

Abstract
The systemic and cerebral microcirculation contribute critically to regulation of local and global blood flow and perfusion pressure. Microvascular dysfunction, commonly seen in numerous cardiovascular pathologies, is associated with alterations in the oxidative environment including potentiated production of reactive oxygen species (ROS) and subsequent activation of redox signaling pathways. NADPH oxidases (Noxs) are a primary source of ROS in the vascular system and play a central role in cardiovascular health and disease. In this review, we focus on the roles of Noxs in ROS generation in resistance arterioles and capillaries, and summarize their contributions to microvascular physiology and pathophysiology in both systemic and cerebral microcirculation. In light of the accumulating evidence that Noxs are pivotal players in vascular dysfunction of resistance arterioles, selectively targeting Nox isozymes could emerge as a novel and effective therapeutic strategy for preventing and treating microvascular diseases.
Keywords: Microcirculation, NADPH oxidase, Reactive oxygen species, Endothelial dysfunction, Arterioles, Remodeling, Myogenic tone, Aging
1. Introduction
1.1. Microcirculation
The microcirculation, comprised of the most distal segments of the vascular system, proceeds from an active network of arterioles to capillaries and venules in the periphery. Together, these vascular beds function to regulate blood flow and perfusion pressure. The microcirculation is responsible for optimizing the supply of nutrients and oxygen to various organs, while simultaneously removing metabolic waste products. Consequently, the rate of blood flow through these beds is tightly controlled by the metabolic demands of the tissue they pervade and is finely tuned by the more proximal small arteries and resistance arterioles. It is widely acknowledged that the small arteries and arterioles account for the majority (80%) of vascular resistance to blood flow [1–3]. They also serve to protect downstream capillaries from the potential damaging effects of high perfusion pressure and, in turn, prevent end-organ damage [4]. Arteriolar lumen size can vary dramatically depending on various species, distinct vascular beds and intrinsic contractile status. However, the common feature shared by arterioles across all vascular beds and species is the fact that they only possess one or two layers of smooth muscle cells in their arterial wall, which play a remarkably effective and fundamental role in regulating vascular contractility and local perfusion. Regulation of arteriolar diameter and blood flow in the microcirculation is a dynamic and well-controlled process that integrates signals from mechanical, chemical, hormonal and neuronal stimuli. This is also achieved by an ensemble of responses elicited by endothelial cells and smooth muscle cells [1,2]. Impaired arteriolar function, as observed in various cardiovascular pathologies such as hypertension, diabetes, stroke and aging, leads to vascular remodeling, tissue ischemia and organ damage [3,5–8]. Emerging evidence has shown that changes in the oxidative environment are often accompanied by the aforementioned vascular dysfunction and cardiovascular diseases in the microcirculation [6,9–12]. As a primary source for oxidative stress and reactive oxygen species (ROS) in vascular cells, the contribution of NADPH oxidases (Noxs) to the cardiovascular system in health and disease has been investigated in a plethora of studies [9–11,13–15]. In the microcirculation, Noxs are implicated in a variety of vascular pathologies including arteriolar remodeling [16,17] and endothelial dysfunction [18,19] in systemic microvessels, as well as impaired neurovascular coupling [20] and disrupted blood-brain barrier (BBB) integrity [21] in the cerebral circulation. The current review aims not to be an exhaustive review on the roles of all sources of oxidases in the microcirculation. Rather, the goal here is to survey what is known of the roles of Noxs in ROS generation in resistance arterioles and to summarize their contributions to microvascular physiology and pathophysiology in both cerebral and systemic microcirculation.
1.2. NADPH oxidases (Noxs) in the microvasculature
The nicotinamide-adenine-dinucleotide phosphate (NADPH) oxidases (Noxs) are an increasingly complex family of proteins whose core components are membrane bound. All Noxs transfer electrons from NADPH to molecular oxygen, thus generating superoxide anion and attendant other downstream ROS with NADP+ as a byproduct [22]. There are seven members of the Nox family characteristically distinguished and referred to by their catalytic core component, namely prototypic Nox2 (previously gp91phox), Nox1, Nox3, Nox4, Nox5, Duox1 and Duox2. These isozymes are heterogeneously distributed across tissue. Their structure and function are complex and have been extensively reviewed elsewhere [9,10,12–14,23]. The focus here will be on their profound implications in vascular function and disease in the microcirculatory system.
Of the seven members of the Nox family, Nox1, Nox2, Nox4 and Nox5 are known to be expressed in human vascular tissue. They are considered the “professional” enzymatic source of ROS in vascular cells, and are found to exert varied and often distinct physiological functions depending on the cell, tissue and/or species (Table 1). For instance, Noxs identified by their core catalytic subunits, i.e. Nox2 and Nox4, are involved in proliferation of endothelial cells [24], whereas Nox1 facilitates smooth muscle cell mitogenesis and proliferation and thus likely aids in cell replenishment under normal conditions [25]. Smooth muscle Nox4 has been implicated widely in the maintenance of a differentiated phenotype [26,27]. Further, Noxs are also involved in regulation of vascular contractility [28–30]. On the other hand, Nox isozymes are also known to be major players in mediating vascular dysfunction and disease [9,11–13,23,31–33].
Table 1.
Nox family of proteins in the microcirculation.
| Nox isozyme* | Canonical subunits/regulators | ROS Generation | Tissue expression in the microvasculaturea |
|---|---|---|---|
| Nox1 | p22phox, NOXO1, NOXA1, Rac1/2 | Superoxide anion. |
Mouse: mesenteric arteries [58], cerebral microvessels [59] Rat: mesenteric arteries [17,60], basilar arterial endothelium [61]; Human: coronary arteriolar endothelium [62], cerebral arteriolar smooth muscle [63] |
| Nox2 | p22phox, p47phox, p67phox, p40phox, Rac1/2 | Inducible; Superoxide anion. |
Mouse: mesenteric arteries [19,58], coronary arteries [64], afferent arterioles [65]; cerebral microvessels [59] Rat: mesenteric arteries [66,67], basilar arterial endothelium [61]; Human: coronary arteriolar endothelium [62], renal arterioles [68], subcutaneous arteriolar smooth muscle [43], cerebral arteriolar smooth muscle [63]. |
| Nox4 | p22phox, Poldip2 [49] | Constitutively active; Capacity for primary production of H2O2 [69]. |
Mouse: mesenteric arteries [19,58], cerebral microvessels [59]; Rat: mesenteric arteries [60], basilar arterial endothelium [61]; Human: renal arterioles [68], coronary arteriolar endothelium [62], subcutaneous arteriolar smooth muscle [43], cerebral arteriolar smooth muscle [63] |
| Nox5 | No subunits identified to date | Superoxide anion; Independent of p22phox; regulated by Ca2+. | Human: coronary arteriolar endothelium and smooth muscle [57,70] |
As primarily identified by its core catalytic subunit comprising cytochrome b558.
Membrane-integrated Nox subunit p22phox is required by Nox1, Nox2 and Nox4 but not the Nox5 isoform for membrane stabilization and activity [23]. The prototypical phagocytic NADPH oxidase is composed of five subunits: p47phox (organizer), p67phox (activator), p40phox, p22phox and catalytic subunit Nox2 (previously gp91phox) [34–37]. p47phox is phosphorylated upon stimulation, and hence the cytosolic units form a complex that includes the small Rho-family GTP-binding protein Rac1/2 [38]. This complex is then well characterized as being translocated to the plasma membrane where it interacts with the heterodimeric flavoprotein (cytochrome b558 complex) formed by p22phox and Nox2. Once assembled, the enzyme is rendered active and generates superoxide anion from molecular oxygen [23]. The Nox2 oxidase complex is not only widely distributed in phagocytes but is ubiquitous in lung, heart and vasculature [23]. In the vasculature, it was first discovered and characterized in fibroblasts [28,36,39,40], vascular smooth muscle cells [41–43], and endothelial cells [44,45]. These findings sparked exploration and discovery of Nox in virtually all cell types and led to the knowledge that similar orthologues are highly conserved in nature dating to some of their earliest evolutionary predecessors in fungi; and resembling the subunit-modulated Noxs that are highly regulated in mammals [46]. Being the first to be discovered, Nox2 remains to be the most extensively studied member of the Nox family in numerous physiological and pathological processes. In the microvasculature, Nox2 appears to be mostly present in the endothelium and adventitia [20,24], responsible for endothelial dysfunction under disease conditions [19,47] but has been shown by Touyz et al. to also be functionally active in human resistance smooth muscle cells [43]. Similar to Nox2, the canonical Nox1 requires binding of p22phox, p47phox homologue NoxO1, p67phox homologue NoxA1 and Rac1/2 for activation [48]. Nox1 oxidase complex is most abundant in colon, prostate, uterus and vascular cells [23]. In contrast to Nox1 and Nox2 complexes, activity of Nox4 was shown to be modulated by the enzyme polymerase delta-interacting protein or Poldip2 [49]. Also distinguishing itself from other Nox complexes, Nox4 appears to be constitutively active and to most favorably produce hydrogen peroxide over superoxide anion [23]. Interestingly, Nox4 is substantially higher in the cerebral circulation and purportedly generates hydrogen peroxide that modulates arteriolar activity as a potent vasodilator under physiological conditions [50,51]. However, the findings in that study using a selective Nox2 inhibitor along with apocynin might challenge the notion of a singular role for Nox4 and perhaps suggest a cross-talk between Nox2 and Nox4 in this vasodilator effect [52]. Nox5 activity does not depend on p22phox, nor cytosolic organizer or activator. Rather, it is regulated by calcium binding through EF-hand motifs [53]. As the most recently discovered Nox member, the expression and functional significance of Nox5 are poorly appreciated since Nox5 is absent in mouse and rat genomes [54]. In the microcirculation, Nox5 isozyme is found in porcine and human coronary resistance arterioles [55–57]. Additionally, its expression is significantly enhanced in these microvessels under disease conditions such as atherosclerosis and myocardial infarction, suggesting that it may be an important mediator of the pathological degeneration of human resistance arteries [56,57].
In this review, we discuss the involvement of Noxs in two distinct microvascular systems: systemic microcirculation and cerebral microcirculation. In each section, we will briefly summarize the expression of Noxs in various vascular beds and cell types. Further, implication of Noxs in both physiological processes such as myogenic tone regulation and vasodilator mechanisms, and cardiovascular pathologies including endothelial dysfunction and vascular remodeling will be discussed in detail. In light of the burgeoning evidence that Noxs are central players in vascular dysfunction of resistance arterioles, targeting Nox could emerge as a novel and effective therapeutic strategy for preventing and treating microvascular diseases.
2. Systemic microcirculation
2.1. Nox expression
In systemic microvessels, expression of Nox isozymes has been described in a number of resistance vascular beds, participating in physiological and pathological processes. For example, mRNA transcript and protein of Nox1, Nox2, Nox4, p22phox, p47phox, p67phox and Rac1/2 subunits were detected in mouse and rat mesenteric resistance arteries [19,58,60,66,67,71], where Noxs are implicated in structural and functional alterations in response to distinct stimuli [17,72,73]. In mouse coronary arteries, protein expression of Nox2 and its regulatory subunits p67phox, p47phox and p22phox were reported [64]. Moreover, immunohistochemical assays illustrate that Nox1, Nox2, Nox4, p22phox, p47phox and p67phox are expressed in human coronary arteriolar endothelium [62]. In addition, Nox5, both at the level of mRNA transcript and protein, was detected in human coronary arterioles. Immunofluorescence staining of Nox5 corroborated its localization in both endothelium and smooth muscle with low expression levels in healthy subjects and high levels in patients with coronary artery disease and myocardial infarction [56,57]. Recently, Nox5 mRNA was also detected in porcine coronary artery smooth muscle cells, mediating growth factor-induced expression and activation of intermediate conductance Ca2+-activated K+ channels (KCNN4) and subsequent smooth muscle cell migration [55]. Noxs are also involved in the microvascular function in the kidney, where Nox2 was found in mouse afferent arteriole, contributing to angiotensin II (AngII)-induced arteriolar contraction [65]. Furthermore, Nox2 and Nox4 transcripts are found in human renal arterioles, and they modulate endothelium-dependent dilation in elderly patients [68]. Other resistance arteries that express functional Nox isozymes or its regulatory subunits include human subcutaneous arterioles from gluteal and abdominal regions [43,74]. Although the molecular evidence for Noxs in cremaster arterioles is lacking, they appear to be present and functionally active in this vascular bed since ROS scavenger Tempol and Nox inhibitor apocynin inhibited ROS production and downstream inward remodeling of rat cremaster arterioles [75]. Overall, accumulating evidence suggest that Nox activity is crucial for modulations in vascular structure and function in the systemic microcirculation.
