

Abstract
Pulmonary fibrosis is a form of lung disease that develops due to aberrant wound-healing following repeated alveoli injury in genetically susceptible individuals, resulting in chronic inflammation, excess deposition of the extracellular matrix components, mainly collagen, and scarring of lung tissue. In addition to irradiation, environmental agents such occupational inhalants, and chemotherapeutic agents, microbial agents also play a role in the etiology of the disease. While viruses have received the most attention, emerging evidence suggest that bacteria and fungi also play a part in the etiology of pulmonary fibrosis. Furthermore, successful use of antibiotics, antiviral and antifungal drugs in several studies to attenuate fibrosis progression is also an indication of microbial involvement in the pathogenesis of the disease and could be a promising therapeutic modality for treating pulmonary fibrosis initiated or exacerbated by infectious agents.
Keywords: Microbial agents, Pulmonary Fibrosis, Idiopathic Pulmonary Fibrosis
Introduction
Pulmonary fibrosis (PF) is a form of progressive lung disease that belongs to a large family of lung diseases called interstitial lung diseases [1]. The most common type of PF is idiopathic pulmonary fibrosis (IPF), and its cause is unknown. IPF is the most severe forms of interstitial lung disease that result in PF, with an annual incidence of 6.8–8.8 per 100,000 and 16.3–17.4 per 100,000 populations, using narrow and broad case definitions respectively in the U.S. [2]. PF develops as a result of a repetitive injury to the alveolar epithelium or endothelium, which triggers elements of the innate and adaptive immune system to restore the tissue architecture of the damaged tissue. Inflammatory mediators such as the profibrotic cytokine transforming growth factor-beta (TGF-β) activate angiogenesis and myofibroblasts to produce extracellular matrix (ECM) components (such as collagen and fibronectin) [3,4]. Failure to inactivate the fibrotic trigger results in exacerbation of inflammatory response, leading to abnormal wound healing response, tissue damage, excess deposition of ECM components, and scarring of the lungs (fibrosis). Lung fibrosis leads to a decrease in oxygen supply to the blood, which results in loss of lung function and respiratory failure [2,4].
The causes of PF are multifactorial. Some of the known causes include exogenous factors such as long-term exposure to environmental and occupational hazards such as asbestos, silica, coal dust, beryllium, hard metals, and radiation treatments. Certain chemotherapy drugs (methotrexate, bleomycin) anti-inflammatory (rituximab, sulfasalazine), heart medications (amiodarone, propranolol), and antibiotics (nitrofurantoin, ethambutol) as well as exposure to animal proteins, molds, and bacteria can also cause PF [2,5]. In addition, medical conditions such as gastrointestinal flux disease, autoimmune diseases (rheumatoid arthritis and systemic lupus erythematosus), sarcoidosis, and certain muscle diseases (such as dermatomyositis, polymyositis, and the anti-synthetase syndrome), can also lead to scarring of the lungs [6-9].
Several studies and reviews support a genetic basis for the development of PF. Familial PF occurs when two or more members of the same biological family are affected by the PF, and approximately 10 to 15 percent of those with an “idiopathic” form of PF have another family member affected by the disease [10-14]. Mutations and polymorphisms in several genes such as telomerase reverse transcriptase gene (TERT), and telomerase RNA component gene (TERC) have been shown to increase susceptibility to IPF [10,11,14-17]. Current therapy for the management of IPF include the use of immunosuppressants (prednisone), chemotherapy drugs (cyclophosphamide), anti-fibrotic drugs (Pirfenidone), proton pump inhibitors, oxygen therapy, and even lung transplant surgery for the management of IPF [18].
In this review, we report experimental findings from several studies with the goal of identifying the role of microbial infectious agents in PF. We provide evidence of antimicrobial drug efficacy in several cases of PF and suggest future experiments that may shed more light on the role of infectious agents in the pathogenicity of PF.
Microbial Agents and Pulmonary Fibrosis
Accumulating evidence suggests that infectious microbial agents (viral, fungal, and bacterial) may play a role in PF. While several studies have identified the presence of infectious agents in induction and exacerbation of PF using animal models, there is a paucity of literature regarding microbial induction of IPF in patients [19-22]. Studies employing antimicrobials such as antivirals, antibiotics, and antifungals also show great promise for the treatment of IPF and strengthen the connection between microbial agents and IPF [20,23-26]. Figure 1 highlights evidence available to support microbial involvement in PF.
Figure 1.
