

Abstract
Inflammatory cytokine IL-6 is elevated in the serum and lungs of patients with pulmonary artery hypertension (PAH). Several animal models of PAH cite the potential role of inflammatory mediators. We investigated IL-6’s role in the pathogenesis of pulmonary vascular disease. Indices of pulmonary vascular remodeling were measured in lung specific IL-6 over expression transgenic mice (Tg(+)) and compared to wild type (Tg(-)) controls in both normoxic and chronic hypoxic conditions. The Tg(+) mice exhibited elevated right ventricular systolic pressures and right ventricular hypertrophy with corresponding pulmonary vasculopathic changes, all of which were exacerbated by chronic hypoxia. IL-6 overexpression increased muscularization of the proximal arterial tree, and hypoxia enhanced this effect. It also reproduced the muscularization and proliferative arteriopathy seen in the distal arteriolar vessels of PAH patients. The latter was characterized by the formation of occlusive neointimal angioproliferative lesions that worsened with hypoxia and were composed of endothelial cells and T-lymphocytes. IL-6-induced arteriopathic changes were accompanied by activation of pro-angiogenic factor, vascular endothelial growth factor, the pro-proliferative kinase, ERK, pro-proliferative transcription factors c-MYC and MAX, the anti-apoptotic proteins survivin and Bcl-2, and down-regulation of the growth inhibitor transforming growth factor-β, and pro-apoptotic kinases JNK and p38. These findings suggest that IL-6 promotes the development and progression of pulmonary vascular remodeling and PAH through pro-proliferative anti-apoptotic mechanisms.
Keywords: Interleukin-6, Pulmonary Artery Hypertension, Proliferation
Pulmonary vascular remodeling is associated with increased pulmonary vascular resistance, pulmonary artery hypertension (PAH) and right heart failure. Advanced PAH is characterized by arteriopathy, which includes muscularization of distal pulmonary arterioles, concentric intimal thickening, and obstruction of the vascular lumen by proliferating endothelial cells to form plexiform lesions.1 Evidence suggests that PAH is associated with genetic perturbations favoring cellular growth, proliferation, and angiogenesis2 and inhibitors of apoptosis, previously thought to be only expressed in cancer cells, promoting a proliferative cellular phenotype resulting in pulmonary vascular remodeling in PAH.3, 4 The histopathological features and known genetic susceptibilities of this condition have led to the hypothesis that PAH arises from hyper-proliferation of pulmonary artery smooth muscle cells (PASMC) and endothelial cells (PAEC).
In addition to the formation of proliferative neointimal lesions and muscularization of the pulmonary vascular bed, perivascular inflammatory cell infiltrates are also present in advanced human cases of PAH. These infiltrates consist of T cells, B cells, and macrophages, suggesting that cytokines and growth factors associated with these inflammatory cells may be promoting PAEC and PASMC hyper-proliferation.5 The pro-inflammatory cytokine, interleukin-6 (IL-6), is consistently increased in the serum and lungs6-8 of patients with idiopathic PAH and in inflammatory diseases6, 9-11 that are associated with PAH. In addition, Kaposi’s sarcoma-associated herpes virus, which may cause PAH in human immunodeficiency virus-negative Castleman’s disease, encodes a constitutively active form of IL-612 resulting in unregulated cell growth and escape from host anti-tumor defenses. Furthermore, unchecked production of IL-6 in tissues leading to chronic inflammation has exhibited a strong association with many cancers.13 Given mounting evidence for the role of inflammation and cancer-like mechanisms in the pathogenesis of PAH, we investigated whether IL-6 promotes the development of pulmonary arteriopathy and consequent PAH. We show that lung-specific overexpression of IL-6 in mice replicates the pathological lesions observed in advanced PAH, including both distal arteriolar muscularization and plexigenic arteriopathy, and leads to increased pulmonary vascular resistance (PVR) and PAH. At the cellular and molecular level, these vasculopathic changes are associated with the activation of vascular endothelial growth factor (VEGF), mitogen activated protein kinase (MAPK) ERK with subsequent increases in proto-oncogene c-MYC /MAX transcription factor complex and the anti-apoptotic proteins survivin and Bcl-2 with down-regulation of the growth inhibitor transforming growth factor (TGF)-β and pro-apoptotic kinases, JNK and p38. This suggests that IL-6 participates in the development of distal pulmonary proliferative arteriopathy and consequent elevation in PVR and development of PAH.
Materials and Methods
For details, see the online supplement material (available online at http://circres.ahajournals.org). Indices of pulmonary vascular remodeling were measured in lung specific IL-6 over expression transgenic mice (Tg(+)) and compared to wild type (Tg(-)) controls in both normoxic and chronic hypoxic conditions.
Results
IL-6 overexpression increases pulmonary artery pressure and ventricular hypertrophy
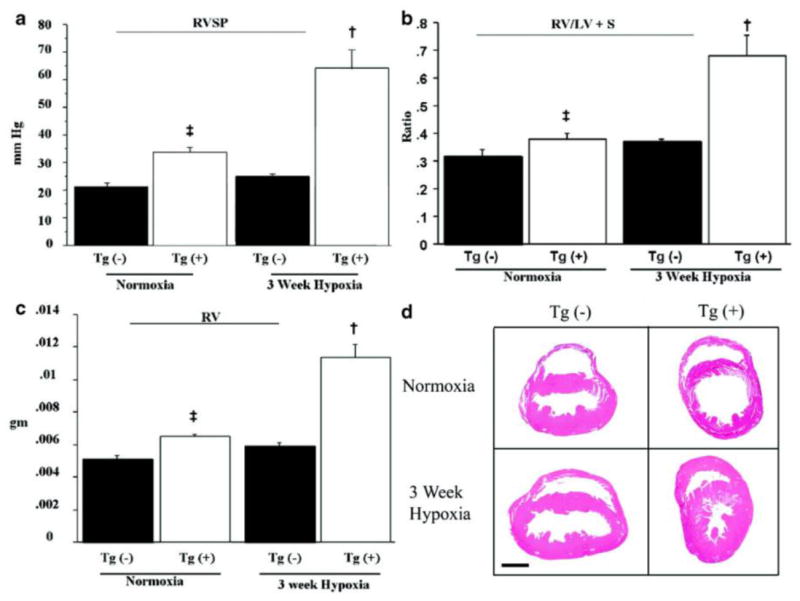
To test the hypothesis that increased IL-6 may cause increased PVR and PAH, we measured the right ventricular systolic pressure (RVSP) in IL-6 Tg(+) and Tg(-) mice. Under normoxic conditions, Tg(+) mice had elevated RVSP compared to Tg(-) mice (Figure 1a). In Tg(+) mice, 3 weeks of exposure to 10% oxygen almost doubled RVSP compared to baseline, and this value was almost 2.6 times higher than the RVSP in hypoxic Tg(-) mice (Figure 1a).
