive complement challenge, to mimic chronic activation, enhanced RPE cell-associated ApoE accumulation and ECM deposition. Thus, under chronic conditions of activation present in AMD,17,67 the accumulation of MAC and ApoE on RPE cells with subsequent release may reflect a source of ApoE–MAC complexes found within sub-RPE deposits. The mechanism by which RPE deposits ApoE/MAC complexes into the ECM is not presently known, but may involve active endocytosis and exocytosis of MAC/ApoE complexes, as previously identified for the removal of MAC from the surface of nucleated cells.68–70
Previously, Johnson and associates38 produced a fetal RPE cell culture model of sub-RPE deposit formation that resembled drusen; in that study, ApoE accumulated, but did not depend on complement activation. ApoE in deposits was sometimes seen adjacent to MAC, and C1q was also associated with ApoE. Furthermore, in C1q-depleted serum, MAC was reduced. Based on these observations, the authors hypothesized that MAC was formed by activation of the classical pathway, and that ApoE-C1q complexes might activate complement leading to MAC formation. In contrast, we found that in RPE cells from aged donors, ApoE accumulation and release was potentiated by alternate complement activation, an effect that depended on MAC formation. The tissue culture models used in these two studies differed, in that we restricted complement activation to the alternative pathway, while in the study by Johnson and associates, complement activation and deposition was dependent on the classical pathway. The data from these two models are not incompatible. As pointed out by Johnson and associates, within an in vivo environment, a mutually reinforcing feedback loop between the classical and alternative pathways may exist. The data from these two reports suggest that this reinforcing feedback loop may also extend to ApoE accumulation. If true, we hypothesize that in the normal aging eye, ApoE accumulates within the sub-RPE space and leads to a low level of complement activation that primarily drives debris clearance through the classical pathway. However, with increased age, decreased complement regulation, or elevated oxidative stress, the process shifts into a pathologic state associated with deregulation of the alternative pathway, and bystander effect on the RPE. With increased stress, RPE becomes susceptible to complement attack,40,71,72 resulting in increased ApoE accumulation and release, which reinforces the feedback loop, produces drusen, and promotes chronic cycles of complement activation and lipid accumulation.
We specifically studied complement-mediated RPE cell ApoE accumulation. However, this phenomenon might not be restricted to RPE cells, and could occur at different time points and in other cell types in response to complement activation. For example, it is possible that a similar process occurs in endothelial cells present within the choriocapillaris. This hypothesis would be consistent with the presence of MAC in the choriocapillaris that accumulates prior to development of clinically evident drusen.10 In contrast, complement-mediated RPE cell-associated ApoE might accumulate in later stages of AMD, in eyes with geographic atrophy and/or choroidal neovascularization, at which time complement is found adjacent to RPE cells.7,10 We are currently exploring in tissue culture and animal models additional mechanisms by which ApoE accumulates in AMD.
The results of our study support the idea that complement-mediated RPE cell-associated ApoE accumulation and subsequent release may be one of mechanisms that account for ApoE in drusen. We continue to investigate in our laboratory the mechanisms by which complement regulates RPE cell ApoE levels and the ability of ApoE to elicit complement activation. We believe that this information will enhance our understanding of the role that complement activation plays to mediate drusen formation and composition, and may elucidate potential therapeutic targets.
Supplementary Material
Acknowledgments
Supported, in part, by National Institutes of Health 5P30EY005722 (Core Grant) and Research to Prevent Blindness.