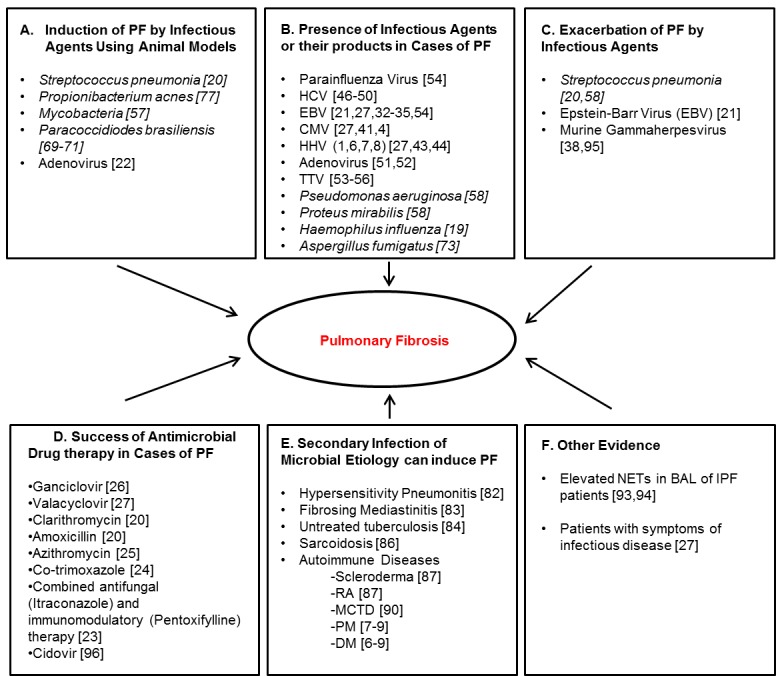
Evidence of Microbial Involvement in Pulmonary Fibrosis. (A) Induction of pulmonary fibrosis by infectious agents using animal models (B) Presence of infectious agents or their products in cases of PF (C) Exacerbation of PF by infectious agents (D) Success of antimicrobial drug therapy in cases of PF (E) Secondary infection of microbial etiology can induce PF (F) other evidence. PF: Pulmonary Fibrosis, HCV: Hepatitis C Virus, EBV: Epstein - Barr virus, CMV: Cytomegalovirus, HHV: Human Herpes Virus, TTV: Torque-Teno Virus, RA: Rheumatoid Arthritis, MCTD: Mixed Connective Tissue Disease, PM: Polymyositis, DM: Dermatomyositis.
Viruses and Pulmonary Fibrosis
IPF has been suggested to have a viral etiology based on the presence of viral signatures in the lungs of IPF patients, and the observation that IPF patients showed improvement when treated with antivirals [26,27]. Viruses detected in IPF patients include members of the Human Herpes Viruses (HHVs) family, such as Epstein-Barr virus (EBV), cytomegalovirus (CMV), Herpes simplex virus (type-1, -6, -7, -8) and Kaposi’s sarcoma herpesvirus [28]. HHVs can induce endoplasmic reticulum stress and apoptosis in epithelial cells in vitro, and the murine herpesvirus type 68, which is very similar to HHVs, can cause IPF by upregulating up-regulating TGF-β [29-31]. All these could be mechanisms of viral involvement in the pathogenesis of IPF.
EBV has received the most attention in relation to IPF. IPF patients have both deoxyribonucleic acid (DNA) and protein of EBV in their bronchoalveolar lavage fluid and lung tissue [32-34]. Tang et al., tested for the presence of eight HHVs in lung specimens of IPF patients and reported that EBV was present in 97 percent of the IPF patients, and 36 percent in the control. Other viruses identified were CMV and HHV-8 [27,35]. Rearrangement in EBV genome associated with productive EBV replication was found in lung tissue biopsies of 61 percent of EBV DNA-positive IPF patients [35]. Also, EBV latent membrane protein 1 is associated with rapid disease progression in IPF [21]. Interestingly although murine herpesvirus type 68 did not induce PF in murine models of PF, it enhanced the progression of fibrosis when administered with fibrotic stimuli [36-38]. Therefore, it can be inferred that viruses may be involved in the progression rather than initiation of IPF.
However, there are conflicting reports that negate the existence of EBV in cases of IPF [39,40]. Most of the studies on EBV and IPF are retrospective and show association and not causation. Also, since infection is a potential complication of therapy, there is also a high prevalence of EBV DNA in the general population it remains unclear how EBV plays a role in IPF.
Cytomegalovirus (CMV) is also present in cases of PF [41,42]. A study found elevated levels (80 percent) of CMV antibodies (anti-CMV) and other HHVs in PF patients compared with 30 percent in the control group [41].
Other HHVs such as HHV-1 and HHV-6 have been shown to have a link to PF. HHV-1 was present in 9 percent of IPF patients and 10 percent in fibrotic idiopathic interstitial pneumonia bronchoalveolar lavage fluid, whereas the control group was negative for the virus [43]. A test for Human Herpes Virus 6 (HHV-6) was 83.3 percent positive in of IPF patients, compared to 30 percent control patients also; HHV-6B antigens were present in mononuclear cells of IPF lung tissue [44]. Herpes viral infections may also contribute to the development of pulmonary hypertension in IPF patients [45].
Variable results have arisen from studies on the relationship between HCV and IPF. An immunological study conducted to evaluate the prevalence of serum antibodies in Japanese IPF patients to HCV showed that 28 percent of IPF patients were seropositive to HCV in contrast to 3.6 percent of the control population [46]. One Italian study showed a higher incidence of IPF in an HCV-positive group compared to an HBV group [47]. HCV infection can also cause active alveolitis, which can lead to IPF [48]. However, HCV is also prevalent in other forms of non-fibrotic respiratory diseases, and some studies have found no correlation between HCV and IPF [49,50].