Figure 1.
IL-6 Tg(+) mice have PAH at baseline that worsens with hypoxia. (a) RVSP is higher in Tg(+) mice (‡ vs. normoxic Tg(-);p<0.05; † vs. hypoxic Tg(-);p<0.05). (b) RV/LV+S are higher in IL-6 Tg(+) mice (‡ vs. normoxic Tg(-);p<0.05; † vs. hypoxic Tg(-);p<0.05). (c) RV weight is higher in IL-6 Tg(+) mice (‡ vs. normoxic Tg(-);p<0.05; † vs. hypoxic Tg(-);p<0.05) and increases further in hypoxia († vs. normoxic Tg(+);p<0.05) (d) Representative photomicrographs of IL-6 Tg(+) and Tg(-) mouse hearts in normoxic and hypoxic conditions. IL-6 Tg(+) mice right ventricles are hypertrophied at baseline and they hypertrophy further with hypoxia (hemotoxylin & eosin staining, magnification x25, bar=0.01mm).
Ventricular wall thickness increased in response to chronic pressure overload, which is a consequence of elevated resistance in the pulmonary artery (PA). Right ventricular hypertrophy (RVH), as measured by right ventricle weight/(left ventricle weight + septum weight; RV/LV+S) and absolute right ventricle weight (RV), were greater in Tg(+) mice than in Tg(-) mice under normoxic conditions (Figure 1b, c). Hypoxia produced even greater RVH in Tg(+) mice while there was no change in ventricular wall thickness in Tg(-) mice (Figure 1b, c). The histological appearance of the hearts was consistent with RVH measurements, showing that right ventricular wall mass was greater in Tg(+) mice in both normoxic and hypoxic conditions(Figure 1d). See online supplement for additional data.
IL-6 overexpression induced muscularization throughout the entire pulmonary vascular bed
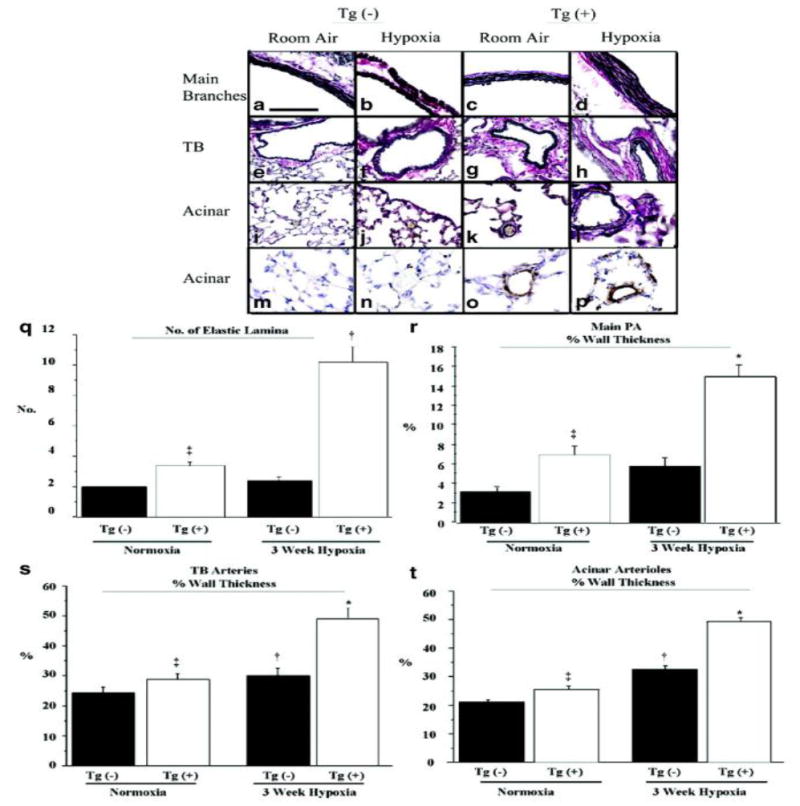
To determine the cause of increased PVR, we examined specific regions of the pulmonary vascular tree for remodeling. Examination of the proximal branches of the main PA revealed that the elastic lamina was increased in normoxic Tg(+) mice compared to their Tg(-) counterparts (Figure 2 c vs. a) and was quantitatively confirmed by counting the number of elastic lamina (Figure 2q). Following hypoxia, the number of elastic lamina in Tg(+) mice more than tripled compared to baseline and exceeded the number in hypoxic Tg(-) littermates by a factor of five. Main PA branches in Tg(+) mice exhibited an increase in not only the number of elastic lamina, but also the percent vessel medial wall thickness, relative to the Tg(-) control, under both normoxic and hypoxic conditions (Figure 2r). The medial wall of the main PA branches in Tg(+) mice more than doubled in thickness in response to hypoxia compared to their Tg(+) normoxic controls, while PA medial wall thickness did not change in hypoxic Tg(-) mice compared to normoxic controls.
Figure 2.
