

Abstract
Borrelia (Borreliella) burgdorferi and closely related genospecies are the causative agents of Lyme disease, the most common tick-borne disease north of the equator. The bacterium, a member of the spirochete phylum, is acquired by a tick vector that feeds on an infected vertebrate host and is transmitted to another vertebrate during subsequent feeding by the next tick stage. The precise navigation of this enzootic cycle entails the regulation of genes required for these two host-specific phases as well as the transitions between them. Recently, an expansive swath of small RNAs has been identified in B. burgdorferi and likely many, if not most, are involved in regulating gene expression. Regardless, with only a few exceptions, the functions of these RNAs are completely unknown. However, several state-of-the-art approaches are available to identify the targets of these RNAs and provide insight into their role in the enzootic cycle and infection.
Keywords: sRNA, RNA-seq, transcriptome, non-coding RNA, Lyme disease, spirochete, gene regulation
Introduction
Lyme disease is an emerging infectious disease that is the most common tick-borne zoonosis in the Northern hemisphere [1,2]. Human infection typically first presents as a skin rash at the site of the tick bite and can disseminate to multiple organ systems resulting in rheumatic, cardiac, and neurologic manifestations. The etiologic agent of Lyme disease is the spirochete Borrelia (Borreliella) burgdorferi, and closely related genospecies, which belong to a morphologically serpentine and deeply branching phylum of bacteria [3-5]. B. burgdorferi is maintained in nature in an enzootic cycle that requires a hard tick vector and a vertebrate host [1,6,7]. Larval ticks acquire the spirochetes by feeding on infected reservoir hosts; the spirochetes persist in the midgut through the molt into nymphs, emigrate to the salivary glands when the nymph feeds, and transmit to a vertebrate. B. burgdorferi adapts to the species-specific environments, including nutrient resources and immune responses, encountered as it traverses its enzootic cycle by sensing its surroundings and substantially shifting its gene expression via several regulatory systems [1,8,9].
The genes of B. burgdorferi constitute a Gordian genome that is highly segmented and predominantly linear [10,11]. A sparse set of recognized orthologs of transcriptional regulators suggests that post-transcriptional mechanisms (including curious post-translational modifications [12]) contribute to controlling gene expression [1,8,10,13]. We identified the first regulatory RNA, DsrABb, and delineated its role in activating the regulon required for transmission and vertebrate infection [14]. Moreover, we demonstrated that this small RNA (sRNA), like most sRNAs, requires the RNA chaperone Hfq for its presumed base-pairing; the B. burgdorferi Hfq is an oddball, but it can complement a heterologous hfq mutant, and it is required for vertebrate infection [15]. Two other RNA-binding proteins have been found in the spirochete: CsrA [16-18] and Bpur [19,20], although neither their function nor RNA targets (other than Bpur binding to its own mRNA) have been determined. The other players in regulating the levels of mRNA are the ribonucleases that degrade RNA and, again, B. burgdorferi encodes a limited quiver, including RNase III, M5, Y, and Z [21,22]; the kinetics of decay have been assayed for several mRNAs and half-lives vary from a minute to almost an hour [22].
The number of putative regulatory sRNAs in B. burgdorferi has exponentially expanded by the publication of several high-throughput transcriptomes [23-25]. The challenge now for molecular borreliologists is to decipher the function of this glut of sRNAs in the physiology of the spirochete and the pathogenesis of Lyme disease. Therefore, we set a course of experimental approaches, including new methodologies to identify potential targets, to reveal the role of sRNAs in gene regulation.