2.2. Systemic microvascular Nox in health
The myogenic response is one of the key means by which resistance arteries autoregulate. This crucial hallmark characteristic of arterioles protects downstream capillaries from potential detrimental effects of high intraluminal pressure, thus preventing end-organ damage due to pressure and perfusion fluctuations. Autoregulation is defined as a process by which resistance arteries constrict and reduce their diameter in response to increased intravascular pressure [76]. Mechanisms of the development and maintenance of pressure-induced vasoconstriction have been rigorously studied for decades. Elevation in intraluminal pressure leads to stimulation of depolarizing influence such as transient receptor potential (TRP) channel opening [77,78], and simultaneous inhibition of hyperpolarizing factors including different types of potassium channels in the smooth muscle cells [79,80]. This results in smooth muscle cell membrane depolarization, Ca2+ influx and smooth muscle contraction [76]. Though several aspects of the mechanism underlying pressure-induced vasoconstriction remain to be elucidated, emerging evidence has pointed to the possible contribution of Nox and ROS in the microvasculature under both physiological and pathological conditions. For example, a study showed that elevated transmural pressure initiated a rapid and transient ROS production in isolated resistance arteries from hamster gracilis muscle [29]. Further, pressure-elicited ROS, enhanced Ca2+ sensitivity and vascular contraction were all attenuated in the presence of diphenylene iodonium (DPI) or Nox2-selective peptide inhibitor Nox2ds-tat (aka gp91ds-tat) demonstrating that Nox2-derived ROS is physiologically critical for enhancing Ca2+ sensitivity and smooth muscle contractility in response to increased intraluminal pressure [29]. Using renal afferent arterioles, another study demonstrated that pathologically enhanced myogenic vasoconstriction is attributed to a 4-fold increase in ROS production in vessels from the spontaneous hypertensive rats compared to control animals [81]. The source of ROS is believed to be Nox2 isozyme, as the superoxide scavenger Tempol and the Nox2 inhibitor gp91ds-tat attenuated pressure-induced vasoconstriction in resistance arterioles [81]. Further, the mechanosensitivity of Nox is strongly suggested as several groups reported that superoxide production is largely stimulated by mechanical stretch in cultured vascular smooth muscle cells [82–84]. Importantly, Nox appears to be the predominant source of ROS in these settings, since Nox inhibitors rather than xanthine oxidase or cyclooxygenase inhibitors effectively attenuated stretch-induced ROS generation [29,82,84]. In agreement with these findings, deleting p47phox as well as reducing Nox1 levels abrogated the response in vascular smooth muscle cells, indicative of a clear role of Nox in stretch-mediated ROS production as well as downstream signaling events [83].
Mechanistically, mechano-activation of Nox is mediated by protein kinase C (PKC), which phosphorylates p47phox and promotes its translocation and interaction with Nox2 [85]. In addition, ROS is found to activate downstream RhoA/Rho kinase signaling in vascular smooth muscle [86,87]. Interestingly, both PKC and Rho kinase pathways are well-established cellular mechanisms that mediate myogenic tone development both at the level of promoting smooth muscle depolarization/Ca2+ entry and enhancing Ca2+ sensitivity [88–91], suggesting possible crosstalk involving Nox2 and the protein kinases to facilitate myogenic responses in vascular smooth muscle cells. Moreover, emerging evidence has pointed to the mechano-sensitivity of Gq protein-coupled receptors such as Ang II receptors and P2Y purinergic receptors in resistance arteries [92,93]. It was suggested that osmotically induced cell membrane stretch initiates a conformational modification in Ang II receptors where transmembrane segment 7 enters the ligand-binding pocket to cause receptor activation [94]. Mechano-activation of Ang II receptors as well as other Gq protein-coupled receptors further stimulate downstream PKC or Rho kinase pathways [91–93,95,96]. Because AngII receptor activation is also strongly associated with Nox activation and ROS production in smooth muscle cells, it is conceivable that complex cellular interactions among AngII receptors, Nox and protein kinases may be essential for developing and maintaining microvascular myogenic tone physiologically, and adversely regulating the contractile responsiveness under disease conditions. Furthermore, since Nox5 possesses EF-hands and is exquisitely sensitive to rises in Ca2+ [53], PKC and Rho kinase-mediated increase in Ca2+ influx and Ca2+ sensitivity may be particularly relevant. In fact, it was demonstrated that PKC phosphorylates Nox5 at key residues, and facilitates enzyme activation as well as ROS production by increasing its sensitivity to Ca2+ [97,98]. However, the physiological significance of this pathway, particularly its participation in vascular function requires further investigation.
2.3. Systemic microvascular Nox in disease
2.3.1. Microvascular remodeling
In addition to rapid contractile and dilatory responses to mechanical or chemical stimuli, resistance vessels also display adaptive structural modifications that alter their diameter and responses to vasoactive molecules [99]. Vascular remodeling is controlled by a number of processes including cytoskeletal remodeling, cell growth, cell proliferation, cell migration, apoptosis, and extracellular matrix modification, all of which are influenced by redox state [2]. The involvement of ROS in different mechanisms of the vascular remodeling process is more thoroughly discussed elsewhere [2]. This section of the review will focus on Nox-mediated inward and outward remodeling at the level of microcirculation. Vascular remodeling can be induced by various factors, such as mechanical and hemodynamic forces, and neurohumoral or paracrine agents. Inward hypertrophic remodeling is believed to commonly occur in resistance arteries that undergo prolonged contraction as reported in cardiovascular diseases such as hypertension and diabetes [100,101]. It involves cell proliferation and alterations in the amount and composition of extracellular matrix components which eventually reduce lumen diameter and increase vascular wall thickness, media/lumen ratio and cross-sectional area [100]. In contrast to inward remodeling that is caused by prolonged contraction as a primary stimulus, outward remodeling happens when vessels are subjected to chronically increased blood flow or shear stress, and they adaptively outward remodel to meet the need for higher metabolic demand [102]. These arterioles exhibit increased wall thickness and vascular diameter as well as an increased luminal size. Both inward and outward hypertrophy are important compensatory mechanisms as they occur when vascular myogenic reactivity is not sufficient to normalize circumferential stress [2]. Also similar to myogenic autoregulation, inward hypertrophy serves to protect downstream capillaries from perfusion pressure fluctuations and prevent end-organ damage, whereas outward remodeling functions to meet higher metabolic demand that is more commonly seen during pregnancy and exercise training [103,104]. Multiple studies have implicated Nox in both types of remodeling in the microvasculature. For example, it was shown that inward hypertrophy of cremaster and mesenteric arterioles induced by prolonged exposure to vasoactive compounds, primarily AngII, is accompanied by increased superoxide anion production [16,17,72,75,105]. Both ROS generation and vascular remodeling could be prevented by the non-specific Nox inhibitor apocynin, suggestive of an important role of Nox in the microvascular remodeling mechanisms [16,17]. In accordance with this notion, Briones et al. inferred that treatment with atorvastatin attenuates AngII-elicited mesenteric resistance artery hypertrophy partially by inhibiting AngII-induced increase in Nox1 expression [17]. Additionally, involvement of Nox in high flow-induced outward remodeling has been evaluated. That is, high flow-mediated ROS production and diameter enlargement of mesenteric resistance arteries were inhibited by Tempol and apocynin [106]. Potentiated p67phox and Nox2 expression levels in these arterioles further suggested that Nox-derived ROS are responsible for flow-initiated outward remodeling in the microcirculation [106]. Similarly, Castier et al. found that prolonged stimulation by high blood flow facilitated artery enlargement and concomitant shear stress reduction from the initial surge [107]. Surprisingly, deleting Nox2 had little effect to reverse these alterations, whereas p47phox deletion conferred significant improvement in artery caliber and shear stress, and elicited pronounced decreases in oxidative stress. Those findings suggested that outward remodeling is mediated by a p47phox-dependent signaling pathway which is independent of Nox2 activation [107]. This could suggest that the hybrid Nox1 system that utilizes p47phox is operant here. In both the inward and outward remodeling processes, it is generally accepted that ROS participates in part via activating matrix metalloproteinases (MMPs), including MMP2 and MMP9 [75,107,108]. Castier et al. proposed that increased superoxide anion and NO and, in turn, peroxynitrite subsequently activate these MMPs and cause vascular remodeling in the microvasculature [107]. However, the specific Nox isozymes or the downstream signaling cascades that facilitate these types of hypertrophy have not yet been identified.
2.3.2. Endothelial dysfunction
Endothelial dysfunction is characterized by a shift in vascular function towards reduced vasodilation, a pro-inflammatory state, and pro-thrombotic properties. This shift is implicated in many types of cardiovascular diseases, such as hypertension, coronary artery disease, chronic heart failure, peripheral vascular disease, diabetes and chronic kidney failure [109]. At the level of microcirculation, endothelial dysfunction is believed to play a key role in the development of atherosclerosis, abnormal angiogenesis, vascular leakage and stroke, all of which are associated with enhanced oxidative stress [110]. Noxs, a primary cause of oxidative stress in the vasculature, have major implications for endothelial dysfunction in the microcirculation by way of producing ROS and subsequently disrupting NO signaling. The diffusion-limited reaction between superoxide anion and NO reduces NO bioavailability and diminishes its anti-proliferative, anti-coagulant, anti-inflammatory and vasodilatory properties. The product of this reaction, peroxynitrite, is in and of itself a powerful oxidant and can exacerbate vascular dysfunction by causing further damage to lipids, proteins and DNA, uncoupling endothelial nitric oxide synthase (eNOS) and diminishing smooth muscle responses to NO [111,112]. In the systemic microvasculature, impaired endothelium-dependent vasodilation and concomitant enhanced Nox-mediated oxidative stress have been observed in microvascular coronary arterioles and mesenteric resistance arteries in a number of cardiovascular diseases, such as obesity [113,114], diabetes [18,19,47,115,116], hypertension [117–119] and ischemia-reperfusion [73]. In these studies, elevated expression of Nox subunits is a common characteristic across different cardiovascular pathologies. For example, ischemia-reperfusion injury induced upregulation of Nox1 and p47phox mRNA transcript levels, which purportedly accounts for superoxide production and potentiation of endothelin 1-induced vasoconstriction [73]. In a mouse model of type 1 diabetes, isolated mesenteric resistance arteries showed a 4-fold increase in Nox activity, and a 4–6 fold enhancement of Nox2 and Nox4 expression compared to vessels from control mice. Moreover, those findings are consistent with Nox being functionally involved in a largely attenuated endothelium-dependent vasodilation [120]. Similarly, mesenteric arterioles obtained from young type 2 diabetic rats showed higher ROS and greater protein expression of Nox2 isozyme compared to young lean rats. ROS production is even more pronounced in aged diabetic rats, in which elevation in p67phox subunit expression was also noted [47]. In other type 2 diabetic mouse models, upregulation of other Nox2 subunits, namely p22phox, p40phox and p47phox, is observed in mesenteric and coronary resistance arteries [18,19,115]. Further corroborating a role of Nox2 in endothelial dysfunction, gp91ds-tat (aka Nox2ds-tat)-treated arterioles exhibited improved vasodilation to acetylcholine compared to untreated vessels. Moreover, evidence that knocking out p47phox prevented disruption of endothelium-dependent vasodilation is consistent with the essential contribution of p47phox to microvascular dysfunction [19]. Several mechanisms underpinning Nox activation and subsequent endothelial dysfunction have been proposed. In diabetic and hypertensive disease models and obese patient samples, TNFα and endothelin-1 were upregulated [114,115,121,122], leading to amplified Nox-mediated oxidative stress and microvascular dysfunction. In a type 2 diabetic mouse model, superoxide anion levels are augmented by the interaction between advanced glycation end products (AGE) and receptor for AGE (RAGE), both of which are heavily involved in pathophysiology of hyperglycemia and diabetes. AGE-RAGE binding further potentiated expression of TNFα, oxidative stress and endothelial dysfunction in coronary microvessels [115]. In another type 2 diabetes study, augmented epidermal growth factor receptor (EGFR) tyrosine kinase was shown to enhance Nox expression and activity, which facilitated an increased vasoconstrictor response and attenuated acetylcholine-induced dilatation [19]. Although the mechanistic implications of ROS signaling pathway in microvascular endothelial dysfunction is not fully understood, it is clear that Nox, particularly Nox2, plays a central role in this pathological process. Therefore, selectively targeting Nox or its stabilizing membrane counterpart p22phox can be a useful strategy in terms of restoring the balance between pro- and anti-oxidants and improving vascular functionality. In fact, one study showed that p22phox small interfering RNA (siRNA) significantly improved endothelium-dependent vasodilator responses in microvessels isolated from diabetic mice [18]. This appears to be mediated by reversal of the augmented phosphorylation of downstream mitogen-activated protein kinases (MAPKs) p38 and extracellular signal-receptor kinase (ERK) following p22phox knockdown [18]. Since it is well established that activation of MAPKs is a fundamental mechanism responsible for cardiovascular dysfunction in diabetes, p22phox siRNA-mediated decrease in phosphorylation of these kinases is expected to effectively ameliorate vascular abnormalities [123,124]. Taken together, the findings support the notion that Nox is a central player and a potential drug target for treating microvascular diseases.
2.3.3. Microvascular angiogenesis
Angiogenesis is a natural process in the microcirculatory system. It is induced by ischemia and is characterized by the sprouting of new blood vessels from existing vessels, as well as the proliferation and migration of microvascular endothelial cells [125]. Several studies have reported that Noxs participate importantly in the regulation of angiogenesis of the microvascular endothelial cells. In particular, Nox4 was found to be a key mediator of pro-angiogenic signaling through distinct cellular pathways [126–128]. For example, blood flow recovery after hind limb ischemia was significantly enhanced in mice overexpressing human Nox4 and was impaired in mice overexpressing a dominant negative form in the endothelium, suggesting that Nox4 is involved in angiogenic response in vivo [129]. In isolated cardiac microvascular endothelial cells, Nox4 expression and activity were substantially increased after cells were subjected to hypoxia/reoxygenation [127]. Transcriptionally downregulating Nox4 exacerbated hypoxia/reoxygenation-induced cell apoptosis and inhibited the expression of hypoxia-inducible factor (HIF-1α) and vascular endothelial growth factor (VEGF). This suggests that Nox4 promotes microvascular endothelial cell survival, migration and angiogenesis after ischemia/reperfusion injury by activating the HIF-1α/VEGF signaling [127]. Other mechanisms underlying Nox4-promoted angiogenic response include activation of receptor tyrosine kinases and the ERK pathway [128], and stimulation of transforming growth factor β1 (TGFβ1) to initiate activation of VEGF receptor 2, p38 MAPK and adenosine monophosphate-activated protein kinase alpha (AMPKα) in microvascular endothelial cells [129]. In addition, there is evidence that Nox2 is an important regulator of angiogenesis in vivo [130]. In particular, Nox2 knockout mice exhibited significantly impaired neovascularization in the ischemic hind limb compared to wild type mice. New blood vessel formation in response to ischemia is an important adaptive response for preserving tissue integrity [131]. Therefore, both Nox4 and Nox2 appear to be protective by orchestrating complex angiogenic signaling pathways in endothelial cells of the microcirculation. However, an exaggerated angiogenic response can be pathological, and be associated with tumor growth and metastasis. A large body of evidence appears to suggest that Nox-derived ROS may function as signaling molecules that mediate growth-related responses and facilitate tumor angiogenesis [132] although much more work appears necessary to test this directly.