Disclosure: P. Yang, None; N.P. Skiba, None; G.M. Tewkesbury, None; V.M. Treboschi, None; P. Baciu, Allergan, Inc. (E); G.J. Jaffe, None
References
- 1. Kawa MP,, Machalinska A,, Roginska D,, et al. Complement system in pathogenesis of AMD: dual player in degeneration and protection of retinal tissue. J Immunol Res. 2014; 2014: 483960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang L,, Clark ME,, Crossman DK,, et al. Abundant lipid and protein components of drusen. PLoS One. 2010; 5: e10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zipfel PF,, Lauer N,, Skerka C. The role of complement in AMD. Adv Exp Med Biol. 2010; 703: 9–24. [DOI] [PubMed] [Google Scholar]
- 4. van der Schaft TL,, Mooy CM,, de Bruijn WC,, et al. Early stages of age-related macular degeneration: an immunofluorescence and electron microscopy study. Br J Ophthalmol. 1993; 77: 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson LV,, Leitner WP,, Staples MK,, et al. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887–896. [DOI] [PubMed] [Google Scholar]
- 6. Mullins RF,, Russell SR,, Anderson DH,, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000; 14: 835–846. [PubMed] [Google Scholar]
- 7. Anderson DH,, Mullins RF,, Hageman GS,, et al. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002; 134: 411–431. [DOI] [PubMed] [Google Scholar]
- 8. Seth A,, Cui J,, To E,, et al. Complement-associated deposits in the human retina. Invest Ophthalmol Vis Sci. 2008; 49: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hageman GS,, Anderson DH,, Johnson LV,, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mullins RF,, Schoo DP,, Sohn EH,, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014; 184: 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mullins RF,, Dewald AD,, Streb LM,, et al. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res. 2011; 93: 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meri S,, Pangburn MK. A mechanism of activation of the alternative complement pathway by the classical pathway: protection of C3b from inactivation by covalent attachment to C4b. Eur J Immunol. 1990; 20: 2555–2561. [DOI] [PubMed] [Google Scholar]
- 13. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009; 104: 115–149. [DOI] [PubMed] [Google Scholar]
- 14. Luo C,, Zhao J,, Madden A,, et al. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp Eye Res. 2013; 112: 93–101. [DOI] [PubMed] [Google Scholar]
- 15. Anderson DH,, Radeke MJ,, Gallo NB,, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010; 29: 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bradley DT,, Zipfel PF,, Hughes AE. Complement in age-related macular degeneration: a focus on function. Eye (Lond). 2011; 25: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholl HP,, Charbel Issa P,, Walier M,, et al. Systemic complement activation in age-related macular degeneration. PLoS One. 2008; 3: e2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loyet KM,, Deforge LE,, Katschke KJ, Jr,, et al. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012; 53: 6628–6637. [DOI] [PubMed] [Google Scholar]
- 19. Liu L,, Qiu W,, Wang H,, et al. Sublytic C5b-9 complexes induce apoptosis of glomerular mesangial cells in rats with Thy-1 nephritis through role of interferon regulatory factor-1-dependent caspase 8 activation. J Biol Chem. 2012; 287: 16410–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lueck K,, Wasmuth S,, Williams J,, et al. Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye (Lond). 2011; 25: 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li W,, Chen S,, Ma M,, et al. Complement 5b-9 complex-induced alterations in human RPE cell physiology. Med Sci Monit. 2010; 16: BR17–BR23. [PubMed] [Google Scholar]
- 22. Muller-Eberhard HJ. The killer molecule of complement. J Invest Dermatol. 1985; 85 1 suppl: 47s–52s. [DOI] [PubMed] [Google Scholar]
- 23. Mahley RW,, Rall SC., Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000; 1: 507–537. [DOI] [PubMed] [Google Scholar]
- 24. Huang Y,, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. 2014; 72 pt A: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abalain JH,, Carre JL,, Leglise D,, et al. Is age-related macular degeneration associated with serum lipoprotein and lipoparticle levels? Clin Chim Acta. 2002; 326: 97–104. [DOI] [PubMed] [Google Scholar]
- 26. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988; 240: 622–630. [DOI] [PubMed] [Google Scholar]