Extensive Uncharacterized sRNAs in B. burgdorferi
Recently, several strand-specific transcriptome-wide RNA-seq studies have reported large numbers of unannotated transcripts in B. burgdorferi [23-25]. Specifically, Lybecker and colleagues identified over 1000 sRNAs with sizes ranging from 50 to 450 nucleotides [25]. These sRNAs were classified based on their genomic location and included sRNAs encoded between (intergenic sRNAs), within (intragenic sRNAs), and opposite (antisense sRNAs) annotated open reading frames (Figure 1). In addition, some sRNAs overlapped the 5′ end of an annotated ORF and were classified as 5′ UTR-associated sRNAs [25]. Small regulatory RNAs influence gene regulation via base-pairing with target mRNAs, affecting their stability, translation, transcription, or processing [26-29]. Trans-encoded sRNAs imperfectly base-pair with target mRNAs, encoded in a different genomic location, while antisense RNAs base-pair with complete complementarity to their cognate mRNA. In addition, some sRNAs regulate cellular processes via binding and sequestering target proteins. While high-throughput RNA-sequencing techniques have identified sRNAs in many different bacteria, determining sRNA function has proven far more challenging. Global transcriptional profiling of sRNA mutant strains has been used to map sRNA regulons, but due to functional redundancy, deleting a single sRNA often does not disturb the transcriptome; furthermore, transcriptional changes may not be a direct target of the sRNA. Therefore, many researchers have focused on identifying the direct protein and RNA targets of sRNAs (the sRNA-targetome). Ascertaining the regulons and functions of the newly identified sRNAs in B. burgdorferi will reveal the mechanisms of post-transcriptional gene regulation as well as the role of sRNAs in bacterial physiology and virulence.
Figure 1.

The genomic locations of different classes of sRNAs. sRNAs were classified based on their relation to annotated open reading frames (ORFs); 5′ UTR, antisense (as), intergenic (inter) and intragenic (intra). ORFs are represented as black arrows and sRNAs are represented by black wavy arrows.
Identifying sRNA-targetomes and -interactomes
Several algorithms predict potential RNA targets of trans-encoded sRNA, although the output can include a plethora of false positives [30]. Therefore, high-throughput techniques are necessary to identify direct sRNA-targets (sRNA-targetome). An in vivo MS2 affinity purification coupled with RNA sequencing (MAPS) technology was developed to globally identify RNA-binding partners of sRNAs [31-33]. The technique is based on the strong interaction between the coat protein of bacteriophage MS2 and the MS2 operator RNA hairpin, an aptamer that can be fused to any sRNA of interest. The sRNA and its binding partners can be co-purified via the MS2 protein and identified by RNA-seq (Figure 2). MAPS has been applied to various classes of RNAs to successfully identify the targetomes of specific RNAs. Recently, two novel RNA-seq-based methods, RIL-seq (RNA interaction by ligation and sequencing) and CLASH (crosslinking, ligation, and sequencing of hybrids), were developed that globally map sRNA-target interactions (sRNA-interactome) [34,35]. However, both methods rely on the interaction of sRNAs with a known RNA-binding protein (RBP). The RNA-binding proteins (Hfq and RNase E) were epitope-tagged and bound RNA was co-immunoprecipitated (co-IP) after in vivo crosslinking. The co-IP RNAs were digested and ligated together to fuse the sRNA and target RNA, generating chimeras, which were subjected to RNA-seq (Figure 3).
Figure 2.

Schematic illustrating affinity purification of MS2 aptamer-tagged sRNAs to identify novel RBPs and RNA targets. UV light crosslinks in vivo-expressed MS2 aptamer-tagged sRNAs to target RNAs and RBPs. Affinity purification is performed using the fusion protein maltose binding protein-MS2 (MBP-MS2) immobilized on amylose resin. Bound RNAs are identified by RNA-seq and RBPs by LC-MS/MS.
Figure 3.