2.3.4. Aging
The cumulative process of diverse deleterious changes in the cells and tissues with advancing age progressively impairs function and eventually causes death [133]. Aging is a major risk factor for cardiovascular diseases [133]. Among multiple other instigators of the aging process, ROS and oxidative stress are often described as a common denominator in aging and age-related cardiovascular diseases [9,11,134–136]. As the “professional” enzymatic source for ROS generation in vascular cells, Nox isozymes appear to play a crucial role in age-associated cardiovascular dysfunction including coronary artery disease, atherosclerosis, hypertension and diabetes via activation of redox signaling pathways [70,137–143]. Multiple studies have identified age-mediated alterations in the activity and/or expression levels of Nox subunits and the pivotal contribution of Noxs to impairment of endothelium-dependent vasodilation, i.e. endothelial dysfunction. For example, aortas from aged hypertensive rats exhibited increased ROS formation compared to age-matched control rats, which was sensitive to broad spectrum Nox inhibitors VAS2870, apocynin and DPI. This was accompanied by enhanced protein levels of Nox1 and Nox2, but not Nox4, in aged aortas. Further, acetylcholine-induced vasodilation was largely attenuated in these aortas compared to control, which was improved by VAS2870 and apocynin [144]. Another study comparing aortas from young and middle aged spontaneously hypertensive rats (SHR) and control Wistar rats showed significantly higher mRNA expression of p22phox and Nox activity in the aortas from older SHR rats, concomitant with lower acetylcholine dilatory responses [145]. Those findings seem to suggest a link between enhanced expression and activity of Noxs and conduit artery endothelial dysfunction associated with aging and hypertension. In the microcirculation, attenuated effects from endothelium-dependent dilatory stimuli were also reported. In aged coronary arterioles, flow and acetylcholine-initiated dilation were significantly diminished [146]. Even though endothelial dysfunction paralleled higher Nox-derived superoxide anion generation in aged coronary microvessels, it appears to be independent of a change in expression of Nox subunits such p47phox, p67phox, Nox1 and p22phox [146]; statistical power could have been an issue for an apparent but nonsignificant rise in p67phox. Nevertheless, the role of Nox-derived ROS in many other manifestations of microvascular dysfunction associated with the aging process is largely unknown. In fact, the microcirculation undergoes significant functional and anatomical changes with age. Apart from declines in endothelium-dependent vasodilation, for instance, age is also associated with Rho kinase signaling-mediated cutaneous vasoconstriction [147]. Since Rho kinase activation has been shown to be regulated by ROS in vascular smooth muscle cells [86], it seems reasonable to propose that age-induced microvascular constriction is also regulated by Nox-mediated ROS signaling. Furthermore, aging is associated with arteriolar stiffness, decreased vascular density and impaired microvascular organization [5]. Nevertheless, signaling mechanisms that underlie the co-existence of Nox-mediated oxidative stress, aging and microvascular pathologies warrant further investigation.
3. Cerebral microcirculation
3.1. Nox expression
In the cerebral vasculature, expression of NADPH oxidases (Nox1, Nox2 and Nox4) at the level of both mRNA and protein has been confirmed by various studies. Specifically, Nox1, Nox2 and Nox4 were detected in human brain vascular smooth muscle cells [63]. In basilar arterial endothelial cells, Nox1 and Nox4 mRNA are highly expressed, whereas Nox2 transcript is expressed to a far lesser extent [61]. The cytosolic components of Nox including p47phox, p67phox, NOXO1 and NOXA1 were also found in these endothelial cells [61]. Furthermore, mRNA of NADPH oxidase subunits Nox1, Nox2, Nox4, p22phox, p47phox and p67phox were also identified in the cerebral neocortical microvessels including cerebral arterioles and capillaries [59]. Using electron microscopy, Nox2 immunoreactivity was detected in the intima and adventitia of cerebral arterioles [20], which may reflect important involvement of ROS-mediated signaling in regulation of cerebral microcirculation.
3.2. Cerebral microvascular Nox in health
Interestingly, cerebral arteries isolated from various healthy animal models presented markedly higher NADPH oxidase activity and ROS generation compared to systemic vessels [50,148], which may be explained at least in part by elevated expression of Nox2 and Nox4 in the cerebral vasculature [50]. It is tempting to speculate that perhaps more significant roles of Nox and ROS are operant in modulating cerebrovascular function and local blood flow under physiological conditions. Indeed, compelling evidence exists for ROS, particularly hydrogen peroxide (H2O2), participating in control of arteriolar reactivity as a potent endogenous vasodilator in the cerebral circulation [50,51,149]. This is supported by the observation that NADPH-induced vasodilation can be abolished by the H2O2 scavenger catalase and Nox inhibitor apocynin, indicating that H2O2, generated secondarily from Nox2 and superoxide anion dismutation, is a potent vasorelaxant. As several endogenous vasoactive stimulants such as AngII and TNFα elicit increases in Nox activity, it was proposed that H2O2-mediated vasodilation in the cerebral microcirculation may play a protective role against endogenous vasoactive hormones, thereby preventing cerebral vasospasm and subsequent neuronal damage. In agreement with this postulate, Miller et al. showed that AngII initiated significantly smaller contractions in cerebral arteries than in systemic vessels, which were potentiated by both catalase and apocynin [50]. In addition, Nox2 was identified to be an important regulator of cerebral blood flow in vivo, as NADPH-induced cerebral vasodilation and increase in blood flow were significantly attenuated by Nox2 inhibitor gp91ds-tat as well as in gp91phox-null mice [150]. Another physiological contribution of Nox-derived ROS was reportedly flow-induced vasodilation in cerebral arteries [151,152]. It was reported in cultured primary cells [153], in isolated arteries [152] as well as in vivo [151] that Nox activity in the endothelium is enhanced by shear stress. Moreover, the evidence that flow-mediated ROS production and vasodilation could be inhibited by catalase and DPI points to the contribution of Nox-generated H2O2 to flow-dependent responses in cerebral circulation [151]. Further mechanistic studies of Nox and ROS-mediated cerebral vascular relaxation revealed mediators both in smooth muscle and endothelium. Sobey et al. reported that dilation of rat cerebral arterioles in response to H2O2 is mediated by activation of large conductance calcium-activated potassium (BKCa) channels in vascular smooth muscle cells. This was proved by selective blockers of these potassium channels (TEA and iberiotoxin) largely inhibiting a putative H2O2-induced vasorelaxation [51]. In agreement with this finding, Cheranov et al. utilized patch clamping to demonstrate that TNFα activates Nox in cerebral smooth muscle cells, leading to production of H2O2 activating BKCa channel currents and resulting in a reduction in global intracellular Ca2+ concentration and vasodilation [154]. In cerebral arterial endothelial cells, Nox-derived ROS were found to propagate the lipid peroxidation metabolite 4-HNE, which further initiated Ca2+ influx through TRPA1 channels, activation of intermediate-conductance Ca2+ channel sparklets and vasodilatation [155].
In addition to facilitating vasodilation under physiological conditions, Nox can mediate cerebral vasoconstriction in response to increases in intraluminal pressure, i.e. myogenic tone. Gebremedhin et al. made key discoveries that increased intraluminal pressure markedly promotes superoxide and H2O2 production in isolated cerebral arteries, which are inhibited by Nox2 inhibitor gp91ds-tat. Elevated ROS levels in smooth muscle cells further evoke redox-sensitive signaling mechanisms involving crosstalk between inactivation of the dual phosphatase PTEN and activation of the PI3K/Akt pathway. This, in turn, inhibits BKCa channels, leading to depolarization and pressure-induced vasoconstriction of cerebral arteries [156]. The seemingly paradoxical involvement of Nox in both dilation and constriction of cerebral arteries in fact appears to highlight the significance of Nox in the fine tuning of cerebral vascular tone and local blood flow. Further, this dichotomy might be attributed to different isoforms of Nox, Nox localization combined with the degree of ROS generated and distinct downstream signaling pathways. Elucidation of these mechanisms will likely require rigorous and directed application of (a) isoform-specific Nox inhibitors; and/or (b) ROS scavengers.
3.3. Cerebral microvascular Nox in disease
3.3.1. Endothelial dysfunction
It is well characterized that ROS impairs cerebral vascular function primarily via inhibition of the endothelium-dependent NO signaling pathway, resulting in dysregulated cerebral microcirculation [157,158]. For example, acetylcholine-induced CBF increase is attenuated by AngII treatment. This process is reversed by apocynin and Nox2 inhibitor, and absent in Nox2 deficient mice, all supportive of the key involvement of Nox2 in AngII-elicited inhibition of NO signaling of the cerebral microvasculature [59]. Indeed, the interaction between superoxide anion and NO not only reduces NO bioavailability, hence diminishing its vasodilatory capability, but also gives rise to peroxynitrite (ONOO−) that leads to damage of DNA, lipids and protein, and more profound vascular pathologies. Several groups have shown that peroxynitrite is implicated in cerebrovascular dysfunction, including impaired NO-dependent vasodilation [159] and augmented vasoconstrictor response during post-ischemic reperfusion [160]. Consistent with the effects on endothelial cells, high concentration of ROS in cerebral arterial smooth muscle also promotes cell contraction and vasoconstriction. Amberg et al. reported that exogenous ROS increases L-type calcium channel activity in isolated myocytes via activation of PKC [161]. As signaling pathways in endothelium and smooth muscle act in concert to facilitate Nox-mediated cerebral vascular dysfunction, Noxs can serve as important targets for therapeutic development against cerebral microvascular diseases.
3.3.2. BBB disruption
Several studies have shown that ROS and Noxs are involved in blood brain barrier (BBB) disruption from various causes. It was reported that Nox inhibition and genetic deletion of Nox2 attenuates stroke-induced BBB disruption and lesions in vivo as well as increases in permeability of brain capillary endothelial cells in vitro [162]. It was also found that apocynin pre-treatment prevents traumatic brain injury-induced ROS production and BBB disruption, thus decreasing neuronal death [21]. Amyloid β (Aβ), whose accumulation in brain capillaries is observed in 40% of the Alzheimer’s disease cases, was found to significantly increase ROS generation and capillary tight junction alterations, both reversed by administration of DPI and an ROS scavenger lipoic acid [163]. Furthermore, increase in BBB permeability in vitro, as determined by FITC-dextran leakage through a monolayer of cultured human brain microvascular endothelial cells (HBMECs), is observed following lipopolysaccharide (LPS) treatment [164]. Concomitantly, LPS potentiated p67phox and p47phox expression, elevated Nox activity and reduced tight junction protein expression. These adverse effects on BBB integrity were all attenuated by the Nox inhibitor apocynin, indicating that Nox, as the primary source of ROS activated by LPS, is a significant regulator of BBB function [164]. Moreover, ROS production and BBB damage were also reported in oxidized low-density lipoprotein-stimulated cerebral arteries [165] and in AngII-treated stroke-prone SHR rats [166]. In both models, Nox inhibition effectively diminished ROS generation and BBB disruption, demonstrating that Nox-dependent oxidative stress represents a common instigator for cerebral microcirculatory BBB dysfunction. Thus, targeting NADPH oxidases may be an effective therapeutic strategy for preventing BBB disruption or restoring its integrity. Although mechanisms underlying Nox-mediated BBB disruption have not been fully elucidated, implications of phosphatidyl inositol 3-kinase, c-Jun N-terminal kinase, Rho kinase in ROS-impaired permeability have been reported [162]. Interestingly, involvement of Rho kinase appears to be both upstream and downstream of Nox and ROS signaling in the cerebral microvasculature, as Rho kinase inhibitor not only prevents ROS-induced BBB damage, but also attenuates oxygen-glucose deprivation-related increases in Nox2 expression and ROS production, restores intercellular tight junctions and preserves BBB integrity [162,167].
3.3.3. Inward remodeling
Another key cerebral vascular abnormality resulting from oxidative stress is alteration in arteriolar structure. It is well acknowledged that hypertension is associated with inward eutrophic remodeling, which is characterized by decreased outer and lumenal diameter and increased media/lumen ratio [168]. This is often observed in resistance-size arteries in the brain [169] as well as peripheral vascular beds [170]. As blood flow is proportional to the fourth power of the vessel’s internal radius, slight changes in lumen diameter can profoundly affect tissue perfusion. Therefore, inward remodeling can exert profound effects on cerebral blood flow. Oxidative stress caused by hypertension is a key mediator of microvascular remodeling in various vascular beds such as mesenteric and subcutaneous arterioles [2]. However, ROS-initiated structural changes in the cerebral vasculature and its potential contribution to vascular function or cerebral blood flow are poorly understood. Chan et al. utilized Nox2 knockout mice to show that superoxide anion generated from the Nox2 isoform oxidase plays a key role in AngII-induced cerebral arteriolar inward remodeling [171]. Nevertheless, the intracellular mechanisms underlying this process require further scrutiny.