Adenovirus, particularly its early region 1 protein, is of interest in lung disease due to its ability to upregulate TGF-β1 expression in bronchiolar epithelial cells and transformation of lung epithelial cells to express mesenchymal markers [51]. In a recent study, intratracheal instillation of high dose adenoviral vectors induced an inflammatory response, lung injury, and PF in a dose-dependent manner [22]. However, contradicting studies show no adenoviral antibodies in the sera of IPF patients, and treatment of patients with corticosteroid may encourage the prevalence of adenovirus [52]. Additional studies employing a larger patient population size is required to quantify messenger RNA in infected lung tissues.
Torque-Teno Virus (TTV), a circular single-stranded virus initially associated with post-transfusion hepatitis infection is significantly more common in IPF patients with acute exacerbation than stable controls [53,54]. Investigators detected TTV DNA in 36.4 percent of IPF patients evaluated, and associated TTV with lower survival rate among the TTV-positive IPF patients evaluated, compared to the TTV-negative IPF patients [55]. In a more recent study by the same group, they suggest that TTV DNA titer may reflect the immunosuppressive state of the host due to treatment [56].
Taken together, the presence of significantly higher viral signatures in patients with IPF may be an indication that viruses may play a role in the pathogenesis of IPF. However, the small sample size of patients and short duration of these studies makes it difficult to draw any bold conclusions. More experiments with adequate controls need to be carried out to elucidate the role that viruses play in cases of IPF.
Bacteria and Pulmonary Fibrosis
One of the earliest evidence of microbial involvement in interstitial lung disease came from studies, which showed that a glycolipid cell-wall component of mycobacteria, trehalose dimycolate induced interstitial pneumonitis and alveolar hemorrhages in mice, with the help T-lymphocytes [57]. Following this discovery, numerous studies have implicated bacteria in the etiology of PF.
A study conducted to culture bronchoalveolar lavage fluid from patients with Granulomatosis with polyangiitis using IPF patients as a control, revealed bacterial colonization of the lungs with species such as Pseudomonas aeruginosa, Streptococcus pneumonia, Moraxella catarrhalis, and Proteus mirabilis in 8 of the 22 IPF patients sampled [58]. Another study found an increase in bacterial burden in BAL of IPF patients particularly with species such as Haemophilus, Streptococcus, Neisseria, and Veillonella which serve as a prediction of decline in lung function and death [59]. Haemophilus influenza and Pneumocystis jirovecii bacteria can also cause chronic pulmonary disease by stimulating inflammation and activating macrophages [19,60].
Two independent lung fibrosis models show that S. pneumonia triggers progression of PF through the action of its pore-forming cytotoxin pneumolysin (ply). Interestingly, antibiotic treatment with clarithromycin or amoxicillin at 24 h and 48 h post-infection led a significant decrease in lung hydroxyproline contents, comparable with mock-infected mice thereby abolishing the infection-induced fibrosis progression [20].
However, it can be argued that since IPF is linked with increased aspiration and gastroesophageal reflux, the increased bacterial burden in IPF patients compared to healthy control groups could be as a result of microaspiration, or lack of the airway mucin gene promoter polymorphism, which is also associated with bacterial burden in BAL [61-66]. In addition, others show using an established murine model of lung fibrosis that P. aeruginosa did not exacerbate bleomycin-induced fibrosis [67].
It remains unclear whether a bacterial infection is a direct cause of IPF, a secondary infection, or only an exacerbating agent, particularly because patients with IPF are susceptible to developing bacterial pneumonia [68].
Fungi and Pulmonary Fibrosis
Inhalation of fungal material can lead to lung fibrosis by infecting the pulmonary tissue, lung cavity, or by triggering an immune reaction. Paracoccidiodes brasiliensis, a fungus involved in the etiology of paracoccidioidomycosis (PCM), can also induce experimental PF [69,70]. PCM is characterized by a chronic inflammatory response, and neutrophil infiltration, leading to lung fibrosis and loss of lung function in most patients [71]. A study conducted on the development of fibrosis in a model of experimental chronic pulmonary PCM, employing a combined antifungal (Itraconazole) and immunomodulatory (Pentoxifylline) therapy revealed that administration of the combined therapy in advanced stage of the disease resulted in a significant reduction of granulomatous inflammation and PF, compared with the results of classical antifungal therapy using itraconazole alone [23]. In addition, Arias and colleagues show that depletion of neutrophils in PCM promotes the resolution of PF and inflammation [72]. The fungus Aspergillus has also been reported to colonize the parenchymal lung cavity in cases of IPF [73]. Untreated chronic pulmonary aspergillosis caused by the fungus A. fumigatus can progress to chronic fibrosing pulmonary aspergillosis, and this can result in chronic scarring of the lungs [74-76].
Success of Antimicrobials and Vaccinations in Murine Models of Pulmonary Fibrosis
Various microbial pathogens such as S. pneumoniae, P. acnes, P. brasiliensis, and proteins from Mycobacteria have been utilized in animal models to induce PF [20,57,69,77]. Several studies have also reported the success of antimicrobials such as ganciclovir, valacyclovir, clarithromycin, amoxicillin, azithromycin, co-trimoxazole (sulfamethoxazole, and trimethoprim), and a combined therapy of antifungal (Itraconazole) and immunomodulatory (Pentoxifylline), in decreasing the microbial load, thereby significantly reducing lung fibrosis by reducing hydroxyl proline levels, restrictive lung function pattern and improving the quality of life of IPF patients [20,23-27]. Others show that protein-based vaccination of mice with established lung fibrosis with the non-cytotoxic S. pneumonia pneumolysin derivative (PdB) provides protection against pneumolysin-induced fibrosis exacerbation [20].