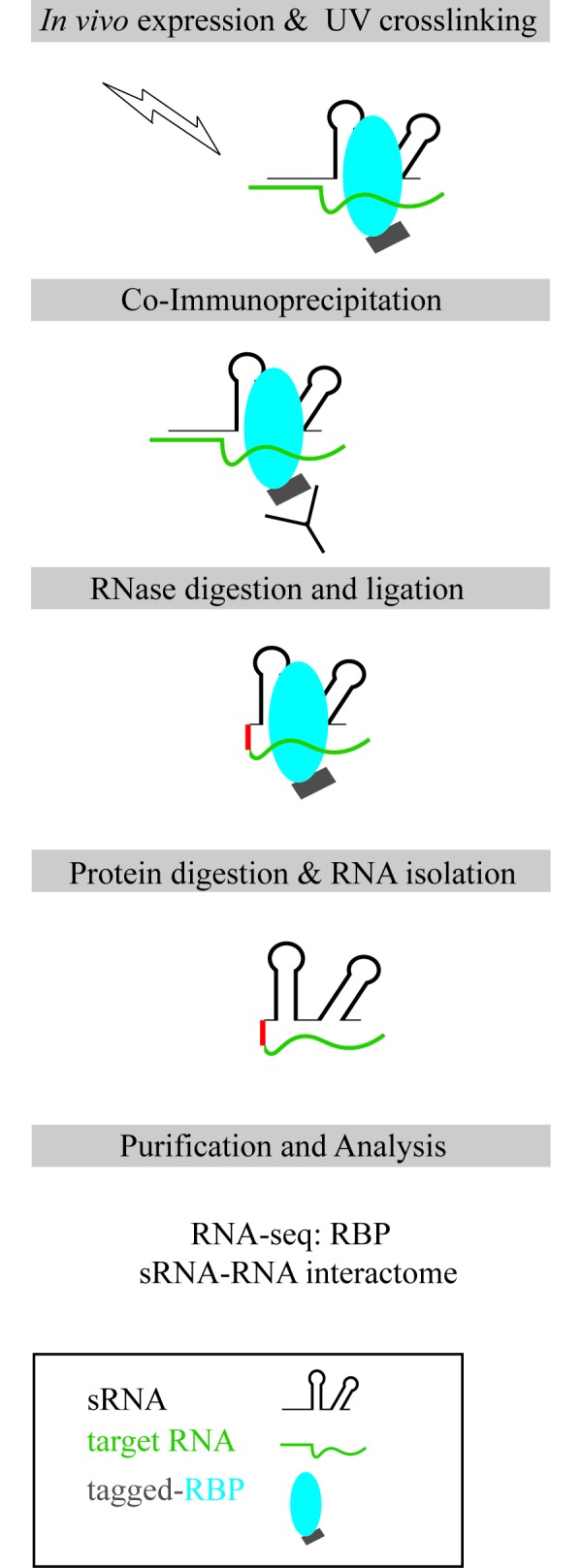
Schematic for global identification of RBP-dependent sRNA-interactomes. UV light crosslinks in vivo-expressed epitope-tagged RBPs with bound RNAs. The RBP-RNA complexes are co-immunoprecipitated and RBP-bound RNAs are partially digested and ligated together. The RBP is digested and the hybrid RNAs are deep-sequenced.
Identifying sRNA-binding Proteins
Many trans-encoded sRNAs require the RNA chaperone Hfq for stability and binding to target RNAs [36], while another class of regulatory RNAs bind and sequester proteins, affecting their availability in the cell. Identifying sRNA-target proteins provides a chart to uncover their regulatory mechanisms and cellular functions. The MS2 tag was also utilized to discern protein-binding partners of specific sRNAs by coupling the sRNA-MS2 affinity purification with liquid chromatography tandem mass spectrometry (LC-MS/MS) (Figure 2) [37,38]. More recently, gradient profiling by sequencing (Grad-seq) was developed as a high-throughput method that globally classifies RNAs by biochemical similarities and RBP interactions across a sedimentation gradient [39]. RNAs and their cognate RBPs are separated by density using a glycerol gradient. Following sedimentation, RNA is extracted from each fraction and subjected to RNA-seq, and proteins are identified via LC-MS/MS. Principle component analyses of the RNA-seq data map biochemically similar collectives of RNAs. The technique also identifies major RNA-binding proteins, including a novel RNA chaperone [39].