3.3.4. Neurovascular uncoupling
Cerebral parenchymal arterioles branch off from upstream arteries on the surface of the brain (pial arteries) and penetrate into the cerebral cortex, where they are encased by astrocytes and neurons. Increased neuronal activity signals through astrocytes, and subsequently leads to arteriolar dilation and an increase in local cerebral blood flow satisfying enhanced glucose and oxygen demand; a process termed neurovascular coupling or functional hyperemia [172]. Neurovascular coupling is disrupted in a variety of pathological conditions, including hypertension, Alzheimer’s disease and ischemic stroke [173], all of which are also associated with elevated levels of arteriolar ROS [20,174,175]. Compelling evidence has shown that Nox2-derived superoxide in the arterioles contributes heavily to impairment of functional hyperemia induced by AngII [20]. AngII evokes significant ROS production in cerebral microvessels and attenuates cerebral blood flow increase from whisker stimulation. These effects were largely inhibited by pegylated-superoxide dismutase (SOD), Tiron, apocynin, and were prevented in mice treated with Nox2 inhibitor gp91ds-tat or in Nox2 deficient mice [20,176]. Aging also represents an important factor for compromised neurovascular coupling, which is believed to cause further age-decline in cortical function and vascular cognitive impairment [177,178]. Further, Toth et al. showed that the cerebral cortex in aged mice had significantly higher mRNA expression of Nox1, Nox2 and Nox4 subunits compared to young mice, also associated with increased level of oxidative stress [179]. Moreover, a whisker stimulation-evoked CBF increase was remarkably lower in aged mice. Both of these effects were inhibited by apocynin, supporting the notion that Nox plays a central role in impairment of neurovascular function and dysregulation of cerebral microcirculation [179]. However, the signaling pathways controlling ROS-mediated damage in neurovascular coupling remain to be unveiled.
3.3.5. Aging
In fact, in addition to compromised functional hyperemia that is briefly described above, it is well documented that aging is associated with diverse cerebrovascular alterations that include reduction in resting CBF, disrupted microvascular integrity, atrophic structural changes and attenuated responses to endothelium-dependent vasodilators [158,180–182]. All these alterations increase the susceptibility of the cerebral vasculature to functional abnormalities, ischemic injury and vascular cognitive impairment [180]. Numerous studies have established a major contribution of Noxs and oxidative stress in age-related vascular dysfunction of the cerebral circulation. For example, aging is associated with increased ROS production in the cerebral cortex and cerebral arteries [158,183], where mRNA transcript levels of Nox subunits such as Nox2, p47phox, and p67phox are elevated in aged groups [158,184]. Further, mice lacking Nox2 do not exhibit cerebrovascular oxidative stress, and are protected from disruption of endothelium-dependent relaxation and impairment of functional hyperemia [158,183]. ROS scavenger Tempol, non-selective Nox inhibitor apocynin and selective Nox2 inhibitor gp91ds-tat all ameliorate aging-induced ROS production and improve vascular function in aged animals [158,183–185]. Recently, the correlation between aging and Alzheimer’s disease was rigorously interrogated, as they, together with hypertension, are purportedly major determinants of cognitive impairment. Aging serves as a powerful risk factor for the cognitive decline caused by hypertension and Alzheimer’s Disease. Therefore, it is not surprising that the contribution of hypertension, Alzheimer’s Disease and aging to cerebrovascular dysfunction and cognitive decline are intertwined and highly interactive. Burgeoning evidence reveals that elevated vascular oxidative stress from Nox appears to be a common thread associated with these conditions [186,187]. In support of this notion, Park et al. utilized aged Tg2576 mice, a mouse line that overexpresses a mutant form of amyloid precursor protein (APP), and is linked to the pathogenesis of Alzheimer’s disease and progressive cognitive deficits. They reported that aging and amyloid deposition, both alone and together, cause cerebrovascular dysfunction including attenuated functional hyperemia and reduced endothelium-dependent dilator responses. Deleting Nox2 not only rescued these abnormal vascular functions in full, but also diminished oxidative stress in aged Tg2576 mice. Similarly, application of Nox2 inhibitor gp91ds-tat largely improved neurovascular coupling and vasodilatory responses in these mice [174]. These reports suggest that Nox2 is a central player in cerebral microcirculatory dysfunction from age-related pathological conditions. In support of these findings, a recent study from Han et al. showed that both apocynin and Tempol reduced ROS levels and improved responses to vasoactive stimuli in aged Tg2576 mice [188]. They also ameliorated vascular smooth muscle-dependent hypercontractile responses observed in aged Tg2576 mice. The authors further noted that the anti-ROS therapy decreased cerebral amyloid angiopathy and micro-hemorrhage that commonly occur in Alzheimer’s Disease [188]. As all these studies point to the key contribution of the Nox, particularly Nox2, to aging-elicited cerebrovascular dysfunction, Nox2 inhibition holds great promise as a therapeutic strategy [52,189–191] for restoring vascular health in cerebral microcirculation, and hopefully delaying the onset of cognitive impairment.
4. Nox inhibitors as therapeutics for microvascular diseases
4.1. Peptidic inhibitors
4.1.1. Nox2ds-tat
Since its initial design and characterization [192], Nox2ds-tat (previously gp91ds-tat) has been one of the most widely used isoform-specific inhibitors of the Nox2 isozyme. It selectively blocks the interaction between Nox2 and p47phox by binding to the latter and preventing p47phox translocation to the plasma membrane. We have previously shown that Nox2ds-tat is a potent inhibitor of the canonical Nox2 oxidase with an IC50 of 0.74 μM, and a selective inhibitor with no effects on Nox1, Nox4 or xanthine oxidase activity [52]. Moreover, the evidence that Nox2ds-tat inhibits neither the canonical Nox1-NoxO1 binding nor the hybrid Nox1-p47phox interaction further demonstrates its specificity on the Nox2 isozyme [52]. Since its development, Nox2ds-tat has proven its value not only in delineating the involvement of Nox2-derived ROS in cardiovascular biology as well as in myriad other organ systems physiology, but also in attenuating Nox2/ROS-mediated cell and tissue damage in vitro and in vivo [193–206]. In resistance arteries from hamster gracilis muscle, the physiological contribution of Nox2 to elevated intraluminal pressure-induced ROS generation, Ca2+ sensitivity and myogenic vasoconstriction was revealed by application of Nox2ds-tat [29]. Moreover, Nox2 was identified as an important source of superoxide anion in the cerebral arteries, as Nox2ds-tat, but not control scrambled peptide, significantly attenuated superoxide anion production in response to increased intraluminal pressure [156]. In addition, Nox2ds-tat was shown to attenuate myogenic tone in afferent arterioles from spontaneous hypertensive rats, indicating that Nox2-derived ROS play a critical role in potentiating the myogenic response in hypertension [81]. Furthermore, since Nox2ds-tat inhibits bradykinin-induced dilation in coronary arteries as well as superoxide anion and H2O2 generation in coronary artery endothelial cells, Nox2 is proposed as an essential enzymatic source of H2O2 that mediates agonist-induced vasodilation in the microcirculation [62]. On the other hand, the therapeutic potential of Nox2ds-tat in the microcirculatory system has also been well described. For example, in a type2 diabetes model, Nox2ds-tat not only was able to attenuate EGF receptor activation-enhanced Nox activity in vascular cells, but also improved EGF-initiated endothelial dysfunction in mesenteric resistance arteries including the rescue of eNOS and Akt signaling [19]. Studies on the functional benefits of Nox2ds-tat are particularly revealing in the cerebral microcirculation. Several reports have shown that Nox2ds-tat mitigates the impairment of neurovascular coupling/functional hyperemia in mice evoked by various pathological stimuli [20,176,183]. These include cerebral flow changes in response to beta amyloid protein [176] and are consistent with the role of Nox2 in behavioral changes resulting from overexpression of beta amyloid [174]. Nevertheless, a commonly raised disadvantage of the inhibitor is its expected poor oral bioavailability. Testing of emergent technologies for better stabilization (i.e. peptide stapling [207,208] and nanoparticle formulations [209–218]) will likely be required to improve the druggability of Nox2ds-tat.
4.1.2. NoxA1ds
NoxA1ds was developed to mimic a putative “activation domain” of Nox1 activator subunit NoxA1 which is homologous to a reported p67phox “activation domain”, thus blocking the Nox1-NoxA1 binding and isozyme activation [219]. Previous work charactering NoxA1ds in our laboratory has corroborated the selectivity of this peptidic inhibitor, which potently inhibited Nox1-derived superoxide anion production in a reconstituted Nox1 cell-free system with an IC50 of 19 nM, and exhibited no inhibitory effects on Nox2-, Nox4-, Nox5-, or xanthine oxidase-derived ROS production [219]. NoxA1ds was shown to inhibit hypoxia-induced ROS generation and endothelial cell migration in human pulmonary arterial endothelial cells [219]. It also reduced uniaxial cyclic stretch-induced Nox1-dependent superoxide anion generation and synthetic phenotypic switch in rat aortic smooth muscle cells [84]. Although no evidence for beneficial effects of NoxA1ds in the microcirculation is yet available, administration of the inhibitor is expected to attenuate microvascular changes potentially mediated by Nox1, such as AngII-elicited vascular hypertrophy [17] and ischemia-reperfusion injury-induced potentiation of vasocontractile response in mesenteric resistance arteries [73].
4.2. Small molecule inhibitors
4.2.1. Diphenylene iodonium (DPI) and apocynin
Both compounds are traditionally and widely utilized as broad spectrum Nox inhibitors to investigate the roles of Noxs in various cardiovascular pathologies. Nevertheless, as both agents generate sizable off-target effects, studies using these inhibitors alone should be subjected to more scrutinized review. Specifically, apart from its inhibitory effects on Noxs, DPI is also demonstrated to be an irreversible and nonselective inhibitor of a wide array of flavin-dependent enzymes, such as nitric oxide synthase and xanthine oxidase [220]. Apocynin was initially believed to inhibit Noxs by specifically preventing p47phox translocation to the plasma membrane [220]. This has been called into question by a reported direct scavenging activity of apocynin of non-radical oxidant species such as HOCl and H2O2 [221,222] as well as its ability to suppress expression of various Nox catalytic subunits and/or inhibit Nox isozymes that do not require p47phox [222,223]. These nonspecific actions of DPI and apocynin largely diminish their value in dissecting the roles of Noxs in redox signaling pathways. Rather, one or more isoform selective inhibitors or siRNAs ought to be employed in addition to DPI and apocynin to clearly validate the participation of Nox. On a positive note, these broad spectrum inhibitors notwithstanding have exhibited protective effects against microvascular diseases. For example, apocynin and DPI suppressed ROS formation and restored endothelial function in aortas as well as cerebral arterioles from aged rats [144,158]. Moreover, apocynin conferred protection against neurovascular uncoupling arising with age [179]. It was also reported that traumatic brain injury-initiated ROS elevation, BBB disruption and neuronal death can all be prevented by apocynin [21]. However, whether these beneficial effects are entirely attributable to Nox blockade warrants further investigation.
4.2.2. VAS2870 and VAS3947
VAS2870 and VAS3947 are triazolopyrimidine derivatives. They have been characterized as promising Nox inhibitors based on the observation that they reduce Nox-derived ROS generation in several cell lines, including primary vascular endothelial and smooth muscle cells, with no antioxidant properties or inhibition of xanthine oxidase or eNOS activity [224–227]. VAS2870 is considered a pan-Nox inhibitor as it suppresses the activity of Nox1, Nox2, Nox4 and Nox5 [224,228]. Its preclinical effectiveness was evaluated by several groups who reported that VAS2870 improved microvascular function in a variety of vascular beds. For instance, experimental ischemia/reperfusion in middle cerebral arteries significantly upregulated Nox2 and Nox4, potentiated ROS production, and caused BBB leakage and brain edema in hyperglycemic rats, which were all markedly blunted by VAS2870 treatment [229]. Moreover, the expression of several key modulators in BBB integrity was decreased following ischemia/reperfusion, and was partially restored by VAS2870 [229]. VAS2870 was shown to reverse attenuated endothelium-dependent vasodilation of mesenteric arteries from type 2 diabetic mice and insulin-resistant rats, as well as arteries treated with amylin, a peptide strongly associated with insulin resistance [230,231]. However, off-target effects of VAS2870 on the cellular thiol redox status, i.e. thiol alkylation, were also reported [232]. In other words, when assessing the roles of Nox in physiological and pathological settings, findings obtained with VAS2870 should be interpreted with further caution. As with all inhibitors, the combination of various pharmacological as well as genetic tools will likely provide more definitive conclusions.
4.2.3. GKT13690 and GKT137831
Both compounds are pyrazolopyridine derivatives. They are Nox inhibitors effective for dual Nox1/Nox4, but not for Nox2, suppression [233]. Moreover, they do not show detectable inhibitory actions against other ROS-generating and redox-sensitive enzymes [233]. Due to high oral bioavailability, ease of synthesis and a reportedly good safety profile, GKT137831 has been proposed as a new therapy for diabetic kidney disease and has undergone phase 2 clinical trials [234]. In the microvasculature, GKT136901 was reported to ameliorate impaired vasodilatation of mesenteric arterioles from obese (db/db) mice displaying enhanced Nox1 expression [235], suggesting that this dual Nox1 and Nox4 inhibitor could be useful therapeutically for treating microvascular dysfunction in a range of cardiovascular diseases.
4.2.4. ML171
ML171 was the first small molecule reported to be an isoform-selective Nox1 inhibitor [236]. ML171 does exhibit inhibitory effects on Nox2, Nox3, Nox4 and xanthine oxidase but with an approximate 20-fold lower potency than that for Nox1. Incidentally, it exerts no apparent inhibitory effects on mitochondrial ROS generation [236]. The relative isoform selectivity renders this compound a useful tool (when a relatively low concentration of ML171 is employed) to elucidate the contribution of Nox1 to redox-signaling. For example, a recent study on the effects of chronic restraint stress on vascular function showed that ML171 (0.5 μM) reduced AngII-initiated vasoconstriction in rat carotid arteries in the presence and absence of endothelium, implying that Nox1-derived ROS positively influence AngII-mediated contraction in carotid arterial smooth muscle cells [237]. Another study comparing endothelial function of coronary arteries from male and female pigs reported that 100 μM ML171 enhanced bradykinin-induced endothelium-dependent relaxation in male rather than female porcine coronary arteries [238]. Nevertheless, it is not clear whether Nox1 alone played a role in suppressing vasodilation, since 100 μM of ML171 is high enough to abolish the activity of other Nox isozymes as well as xanthine oxidase [236].