It is noteworthy that antibiotics in addition to their traditional antimicrobial effects also possess immunomodulatory and anti-inflammatory properties [25,78]. Macrolide antibiotics such as clarithromycin, azithromycin, and erythromycin can prevent the production of pro-inflammatory cytokines and immune mediators [79-81].
Therefore, additional studies need to be carried out to elucidate the precise mechanism of action underlying the therapeutic effects of antibiotics in treatment of PF.
Pulmonary Fibrosis Caused as a Complication of a Disease with Microbial Etiology
PF may occur as a secondary complication due to diseases of microbial etiology. Such diseases include hypersensitivity pneumonitis (also called “hot tub lung”), fibrosing mediastinitis, sarcoidosis, and untreated tuberculosis [82-86]. Autoimmune diseases with infectious etiology such as scleroderma, systemic lupus erythematosus, rheumatoid arthritis, and mixed connective tissue diseases can also induce PF [87-92].
Other evidence such as increase in activated neutrophils in BAL fluid from IPF patients associated with early mortality, and raised plasma concentrations of alpha-defensins could indicate the presence of microbial agents [93-94]. Table 1 highlights all studies on microbial agents and antimicrobial therapy in PF.
Table 1. Studies on Microbial Agents and Antimicrobial Therapy in Pulmonary Fibrosis.
| A. Microbial Agent implicated in Pulmonary Fibrosis | ||
| Viruses | References | |
| Hepatitis C Virus (HCV) | [46-50] | |
| Epstein-Barr Virus (EBV) | [21,27,32-35,54] | |
| Cytomegalovirus (CMV) | [27,41,42] | |
| Human Herpes Virus 1 (HHV-1) | [43] | |
| Human Herpes Virus 6 (HHV-6) | [44] | |
| Human Herpes Virus 7 (HHV-7) | [27] | |
| Human Herpes Virus 8 (HHV-8) | [27] | |
| Adenovirus | [22,51,52] | |
| Torque-Teno Virus (TTV) | [53-56] | |
| Murine gammaherpes virus type 68 (MHV-68) | [29,36-38,67,95] | |
| Parainfluenza Virus | [54] | |
| Parvovirus B19 | [42] | |
| Bacteria | ||
| Pseudomonas aeruginosa | [58] | |
| Streptococcus pneumonia | [20,58] | |
| Moraxella catarrhalis | [58] | |
| Proteus mirabilis | [58] | |
| Propionibacterium acnes | [77] | |
| Staphylococcus aureus | [28] | |
| Pneumocystis jirovecii | [60] | |
| Haemophilus influenza | [19] | |
| Mycobacteria | [57] | |
| Fungi | ||
| Aspergillus fumigatus | [73] | |
| Paracoccidiodes brasiliensis | [69-71] | |
| B. Antimicrobial Studies on infectious Agents involved in Pulmonary Fibrosis | ||
| Antivirals | Findings | |
| Ganciclovir | 2-week course of ganciclovir may attenuate disease progression in a subgroup of advanced IPFa patients [26]. | |
| Valacyclovir | Treatment with this antiviral led to a decrease in sputum viral load in lungs of patients with IPF [27]. | |
| Cidovir | Antiviral treatment administered to symptomatic animals, improved survival from 20 to 80% compared with untreated symptomatic animals, but lung fibrosis persisted in 60% of the mice [96]. | |
| Antibiotics | ||
| Clarithromycin | Antibiotic treatment with clarithromycin or amoxicillin led to significantly decreased lung hydroxyproline contents, thereby in blocking S. pneumoniae-induced fibrosis exacerbation in mice [20]. | |
| Amoxicillin | Antibiotic treatment with clarithromycin or amoxicillin led to significantly decreased lung hydroxyproline contents, thereby in blocking S. pneumoniae-induced fibrosis exacerbation in mice [20]. | |
| Azithromycin | This antibiotic showed a significant reduction in both fibrosis and restrictive lung function pattern in a bleomycin-induced PFb mouse model [25]. | |
| Co-trimoxazole | Treating IPF patients with the addition of co-trimoxazole 960mg twice daily had no effect on lung function but resulted in improved quality of life and a reduction in mortality in those adhering to treatment [24]. | |
| Antifungals | ||
| Combined antifungal (Itraconazole) and immunomodulatory (Pentoxifylline) therapy | A study conducted on the development of fibrosis in a model of experimental chronic pulmonary PCMc, employing a combined antifungal (Itraconazole) and immunomodulatory (Pentoxifylline) therapy resulted in a significant reduction of granulomatous inflammation and PF, when compared with the results of classical antifungal therapy using itraconazole alone [23]. | |
aIPF: Idiopathic pulmonary fibrosis, bPF: Pulmonary fibrosis, cPCM: Paracoccidioidomycosis
Conclusion
Evidence from the literature show that infectious pathogens exist in some cases of PF, can induce and exacerbate the disease, and antimicrobials may be useful in improving the quality of life of patients with PF. Experimental animal models of PF show that infectious agents play a role in the initiation of PF, however, whether this is true in patients is yet to be reported. The possibility of utilizing antimicrobials as a potential therapeutic strategy administered early in the course of the disease to improve the quality of life needs to be critically explored. More studies need to be designed and executed with a larger population size to investigate the efficacy of antimicrobials to either halt or to slow the progression of PF.