- 27. Wasan KM,, Brocks DR,, Lee SD,, et al. Impact of lipoproteins on the biological activity and disposition of hydrophobic drugs: implications for drug discovery. Nat Rev Drug Discov. 2008; 7: 84–99. [DOI] [PubMed] [Google Scholar]
- 28. Anderson DH,, Ozaki S,, Nealon M,, et al. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: implications for the process of drusen formation. Am J Ophthalmol. 2001; 131: 767–781. [DOI] [PubMed] [Google Scholar]
- 29. Ishida BY,, Bailey KR,, Duncan KG,, et al. Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J Lipid Res. 2004; 45: 263–271. [DOI] [PubMed] [Google Scholar]
- 30. Klaver CC,, Kliffen M,, van Duijn CM,, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet. 1998; 63: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malek G,, Li CM,, Guidry C,, et al. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am J Pathol. 2003; 162: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shanmugaratnam J,, Berg E,, Kimerer L,, et al. Retinal Muller glia secrete apolipoproteins E and J which are efficiently assembled into lipoprotein particles. Brain Res Mol Brain Res. 1997; 50: 113–120. [DOI] [PubMed] [Google Scholar]
- 33. Dentchev T,, Milam AH,, Lee VM,, et al. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis. 2003; 9: 184–190. [PubMed] [Google Scholar]
- 34. Machalinska A,, Kawa MP,, Marlicz W,, et al. Complement system activation and endothelial dysfunction in patients with age-related macular degeneration (AMD): possible relationship between AMD and atherosclerosis. Acta Ophthalmol. 2012; 90: 695–703. [DOI] [PubMed] [Google Scholar]
- 35. Sivak JM. The aging eye: common degenerative mechanisms between the Alzheimer's brain and retinal disease. Invest Ophthalmol Vis Sci. 2013; 54: 871–880. [DOI] [PubMed] [Google Scholar]
- 36. Curcio CA,, Johnson M,, Huang JD,, et al. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010; 51: 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li CM,, Clark ME,, Chimento MF,, et al. Apolipoprotein localization in isolated drusen and retinal apolipoprotein gene expression. Invest Ophthalmol Vis Sci. 2006; 47: 3119–3128. [DOI] [PubMed] [Google Scholar]
- 38. Johnson LV,, Forest DL,, Banna CD,, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2011; 108: 18277–18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaffe GJ,, Earnest K,, Fulcher S,, et al. Antitransferrin receptor immunotoxin inhibits proliferating human retinal pigment epithelial cells. Arch Ophthalmol. 1990; 108: 1163–1168. [DOI] [PubMed] [Google Scholar]
- 40. Yang P,, Baciu P,, Kerrigan BC,, et al. Retinal pigment epithelial cell death by the alternative complement cascade: role of membrane regulatory proteins, calcium, PKC, and oxidative stress. Invest Ophthalmol Vis Sci. 2014; 55: 3012–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim SW,, Heo JH,, Kim CH,, et al. Rapid and direct detection of apolipoprotein E genotypes using whole blood from humans. J Toxicol Environ Health A. 2010; 73: 1502–1510. [DOI] [PubMed] [Google Scholar]
- 42. Song F,, Poljak A,, Crawford J,, et al. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS One. 2012; 7: e34078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mooijaart SP,, Berbee JF,, van Heemst D,, et al. ApoE plasma levels and risk of cardiovascular mortality in old age. PLoS Med. 2006; 3: e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zybailov B,, Mosley AL,, Sardiu ME,, et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006; 5: 2339–2347. [DOI] [PubMed] [Google Scholar]
- 45. Ishihama Y,, Oda Y,, Tabata T,, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005; 4: 1265–1272. [DOI] [PubMed] [Google Scholar]
- 46. Yang P,, Wiser JL,, Peairs JJ,, et al. Human RPE expression of cell survival factors. Invest Ophthalmol Vis Sci. 2005; 46: 1755–1764. [DOI] [PubMed] [Google Scholar]
- 47. Medearis S,, Han IC,, Huang JK,, et al. The role of Bcl-xL in mouse RPE cell survival. Invest Ophthalmol Vis Sci. 2011; 52: 6545–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hed J,, Johansson M,, Lindroth M. Complement activation according to the alternate pathway by glass and plastic surfaces and its role in neutrophil adhesion. Immunol Lett. 1984; 8: 295–299. [DOI] [PubMed] [Google Scholar]