4.2.5. Ebselen
Ebselen was initially characterized as a glutathione peroxidase mimetic [239] and a scavenger of peroxynitrite [240]. Later, inhibitory actions of ebselen congener JM-77b on Noxs were reported with relatively higher selectivity for Nox2 (IC50=0.4 μM) compared to Nox1 (IC50=6.3 μM), Nox4 (no detectable inhibition) and Nox5 (IC50=17 μM) [241]. Its ability to inhibit Nox2 notwithstanding, the antioxidant properties of ebselen, per se, were favorably utilized in different disease models to improve vascular reactivity. In a study on hypoxia/reoxygenation-initiated brain damage, ebselen ameliorated endothelial dysfunction in rat posterior cerebral arteries following hypoxia/reoxygenation insults [242]. Additionally, administering ebselen for 7 days significantly reduced vasospasm in right internal carotid arteries and middle cerebral arteries after subarachnoid hemorrhage in primates [243]. It was also reported to restore voltage-gated potassium (Kv) channel expression and activity in coronary arteriolar smooth muscle cells where channels were suppressed in diabetic rats. Consequently, ebselen potentiated Kv channel-dependent vasodilation in small coronary arteries [244]. However, whether or not any of these protective functions is associated with Nox inhibition is not entirely clear.
4.2.6. Tetrahydroquinolines\
In light of the profound implication of Nox2 in myriad macro- and microvascular pathologies, and perception of poor clinical applicability of peptidic Nox2ds-tat, development of small molecule Nox2-specific inhibitors has long been a quest for use as both an investigative tool and a therapeutic strategy. Using high throughput screening and rational design, our laboratory identified bridged tetrahydroquinolines as specific Nox2 inhibitors [191]. Among a group of structurally related molecules, compounds 11 g [(±)-(1S,4R,9S)-5-bromo-3,3-dimethyl-9-(2-methylallyl)−10-pentyl-1,2,3,4-tetrahydro-1,3-(epiminomehono) naphthalene] and 11 h [(±)-(1S,4R,9S)-5-bromo-3,3-dimethyl-9-(2-methyallyl)-10-(thiophen-2-ylmethyl)-1,2,3,4-tetrahydro-1,4-(epino-methano)naphthalene] displayed isoform-selective inhibition on Nox2 in intact COS-Nox2 cells with IC50 of 20 μM and 32 μM, respectively. Importantly, 11 g and 11 h had no inhibitory actions on ROS production by Nox1-, Nox4-, Nox5-expressing systems or xanthine oxidase, and showed no free radical scavenging activity [191]. Unpublished work from our group suggests that these inhibitors likely interfere with Nox2-p47phox binding via blockade of the p22phox-p47phox interface. Functionally, 11 g and 11 h significantly inhibit TNFα-induced inflammatory activation in aortic endothelial cells and endothelial dysfunction in mouse aorta (unpublished). Although they have not been evaluated on the microcirculation, these inhibitors hold great promise in improving Nox2-mediated microvascular complications.
Other Nox inhibitors include S17834 [245] and AEBSF [246], both of which decrease superoxide formation by Nox oxidases but also display multiple off-target effects [220]. Fulvene-5 [247] was also shown to reduce Nox2 and Nox4 activity, however, further interrogation on its specificity for Nox isozymes, mechanism of action, and effectiveness on the cardiovascular system is not reported. In summary, even though important strides in discovering Nox inhibitors have been made during the past decade, selective and clinically applicable Nox inhibitors are scarce and require further assessment of their mechanisms of action, pharmacokinetic properties and in vivo efficacy. Inhibitors specifically for Nox3, Nox4, Nox5, DUOX1/2 are not yet available, which may be a consequence of a poor understanding of interactions of this subset of Noxs with intracellular subunits. Indeed, further research developing more Nox modulators is warranted as they will not only advance the field of Nox research but also provide novel hit compounds that might be manipulated in clinical settings.
5. Conclusions and future perspectives
The dynamic network of the microcirculatory system is of vital importance to the modulation of blood flow and perfusion pressure locally and systemically. Impaired microvascular function often leads to vascular remodeling, tissue ischemia and organ damage. Structural and functional abnormalities of the microcirculation, as observed in various cardiovascular diseases, are strongly associated with an imbalance of the redox environment. As the likely major source of ROS in the vascular system, Noxs are important signaling molecules that participate physiologically in the regulation of microvascular tone. All the more crucial, excessive production of ROS (resulting from increased Nox expression or activity) plays a central role in triggering microvascular remodeling, endothelial dysfunction, increased BBB permeability and impaired neurovascular coupling in both the systemic and cerebral microcirculations. These manifestations are all hallmarks of cardiovascular disease including those evident in hypertension, diabetes, stroke and aging. The central involvement of Nox-derived ROS signaling in microvascular function and pathology is depicted in Fig. 1. Despite their importance, understanding of Nox and ROS as cellular signaling agents at the level of the microcirculation is far from sufficient. One important limiting factor lies in the paucity of a broad array of specific tools to delineate the individual roles of Nox isoforms. With the development of selective Nox1 and 2 inhibitors, the roles of the Nox1 and Nox2 isozymes are poised to be the best characterized in the vasculature in coming years. More work is needed, however, to develop novel, isoform-selective inhibitors (peptidic or small molecule) with optimal pharmacokinetic and pharmacodynamic profiles for each of the Nox isozymes. This is particularly critical for Nox5, both owing to its absence of expression in rats and mice, rendering it impossible to utilize conventional gene deletion models, and because Nox5 is abundantly expressed in human vascular cells and implicated in a growing number of human cardiovascular pathologies. Moreover, considerably more work is required to gain mechanistic insights into the regulation of Nox-mediated redox signaling pathways in the microcirculatory system. In particular, questions related to what specific Nox isoforms are involved, how signaling cascades are differentially regulated, how Noxs integrate the complex network of signaling and govern cellular and vascular function and whether these experimental findings can be translated into human diseases, require immediate attention. It is also noteworthy that Nox/ROS-implicated redox pathways remain far less interrogated in cerebral vs. other microvascular cells, which may be a consequence of limited tissue mass and low yield of signaling proteins. While most research focuses on the functional aspects of Noxs in arteriolar activity, more mechanistic studies of the Nox in cultured primary microvascular cells is critical. Finally, a better appreciation of the complexity of these signaling pathways will lead to the development of novel and effective therapeutic strategies aimed at preventing and treating microvascular pathologies in humans.
Fig. 1.
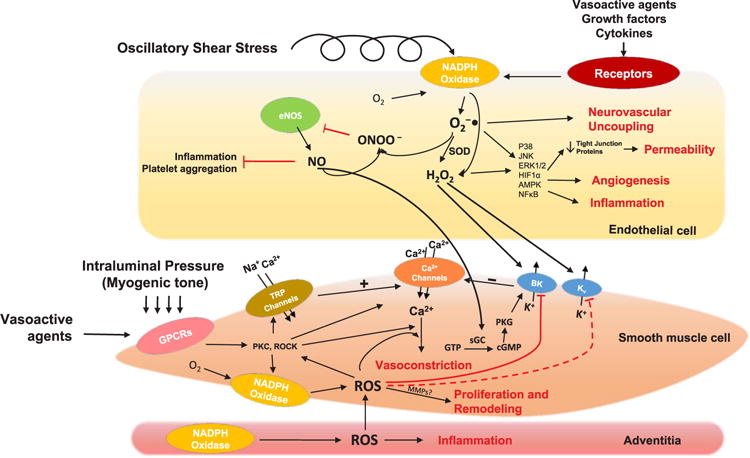
Major Nox-mediated redox-sensitive signaling pathways involved in microvascular physiology and pathophysiology. Schematic diagram illustrating known Nox and ROS-mediated signaling pathways in cerebral and systemic microvascular endothelium and smooth muscle. Arrows denote positive activation of downstream targets. Blunt-ended arrows indicate inhibitory effects on targets. Dashed arrows denote pathways that are active in other vascular systems but have not been reported in the microcirculation. In endothelial cells, Noxs can be activated by hormones, cytokines, growth factors and oscillatory shear stress [248,249], which stimulate ROS-dependent pathways that eventually lead to increased angiogenesis, inflammation, BBB permeability and impaired neurovascular coupling. Interaction between superoxide anion and NO yields peroxynitrite, which reduces NO bioavailability and causes endothelial dysfunction. In smooth muscle cells of cerebral and systemic arterioles, Noxs can be stimulated by mechanical stretch as well as vasoactive agents, e.g. AngII. Nox-derived ROS are centrally involved in inhibiting potassium channels, activating protein kinases and increasing Ca2+ sensitivity of the contractile apparatus. These effects are physiologically important for myogenic tone regulation, but may also lead to augmented vasoconstriction and diminished vasorelaxation. Microvascular remodeling is also dependent on Nox-mediated ROS production through putative stimulation of MMPs. Abbreviations: O2−•: superoxide anion; H2O2: hydrogen peroxide; SOD: superoxide dismutase; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; ONOO−: peroxynitrite; p38: p38 mitogen-activated protein kinases; JNK: c-Jun N-terminal kinases; ERK: extracellular signal-regulated kinases; HIF1α: hypoxia-inducible factor 1-alpha; AMPK: 5′AMP-activated protein kinases; NFκB: nuclear factor-κB; GPCRs: G protein-coupled receptors; TRP channels: transient receptor potential channels; PKC: protein kinase C; ROCK: rho-associated protein kinase; ROS: reactive oxygen species; BK: big-conductance Ca2+-activated K+ channels; Kv: voltage-gated K+ channels; PKG: cGMP-dependent protein kinase; GTP: guanosine triphosphate; cGMP: cyclic guanosine monophosphate; sGC: soluble guanylyl cyclase; MMP: matrix metalloproteinases.
Acknowledgments
This work was supported by National Institutes of Health grants R01-HL112914, R01HL079207 and P01-HL103455 and by the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania. The authors would like to thank Ms. Cynthia Hatfield and Dr. Eugenia Cifuentes-Pagano for their assistance with the editing of this manuscript.