Taken together these studies provide evidence to show that infectious agents can either induce or exacerbate PF, however, additional studies to assess the viability of infectious agents after induction of PF will delineate the role of antimicrobials in the treatment of PF.
Glossary
- PF
Pulmonary Fibrosis
- IPF
Idiopathic Pulmonary Fibrosis
- AE-IPF
Acute Exacerbated Idiopathic Pulmonary Fibrosis
- BAL
Bronchioalveolar Lavage
- NETs
Neutrophil Extracellular Traps
Author Contributions
Ozioma S. Chioma & Wonder P. Drake contributed to drafting the manuscript. This work was supported by National Institute of Health grants (5R01HL117074-05, 6K24HL127301-02) to Wonder P. Drake.
References
- American Lung Association: Pulmonary Fibrosis [Internet] [cited 2016. Nov 18]. Available from: http://www.lung.org/lung-health-and-diseases/lung-disease-lookup/pulmonary-fibrosis/ .
- Nalysnyk L, Cid-Ruzafa J, Rotella P. et al. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–361. doi: 10.1183/09059180.00002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13(22):3056–3062. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- Takizawa H, Shiga J, Moroi Y. et al. Interstitial lung disease in dermatomyositis: clinicopathological study. J Rheumatol. 1987;14(1):102–107. [PubMed] [Google Scholar]
- Pulmonary Fibrosis Foundation: Causes and Symptoms [Internet] [cited 2016. Nov 18]. Available from: http://www.pulmonaryfibrosis.org/life-with-pf/about-pf/
- Kang EH, Lee EB, Shin KC. et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology. 2005;44(10):1282–1286. doi: 10.1093/rheumatology/keh723. [DOI] [PubMed] [Google Scholar]
- Lakhanpal S, Lie JT, Conn DL. et al. Pulmonary disease in polymyositis/dermatomyositis: a clinicopathological analysis of 65 autopsy cases. Ann Rheum Dis. 1987;46(1):23–29. doi: 10.1136/ard.46.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai SK, Yang IV, Schwarz MI. et al. Incorporating genetics into the identification and treatment of Idiopathic Pulmonary Fibrosis. BMC Med. 2015;13:191. doi: 10.1186/s12916-015-0434-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 2002;57(4):338–342. doi: 10.1136/thorax.57.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam JS, Limper AH. Idiopathic pulmonary fibrosis: is it a familial disease? Curr Opin Pulm Med. 2006;12(5):312–317. doi: 10.1097/01.mcp.0000239546.24831.61. [DOI] [PubMed] [Google Scholar]
- Lee HL, Ryu JH, Wittmer MH. et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest. 2005;127(6):2034–2041. doi: 10.1378/chest.127.6.2034. [DOI] [PubMed] [Google Scholar]
- van Moorsel CH, Hoffman TW, van Batenburg AA. et al. Understanding Idiopathic Interstitial Pneumonia: A Gene-Based Review of Stressed Lungs. Biomed Res Int. 2015:304186. doi: 10.1155/2015/304186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Moorsel CH, van Oosterhout MF, Barlo NP. et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182(11):1419–1425. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- Aquino-Galvez A, Perez-Rodriguez M, Camarena A. et al. MICA polymorphisms and decreased expression of the MICA receptor NKG2D contribute to idiopathic pulmonary fibrosis susceptibility. Hum Genet. 2009;125(5-6):639–648. doi: 10.1007/s00439-009-0666-1. [DOI] [PubMed] [Google Scholar]
- Hodgson U, Pulkkinen V, Dixon M. et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79(1):149–154. doi: 10.1086/504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez MM, Chan AL, Norris AG. et al. Acute exacerbation of idiopathic pulmonary fibrosis-a review of current and novel pharmacotherapies. J Thorac Dis. 2015;7(3):499–519. doi: 10.3978/j.issn.2072-1439.2015.01.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie TJ, Olson AL, Cosgrove GP. et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology. 2010;15(6):909–917. doi: 10.1111/j.1440-1843.2010.01774.x. [DOI] [PubMed] [Google Scholar]
- Knippenberg S, Ueberberg B, Maus R. et al. Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax. 2015;70(7):636–645. doi: 10.1136/thoraxjnl-2014-206420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto K, Hayakawa H, Sato A. et al. Involvement of Epstein-Barr virus latent membrane protein 1 in disease progression in patients with idiopathic pulmonary fibrosis. Thorax. 2000;55(11):958–961. doi: 10.1136/thorax.55.11.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Chen T, Bozkanat M. et al. Intratracheal instillation of high dose adenoviral vectors is sufficient to induce lung injury and fibrosis in mice. PLoS One. 2014;9(12):e116142. doi: 10.1371/journal.pone.0116142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranjo TW, Lopera DE, Diaz-Granados LR. et al. Combined itraconazole-pentoxifylline treatment promptly reduces lung fibrosis induced by chronic pulmonary paracoccidioidomycosis in mice. Pulm Pharmacol Ther. 2011;24(1):81–91. doi: 10.1016/j.pupt.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Shulgina L, Cahn AP, Chilvers ER. et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax. 2013;68(2):155–162. doi: 10.1136/thoraxjnl-2012-202403. [DOI] [PubMed] [Google Scholar]