Identifying Functional Antisense sRNAs
Antisense sRNAs (asRNAs) are transcribed opposite to protein-coding strands and have complete complementarity to the corresponding sense RNA. Although asRNAs are ubiquitously found in all organisms, their functions remain mostly elusive [40-42]. The majority of functionally characterized asRNAs are encoded on plasmids and opposite phage, transposon and toxin genes. Antisense transcripts regulate gene expression via base-pairing with their cognate sense RNA and can affect transcription, transcript stability, and translation. asRNAs were initially considered to be transcriptional noise. However, several high-throughput approaches have demonstrated a function for many asRNAs throughout the bacterial world. Sense/antisense RNA base-pairing leads to the formation of double-stranded RNA (dsRNA), which is the substrate for endoribonuclease III (RNase III). Lybecker et al. identified the dsRNA transcriptome of Escherichia coli by immunoprecipitation with a dsRNA-specific antibody from both wild-type and RNase III-deficient cells followed by RNA-seq [43]. In addition, substantial dsRNA formation was found in Staphylococcus aureus: genome-wide RNase III-dependent short RNA fragments were observed in regions of overlapping sense/antisense transcription [44]. Furthermore, Lioliou et al. co-immunoprecipitated RNAs with a catalytically inactive RNase III revealing numerous asRNAs associated with RNase III [45]. Taken together, the research indicates that asRNAs regulate gene expression via a dsRNA intermediate. Recent evidence suggests that asRNA may also act in trans to regulate target RNAs encoded from different genomic locations [35].
Elucidating the Function of Antisense sRNAs and Intragenic sRNAs
The reverse genetic approach is commonly wielded to dissect gene function in B. burgdorferi [11,46]; however, mutating asRNAs or intragenic sRNAs (intraRNAs), which are encoded opposite to or within an annotated ORF, respectively, is technically challenging: deleting the sRNA gene will have a severe polar effect on the overlapping ORF. Several groups have overexpressed asRNAs and intraRNAs to study their function in gene regulation. However, there are inauspicious caveats to this approach, particularly related to overproducing the sRNA, which can sequester crucial RNA-binding proteins, such as Hfq, resulting in pleiotropic dysregulation of gene expression. To generate asRNA-deficient strains, we are currently characterizing the asRNA promoters and then mutating a few nucleotides in the −35 and −10 regions to inhibit transcription of the asRNA while maintaining the coding region of the complementary sense RNA. IntraRNAs can be generated either by processing the full-length mRNA or from an internal promoter within the ORF. Adams et al. globally identified transcriptional start sites and processed ends of transcripts in B. burgdorferi [24]. IntraRNAs with internal promoters can be mutated via the same site-directed mutagenesis as the asRNA promoters, which will deplete the cell of only the intraRNA. However, a different approach is required to study intraRNAs that are generated by RNA processing of the mRNA. Chao et al. successfully uncoupled the function of a 3′ UTR intraRNA from its cognate gene by mutating the start codon, effectively blocking translation of the protein, but allowing for sRNA production [47]. Of crucial importance for employing a reverse genetic approach to elucidate the function of genes in B. burgdorferi microbiology and Lyme disease pathogenesis, sRNAs that are encoded within an ORF targeted for disruption must be considered in the mutagenesis strategy, as inadvertently deleting an sRNA may have profound pleiotropic consequences.
Role of sRNAs in Pathogenesis
Non-coding RNAs are key components in intricate networks regulating the development of bacterial disease [29,48-52]. Discovering the sRNAs that are important in the enzootic life cycle of B. burgdorferi and the pathogenesis of Lyme disease may reveal novel virulence factors and, potentially, new therapeutic targets. Several culture conditions have been used to simulate the environments of the tick vector and vertebrate host, as well as transmission and acquisition between the two; however, Iyer et al. clearly demonstrated that these conditions are poor mimics of the physiologically relevant milieu [53]. Therefore, a more direct approach to identifying sRNAs involved in virulence or required for the enzootic cycle would be to apply RNA-sequencing of B. burgdorferi in both the tick vector and the vertebrate host. Moreover, recent studies have used dual RNA-sequencing to simultaneously sequence host and pathogen transcriptomes in co-culture and from tissue [54-57]. However, there are technical limitations of applying these RNA-seq methods to the tick-mouse experimental model of B. burgdorferi infection, particularly due to the low numbers of spirochetes in the vertebrate.