Pulmonary artery (PA) tree of IL-6 Tg(+) mice has increased muscularization that worsens with hypoxia. (a-l) Representative photomicrographs of the elastic lamina of the PA vasculature of IL-6 Tg(+) and Tg(-) mice in normoxic and hypoxic conditions. Main PA branches (Tg(-) a, b vs. Tg(+) c, d), PA at the level of the terminal bronchioles (TB) (Tg(-) e, f, vs. Tg(+) g, h), PA distal to TB (acinar) (Tg(-) i, j vs. Tg(+) k, l). Elastic tissue stain, magnification x400, bar=0.001mm. (m-p) Representative photomicrographs of smooth muscle hypertrophy of distal acinar arterioles of the PA vasculature of IL-6 Tg(+) in normoxic and hypoxic conditions (Tg(-) m, n vs. Tg(+) o, p). Immunohistochemistry with α-smooth muscle actin, magnification x400, bar=0.001mm. (q-t) Thickness of the medial wall is increased at all levels of the PA tree of IL-6 Tg(+) mice compared to Tg(-) mice at baseline and worsens with hypoxia. (q) Number of elastic lamina of main PA branches (‡ vs. normoxic Tg(-);p<0.05, † vs. normoxic Tg(+);p<0.05, and † vs. hypoxic Tg(-);p<0.05). (r) Percent wall thickness (%WT) of the main PA branches (* vs. hypoxic Tg(-);p<0.05 and * vs. normoxic Tg(+);p<0.05. ‡ vs. normoxic Tg(-);p<0.05). (s) %WT of the TB PA vessels (* vs. hypoxic Tg(-);p<0.05, * vs. normoxic Tg(+);p<0.05, † vs. normoxic Tg(-);p<0.05, ‡ vs. normoxic Tg(-);p<0.05). (t) %WT of the acinar pulmonary arteriolar vessels († vs. normoxic Tg(-);p<0.05, ‡ vs. normoxic Tg(-);p<0.05, * vs. hypoxic Tg(-);p<0.05 and * vs. normoxic Tg(+);p<0.05).
The terminal bronchioles (TB) and distal acinar arterioles were examined for evidence of muscularization. The most notable findings were that the distal acinar arterioles of Tg(+) mice were muscularized at baseline and became more thickly muscularized in hypoxia unlike the Tg(-) mice arterioles as shown by elastic staining (Figure 2 Tg(+) k, l vs. Tg(-) i, j) and by immunohistochemistry with α-smooth muscle actin (Figure 2 Tg(+) o, p vs. Tg(-) m, n). See online supplement for detailed results together with the quantitative results of the medial wall thickness of both the TB and acinar vessels.
Il-6 overexpression induced arteriolar neointimal occlusive lesions
We examined the vascular bed for occlusive neointimal lesions that, like arteriole muscularization, may contribute to increased PVR. We found that the arterioles in Tg(+) mice had thick intimal walls, with many of the arteriolar lumens being partially (27±5%) or completely occluded (4±2%). Hypoxia increased the number of partially (55±3%) or completely occluded arterioles (14±1%, Figure 3b, d, e). In contrast, all arterioles in the lungs of Tg(-) normoxic mice were patent, and the intima was not thickened. Only after exposure to hypoxia, did the arteriolar lumens become partially occluded, with the number of these vessels reaching 3±2% (Figure 3a, c, e).
Figure 3.
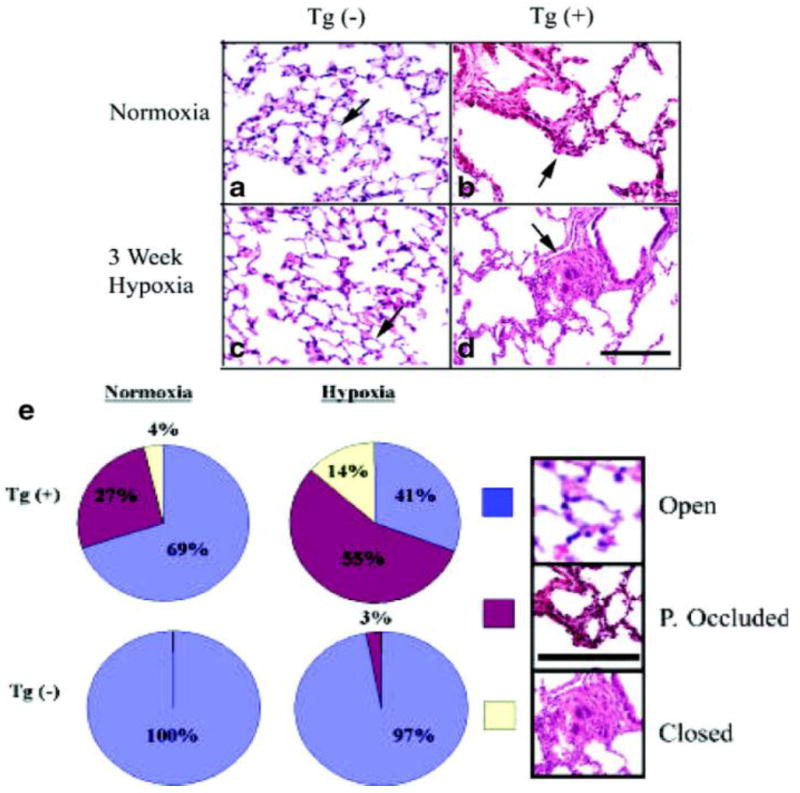
Arterioles of IL-6 Tg(+) mice have both neointimal occlusive lesions and a reduction in the total number of distal arterioles. (a-d) Representative photomicrographs of the lung parenchyma of IL-6 Tg(+) and Tg(-) mice showing neointimal hyperplasia of acinar arterioles in IL-6 Tg(+) mice in normoxic conditions (b) and occlusive arteriopathy in hypoxic conditions (d). No neointimal hyperplastic or occlusive lesions are seen in Tg(-) mice (a and c); (hematoxylin & eosin staining, magnification x400, bar=0.001mm). (e) Pie chart demonstrates that IL-6 Tg(+) mice have a higher percentage (%) of partially (P.) occluded and closed luminal acinar arterioles at baseline that worsens with hypoxia compared with Tg(-) mice. Hematoxylin & eosin stain included in key to demonstrate representative examples of an open (blue), partially occluded (red) and closed acinar arteriole (yellow), magnification x40, bar=0.001mm, arrows=arterioles.
Hypoxia induced loss of pulmonary arterioles in IL-6 overexpressing mice
See online supplement and Online Figure I for results.