- Wuyts WA, Willems S, Vos R. et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res. 2010;36(10):602–614. doi: 10.3109/01902148.2010.492895. [DOI] [PubMed] [Google Scholar]
- Egan JJ, Adamali HI, Lok SS. et al. Ganciclovir antiviral therapy in advanced idiopathic pulmonary fibrosis: an open pilot study. Pulm Med. 2011:240805. doi: 10.1155/2011/240805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YW, Johnson JE, Browning PJ. et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41(6):2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MK, Zhou Y, Murray S. et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2(7):548–556. doi: 10.1016/S2213-2600(14)70069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik PN, Horowitz JC, Moore TA. et al. Pulmonary fibrosis induced by gamma-herpesvirus in aged mice is associated with increased fibroblast responsiveness to transforming growth factor-beta. J Gerontol A Biol Sci Med Sci. 2012;67(7):714–725. doi: 10.1093/gerona/glr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79(11):6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson WE, Crossno PF, Polosukhin VV. et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- Manika K, Alexiou-Daniel S, Papakosta D. et al. Epstein-Barr virus DNA in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(2):134–140. [PubMed] [Google Scholar]
- Egan JJ, Stewart JP, Hasleton PS. et al. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax. 1995;50(12):1234–1249. doi: 10.1136/thx.50.12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JP, Egan JJ, Ross AJ. et al. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1336–1341. doi: 10.1164/ajrccm.159.4.9807077. [DOI] [PubMed] [Google Scholar]
- Kelly BG, Lok SS, Hasleton PS. et al. A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002;166(4):510–513. doi: 10.1164/rccm.2103058. [DOI] [PubMed] [Google Scholar]
- Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. 2011;29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- Lok SS, Haider Y, Howell D. et al. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J. 2002;5(1228):1232. doi: 10.1183/09031936.02.00272902. [DOI] [PubMed] [Google Scholar]
- Mora AL, Woods CR, Garcia A. et al. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L711–L721. doi: 10.1152/ajplung.00007.2005. [DOI] [PubMed] [Google Scholar]
- Zamo A, Poletti V, Reghellin D. et al. HHV-8 and EBV are not commonly found in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(2):123–128. [PubMed] [Google Scholar]
- Wangoo A, Shaw RJ, Diss TC. et al. Cryptogenic fibrosing alveolitis: lack of association with Epstein-Barr virus infection. Thorax. 1997;52(10):888–891. doi: 10.1136/thx.52.10.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemaru M, Kasuga I, Kusumoto H. et al. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur Respir J. 1997;10(9):2040–2045. doi: 10.1183/09031936.97.10092040. [DOI] [PubMed] [Google Scholar]
- Magro CM, Allen J, Pope-Harman A. et al. The role of microvascular injury in the evolution of idiopathic pulmonary fibrosis. Am J Clin Pathol. 2003;119(4):556–567. doi: 10.1309/0B06-Y93E-GE6T-Q36Y. [DOI] [PubMed] [Google Scholar]
- Lasithiotaki I, Antoniou KM, Vlahava VM. et al. Detection of herpes simplex virus type-1 in patients with fibrotic lung diseases. PLoS One. 2011;6(12):e27800. doi: 10.1371/journal.pone.0027800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen V, Salmenkivi K, Kinnula VL. et al. A novel screening method detects herpesviral DNA in the idiopathic pulmonary fibrosis lung. Ann Med. 2012;44(2):178–186. doi: 10.3109/07853890.2010.532151. [DOI] [PubMed] [Google Scholar]
- Calabrese F, Kipar A, Lunardi F. et al. Herpes virus infection is associated with vascular remodeling and pulmonary hypertension in idiopathic pulmonary fibrosis. PLoS One. 2013;8(2):e55715. doi: 10.1371/journal.pone.0055715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Ohta K, Suzuki N. et al. Idiopathic pulmonary fibrosis and high prevalence of serum antibodies to hepatitis C virus. Am Rev Respir Dis. 1992;146(1):266–268. doi: 10.1164/ajrccm/146.1.266. [DOI] [PubMed] [Google Scholar]