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Martinez-Lemus LA. The dynamic structure of arterioles. Basic Clin Pharmacol Toxicol. 2012;110(1):5–11. doi: 10.1111/j.1742-7843.2011.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staiculescu MC, Foote C, Meininger GA, Martinez-Lemus LA. The role of reactive oxygen species in microvascular remodeling. Int J Mol Sci. 2014;15(12):23792–23835. doi: 10.3390/ijms151223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation. 2001;104(6):735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- 4.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66(1):8–17. doi: 10.1161/01.res.66.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Bentov I, Reed MJ. The effect of aging on the cutaneous microvasculature. Microvasc Res. 2015;100:25–31. doi: 10.1016/j.mvr.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Silva TM, Miller AA. Cerebral small vessel disease: targeting oxidative stress as a novel therapeutic strategy? Front Pharmacol. 2016;7 doi: 10.3389/fphar.2016.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrick DM, Sharpe GM, Sun H, Mayhan WG. Diabetes-induced cerebrovascular dysfunction: role of poly(ADP-ribose) polymerase. Microvasc Res. 2007;73(1):1–6. doi: 10.1016/j.mvr.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Didion SP, Lynch CM, Baumbach GL, Faraci FM. Impaired endothelium-dependent responses and enhanced influence of Rho-kinase in cerebral arterioles in type II diabetes. Stroke; J Cereb Circ. 2005;36(2):342–347. doi: 10.1161/01.STR.0000152952.42730.92. [DOI] [PubMed] [Google Scholar]
- 9.Csanyi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med. 2009;47(9):1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51(7):1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahoo S, Meijles DN, Pagano PJ. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin Sci. 2016;130(5):317–335. doi: 10.1042/CS20150087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 13.Frazziano G, Champion HC, Pagano PJ. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am J Physiol Heart Circ Physiol. 2012;302(11):H2166–H2177. doi: 10.1152/ajpheart.00780.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, Janssen B, Gorlach A, Schmidt HH. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med. 2012;90(12):1391–1406. doi: 10.1007/s00109-012-0963-3. [DOI] [PubMed] [Google Scholar]
- 15.McCarty MF. NADPH oxidase activity in cerebral arterioles is a key mediator of cerebral small vessel disease-implications for prevention. Healthcare. 2015;3(2):233–251. doi: 10.3390/healthcare3020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22(3):535–542. doi: 10.1097/00004872-200403000-00016. [DOI] [PubMed] [Google Scholar]
- 17.Briones AM, Rodríguez-Criado N, Hernanz R, García-Redondo AB, Rodrigues-Díez RR, Alonso MJ, Egido J, Ruiz-Ortega M, Salaices M. Atorvastatin prevents angiotensin II–induced vascular remodeling and oxidative stress. Hypertension. 2009;54(1):142–149. doi: 10.1161/HYPERTENSIONAHA.109.133710. [DOI] [PubMed] [Google Scholar]
- 18.Kassan M, Choi S-K, Galán M, Lee Y-H, Trebak M, Matrougui K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 MAP kinase-dependent mechanisms in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2014;306(7):H972–H980. doi: 10.1152/ajpheart.00872.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassan M, Ait-Aissa K, Ali M, Trebak M, Matrougui K. Augmented EGF receptor tyrosine kinase activity impairs vascular function by NADPH oxidase-dependent mechanism in type 2 diabetic mouse. BBA - Mol Cell Res. 2015;1853(10):2404–2410. doi: 10.1016/j.bbamcr.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95(10):1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 21.Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK, Suh SW. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012;1481:49–58. doi: 10.1016/j.brainres.2012.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283(25):16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 24.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, Gorlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006;8(9–10):1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 25.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401(6748):79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 26.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27(1):42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deliri H, McNamara CA. Nox 4 regulation of vascular smooth muscle cell differentiation marker gene expression. Arterioscler Thromb Vasc Biol. 2007;27(1):12–14. doi: 10.1161/01.ATV.0000254154.43871.50. [DOI] [PubMed] [Google Scholar]
- 28.Di Wang H, Hope S, Du Y, Quinn MT, Cayatte A, Pagano PJ, Cohen RA. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension. 1999;33(5):1225–1232. doi: 10.1161/01.hyp.33.5.1225. [DOI] [PubMed] [Google Scholar]
- 29.Keller M, Lidington D, Vogel L, Peter BF, Sohn HY, Pagano PJ, Pitson S, Spiegel S, Pohl U, Bolz SS. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J: Off Publ Fed Am Soc Exp Biol. 2006;20(6):702. doi: 10.1096/fj.05-4075fje. [DOI] [PubMed] [Google Scholar]
- 30.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb, Vasc Biol. 2011;31(6):1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 31.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86(5):494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 32.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86(9):E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 33.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44(3):248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 34.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins, J. Leukoc Biol. 1996;60(6):677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 35.Zhan S, Vazquez N, Zhan S, Wientjes FB, Budarf ML, Schrock E, Ried T, Green ED, Chanock SJ. Genomic structure, chromosomal localization, start of transcription, and tissue expression of the human p40-phox, a new component of the nicotinamide adenine dinucleotide phosphate-oxidase complex. Blood. 1996;88(7):2714–2721. [PubMed] [Google Scholar]
- 36.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci USA. 1997;94(26):14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Babior BM. NADPH oxidase: an update. Blood. 1999;93(5):1464–1476. [PubMed] [Google Scholar]
- 38.Miyano K, Sumimoto H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie. 2007;89(9):1133–1144. doi: 10.1016/j.biochi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Meier B, Jesaitis AJ, Emmendorffer A, Roesler J, Quinn MT. The cytochrome b-558 molecules involved in the fibroblast and polymorphonuclear leucocyte superoxide-generating NADPH oxidase systems are structurally and genetically distinct. Biochem J. 1993;289(Pt 2):481–486. doi: 10.1042/bj2890481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA. An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol. 1995;268(6 Pt 2):H2274–H2280. doi: 10.1152/ajpheart.1995.268.6.H2274. [DOI] [PubMed] [Google Scholar]
- 41.Mohazzab KM, Wolin MS. Sites of superoxide anion production detected by lucigenin in calf pulmonary artery smooth muscle. Am J Physiol. 1994;267(6 Pt 1):L815–L822. doi: 10.1152/ajplung.1994.267.6.L815. [DOI] [PubMed] [Google Scholar]
- 42.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 43.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res: J Am Heart Assoc. 2002;90(11):1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 44.Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol. 1994;266(6 Pt 2):H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- 45.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271(4 Pt 2):H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 46.Kawahara T, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol. 2007;7:109. doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vessières E, Guihot A-L, Toutain B, Maquigneau M, Fassot C, Loufrani L, Henrion D. COX-2-derived prostanoids and oxidative stress additionally reduce endothelium-mediated relaxation in old type 2 diabetic rats. PLoS One. 2013;8(7):e68217. doi: 10.1371/journal.pone.0068217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal. 2009;11(6):1279–1287. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 49.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105(3):249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller AA, Drummond GR, Schmidt HH, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97(10):1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 51.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke; J Cereb Circ. 1997;28(11):2290–2294. doi: 10.1161/01.str.28.11.2290. discussion 2295. [DOI] [PubMed] [Google Scholar]
- 52.Csanyi G, Cifuentes-Pagano E, Ghouleh I Al, Ranayhossaini DJ, Egana L, Lopes LR, Jackson HM, Kelley EE, Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51(6):1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) J Biol Chem. 2004;279(18):18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 54.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11(10):2443–2452. doi: 10.1089/ars.2009.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gole HK, Tharp DL, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channels (KCNN4) in porcine coronary smooth muscle requires NADPH oxidase 5 (NOX5) PLoS One. 2014;9(8):e105337. doi: 10.1371/journal.pone.0105337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guzik B, Guzik TJ, Chen W, Gongora MC, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52(22):1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hahn NE, Meischl C, Kawahara T, Musters RJ, Verhoef VM, van der Velden J, Vonk AB, Paulus WJ, van Rossum AC, Niessen HW, Krijnen PA. NOX5 expression is increased in intramyocardial blood vessels and cardiomyocytes after acute myocardial infarction in humans. Am J Pathol. 2012;180(6):2222–2229. doi: 10.1016/j.ajpath.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 58.Bruder-Nascimento T, Callera GE, Montezano AC, He Y, Antunes TT, Nguyen Dinh Cat A, Tostes RC, Touyz RM. Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: role of Rac1/2-sensitive Nox-dependent pathways. Clin Sci. 2015;128(7):411–423. doi: 10.1042/CS20140456. [DOI] [PubMed] [Google Scholar]
- 59.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26(4):826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 60.Androwiki AC, Camargo Lde L, Sartoretto S, Couto GK, Ribeiro IM, Verissimo-Filho S, Rossoni LV, Lopes LR. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front Chem. 2015;3:24. doi: 10.3389/fchem.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, Ibayashi S, Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke; J Cereb Circ. 2005;36(5):1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 62.Larsen BT, Bubolz AH, Mendoza SA, Pritchard JKA, Gutterman DD. Bradykinin-induced dilation of human coronary arterioles requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol. 2009;29(5):739. doi: 10.1161/ATVBAHA.108.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coucha M, Abdelsaid M, Li W, Johnson MH, Orfi L, El-Remessy AB, Fagan SC, Ergul A. Nox4 contributes to the hypoxia-mediated regulation of actin cytoskeleton in cerebrovascular smooth muscle. Life Sci. 2016;163:46–54. doi: 10.1016/j.lfs.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park Y, Yang J, Zhang H, Chen X, Zhang C. Effect of PAR2 in regulating TNF-α and NAD(P)H oxidase in coronary arterioles in type 2 diabetic mice. Basic Res Cardiol. 2011;106(1):111–123. doi: 10.1007/s00395-010-0129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlström M, Lai EY, Ma Z, Patzak A, Brown RD, Persson AEG. Role of NOX2 in the regulation of afferent arteriole responsiveness. Am J Physiol - Regul Integr Comp Physiol. 2009;296(1):R72–R79. doi: 10.1152/ajpregu.90718.2008. [DOI] [PubMed] [Google Scholar]
- 66.Franco Mdo C, Akamine EH, Marco GS Di, Casarini DE, Fortes ZB, Tostes RC, Carvalho MH, Nigro D. NADPH oxidase and enhanced superoxide generation in intrauterine undernourished rats: involvement of the renin-angiotensin system. Cardiovasc Res. 2003;59(3):767–775. doi: 10.1016/s0008-6363(03)00461-9. [DOI] [PubMed] [Google Scholar]
- 67.Tarhouni K, Freidja ML, Guihot AL, Vessieres E, Grimaud L, Toutain B, Lenfant F, Arnal JF, Loufrani L, Henrion D. Role of estrogens and age in flow-mediated outward remodeling of rat mesenteric resistance arteries. Am J Physiol Heart Circ Physiol. 2014;307(4):H504–H514. doi: 10.1152/ajpheart.00986.2013. [DOI] [PubMed] [Google Scholar]
- 68.Schlüter T, Zimmermann U, Protzel C, Miehe B, Klebingat K-J, Rettig R, Grisk O. Intrarenal artery superoxide is mainly NADPH oxidase-derived and modulates endothelium-dependent dilation in elderly patients. Cardiovasc Res. 2010;85(4):814–824. doi: 10.1093/cvr/cvp346. [DOI] [PubMed] [Google Scholar]
- 69.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286(15):13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52(22):1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeLano FA, Parks DA, Ruedi JM, Babior BM, Schmid-Schonbein GW. Microvascular display of xanthine oxidase and NADPH oxidase in the spontaneously hypertensive rat. Microcirculation. 2006;13(7):551–566. doi: 10.1080/10739680600885152. [DOI] [PubMed] [Google Scholar]
- 72.Amiri F, Virdis A, Neves MF, Iglarz M, Seidah NG, Touyz RM, Reudelhuber TL, Schiffrin EL. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation. 2004;110(15):2233–2240. doi: 10.1161/01.CIR.0000144462.08345.B9. [DOI] [PubMed] [Google Scholar]
- 73.Martinez-Revelles S, Caracuel L, Marquez-Martin A, Dantas A, Oliver E, D’Ocon P, Vila E. Increased endothelin-1 vasoconstriction in mesenteric resistance arteries after superior mesenteric ischaemia-reperfusion. Br J Pharmacol. 2012;165(4):937–950. doi: 10.1111/j.1476-5381.2011.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Virdis A, Duranti E, Colucci R, Ippolito C, Tirotta E, Lorenzini G, Bernardini N, Blandizzi C, Taddei S. Ghrelin restores nitric oxide availability in resistance circulation of essential hypertensive patients: role of NAD(P)H oxidase. Eur Heart J. 2015;36(43):3023–3030. doi: 10.1093/eurheartj/ehv365. [DOI] [PubMed] [Google Scholar]
- 75.Martinez-Lemus LA, Zhao G, Galiñanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol - Heart Circ Physiol. 2011;300(6):2005–2015. doi: 10.1152/ajpheart.01066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508(Pt 1):199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Baylie RL, Tavares MJ, Brayden JE. TRPM4 channels couple purinergic receptor mechanoactivation and myogenic tone development in cerebral parenchymal arterioles. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2014;34(10):1706–1714. doi: 10.1038/jcbfm.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95(9):922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 79.Amberg GC, Santana LF. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291(2):C348–C356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- 80.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256(5056):532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 81.Ren Y, D’Ambrosio MA, Liu R, Pagano PJ, Garvin JL, Carretero OA. Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;298(6):H1769–H1775. doi: 10.1152/ajpheart.00537.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hishikawa K, Oemar BS, Yang Z, Lüscher TF. Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ Res. 1997;81(5):797–803. doi: 10.1161/01.res.81.5.797. [DOI] [PubMed] [Google Scholar]
- 83.Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res. 2003;92(11):1–7. doi: 10.1161/01.RES.0000077044.60138.7C. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez AI, Csanyi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, Vorp DA, Pagano PJ. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2015;35(2):430–438. doi: 10.1161/ATVBAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108(10):1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 86.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol - Lung Cell Mol Physiol. 2008;295(3):515–529. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4(11):e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Osol G, Laher I, Cipolla M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ Res. 1991;68(2):359–367. doi: 10.1161/01.res.68.2.359. [DOI] [PubMed] [Google Scholar]
- 89.Jarajapu YP, Knot HJ. Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am J Physiol Heart Circ Physiol. 2005;289(5):H1917–H1922. doi: 10.1152/ajpheart.01012.2004. [DOI] [PubMed] [Google Scholar]