IL-6-induced distal pulmonary vascular wall endothelial cellular growth and proliferation
To determine if PAEC types contribute to intimal wall thickening in arterioles of Tg(+) mice, we performed immunohistochemical analysis of factor VIII, an endothelial cell marker. We found that the arteriole walls were thickened, in part, by multiple layers of PAEC. Under both normoxic and hypoxic conditions, the PAEC layers either formed smooth and thick concentric occlusive lesions or plexigenic-like occlusive lesions due to a piling up of PAEC in a non-uniform fashion (Figure 4c, d). While arteriolar walls in Tg(-) mice had a normal appearance under both normoxic and hypoxic conditions (Figure 4a, b). At baseline and in hypoxic conditions, PAEC lining the arterioles of Tg(-) mice exhibited minimal expression of vascular endothelial growth factor receptor 2 (VEGFR2) (Figure 4e, f), while PAEC had elevated VEGFR2 expression in Tg(+) mice (Figure 4g, h), suggesting that the growth potential of these cells was increased and confirming that the plexigenic-like lesions in Tg(+) mice consisted predominately of PAEC. In Tg(+) mice, increases in PAEC and VEGFR2 expression were associated with increased proliferation within the intimal wall of arterioles at baseline and in hypoxic conditions, as assessed by staining for the proliferation marker, PCNA (Figure 4k, l). No change in PCNA levels were detected in the Tg(-) lungs (Figure 4i, j).
Figure 4.
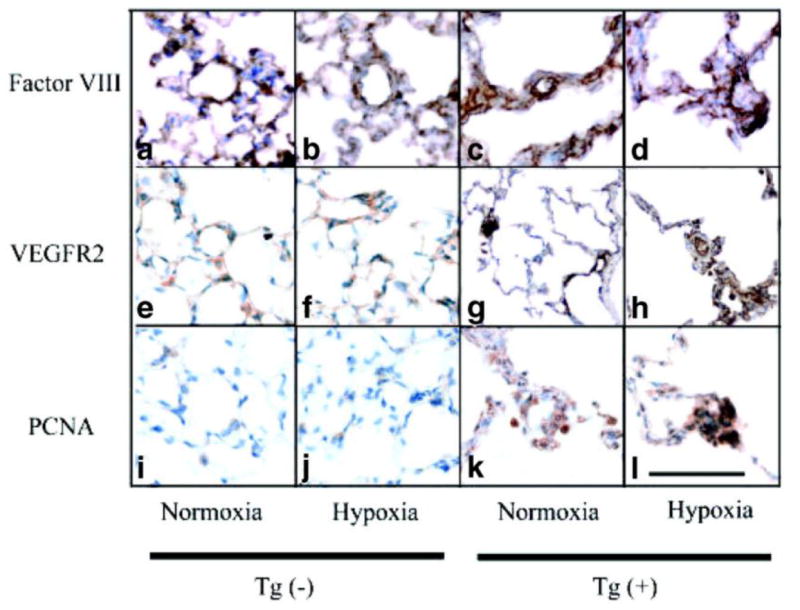
Angioproliferative lesions are present in Tg(+) mice distal arterioles. (a-l) Representative photomicrographs showing the formation of thick occlusive neointimal lesions. Endothelial cells (Factor VIII) are forming thick layers in the distal acinar arterioles of Tg(+) mice (c and d) and have increased expression of vascular endothelial growth factor receptor 2 (VEGFR2: g and h) which was not seen in Tg(-) mice (a, b or e, f). There is increased cellular proliferation in the walls of the distal arterioles of IL-6 Tg(+) mice (PCNA: k and l) in normoxic and hypoxic conditions, which was not seen in Tg(-) mice (i and j). Immunohistochemistry staining, magnification x400, bar=0.001mm.
Characterization of Pulmonary Vasculopathy: Pro-Growth/Pro-Proliferative Factors and Pro-survival/Anti-apoptotic Mediators are activated in IL-6 Tg(+) Mice
We determined whether factors that stimulate growth, proliferation and survival and inhibit apoptosis of PAEC and PASMC were underlying the mechanism through which IL-6 promotes the characteristic pathophysiologic phenotype of PAH. This was preformed by investigating a number of key modulators that may be involved, at the level of the protein in immunoblot analysis of whole lung lysates (Online Figure IIa, b, c, d, e). See the online supplement for the results.
Inflammatory cells contribute to IL-6-induced neointimal lesions in arterioles
Given that lymphocytes form conglomerates near major airways in this mouse model14, we examined the pulmonary vascular bed for evidence of a similar cellular inflammatory response at sites of arteriolar occlusive lesions. Under normoxic conditions, the number of periarteriolar lymphocytes (determined by the high nuclear to cytoplasmic ratio) was greater in Tg(+) mice than in Tg(-) mice (Figure 5c), with T cells, determined by immunohistochemistry (Figure 5g), but not B cells (Figure 5k) being increased within the pulmonary vascular bed of Tg(+) animals. Other inflammatory cells were not seen (Figure 5a-d). Following hypoxia, these T cells (Figure 5d) contributed to the obstruction of the arteriolar lumen. This was confirmed by immunoblot analysis of whole lung lysates (Online Figure III). Taken together, IL-6 mainly recruits lymphocytes, and the lymphocytes that are recruited to the pulmonary vascular bed are predominately T cells, not B cells. Further characterization of lymphocyte recruitment and function was determined in IL-6 Tg(+) mice. The data can be seen on the online supplement together with Online Figure III
Figure 5.
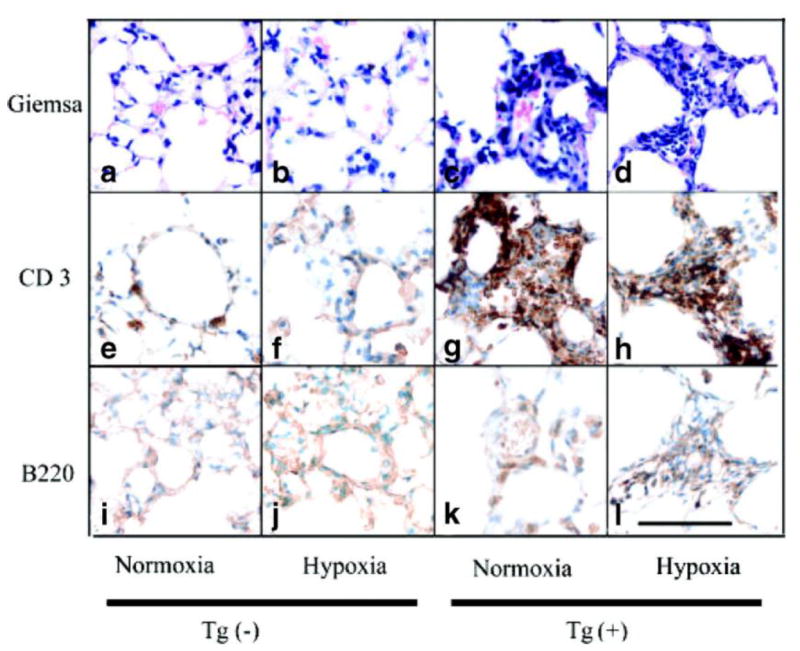
Inflammatory cells present in the neointimal lesions of the arterioles of Tg(+) mice. (a-l) Representative photomicrographs showing an increase in periarteriolar lymphocytes (Giemsa stain: a-d), specifically T cells (CD3: e-h) compared to B cells (B220: i-l) in IL-6 Tg(+) mice with little change in Tg(-) mice at baseline. In hypoxic conditions, CD3+ T cells are obliterating the arteriolar lumen of IL-6 Tg(+) mice (d and h). Magnification x400, bar=0.001 mm.