- Arase Y, Suzuki F, Suzuki Y. et al. Hepatitis C virus enhances incidence of idiopathic pulmonary fibrosis. World J Gastroenterol. 2008;14(38):5880–5886. doi: 10.3748/wjg.14.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K, Yamaguchi S, Fujimoto K. et al. Bronchoalveolar lavage fluid findings in patients with chronic hepatitis C virus infection. Thorax. 1996;51(3):312–314. doi: 10.1136/thx.51.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meliconi R, Andreone P, Fasano L. et al. Incidence of hepatitis C virus infection in Italian patients with idiopathic pulmonary fibrosis. Thorax. 1996;51(3):315–317. doi: 10.1136/thx.51.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving WL, Day S, Johnston ID. Idiopathic pulmonary fibrosis and hepatitis C virus infection. Am Rev Respir Dis. 1993;148(6 Pt 1):1683–1684. doi: 10.1164/ajrccm/148.6_Pt_1.1683. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Hogg JC. Adenovirus infections and lung disease. Curr Opin Pharmacol. 2007;7(3):237–243. doi: 10.1016/j.coph.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano K, Nomoto Y, Kunitake R. et al. Detection of adenovirus E1A DNA in pulmonary fibrosis using nested polymerase chain reaction. Eur Respir J. 1997;10(7):1445–1449. doi: 10.1183/09031936.97.10071445. [DOI] [PubMed] [Google Scholar]
- Hino S, Miyata H. Torque teno virus (TTV): current status. Rev Med Virol. 2007;17(1):45–57. doi: 10.1002/rmv.524. [DOI] [PubMed] [Google Scholar]
- Wootton SC, Kim DS, Kondoh Y. et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1698–1702. doi: 10.1164/rccm.201010-1752OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando M, Ohno S, Oshikawa K. et al. Infection of TT virus in patients with idiopathic pulmonary fibrosis. Respir Med. 2001;95(12):935–942. doi: 10.1053/rmed.2001.1151. [DOI] [PubMed] [Google Scholar]
- Bando M, Nakayama M, Takahashi M. et al. Serum torque teno virus DNA titer in idiopathic pulmonary fibrosis patients with acute respiratory worsening. Intern Med. 2015;54(9):1015–1019. doi: 10.2169/internalmedicine.54.3610. [DOI] [PubMed] [Google Scholar]
- Seggev JS, Goren MB, Kirkpatrick CH. The pathogenesis of trehalose dimycolate-induced interstitial pneumonitis. III. Evidence for a role for T lymphocytes. Cell Immunol. 1984;85(2):428–435. doi: 10.1016/0008-8749(84)90256-9. [DOI] [PubMed] [Google Scholar]
- Richter AG, Stockley RA, Harper L. et al. Pulmonary infection in Wegener granulomatosis and idiopathic pulmonary fibrosis. Thorax. 2009;64(8):692–697. doi: 10.1136/thx.2008.110445. [DOI] [PubMed] [Google Scholar]
- Molyneaux PL, Cox MJ, Willis-Owen SA. et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–913. doi: 10.1164/rccm.201403-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CE, Yi ES, Ryu JH. Autopsy findings in 42 consecutive patients with idiopathic pulmonary fibrosis. Eur Respir J. 2008;32(1):170–174. doi: 10.1183/09031936.00176307. [DOI] [PubMed] [Google Scholar]
- Seibold MA, Wise AL, Speer MC. et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerlin TE, Murphy E, Zhang W. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy MG, Livraghi-Butrico A, Fletcher AA. et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin RW, Pope CE, Pellegrini CA. et al. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158(6):1804–1808. doi: 10.1164/ajrccm.158.6.9804105. [DOI] [PubMed] [Google Scholar]
- Lee JS, Ryu JH, Elicker BM. et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390–1394. doi: 10.1164/rccm.201101-0138OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino E, Carbone R, Marabotto E. et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J. 2013;42(5):1322–1331. doi: 10.1183/09031936.00101212. [DOI] [PubMed] [Google Scholar]
- Ashley SL, Jegal Y, Moore TA. et al. gamma-Herpes virus-68, but not Pseudomonas aeruginosa or influenza A (H1N1), exacerbates established murine lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;307(3):L219–L230. doi: 10.1152/ajplung.00300.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux PL, Maher TM. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir Rev. 2013;22(129):376–381. doi: 10.1183/09059180.00000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco L, Najvar L, Gomez BL. et al. Experimental pulmonary fibrosis induced by Paracoccidioides brasiliensis conidia: measurement of local host responses. Am J Trop Med Hyg. 1998;58(4):424–430. doi: 10.4269/ajtmh.1998.58.424. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Restrepo A, Cano LE. Pulmonary immune responses induced in BALB/c mice by Paracoccidioides brasiliensis conidia. Mycopathologia. 2008;165(4-5):313–330. doi: 10.1007/s11046-007-9072-1. [DOI] [PubMed] [Google Scholar]
- Restrepo A, Benard G, de Castro CC. et al. Pulmonary paracoccidioidomycosis. Semin Respir Crit Care Med. 2008;29(2):182–197. doi: 10.1055/s-2008-1063857. [DOI] [PubMed] [Google Scholar]
- Puerta-Arias JD, Pino-Tamayo PA, Arango JC. et al. Depletion of Neutrophils Promotes the Resolution of Pulmonary Inflammation and Fibrosis in Mice Infected with Paracoccidioides brasiliensis. PLoS One. 2016;11(9):e0163985. doi: 10.1371/journal.pone.0163985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Mishra M, Singhal A. et al. Aspergilloma coexisting with idiopathic pulmonary fibrosis: a rare occurrence. J Postgrad Med. 2013;59(2):145–148. doi: 10.4103/0022-3859.113841. [DOI] [PubMed] [Google Scholar]