The function of an sRNA in the enzootic cycle of B. burgdorferi, including persistence in the tick vector and transmission to the vertebrate host, can be assayed using sRNA mutant strains in the tick-mouse model. However, the deletion of a single sRNA in other pathogenic microorganisms often has a modest phenotype, likely because most sRNAs serve as fine-tuners of gene regulation [58]. In addition, mRNAs can be redundantly regulated by multiple sRNAs, so the mutation of a single sRNA may not have a drastic effect. Therefore, identifying and characterizing sRNAs and their interactomes prior to employing animal models is paramount toward understanding their roles in infection and disease pathogenesis.
Conclusion
B. burgdorferi, the etiologic agent of Lyme disease, encodes a vast sRNA transcriptome [25] that the spirochete almost certainly avails to regulate gene expression during the course of its life cycle as it encounters disparate environmental conditions and host-specific responses [1,8,9]. Adaptation to each phase of the enzootic cycle is associated with a sea change in the expressed regulon in response to environmental and metabolic cues; one signal transduction pathway features DsrABb, the first sRNA characterized in B. burgdorferi [14]. Invariably, the levels of gene expression must be optimized for persistence in the tick vector, transmission to a vertebrate host, establishment of infection in the vertebrate, and acquisition back to the tick, yet the repertoire of known regulatory factors in B. burgdorferi is rather small, consisting of a couple alternative sigma factors, a couple two-component systems, a couple purine second messengers, and a couple or so transcription factors [1,8,9,13]. We hypothesize that sRNAs are responsible for fine-tuning gene regulation throughout the enzootic cycle of the spirochete and we hope we have provided a beneficial guide to tease out the functional regulators from the sea of sRNAs.
Acknowledgments
We thank Dan Drecktrah and Kelley Henderson for thoughtful and critical reading of the manuscript.
Glossary
- sRNA
small RNA
- RNA-seq
RNA sequencing
- UTR
untranslated region
- RBP
RNA-binding protein
- asRNA
antisense RNA
- dsRNA
double-stranded RNA
- intraRNA
intragenic RNA
Author Contributions
M.C.L. and D.S.S. wrote the manuscript. Research in their laboratories on sRNAs of B. burgdorferi is supported by National Institutes of Health grants AI123672 (to M.C.L. with J.T. Skare and J. Hyde) and AI051486 (to D.S.S.).
References
- Radolf JD, Caimano MJ, Stevenson B. et al. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10:87–99. doi: 10.1038/nrmicro2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Strle F, Wormser GP. et al. Lyme borreliosis. Nat Rev Dis Primers. 2016;2:16090. doi: 10.1038/nrdp.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Grodzicki RL, Kornblatt AN. et al. The spirochetal etiology of Lyme disease. N Engl J Med. 1983;308:733–740. doi: 10.1056/NEJM198303313081301. [DOI] [PubMed] [Google Scholar]
- Burgdorfer W, Barbour AG, Hayes SF. et al. Lyme disease-a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- Benach JL, Bosler EM, Hanrahan JP. et al. Spirochetes isolated from the blood of two patients with Lyme disease. N Engl J Med. 1983;308:740–742. doi: 10.1056/NEJM198303313081302. [DOI] [PubMed] [Google Scholar]
- Lane RS, Piesman J, Burgdorfer W. Lyme borreliosis: relation of its causative agent to its vectors and hosts in North America and Europe. Annu Rev Entomol. 1991;36:587–609. doi: 10.1146/annurev.en.36.010191.003103. [DOI] [PubMed] [Google Scholar]
- Piesman J, Schwan T. Ecology of borreliae and their arthropod vectors. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk UK: Caister Academic Press; 2010. [Google Scholar]
- Samuels DS. Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol. 2011;65:479–499. doi: 10.1146/annurev.micro.112408.134040. [DOI] [PubMed] [Google Scholar]