Discussion
Patients with severe PAH and animal models of PAH15-21 exhibit increases in inflammatory cells, growth factors, and cytokines. IL-6, a pleiotropic cytokine, is frequently elevated, suggesting that PAH development is associated with IL-6-induced inflammation. Our results demonstrate that IL-6 lung-specific overexpression produces distal arteriolar-occlusive plexigenic lesions and arteriolar wall muscularization. These changes in the distal vascular bed are associated with and may lead to proximal pulmonary artery wall hypertrophy and RVH as well as increased RVSP and PVR. Injection of recombinant human IL-6 (rhIL-6) also produces RVH in rats22 and mice18. However, they lack the associated distal obliterative muscularized vascular lesions observed in the transgenic mice that constitutively overexpress IL-6. Importantly, however, IL-6 knock out mice exposed to hypoxia are resistant to the development of increased RVSP.23 The lack of correlation between pulmonary vascular remodeling and the presence of elevated pulmonary artery pressures in other murine models24-27 has slowed our understanding of the pathobiology of PAH. IL-6 Tg(+) mice, where the pathological and physiological changes observed in the pulmonary artery bed correlate with the severity of PAH, may enable a better understanding of PAH pathobiology, including the role of increased IL-6 in the development of PAH in humans.
A major finding in this study is that distal vascular remodeling in Tg(+) mice is similar to that seen in patients with severe PAH,5 with (1) concentric intimal wall thickening, (2) plexigenic lesions, (3) recruitment of inflammatory cells, and (4) distal arteriolar wall muscularization. These features occurred de novo under normoxic conditions and worsened with hypoxia. In other rodent models,15-21 vascular remodeling is limited to either dysregulated PAEC or PASMC, but not both. The Tg(+) mouse, which exhibits all four main pathological features of PAH, is, to our knowledge, the only in vivo model that recapitulates the pathological features of PAH in humans. Therefore, this model may reveal how interactions between hyper-proliferative PASMC and PAEC and inflammatory cells as well as the pathobiological phenotypes of these cells contribute to PAH development.
Disorganized PAEC proliferation leading to the formation of neointimal obliterative lesions is described in many cases of idiopathic PAH5 or associated PAH, and may be why the human form of severe PAH is difficult to treat with the current available drugs.28-30 This has lead to the search for newer models of PAH in which a neointima is formed and occludes the vascular lumen. A number of 2 hit injurious murine models have been able to reproduce neointimal occlusive lesions,15, 31-33 as well as a genetically altered model,34 however less than 5% of these mice developed these lesions. Our study is of interest because we show that by solely overexpressing IL-6 without an additional stress, PAEC are stimulated to either form smooth concentric multilayers leading to thickening of the intimal wall or to pile on top of one another, narrowing the distal arteriolar lumen and forming a plexiform lesion. Both features were present in all mice under normoxic conditions, when PAH is mild, and increased under hypoxic conditions, when RVSP is maximal and the disease is severe. This suggests that aberrant PAEC proliferation and lesion formation are pathologically relevant, are useful markers of disease progression, and that overexpression of IL-6, a single genetic perturbation, is able to reproduce the characteristic obliterative lesions seen in the aforementioned models and replicate that of human disease.
Distal extension of smooth muscle into small peripheral, normally nonmuscular, pulmonary arteries within the respiratory acinous is notable in all forms of PAH. The cellular processes underlying muscularization of this distal part of the pulmonary vascular bed are incompletely understood but are thought to result from the abnormal growth of PASMC, which have impaired responses to anti-proliferative pro-apoptotic stimuli such as bone morphogenic protein (BMP) and TGF-β.2, 35-37 We show that lung specific overexpression of IL-6 induces 3 forms of muscularization. First, IL-6 results in distal extension of smooth muscle into the small peripheral pulmonary arteries at the level of the acinous and with the added insult of hypoxia, the medial wall further hypertrophies. Secondly, IL-6 results in an increase in the medial wall thickness of the main and bronchial level pulmonary arteries and thirdly, there is an increase in the number of layers of elastic lamella, both of which increase further in hypoxic conditions. The combination of these striking changes in muscularization have not been observed in other pulmonary artery hypertension murine models. However, in the spontaneously hypertensive rat,38 increased arterial medial wall thickness is associated with increased number of lamina in major blood vessels as well as increased wall thickness, although less striking than in hypoxic IL-6 Tg(+) mice. Furthermore, the hypertrophic changes observed are augmented under increased pressure, suggesting that secondary structural adaptations become superimposed upon primary genetic ones. In IL-6 Tg(+) mice, where growth development is altered,14 as in the fawn-hooded rat,39 early genetic abnormalities in pulmonary vascular development may contribute to the progression of PAH in the adult Tg(+) mice with and without a hypoxic injurious stimulus.
It is unclear what triggers PAEC and PASMC to have a pro-proliferative phenotype while maintaining an insensitivity towards growth inhibitory stimuli in patients with PAH. In humans, plexiform lesions express angiogenic factors including VEGF and its receptor VEGFR2,40 suggesting that VEGF may play a pro-angioproliferative role in the development of plexiform lesions, a growth factor shared by the plexiform lesions observed in IL-6 Tg(+) mice as well as other animal models with angioproliferative lesions.33 VEGF may also be an important survival factor for PASMC in the presence of IL-6. IL-6 triggers cultured smooth muscle cell proliferation both directly, through up-regulation of VEGFR2 expression and phosphorylation, and indirectly, through up-regulation of matrix metalloproteinase-9.41 IL-6-induced VEGF expression may also indirectly increase the number of PASMC by transforming PAEC into smooth muscle-like cells, as observed in cultured human PAEC.42 These findings, taken with our results, suggest that the presence of abnormal levels of IL-6 may activate, amplify, and maintain the growth and proliferation of PAEC and PASMC by up-regulating VEGF expression.