- 90.Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol. 2007;292(6):H2613–H2622. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- 91.Li Y, Brayden JE. Rho kinase activity governs arteriolar myogenic depolarization. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2015 doi: 10.1177/0271678X15621069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schleifenbaum J, Kassmann M, Szijártó IA, Hercule HC, Tano J-Y, Weinert S, Heidenreich M, Pathan AR, Anistan Y-M, Alenina N, Rusch NJ, Bader M, Jentsch TJ, Gollasch M. Stretch–activation of angiotensin II Type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res. 2014;115(2):263–272. doi: 10.1161/CIRCRESAHA.115.302882. [DOI] [PubMed] [Google Scholar]
- 93.Brayden JE, Li Y, Tavares MJ. Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2013;33(2):293–299. doi: 10.1038/jcbfm.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S, Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Fukuzaki K, Sato T, Ge J, Mochizuki N, Nakaya H, Saku K, Komuro I. Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep. 2008;9(2):179–186. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008;27(23):3092–3103. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, Davis MJ, Meininger GA, Hill MA. Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. J Physiol. 2016 doi: 10.1113/JP272834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem. 2007;282(9):6494–6507. doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 98.Chen F, Yu Y, Haigh S, Johnson J, Lucas R, Stepp DW, Fulton DJ. Regulation of NADPH oxidase 5 by protein kinase C isoforms. PLoS One. 2014;9(2):e88405. doi: 10.1371/journal.pone.0088405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology. 2009;24(1):45. doi: 10.1152/physiol.00029.2008. [DOI] [PubMed] [Google Scholar]
- 100.Rizzoni D, Porteri E, Guefi D, Piccoli A, Castellano M, Pasini G, Muiesan ML, Mulvany MJ, Rosei EA. Cellular hypertrophy in subcutaneous small arteries of patients with renovascular hypertension. Hypertens: J Am Heart Assoc. 2000;35(4):931–935. doi: 10.1161/01.hyp.35.4.931. [DOI] [PubMed] [Google Scholar]
- 101.Rizzoni D, Porteri E, Castellano M, Bettoni G, Muiesan ML, Muiesan P, Giulini SM, Agabiti-Rosei E. Vascular hypertrophy and remodeling in secondary hypertension. Hypertension. 1996;28(5):785–790. doi: 10.1161/01.hyp.28.5.785. [DOI] [PubMed] [Google Scholar]
- 102.De Mey JGR, Schiffers PM, Hilgers RHP, Sanders MMW. Toward functional genomics of flow-induced outward remodeling of resistance arteries. Am J Physiol - Heart Circ Physiol. 2005;288(3):1022–1027. doi: 10.1152/ajpheart.00800.2004. [DOI] [PubMed] [Google Scholar]
- 103.Cipolla MJ, Sweet JG, Chan SL. Cerebral vascular adaptation to pregnancy and its role in the neurological complications of eclampsia. J Appl Physiol. 2011;110(2):329–339. doi: 10.1152/japplphysiol.01159.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amaral SL, Michelini LC. Effect of gender on training-induced vascular remodeling in SHR. Braz J Med Biol Res. 2011;44(9):814–826. doi: 10.1590/s0100-879x2011007500055. [DOI] [PubMed] [Google Scholar]
- 105.Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertens: J Am Heart Assoc. 2002;40(4):504–510. doi: 10.1161/01.hyp.0000034738.79310.06. [DOI] [PubMed] [Google Scholar]
- 106.Chantemèle EJ Belin De, Vessières E, Dumont O, Guihot A-L, Toutain B, Loufrani L, Henrion D. Reactive oxygen species are necessary for high flow (shear stress)-induced diameter enlargement of rat resistance arteries. Microcirculation. 2009;16(5):391–402. doi: 10.1080/10739680902816301. [DOI] [PubMed] [Google Scholar]
- 107.Castier Y, Brandes RP, Leseche G, Tedgui A, Lehoux S. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res. 2005;97(6):533–540. doi: 10.1161/01.RES.0000181759.63239.21. [DOI] [PubMed] [Google Scholar]
- 108.Haas TL, Doyle JL, Distasi MR, Norton LE, Sheridan KM, Unthank JL. Involvement of MMPs in the outward remodeling of collateral mesenteric arteries. Am J Physiol - Heart Circ Physiol. 2007;293(4):2429–2437. doi: 10.1152/ajpheart.00100.2007. [DOI] [PubMed] [Google Scholar]
- 109.Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057–1069. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Griendling KK, Sobey CG, Selemidis S, Drummond GR. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103(9):1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 112.Weber M, Lauer N, Mülsch A, Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med. 2001;31(11):1360–1367. doi: 10.1016/s0891-5849(01)00706-7. [DOI] [PubMed] [Google Scholar]
- 113.Galili O, Versari D, Sattler KJ, Olson ML, Mannheim D, McConnell JP, Chade AR, Lerman LO, Lerman A. Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. Am J Physiol - Heart Circ Physiol. 2007;292(2):904–911. doi: 10.1152/ajpheart.00628.2006. [DOI] [PubMed] [Google Scholar]
- 114.Virdis A, Santini F, Colucci R, Duranti E, Salvetti G, Salvetti A, Rugani I, Segnani C, Anselmino M, Bernardini N, Blandizzi C, Pinchera A, Taddei S. Vascular generation of tumor necrosis factor-α reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol. 2011;58(3):238–247. doi: 10.1016/j.jacc.2011.01.050. [DOI] [PubMed] [Google Scholar]
- 115.Gao X, Zhang H, Schmidt AM, Zhang C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am J Physiol - Heart Circ Physiol. 2008;295(2):491–498. doi: 10.1152/ajpheart.00464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Galán M, Kassan M, Choi S-K, Partyka M, Trebak M, Henrion D, Matrougui K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60(1):71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Virdis A, Colucci R, Versari D, Ghisu N, Fornai M, Antonioli L, Duranti E, Daghini E, Giannarelli C, Blandizzi C, Taddei S, Del Tacca M. Atorvastatin prevents endothelial dysfunction in mesenteric arteries from spontaneously hypertensive rats: role of cyclooxygenase 2–derived contracting prostanoids. Hypertension. 2009;53(6):1008–1016. doi: 10.1161/HYPERTENSIONAHA.109.132258. [DOI] [PubMed] [Google Scholar]
- 118.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4+CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler, Thromb, Vasc Biol. 2011;31(11):2534–2542. doi: 10.1161/ATVBAHA.111.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/cSRC pathway. J Hypertens. 2014;32(3):542–554. doi: 10.1097/HJH.0000000000000059. [DOI] [PubMed] [Google Scholar]
- 120.Galan M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, Matrougui K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60(1):71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Amiri F, Ko EA, Javeshghani D, Reudelhuber TL, Schiffrin EL. Deleterious combined effects of salt-loading and endothelial cell restricted endothelin-1 overexpression on blood pressure and vascular function in mice. J Hypertens. 2010;28(6):1243. doi: 10.1097/HJH.0b013e328338bb8b. [DOI] [PubMed] [Google Scholar]
- 122.Thengchaisri N, Hein TW, Ren Y, Kuo L. Endothelin-1 impairs coronary arteriolar dilation: role of p38 kinase-mediated superoxide production from NADPH oxidase. J Mol Cell Cardiol. 2015;86:75–84. doi: 10.1016/j.yjmcc.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu Z, Cao W. p38 mitogen-activated protein kinase: a critical node linking insulin resistance and cardiovascular diseases in type 2 diabetes mellitus. Endocr Metab Immune Disord Drug Targets. 2009;9(1):38–46. doi: 10.2174/187153009787582397. [DOI] [PubMed] [Google Scholar]
- 124.Huang A, Yang YM, Yan C, Kaley G, Hintze TH, Sun D. Altered MAPK signaling in progressive deterioration of endothelial function in diabetic mice. Diabetes. 2012;61(12):3181–3188. doi: 10.2337/db12-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49(3):507–521. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 126.Di Bartolo BA, Cartland SP, Prado-Lourenco L, Griffith TS, Gentile C, Ravindran J, Azahri NS, Thai T, Yeung AW, Thomas SR, Kavurma MM. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) promotes angiogenesis and ischemia-induced neovascularization via NADPH oxidase 4 (NOX4) and nitric oxide-dependent mechanisms. J Am Heart Assoc. 2015;4(11) doi: 10.1161/JAHA.115.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang J, Hong Z, Zeng C, Yu Q, Wang H. NADPH oxidase 4 promotes cardiac microvascular angiogenesis after hypoxia/reoxygenation in vitro. Free Radic Biol Med. 2014;69:278–288. doi: 10.1016/j.freeradbiomed.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 128.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2007;27(11):2319–2324. doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 129.Chen L, Xiao J, Kuroda J, Ago T, Sadoshima J, Cohen RA, Tong X. Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim Et Biophys Acta. 2014;1842(12 Pt A):2489–2499. doi: 10.1016/j.bbadis.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 130.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. 2005;111(18):2347–2355. doi: 10.1161/01.CIR.0000164261.62586.14. [DOI] [PubMed] [Google Scholar]
- 131.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 132.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266(1):37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lakatta EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev. 2002;7(1):29–49. doi: 10.1023/a:1013797722156. [DOI] [PubMed] [Google Scholar]
- 134.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108(16):1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 135.Zalba G, Beaumont J, San Jose G, Fortuno A, Fortuno MA, Diez J. Vascular oxidant stress: molecular mechanisms and pathophysiological implications. J Physiol Biochem. 2000;56(1):57–64. doi: 10.1007/BF03179777. [DOI] [PubMed] [Google Scholar]
- 136.Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6(2):109–120. doi: 10.14336/AD.2014.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cifuentes ME, Rey FE, Carretero OA, Pagano PJ. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol. 2000;279(5):H2234–H2240. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 138.Frazziano G, Ghouleh I Al, Baust J, Shiva S, Champion HC, Pagano PJ. Nox-derived, ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. Am J Physiol Heart Circ Physiol. 2014;306(2):H197–H205. doi: 10.1152/ajpheart.00977.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Al Ghouleh I, Frazziano G, Rodriguez AI, Csanyi G, Maniar S, St Croix CM, Kelley EE, Egana LA, Song GJ, Bisello A, Lee YJ, Pagano PJ. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res. 2013;97(1):134–142. doi: 10.1093/cvr/cvs295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105(12):1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 141.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE (−/−) mice. J Clin Investig. 2001;108(10):1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ., Jr Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. 2011;216(2):321–326. doi: 10.1016/j.atherosclerosis.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112(17):2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 144.Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56(3):490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 145.Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, Etayo JC, Diez J. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension. 2000;35(5):1055–1061. doi: 10.1161/01.hyp.35.5.1055. [DOI] [PubMed] [Google Scholar]
- 146.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90(11):1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 147.Thompson-Torgerson CS, Holowatz LA, Flavahan NA, Kenney WL. Rho kinase-mediated local cold-induced cutaneous vasoconstriction is augmented in aged human skin. Am J Physiol Heart Circ Physiol. 2007;293(1):H30–H36. doi: 10.1152/ajpheart.00152.2007. [DOI] [PubMed] [Google Scholar]
- 148.Miller AA, Drummond GR, De Silva TM, Mast AE, Hickey H, Williams JP, Broughton BR, Sobey CG. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: role of Nox2. Am J Physiol Heart Circ Physiol. 2009;296(1):H220–H225. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- 149.Sobey CG, Heistad DD, Faraci FM. Potassium channels mediate dilatation of cerebral arterioles in response to arachidonate. Am J Physiol. 1998;275(5 Pt 2):H1606–H1612. doi: 10.1152/ajpheart.1998.275.5.H1606. [DOI] [PubMed] [Google Scholar]
- 150.Park L, Anrather J, Zhou P, Frys K, Wang G, Iadecola C. Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -indepen dent mechanisms. Arterioscler Thromb, Vasc Biol. 2004;24(10):1860–1865. doi: 10.1161/01.ATV.0000142446.75898.44. [DOI] [PubMed] [Google Scholar]
- 151.Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2006;26(6):836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- 152.Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke; J Cereb Circ. 2009;40(5):1827–1833. doi: 10.1161/STROKEAHA.108.536805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, Griendling KK. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ Res. 1998;82(10):1094–1101. doi: 10.1161/01.res.82.10.1094. [DOI] [PubMed] [Google Scholar]
- 154.Cheranov SY, Jaggar JH. TNF-α dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am J Physiol - Cell Physiol. 2006;290(4):964–971. doi: 10.1152/ajpcell.00499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal. 2015;8(358):ra2. doi: 10.1126/scisignal.2005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gebremedhin D, Terashvili M, Wickramasekera N, Zhang DX, Rau N, Miura H, Harder DR. Redox signaling via oxidative inactivation of PTEN modulates pressure-dependent myogenic tone in rat middle cerebral arteries. PLoS One. 2013;8(7):e68498. doi: 10.1371/journal.pone.0068498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Miller AA, De Silva TM, Judkins CP, Diep H, Drummond GR, Sobey CG. Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke; J Cereb Circ. 2010;41(4):784–789. doi: 10.1161/STROKEAHA.109.575365. [DOI] [PubMed] [Google Scholar]
- 158.Mayhan WG, Arrick DM, Sharpe GM, Sun H. Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation. 2008;15(3):225–236. doi: 10.1080/10739680701641421. [DOI] [PubMed] [Google Scholar]
- 159.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10(11):1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 160.Cipolla MJ, Sweet JG, Gokina NI, White SL, Nelson MT. Mechanisms of enhanced basal tone of brain parenchymal arterioles during early postischemic reperfusion: role of ET-1-induced peroxynitrite generation. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2013;33(10):1486–1492. doi: 10.1038/jcbfm.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Amberg GC, Earley S, Glapa SA. Local regulation of arterial L-type calcium channels by reactive oxygen species. Circ Res. 2010;107(8):1002–1010. doi: 10.1161/CIRCRESAHA.110.217018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke; a J Cereb Circ. 2007;38(11):3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 163.Carrano A, Hoozemans JJM, van der Vies SM, Rozemuller AJM, van Horssen J, de Vries HE. Amyloid beta induces oxidative stress-mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15(5):1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 164.Zhao Z, Hu J, Gao X, Liang H, Liu Z. Activation of AMPK attenuates lipopolysaccharide-impaired integrity and function of blood-brain barrier in human brain microvascular endothelial cells. Exp Mol Pathol. 2014;97(3):386–392. doi: 10.1016/j.yexmp.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 165.Schreurs MPH, Cipolla MJ. Cerebrovascular dysfunction and blood–brain barrier permeability induced by oxidized LDL are prevented by apocynin and magnesium sulfate in female rats. J Cardiovasc Pharmacol. 2014;63(1):33–39. doi: 10.1097/FJC.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim-Mitsuyama S, Yamamoto E, Tanaka T, Zhan Y, Izumi Y, Izumiya Y, Ioroi T, Wanibuchi H, Iwao H. Critical role of angiotensin II in excess salt-induced brain oxidative stress of stroke-prone spontaneously hypertensive rats. Stroke; a J Cereb Circ. 2005;36(5):1082–1083. doi: 10.1161/01.STR.0000163084.16505.e3. [DOI] [PubMed] [Google Scholar]
- 167.Gibson CL, Srivastava K, Sprigg N, Bath PMW, Bayraktutan U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J Neurochem. 2014;129(5):816–826. doi: 10.1111/jnc.12681. [DOI] [PubMed] [Google Scholar]
- 168.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertens: J Am Heart Assoc. 2001;38(3, Part 2 Suppl):581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 169.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension. 1989;13(6, Part2):968–972. doi: 10.1161/01.hyp.13.6.968. [DOI] [PubMed] [Google Scholar]