- Caimano MJ, Drecktrah D, Kung F. et al. Interaction of the Lyme disease spirochete with its tick vector. Cell Microbiol. 2016;18:919–927. doi: 10.1111/cmi.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM. et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Brisson D, Drecktrah D, Eggers CH. et al. Genetics of Borrelia burgdorferi. Annu Rev Genet. 2012;46:515–536. doi: 10.1146/annurev-genet-011112-112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Miller KA, Bian J. et al. Spirochaete flagella hook proteins self-catalyse a lysinoalanine covalent crosslink for motility. Nat Microbiol. 2016;1:16134. doi: 10.1038/nmicrobiol.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS, Radolf JD. Who is the BosR around here anyway? Mol Microbiol. 2009;74:1295–1299. doi: 10.1111/j.1365-2958.2009.06971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker MC, Samuels DS. Temperature-induced regulation of RpoS by a small RNA in Borrelia burgdorferi. Mol Microbiol. 2007;64:1075–1089. doi: 10.1111/j.1365-2958.2007.05716.x. [DOI] [PubMed] [Google Scholar]
- Lybecker MC, Abel CA, Feig AL. et al. Identification and function of the RNA chaperone Hfq in the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2010;78:622–635. doi: 10.1111/j.1365-2958.2010.07374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan E, Esteve-Gassent MD, Maruskova M. et al. Overexpression of CsrA (BB0184) alters the morphology and antigen profiles of Borrelia burgdorferi. Infect Immun. 2009;77:5149–5162. doi: 10.1128/IAI.00673-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CW, Li C. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect Immun. 2011;79:1270–1279. doi: 10.1128/IAI.00871-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Zhou J, Norgard MV. CsrA (BB0184) is not involved in activation of the RpoN-RpoS regulatory pathway in Borrelia burgdorferi. Infect Immun. 2014;82:1511–1522. doi: 10.1128/IAI.01555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras BL, Chenail AM, Carroll DW. et al. Bpur, the Lyme disease spirochete's PUR domain protein: identification as a transcriptional modulator and characterization of nucleic acid interactions. J Biol Chem. 2013;288:26220–26234. doi: 10.1074/jbc.M113.491357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras BL, Jones GS, Verma A. et al. Posttranscriptional self-regulation by the Lyme disease bacterium's BpuR DNA/RNA-binding protein. J Bacteriol. 2013;195:4915–4923. doi: 10.1128/JB.00819-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves M. Novel ribosome biogenesis in the Lyme disease spirochete Borrelia burgdorferi. [Missoula, MT]: The University of Montana; 2013. [Dissertation] [Google Scholar]
- Archambault L, Borchert JS, Bergeron J. et al. Measurements of mRNA degradation in Borrelia burgdorferi. J Bacteriol. 2013;195:4879–4887. doi: 10.1128/JB.00659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WK, Savage CR, Brissette CA. et al. RNA-seq of Borrelia burgdorferi in multiple phases of growth reveals insights into the dynamics of gene expression, transcriptome architecture, and noncoding RNAs. PLoS One. 2016;11:e0164165. doi: 10.1371/journal.pone.0164165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PP, Flores Avile C, Popitsch N. et al. In vivo expression technology and 5′ end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Res. 2017;45:775–792. doi: 10.1093/nar/gkw1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popitsch N, Bilusic I, Rescheneder P. et al. Temperature-dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genomics. 2017;18:28. doi: 10.1186/s12864-016-3398-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel CL, Storz G. Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev. 2010;34:866–882. doi: 10.1111/j.1574-6976.2010.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldelari I, Chao Y, Romby P. et al. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med. 2013;3:a010298. doi: 10.1101/cshperspect.a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Ying X, Lu Q. et al. Predicting sRNAs and their targets in bacteria. Genomics Proteomics Bioinformatics. 2012;10:276–284. doi: 10.1016/j.gpb.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier MC, Lalaouna D, Massé E. A game of tag: MAPS catches up on RNA interactomes. RNA Biol. 2016;13:473–476. doi: 10.1080/15476286.2016.1156830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers G, Massé E. Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev. 2012;26:726–739. doi: 10.1101/gad.182493.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D, Prévost K, Eyraud A. et al. Identification of unknown RNA partners using MAPS. Methods. 2017;117:28–34. doi: 10.1016/j.ymeth.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Waters SA, McAteer SP, Kudla G. et al. Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. EMBO J. 2017;36:374–387. doi: 10.15252/embj.201694639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S, Peer A. Global mapping of small RNA-target interactions in bacteria. Mol Cell. 2016;63:884–897. doi: 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran CP, Rieder R, Podkaminski D. et al. Use of aptamer tagging to identify in vivo protein binding partners of small regulatory RNAs. Methods Mol Biol. 2012;905:177–200. doi: 10.1007/978-1-61779-949-5_11. [DOI] [PubMed] [Google Scholar]