TGF-β/Bone morphogenic protein(BMP) signaling, a network of proteins that control cell growth, is impaired and the TGF-β receptor is absent in the PAEC in the core of plexiform lesions in PAH.43, 44 This suggests that PAEC in plexiform lesions have lost their check and balance system to control PAEC growth giving rise to a hyper-proliferative PAEC phenotype. In addition, PASMC from patients with PAH are resistant to the anti-proliferative effects of TGF-β,2 suggesting that the failure of TGF-β to suppress PASMC growth in PAH, may in part, underlie the increased muscularization of normally non-muscularized distal pulmonary arteries of patients with PAH. IL-6 has recently been found to negatively regulate the TGF-β/BMP signaling cascade.45 In the IL-6 Tg(+) mouse model, where both muscularization and angioproliferative lesions are abundant and PAH is present, we show that the expression of TGF-β is reduced, in a rich milieu of angioproliferative growth factor VEGF and its receptor. Taken together, the IL-6 Tg(+) mouse model shares similar growth factor characteristics to that of patients with PAH and thus this model may enable investigators to delineate the trigger behind the molecular imbalance which favors the increased expression of proliferative growth factors.
IL-6 overexpression may predispose to proliferative cellular phenotypes and exaggerated PAH as a result of unopposed mitogen activated protein kinase intracellular signaling, normally countered by anti-proliferative TGF-β mediated signaling. Both p38MAPK and ERK are noted to be unopposed in PASMC from patients with mutations in the TGF-β/BMP pathway, resulting in a proliferative apoptotic resistant phenotype.46 The IL-6 Tg(+) mice share a similar biology to patient’s PASMC whereby ERKs activity also is up-regulated in an unopposed environment, which is in part due to the lack of TGF-β and in part due to the lack of pro-apoptotic MAPKinases p38 and pJNK. These findings are also consistent with in vitro work where IL-6 stimulated human endothelial cells47 also have reduced p38 and pJNK. In other cell systems, IL-6 activates the MAPK signaling pathway via ERK and in turn, blocks the TGF-β/BMP pathway by preventing the nuclear translocation of Smad, a downstream BMP signaling protein,48, 49 resulting in cellular proliferation. Given that this mouse model and the vasculature of PAH patients are deficient in growth controlling TGF-β/BMP proteins46 in the setting of elevated IL-6 levels and both share ERK activation, further investigation of unopposed IL-6-ERK signaling may uncover the mechanism by which vascular cells switch from a balanced growth controlled state to an excessively pro-proliferative one.
IL-6 overexpression may predispose to exaggerated PAH as a result of coordinating a number of downstream pro-proliferative, pro-survival, and anti-apoptotic signaling pathways (Figure 6). We found that c-myc, and its obligate binding partner, MAX may be key in IL-6’s downstream proliferative signal cascade, promoting cellular proliferative phenotypes in PAH. While it is also clear from our work that IL-6 selectively blocks apoptosis within the pulmonary vascular bed by down regulating TGF-β and pro-apoptotic MAPKs, pJNK and p38, while upregulating prosurvival factors survivin and Bcl-2. Taken together, these complex obliterative vascular lesions are likely forming because IL-6 is favoring a pro-proliferative anti-apoptotic cell state, as observed in angioproliferative lesions of patients with PAH. (See online supplement for an expanded discussion regarding IL-6 and its role in regulating proliferative and antiapoptotic pathways.)
Figure 6.
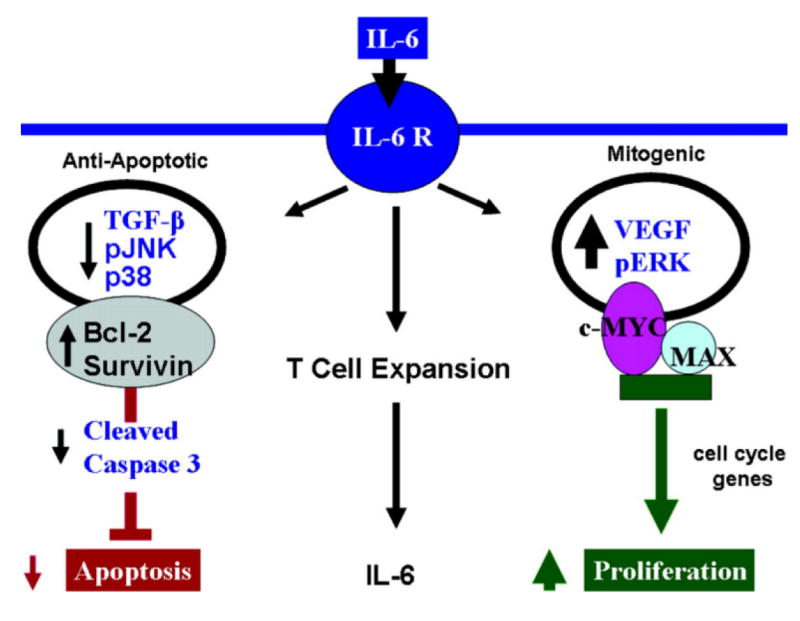
Hypothetical mechanisms leading to hyper-proliferative apoptotic resistant PASMC and PAEC phenotypes that result in angio-proliferative occlusive lesions and muscularization of distal vessels resulting in PAH. IL-6 induces angio-proliferative growth factor VEGF and intracellular MAPK ERK to activate pro-proliferative transcription factor complex c-MYC and MAX resulting in an increase in cell cycle genes to promote PASMC and PAEC proliferation. Concurrently, IL-6 down regulates growth inhibitor TGF-β and other MAPK that are pro-apoptotic (p38 and JNK1) while up-regulating apoptotic inhibitors survivin and Bcl-2 resulting in apoptotic resistant PASMC and PAEC phenotypes. Inflammatory cells, specifically T cells may have an important role in maintaining the secretion of the pro-proliferative cytokine IL-6.
Elevation of IL-6 in the serum and inflammatory cellular infiltrate in plexiform lesions in PAH patients and now IL-6 Tg(+) mice with replicative pathophysiological changes of PAH suggest that cellular immunity may play an active role in the dysregulation of PAEC and PASMC and the development of PAH. Further discussion regarding our findings and controversies of the role inflammation may play in this disease is highlighted in the online supplement material.