- 49. Gorham RD, Jr,, Forest DL,, Tamamis P,, et al. Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp Eye Res. 2013; 116: 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kay JE,, Korner A. Effect of cycloheximide on protein and ribonucleic acid synthesis in cultured human lymphocytes. Biochem J. 1966; 100: 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Castoralova M,, Brezinova D,, Sveda M,, et al. SUMO-2/3 conjugates accumulating under heat shock or MG132 treatment result largely from new protein synthesis. Biochim Biophys Acta. 2012; 1823: 911–919. [DOI] [PubMed] [Google Scholar]
- 52. Piehowski PD,, Petyuk VA,, Orton DJ,, et al. Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J Proteome Res. 2013; 12: 2128–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moore DJ,, Clover GM. The effect of age on the macromolecular permeability of human Bruch's membrane. Invest Ophthalmol Vis Sci. 2001; 42: 2970–2975. [PubMed] [Google Scholar]
- 54. Jeon H,, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005; 74: 535–562. [DOI] [PubMed] [Google Scholar]
- 55. Blacklow SC. Versatility in ligand recognition by LDL receptor family proteins: advances and frontiers. Curr Opin Struct Biol. 2007; 17: 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bu G., Apolipoprotein E. and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009; 10: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Andersen OM,, Vorum H,, Honore B,, et al. Ca2+ binding to complement-type repeat domains 5 and 6 from the low-density lipoprotein receptor-related protein. BMC Biochem. 2003; 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guttman M,, Prieto JH,, Croy JE,, et al. Decoding of lipoprotein-receptor interactions: properties of ligand binding modules governing interactions with apolipoprotein E. Biochemistry. 2010; 49: 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hadders MA,, Bubeck D,, Roversi P,, et al. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep. 2012; 1: 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scibek JJ,, Plumb ME,, Sodetz JM. Binding of human complement C8 to C9: role of the N-terminal modules in the C8 alpha subunit. Biochemistry. 2002; 41: 14546–14551. [DOI] [PubMed] [Google Scholar]
- 61. Musingarimi P,, Plumb ME,, Sodetz JM. Interaction between the C8 alpha-gamma and C8 beta subunits of human complement C8: role of the C8 beta N-terminal thrombospondin type 1 module and membrane attack complex/perforin domain. Biochemistry. 2002; 41: 11255–11260. [DOI] [PubMed] [Google Scholar]
- 62. Daly NL,, Scanlon MJ,, Djordjevic JT,, et al. Three-dimensional structure of a cysteine-rich repeat from the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 1995; 92: 6334–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DiScipio RG,, Berlin C. The architectural transition of human complement component C9 to poly(C9). Mol Immunol. 1999; 36: 575–585. [DOI] [PubMed] [Google Scholar]
- 64. Aleshin AE,, Schraufstatter IU,, Stec B,, et al. Structure of complement C6 suggests a mechanism for initiation and unidirectional, sequential assembly of membrane attack complex (MAC). J Biol Chem. 2012; 287: 10210–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kockx M,, Guo DL,, Huby T,, et al. Secretion of apolipoprotein E from macrophages occurs via a protein kinase A and calcium-dependent pathway along the microtubule network. Circ Res. 2007; 101: 607–616. [DOI] [PubMed] [Google Scholar]
- 66. Stanzel BV,, Blumenkranz MS,, Binder S,, et al. Longterm cultures of the aged human RPE do not maintain epithelial morphology and high transepithelial resistance. Graefes Arch Clin Exp Ophthalmol. 2012; 250: 313–315. [DOI] [PubMed] [Google Scholar]
- 67. Sohn JH,, Kaplan HJ,, Suk HJ,, et al. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest Ophthalmol Vis Sci. 2000; 41: 3492–3502. [PMC free article] [PubMed] [Google Scholar]
- 68. Cole DS,, Morgan BP. Beyond lysis: how complement influences cell fate. Clin Sci (Lond). 2003; 104: 455–466. [DOI] [PubMed] [Google Scholar]
- 69. Pilzer D,, Gasser O,, Moskovich O,, et al. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005; 27: 375–387. [DOI] [PubMed] [Google Scholar]
- 70. Moskovich O,, Fishelson Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem. 2007; 282: 29977–29986. [DOI] [PubMed] [Google Scholar]
- 71. Thurman JM,, Renner B,, Kunchithapautham K,, et al. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009; 284: 16939–16947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berchuck JE,, Yang P,, Toimil BA,, et al. All-trans-retinal sensitizes human RPE cells to alternative complement pathway-induced cell death. Invest Ophthalmol Vis Sci. 2013; 54: 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.