- Said N, Rieder R, Hurwitz R. et al. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009;37:e133. doi: 10.1093/nar/gkp719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Förstner KU, Holmqvist E. et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci U S A. 2016;113:11591–11596. doi: 10.1073/pnas.1609981113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason MK, Storz G. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet. 2010;44:167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georg J, Hess WR. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker M, Bilusic I, Raghavan R. Pervasive transcription: detecting functional RNAs in bacteria. Transcription. 2014;5:e944039. doi: 10.4161/21541272.2014.944039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybecker M, Zimmermann B, Bilusic I. et al. The double-stranded transcriptome of Escherichia coli. Proc Natl Acad Sci U S A. 2014;111:3134–3139. doi: 10.1073/pnas.1315974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa I, Toledo-Arana A, Dobin A. et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A. 2011;108:20172–20177. doi: 10.1073/pnas.1113521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioliou E, Sharma CM, Caldelari I. et al. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012;8:e1002782. doi: 10.1371/journal.pgen.1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groshong AM, Blevins JS. Insights into the biology of Borrelia burgdorferi gained through the application of molecular genetics. Adv Appl Microbiol. 2014;86:41–143. doi: 10.1016/B978-0-12-800262-9.00002-0. [DOI] [PubMed] [Google Scholar]
- Chao Y, Papenfort K, Reinhardt R. et al. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 2012;31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Reytor D, Plaza N, Espejo RT. et al. Role of non-coding regulatory RNA in the virulence of human pathogenic vibrios. Front Microbiol. 2016;7:2160. doi: 10.3389/fmicb.2016.02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman S, Cho KH. The mechanisms of virulence regulation by small noncoding RNAs in low GC gram-positive pathogens. Int J Mol Sci. 2015;16:29797–29814. doi: 10.3390/ijms161226194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva G, Sahr T, Buchrieser C. Small RNAs, 5′ UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol Rev. 2015;39:331–349. doi: 10.1093/femsre/fuv022. [DOI] [PubMed] [Google Scholar]
- Papenfort K, Vogel J. Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol. 2014;4:91. doi: 10.3389/fcimb.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Repoila F, Cossart P. Small noncoding RNAs controlling pathogenesis. Curr Opin Microbiol. 2007;10:182–188. doi: 10.1016/j.mib.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Iyer R, Caimano MJ, Luthra A. et al. Stage-specific global alterations in the transcriptomes of Lyme disease spirochetes during tick feeding and following mammalian host adaptation. Mol Microbiol. 2015;95:509–538. doi: 10.1111/mmi.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss AM, Beckstette M, Pimenova M. et al. Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc Natl Acad Sci U S A. 2017;114:E791–E800. doi: 10.1073/pnas.1613405114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol. 2012;10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- Baddal B, Muzzi A, Censini S. et al. Dual RNA-seq of nontypeable Haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. MBio. 2015;6:e01765. doi: 10.1128/mBio.01765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienksma RA, Suarez-Diez M, Mollenkopf HJ. et al. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics. 2015;16:34. doi: 10.1186/s12864-014-1197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barquist L, Vogel J. Accelerating discovery and functional analysis of small RNAs with new technologies. Annu Rev Genet. 2015;49:367–394. doi: 10.1146/annurev-genet-112414-054804. [DOI] [PubMed] [Google Scholar]