The importance of the loss of small pulmonary arteriolar vessels which may contribute to the development of PVR is controversial and for further discussion regarding our data see the online supplement.
In summary, our work supports the hypothesis that IL-6 directly promotes a pro-proliferative apoptotic resistant milieu within the PA wall, resulting in a vasculopathy mirroring that seen in patients with severe PAH. Unlike other murine models, this transgenic mouse model replicates the pathophysiology of human PAH, with muscularization and arteriolar-occlusive changes occurring in the distal vascular bed leading to elevated PVR and secondary chronic pressure overload occurring in the main PA and right ventricle. Our work demonstrates that, in the pulmonary vascular bed, regulation of inflammatory cytokine IL-6 is tightly linked with cellular growth and proliferation through a number of proliferative apoptotic resistant downstream pathways. Future work to establish the underlying mechanism of these complicated signaling systems in the lung specific IL-6 Tg(+) mice, and to determine several of the key molecules necessary to inhibit the excessive proliferative cellular state will improve our understanding of the disease and thereby develop new therapies that target the angioproliferative lesions in patients with refractory PAH. The development of PAH in mice overexpressing IL-6 in the lung, taken with the presence of increased IL-6 in PAH patients, suggests that IL-6 is integral to the development and progression of pulmonary vascular remodeling, PVR, and PAH.
Supplementary Material
Acknowledgments
David A. Schoenfeld, PhD, Biostatistical Center, Harvard Medical School.
Sources of Funding
Supported by NHLBI grants HL074859 (to A.B.W); HL007874 and HL039150 (to C.A.H).
Footnotes
Disclosures
None
References
- 1.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered Growth Responses of Pulmonary Artery Smooth Muscle Cells From Patients With Primary Pulmonary Hypertension to Transforming Growth Factor-{beta}1 and Bone Morphogenetic Proteins. Circulation. 2001;104:790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 3.Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat Med. 2002;8:987–994. doi: 10.1038/nm750. [DOI] [PubMed] [Google Scholar]
- 4.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. Journal of Clinical Investigation. 2005;115:1479–1491. doi: 10.1172/JCI23203. [see comment] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [see comment] [PMC free article] [PubMed] [Google Scholar]
- 6.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. American Journal of Respiratory & Critical Care Medicine. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 7.Katsushi H, Kazufumi N, Hideki F, Katsumasa M, Hiroshi M, Kengo K, Hiroshi D, Nobuyoshi S, Tetsuro E, Hiromi M, Tohru O. Epoprostenol therapy decreases elevated circulating levels of monocyte chemoattractant protein-1 in patients with primary pulmonary hypertension. Circ J. 2004;68:227–231. doi: 10.1253/circj.68.227. [DOI] [PubMed] [Google Scholar]
- 8.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 9.Yoshio T, Masuyama JI, Kohda N, Hirata D, Sato H, Iwamoto M, Mimori A, Takeda A, Minota S, Kano S. Association of interleukin 6 release from endothelial cells and pulmonary hypertension in SLE. Journal of Rheumatology. 1997;24:489–495. [PubMed] [Google Scholar]
- 10.Lesprit P, Godeau B, Authier FJ, Soubrier M, Zuber M, Larroche C, Viard JP, Wechsler B, Gherardi R. Pulmonary hypertension in POEMS syndrome: a new feature mediated by cytokines. American Journal of Respiratory & Critical Care Medicine. 1998;157:907–911. doi: 10.1164/ajrccm.157.3.9707095. [DOI] [PubMed] [Google Scholar]
- 11.Nishimaki T, Aotsuka S, Kondo H, Yamamoto K, Takasaki Y, Sumiya M, Yokohari R. Immunological analysis of pulmonary hypertension in connective tissue diseases. Journal of Rheumatology. 1999;26:2357–2362. [PubMed] [Google Scholar]
- 12.Parravinci C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. Am J Pathol. 1997;151:1517–1522. [PMC free article] [PubMed] [Google Scholar]
- 13.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 14.DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR, Whitsett JA, Flavell RA. Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. Journal of Clinical Investigation. 1994;94:2028–2035. doi: 10.1172/JCI117556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB Journal. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 16.Wang GS, Qian GS, Mao BL, Cai WQ, Chen WZ, Chen Y. Changes of interleukin-6 and Janus kinases in rats with hypoxic pulmonary hypertension. Chung Hua Chieh Ho Ho Hu Hsi Tsa Chih. 2003;26:664–667. [PubMed] [Google Scholar]
- 17.Kubo K, Hanaoka M, Hayano T, Miyahara T, Hachiya T, Hayasaka M, Koizumi T, Fujimoto K, Kobayashi T, Honda T. Inflammatory cytokines in BAL fluid and pulmonary hemodynamics in high-altitude pulmonary edema. Respir Physiol. 1998;111:301–310. doi: 10.1016/s0034-5687(98)00006-1. [DOI] [PubMed] [Google Scholar]
- 18.Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest. 2005;128:572S–573S. doi: 10.1378/chest.128.6_suppl.572S-a. [DOI] [PubMed] [Google Scholar]
- 19.Kasahara Y, Kimura H, Kurosu K, Sugito K, Mukaida N, Matsushima K, Kuriyama T. MCAF/MCP-1 protein expression in a rat model for pulmonary hypertension induced by monocrotaline. Chest. 1998;114:67S. doi: 10.1378/chest.114.1_supplement.67s. [DOI] [PubMed] [Google Scholar]
- 20.Faul JL, Nishimura T, Berry GJ, Benson GV, Pearl RG, Kao PN. Triptolide attenuates pulmonary arterial hypertension and neointimal formation in rats. American Journal of Respiratory & Critical Care Medicine. 2000;162:2252–2258. doi: 10.1164/ajrccm.162.6.2002018. [see comment] [DOI] [PubMed] [Google Scholar]
- 21.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. American Journal of Respiratory & Critical Care Medicine. 2007;175:1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyata M, Sakuma F, Yoshimura A, Ishikawa H, Nishimaki T, Kasukawa R. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int Arch Allergy Immunol. 1995;108:287–291. doi: 10.1159/000237166. [DOI] [PubMed] [Google Scholar]