- Denning DW, Riniotis K, Dobrashian R. et al. Chronic cavitary and fibrosing pulmonary and pleural aspergillosis: case series, proposed nomenclature change, and review. Clin Infect Dis. 2003;37(Suppl 3):S265–S280. doi: 10.1086/376526. [DOI] [PubMed] [Google Scholar]
- Denning DW, Cadranel J, Beigelman-Aubry C. et al. Chronic pulmonary aspergillosis: rationale and clinical guidelines for diagnosis and management. Eur Respir J. 2016;47(1):45–68. doi: 10.1183/13993003.00583-2015. [DOI] [PubMed] [Google Scholar]
- Kosmidis C, Newton P, Muldoon EG. et al. Chronic fibrosing pulmonary aspergillosis: a cause of ‘destroyed lung’ syndrome. Infect Dis. 2016:1–6. doi: 10.1080/23744235.2016.1232861. [DOI] [PubMed] [Google Scholar]
- Jiang D, Huang X, Geng J. et al. Pulmonary fibrosis in a mouse model of sarcoid granulomatosis induced by booster challenge with Propionibacterium acnes. Oncotarget. 2016;7(23):33703–33714. doi: 10.18632/oncotarget.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buret AG. Immuno-modulation and anti-inflammatory benefits of antibiotics: the example of tilmicosin. Can J Vet Res. 2010;74(1):1–10. [PMC free article] [PubMed] [Google Scholar]
- Labro MT. Antibiotics as anti-inflammatory agents. Curr Opin Investig Drugs. 2002;3(1):61–68. [PubMed] [Google Scholar]
- Pukhalsky AL, Shmarina GV, Kapranov NI. et al. Anti-inflammatory and immunomodulating effects of clarithromycin in patients with cystic fibrosis lung disease. Mediators Inflamm. 2004;13(2):111–117. doi: 10.1080/09629350410001688495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A, Bush A. Anti-inflammatory effects of macrolides in lung disease. Pediatr Pulmonol. 2001;31(6):464–473. doi: 10.1002/ppul.1076. [DOI] [PubMed] [Google Scholar]
- Embil J, Warren P, Yakrus M. et al. Pulmonary illness associated with exposure to Mycobacterium-avium complex in hot tub water. Hypersensitivity pneumonitis or infection? Chest. 1997;111(3):813–816. doi: 10.1378/chest.111.3.813. [DOI] [PubMed] [Google Scholar]
- Loyd JE, Tillman BF, Atkinson JB. et al. Mediastinal fibrosis complicating histoplasmosis. Medicine. 1988;67(5):295–310. doi: 10.1097/00005792-198809000-00002. [DOI] [PubMed] [Google Scholar]
- Patz EF, Jr., Swensen SJ, Erasmus J. Pulmonary manifestations of nontuberculous Mycobacterium. Radiol Clin North Am. 1995;33(4):719–729. [PubMed] [Google Scholar]
- Nardi A, Brillet PY, Letoumelin P. et al. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J. 2011;38(6):1368–1373. doi: 10.1183/09031936.00187410. [DOI] [PubMed] [Google Scholar]
- Bonham CA, Strek ME, Patterson KC. From granuloma to fibrosis: sarcoidosis associated pulmonary fibrosis. Curr Opin Pulm Med. 2016;22(5):484–491. doi: 10.1097/MCP.0000000000000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman C, Dovrish Z, Shoenfeld Y. et al. Do infections facilitate the emergence of systemic sclerosis? Autoimmun Rev. 2011;10(5):244–247. doi: 10.1016/j.autrev.2010.09.010. [DOI] [PubMed] [Google Scholar]
- James JA, Kaufman KM, Farris AD. et al. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997;100(12):3019–3026. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhary VR, Tilahun AY, Clark CR. et al. Chronic exposure to staphylococcal superantigen elicits a systemic inflammatory disease mimicking lupus. J Immunol. 2012;189(4):2054–2062. doi: 10.4049/jimmunol.1201097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsson R, Aalokken TM, Molberg O. et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012;71(12):1966–1972. doi: 10.1136/annrheumdis-2011-201253. [DOI] [PubMed] [Google Scholar]
- Suda T. Up-to-Date Information on Rheumatoid Arthritis-Associated Interstitial Lung Disease. Clin Med Insights Circ Respir Pulm Med. 2015;9(Suppl 1):155–162. doi: 10.4137/CCRPM.S23289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, He B, Liu J. et al. Molecular Insight into Gut Microbiota and Rheumatoid Arthritis. Int J Mol Sci. 2016;17(3):431. doi: 10.3390/ijms17030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder BW, Brown KK, Schwarz MI. et al. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133(1):226–232. doi: 10.1378/chest.07-1948. [DOI] [PubMed] [Google Scholar]
- Mukae H, Iiboshi H, Nakazato M. et al. Raised plasma concentrations of alpha-defensins in patients with idiopathic pulmonary fibrosis. Thorax. 2002;57(7):623–628. doi: 10.1136/thorax.57.7.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan TR, Moore BB, Weinberg JB. et al. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am J Respir Crit Care Med. 2008;177(7):771–780. doi: 10.1164/rccm.200708-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora AL, Torres-Gonzalez E, Rojas M. et al. Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am J Respir Crit Care Med. 2007;175(11):1139–1150. doi: 10.1164/rccm.200610-1426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]