- 170.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension: dual processes of remodeling and growth. Hypertension. 1993;21(4):391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 171.Chan SL, Baumbach GL. Deficiency of Nox2 prevents angiotensin II-induced inward remodeling in cerebral arterioles. Front Physiol. 2013;4:133. doi: 10.3389/fphys.2013.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9(11):1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 173.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100(1):328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 174.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105(4):1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 176.Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cere- brovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25(7):1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher NDL. Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology. 2013;81(10):904–909. doi: 10.1212/WNL.0b013e3182a351aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fabiani M, Gordon BA, Maclin EL, Pearson MA, Brumback-Peltz CR, Low KA, McAuley E, Sutton BP, Kramer AF, Gratton G. Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. Neuroimage. 2014;85(Pt 1):592–607. doi: 10.1016/j.neuroimage.2013.04.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am J Physiol Heart Circ Physiol. 2014;306(3):H299–H308. doi: 10.1152/ajpheart.00744.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Farkas E, Luiten PGM. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64(6):575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 181.Hajdu MA, Heistad DD, Siems JE, Baumbach GL. Effects of aging on mechanics and composition of cerebral arterioles in rats. Circ Res. 1990;66(6):1747–1754. doi: 10.1161/01.res.66.6.1747. [DOI] [PubMed] [Google Scholar]
- 182.Mayhan WG, Faraci FM, Baumbach GL, Heistad DD. Effects of aging on responses of cerebral arterioles. AJP - Heart Circ Physiol. 1990;258(4):H1138. doi: 10.1152/ajpheart.1990.258.4.H1138. [DOI] [PubMed] [Google Scholar]
- 183.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27(12):1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 184.Peña Silva RA, Chu Y, Miller JD, Mitchell IJ, Penninger JM, Faraci FM, Heistad DD. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke; J Cereb Circ. 2012;43(12):3358–3363. doi: 10.1161/STROKEAHA.112.667063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Modrick ML, Didion SP, Sigmund CD, Faraci FM. Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol - Heart Circ Physiol. 2009;296(6):1914–1919. doi: 10.1152/ajpheart.00300.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7(6):476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke; J Cereb Circ. 2009;40(3 Suppl):S40–S44. doi: 10.1161/STROKEAHA.108.533638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Han BH, Zhou M-l, Johnson AW, Singh I, Liao F, Vellimana AK, Nelson JW, Milner E, Cirrito JR, Basak J, Yoo M, Dietrich HH, Holtzman DM, Zipfel GJ. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci. 2015;112(8):E881–E890. doi: 10.1073/pnas.1414930112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rey FE, Li XC, Carretero OA, Garvin JL, Pagano PJ. Perivascular superoxide anion contributes to impairment of endothelium-dependent relaxation: role of gp91(phox) Circulation. 2002;106(19):2497–2502. doi: 10.1161/01.cir.0000038108.71560.70. [DOI] [PubMed] [Google Scholar]
- 190.Cifuentes-Pagano E, Csanyi G, Pagano PJ. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci: CMLS. 2012;69(14):2315–2325. doi: 10.1007/s00018-012-1009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cifuentes-Pagano E, Saha J, Csanyi G, Ghouleh IA, Sahoo S, Rodriguez A, Wipf P, Pagano PJ, Skoda EM. Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors. MedChemComm. 2013;4(7):1085–1092. doi: 10.1039/C3MD00061C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res. 2001;89(5):408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 193.Al-Shabrawey M, Bartoli M, El-Remessy AB, Platt DH, Matragoon S, Behzadian MA, Caldwell RW, Caldwell RB. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascu-larization during ischemic retinopathy. Am J Pathol. 2005;167(2):599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J Physiol. 2006;576(Pt 2):557–567. doi: 10.1113/jphysiol.2006.111070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, Pagano PJ. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol. 2005;288(2):H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- 196.Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109(14):1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 197.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23(5):776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 198.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel, NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92(6):637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 199.Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res. 2004;95(6):587–594. doi: 10.1161/01.RES.0000142317.88591.e6. [DOI] [PubMed] [Google Scholar]
- 200.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290(5):H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 201.Sukumar P, Viswambharan H, Imrie H, Cubbon RM, Yuldasheva N, Gage M, Galloway S, Skromna A, Kandavelu P, Santos CX, Gatenby VK, Smith J, Beech DJ, Wheatcroft SB, Channon KM, Shah AM, Kearney MT. Nox2 NADPH oxidase has a critical role in insulin resistance-related endothelial cell dysfunction. Diabetes. 2013;62(6):2130–2134. doi: 10.2337/db12-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal. 2013;18(13):1537–1548. doi: 10.1089/ars.2012.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cangemi R, Celestini A, Del Ben M, Pignatelli P, Carnevale R, Proietti M, Calabrese CM, Basili S, Violi F. Role of platelets in NOX2 activation mediated by TNFalpha in heart failure. Intern Emerg Med. 2014;9(2):179–185. doi: 10.1007/s11739-012-0837-2. [DOI] [PubMed] [Google Scholar]
- 204.Yamamoto Y, Saito T, Feng GG, Li J, Yasuda Y, Kazaoka Y, Fujiwara Y, Kinoshita H. Intermittent local periodontal inflammation causes endothelial dysfunction of the systemic artery via increased levels of hydrogen peroxide concomitantly with overexpression of superoxide dismutase. Int J Cardiol. 2016;222:901–907. doi: 10.1016/j.ijcard.2016.08.099. [DOI] [PubMed] [Google Scholar]
- 205.Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun. 2016;58:291–309. doi: 10.1016/j.bbi.2016.07.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, McNeil C, Wheatcroft S, Yuldasheva N, Febbriao M, Kearney M, Naseem KM. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125(17):2693–2703. doi: 10.1182/blood-2014-05-574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D-amino acid substitution: improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc Natl Acad Sci USA. 2005;102(2):413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305(5689):1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lakkireddy HR, Urmann M, Besenius M, Werner U, Haack T, Brun P, Alie J, Illel B, Hortala L, Vogel R, Bazile D. Oral delivery of diabetes peptides -comparing standard formulations incorporating functional excipients and nanotechnologies in the translational context. Adv Drug Deliv Rev. 2016;106(Pt B):196–222. doi: 10.1016/j.addr.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 210.Li X, Wang C, Liang R, Sun F, Shi Y, Wang A, Liu W, Sun K, Li Y. The glucose-lowering potential of exenatide delivered orally via goblet cell-targeting nano-particles. Pharm Res. 2015;32(3):1017–1027. doi: 10.1007/s11095-014-1513-1. [DOI] [PubMed] [Google Scholar]
- 211.Patil NH, Devarajan PV. Insulin-loaded alginic acid nanoparticles for sublingual delivery. Drug Deliv. 2016;23(2):429–436. doi: 10.3109/10717544.2014.916769. [DOI] [PubMed] [Google Scholar]
- 212.Singh G, Pai RS. Atazanavir-loaded Eudragit, RL 100 nanoparticles to improve oral bioavailability: optimization and in vitro/in vivo appraisal. Drug Deliv. 2016;23(2):532–539. doi: 10.3109/10717544.2014.930760. [DOI] [PubMed] [Google Scholar]
- 213.Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65(6):822–832. doi: 10.1016/j.addr.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Agrawal AK, Harde H, Thanki K, Jain S. Improved stability and antidiabetic potential of insulin containing folic acid functionalized polymer stabilized multilayered liposomes following oral administration. Biomacromolecules. 2014;15(1):350–360. doi: 10.1021/bm401580k. [DOI] [PubMed] [Google Scholar]
- 215.Araujo F, Shrestha N, Shahbazi MA, Fonte P, Makila EM, Salonen JJ, Hirvonen JT, Granja PL, Santos HA, Sarmento B. The impact of nanoparticles on the mucosal translocation and transport of GLP-1 across the intestinal epithelium. Biomaterials. 2014;35(33):9199–9207. doi: 10.1016/j.biomaterials.2014.07.026. [DOI] [PubMed] [Google Scholar]
- 216.Bouttefeux O, Beloqui A, Preat V. Delivery of peptides via the oral route: diabetes treatment by peptide-loaded nanoparticles. Curr Pharm Des. 2016;22(9):1161–1176. doi: 10.2174/1381612822666151216150238. [DOI] [PubMed] [Google Scholar]
- 217.Aguirre TA, Teijeiro-Osorio D, Rosa M, Coulter IS, Alonso MJ, Brayden DJ. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. 2016;106(Pt B):223–241. doi: 10.1016/j.addr.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 218.Krol S, Ellis-Behnke R, Marchetti P. Nanomedicine for treatment of diabetes in an aging population: state-of-the-art and future developments. Nanomedicine. 2012;8(Suppl 1):S69–S76. doi: 10.1016/j.nano.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 219.Ranayhossaini DJ, Rodriguez AI, Sahoo S, Chen BB, Mallampalli RK, Kelley EE, Csanyi G, Gladwin MT, Romero G, Pagano PJ. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J Biol Chem. 2013;288(51):36437–36450. doi: 10.1074/jbc.M113.521344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cifuentes-Pagano E, Meijles DN, Pagano PJ. The quest for selective nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid Redox Signal. 2014;20(17):2741–2754. doi: 10.1089/ars.2013.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Petronio MS, Zeraik ML, Fonseca LM, Ximenes VF. Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules. 2013;18(3):2821–2839. doi: 10.3390/molecules18032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51(2):211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 223.El-Naga RN. Apocynin protects against ethanol-induced gastric ulcer in rats by attenuating the upregulation of NADPH oxidases 1 and 4. Chem Biol Interact. 2015;242:317–326. doi: 10.1016/j.cbi.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 224.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71(2):331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 225.Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH, Morawietz H. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun. 2006;344(1):200–205. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 226.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161(4):885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lange S, Heger J, Euler G, Wartenberg M, Piper HM, Sauer H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic stem cell-derived endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovasc Res. 2009;81(1):159–168. doi: 10.1093/cvr/cvn258. [DOI] [PubMed] [Google Scholar]
- 228.Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci: CMLS. 2012;69(14):2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tuo YH, Liu Z, Chen JW, Wang QY, Li SL, Li MC, Dai G, Wang JS, Zhang YL, Feng L, Shi ZS. NADPH oxidase inhibitor improves outcome of mechanical reperfusion by suppressing hemorrhagic transformation. J Neurointerventional Surg. 2016 doi: 10.1136/neurintsurg-2016-012377. [DOI] [PubMed] [Google Scholar]
- 230.El Assar M, Angulo J, Santos-Ruiz M, Moreno P, Novials A, Villanueva-Penacarrillo ML, Rodriguez-Manas L. Differential effect of amylin on endothelial-dependent vasodilation in mesenteric arteries from control and insulin resistant rats. PLoS One. 2015;10(3):e0120479. doi: 10.1371/journal.pone.0120479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Huang A, Yang YM, Yan C, Kaley G, Hintze TH, Sun D. Altered MAPK signaling in progressive deterioration of endothelial function in diabetic mice. Diabetes. 2012;61(12):3181–3188. doi: 10.2337/db12-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sun QA, Hess DT, Wang B, Miyagi M, Stamler JS. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo [4,5-d]pyrimidine (VAS2870) Free Radic Biol Med. 2012;52(9):1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53(21):7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 234.Genkyotex Innovation SAS. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); Safety and efficacy of oral GKT137831 in patients with type 2 diabetes and albuminura. Available from: 〈 https://www.clinicaltrials.gov/ct2/show/record/NCT02010242〉, 2000 (cited 2017Feb13), NLM Identifier: NCT02010242. [Google Scholar]
- 235.Qiu S, Mintz JD, Salet CD, Han W, Giannis A, Chen F, Yu Y, Su Y, Fulton DJ, Stepp DW. Increasing muscle mass improves vascular function in obese (db/db) mice. J Am Heart Assoc. 2014;3(3):e000854. doi: 10.1161/JAHA.114.000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5(10):981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Coco H, Pernomian L, Pereira PC, Gomes MS, Marchi KC, Lopes AH, Cunha TM, Tirapelli CR, de Oliveira AM. Chronic restraint stress increases angiotensin II potency in the rat carotid: role of cyclooxygenases and reactive oxygen species. J Pharm Pharmacol. 2017;69(1):52–65. doi: 10.1111/jphp.12659. [DOI] [PubMed] [Google Scholar]
- 238.Wong PS, Randall MD, Roberts RE. Sex differences in the role of NADPH oxidases in endothelium-dependent vasorelaxation in porcine isolated coronary arteries. Vasc Pharmacol. 2015;72:83–92. doi: 10.1016/j.vph.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 239.Sies H. Ebselen, a selenoorganic compound as glutathione peroxidase mimic. Free Radic Biol Med. 1993;14(3):313–323. doi: 10.1016/0891-5849(93)90028-s. [DOI] [PubMed] [Google Scholar]
- 240.Sies H, Masumoto H. Ebselen as a glutathione peroxidase mimic and as a scavenger of peroxynitrite. Adv Pharmacol. 1997;38:229–246. doi: 10.1016/s1054-3589(08)60986-2. [DOI] [PubMed] [Google Scholar]
- 241.Smith SM, Min J, Ganesh T, Diebold B, Kawahara T, Zhu Y, McCoy J, Sun A, Snyder JP, Fu H, Du Y, Lewis I, Lambeth JD. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem Biol. 2012;19(6):752–763. doi: 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Xie H, Ray PE, Short BL. NF-kappaB activation plays a role in superoxide-mediated cerebral endothelial dysfunction after hypoxia/reoxygenation. Stroke; J Cereb Circ. 2005;36(5):1047–1052. doi: 10.1161/01.STR.0000157664.34308.cc. [DOI] [PubMed] [Google Scholar]
- 243.Handa Y, Kaneko M, Takeuchi H, Tsuchida A, Kobayashi H, Kubota T. Effect of an antioxidant, ebselen, on development of chronic cerebral vasospasm after subarachnoid hemorrhage in primates. Surg Neurol. 2000;53(4):323–329. doi: 10.1016/s0090-3019(00)00168-3. [DOI] [PubMed] [Google Scholar]
- 244.Bubolz AH, Wu Q, Larsen BT, Gutterman DD, Liu Y. Ebselen reduces nitration and restores voltage-gated potassium channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293(4):H2231–H2237. doi: 10.1152/ajpheart.00717.2007. [DOI] [PubMed] [Google Scholar]
- 245.Cayatte AJ, Rupin A, Oliver-Krasinski J, Maitland K, Sansilvestri-Morel P, Boussard MF, Wierzbicki M, Verbeuren TJ, Cohen RA. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets nadph oxidase. Arterioscler Thromb Vasc Biol. 2001;21(10):1577–1584. doi: 10.1161/hq1001.096723. [DOI] [PubMed] [Google Scholar]
- 246.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272(20):13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 247.Bhandarkar SS, Jaconi M, Fried LE, Bonner MY, Lefkove B, Govindarajan B, Perry BN, Parhar R, Mackelfresh J, Sohn A, Stouffs M, Knaus U, Yancopoulos G, Reiss Y, Benest AV, Augustin HG, Arbiser JL. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J Clin Investig. 2009;119(8):2359–2365. doi: 10.1172/JCI33877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res. 2003;93(12):1225–1232. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Siu KL, Gao L, Cai H. Differential roles of protein complexes NOX1-NOXO1 and NOX2-p47phox in mediating endothelial redox responses to oscillatory and unidirectional laminar shear stress. J Biol Chem. 2016;291(16):8653–8662. doi: 10.1074/jbc.M115.713149. [DOI] [PMC free article] [PubMed] [Google Scholar]