- 23.Savale L, Izikki M, Tu L, Rideau D, Raffestin B, Naitre B, Adnot S. Attenuated Hypoxic Pulmonary Hypertension in Mice lacking the Interleukin-6 Gene. American Journal of Respiratory & Critical Care Medicine. 2007;175:A42. [Google Scholar]
- 24.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circulation Research. 2004;94:1109–1114. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 25.Merklinger SL, Wagner RA, Spiekerkoetter E, Hinek A, Knutsen RH, Kabir MG, Desai K, Hacker S, Wang L, Cann GM, Ambartsumian NS, Lukanidin E, Bernstein D, Husain M, Mecham RP, Starcher B, Yanagisawa H, Rabinovitch M. Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary hypertension. Circulation Research. 2005;97:596–604. doi: 10.1161/01.RES.0000182425.49768.8a. [DOI] [PubMed] [Google Scholar]
- 26.Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 27.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup V, Hogaboam C, Taraseviciene-Stewart L, Voelkel N, Rabinovitch M, Grunig E, Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. The Journal of Experimental Medicine. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rai PR, Cool C, King J, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The Cancer Paradigm of Severe Angioproliferative Pulmonary Hypertension. Am J Respir Crit Care Medicine. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagenvoort C, Wagenvoort N. Primary Pulmonary Hypertension:A Pathologic Study of the Lung Vessels in 156 Clinically Diagnosed Cases. Circulation. 1970;42:1163–1184. [Google Scholar]
- 30.Palevsky HI, Schloo BL, Pietra GG, Weber KT, Janicki JS, Rubin E, Fishman AP. Primary pulmonary hypertension. Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation. 1989;80:1207–1221. doi: 10.1161/01.cir.80.5.1207. [DOI] [PubMed] [Google Scholar]
- 31.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol. 1997;151:1019–1025. [PMC free article] [PubMed] [Google Scholar]
- 32.Okada K, Bernstein ML, Zhang W, Schuster DP, Botney MD. Angiotensin-converting enzyme inhibition delays pulmonary vascular neointimal formation. American Journal of Respiratory & Critical Care Medicine. 1998;158:939–950. doi: 10.1164/ajrccm.158.3.9710007. [DOI] [PubMed] [Google Scholar]
- 33.Ivy DD, McMurtry IF, Colvin K, Imamura M, Oka M, Lee DS, Gebb S, Jones PL. Development of occlusive neointimal lesions in distal pulmonary arteries of endothelin B receptor-deficient rats: a new model of severe pulmonary arterial hypertension. Circulation. 2005;111:2988–2996. doi: 10.1161/CIRCULATIONAHA.104.491456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagna G, Nguyen PH, Ni W, Hata A. BMP-dependent activation of caspase-9 and caspase-8 mediates apoptosis in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1059–1067. doi: 10.1152/ajplung.00180.2006. [DOI] [PubMed] [Google Scholar]
- 36.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L740–754. doi: 10.1152/ajplung.00284.2002. [DOI] [PubMed] [Google Scholar]
- 37.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 38.Eccleston-Joyner CA, Gray SD. Arterial hypertrophy in the fetal and neonatal spontaneously hypertensive rat. Hypertension. 1988;12:513–518. doi: 10.1161/01.hyp.12.5.513. [DOI] [PubMed] [Google Scholar]
- 39.Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, McMurtry IF, Stelzner TJ. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145:793–797. doi: 10.1164/ajrccm/145.4_Pt_1.793. [DOI] [PubMed] [Google Scholar]
- 40.Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Human pathology. 1997;28:434–442. doi: 10.1016/s0046-8177(97)90032-0. [DOI] [PubMed] [Google Scholar]
- 41.Yao JS, Zhai W, Fan Y, Lawton MT, Barbaro NM, Young WL, Yang GY. Interleukin-6 upregulates expression of KDR and stimulates proliferation of human cerebrovascular smooth muscle cells. J Cereb Blood Flow Metab. 2007;27:510–520. doi: 10.1038/sj.jcbfm.9600365. [DOI] [PubMed] [Google Scholar]
- 42.Sakao S, Taraseviciene-Stewart L, Cool CD, Tada Y, Kasahara Y, Kurosu K, Tanabe N, Takiguchi Y, Tatsumi K, Kuriyama T, Voelkel NF. VEGF-R blockade causes endothelial cell apoptosis, expansion of surviving CD34+ precursor cells and transdifferentiation to smooth muscle-like and neuronal-like cells. FASEB Journal. 2007;21:3640–3652. doi: 10.1096/fj.07-8432com. [DOI] [PubMed] [Google Scholar]
- 43.Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth factor-beta signaling in idiopathic pulmonary arterial hypertension. American Journal of Respiratory & Critical Care Medicine. 2004;170:1340–1348. doi: 10.1164/rccm.200311-1602OC. [DOI] [PubMed] [Google Scholar]
- 44.Yeager ME, Golpon HA, Voelkel NF, Tuder RM. Microsatellite mutational analysis of endothelial cells within plexiform lesions from patients with familial, pediatric, and sporadic pulmonary hypertension. Chest. 2002;121:61S. [PubMed] [Google Scholar]
- 45.Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, West J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–1479. doi: 10.1152/ajplung.00197.2006. [DOI] [PubMed] [Google Scholar]
- 46.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circulation Research. 2005;96:1053–1063. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 47.Waxman AB, Mahboubi K, Knickelbein RG, Mantell LL, Manzo N, Pober JS, Elias JA. Interleukin-11 and Interleukin-6 Protect Cultured Human Endothelial Cells from H2O2-Induced Cell Death. Am J Respir Cell Mol Biol. 2003;29:513–522. doi: 10.1165/rcmb.2002-0044OC. [DOI] [PubMed] [Google Scholar]
- 48.Shi W, Chen H, Sun J, Chen C, Zhao J, Wang YL, Anderson KD, Warburton D. Overexpression of Smurf1 negatively regulates mouse embryonic lung branching morphogenesis by specifically reducing Smad1 and Smad5 proteins. Am J Physiol Lung Cell Mol Physiol. 2004;286:L293–300. doi: 10.1152/ajplung.00228.2003. [DOI] [PubMed] [Google Scholar]
- 49.Chen HB, Shen J, Ip YT, Xu L. Identification of phosphatases for Smad in the BMP/DPP pathway. Genes Dev. 2006;20:648–653. doi: 10.1101/gad.1384706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.