

Abstract
Background
The genome of Protochlamydia amoebophila UWE25, a Parachlamydia-related endosymbiont of free-living amoebae, was recently published, providing the opportunity to search for genomic islands (GIs).
Results
On the residual cumulative G+C content curve, a G+C-rich 19-kb region was observed. This sequence is part of a 100-kb chromosome region, containing 100 highly co-oriented ORFs, flanked by two 17-bp direct repeats. Two identical gly-tRNA genes in tandem are present at the proximal end of this genetic element. Several mobility genes encoding transposases and bacteriophage-related proteins are located within this chromosome region. Thus, this region largely fulfills the criteria of GIs. The G+C content analysis shows that several modules compose this GI. Surprisingly, one of them encodes all genes essential for F-like conjugative DNA transfer (traF, traG, traH, traN, traU, traW, and trbC), involved in sex pilus retraction and mating pair stabilization, strongly suggesting that, similarly to the other F-like operons, the parachlamydial tra unit is devoted to DNA transfer. A close relatedness of this tra unit to F-like tra operons involved in conjugative transfer is confirmed by phylogenetic analyses performed on concatenated genes and gene order conservation. These analyses and that of gly-tRNA distribution in 140 GIs suggest a proteobacterial origin of the parachlamydial tra unit.
Conclusions
A GI of the UWE25 chromosome encodes a potentially functional F-like DNA conjugative system. This is the first hint of a putative conjugative system in chlamydiae. Conjugation most probably occurs within free-living amoebae, that may contain hundreds of Parachlamydia bacteria tightly packed in vacuoles. Such a conjugative system might be involved in DNA transfer between internalized bacteria. Since this system is absent from the sequenced genomes of Chlamydiaceae, we hypothesize that it was acquired after the divergence between Parachlamydiaceae and Chlamydiaceae, when the Parachlamydia-related symbiont was an intracellular bacteria. It suggests that this heterologous DNA was acquired from a phylogenetically-distant bacteria sharing an amoebal vacuole. Since Parachlamydiaceae are emerging agents of pneumonia, this GI might be involved in pathogenicity. In future, conjugative systems might be developed as genetic tools for Chlamydiales.
Background
First described in 1997, Parachlamydia acanthamoebae is an obligate intracellular bacterium naturally infecting free-living amoebae [1,2]. It was isolated from Acanthamoeba spp. recovered from the nasal mucosa of healthy volunteers [1]. Later, additional strains of Parachlamydiaceae have been found within about 5% of Acanthamoeba spp. and once within Hartmanella vermiformis [2,3]. The 16S rRNA sequences of these Parachlamydiaceae are about 14% different from those of both genera Chlamydophila and Chlamydia [2,3]. Since the 16S rRNA sequence difference between Chlamydophila sp. and Chlamydia sp. is 6% only, it clearly appears that the speciation between the two latter occurred after the divergence between Parachlamydiaceae and Chlamydiaceae.
Like other Chlamydiales, Parachlamydiaceae can present two developmental stages: the reticulate body, a metabolically active dividing form, and the elementary body, an infective stage; the crescent body is another infective form, not observed in Chlamydiaceae [4]. Differentiation of the infective stages in reticulate bodies and multiplication of the latter were recently shown to occur within amoebal vacuoles, that may contain hundreds of bacteria [4]. Depending on the symbiotic/pathogenic relationships prevailing between both organisms, the escape of the bacteria from the amoeba may occur either by the release of secreted vesicles or by the lysis of the host [4].
There is a growing evidence of the human pathogenicity of Parachlamydiaceae [2]. For instance, positive Parachlamydia serologies were shown to be associated with a febrile epidemic [5], community-acquired pneumonia [6], and inhalation pneumonia [7]. The role of Parachlamydia-related bacteria as agents of inhalation pneumonia is further suggested by the temperature-dependent release of the bacteria from their amoebal reservoir [8]. PCR amplification of parachlamydial DNA from monocytes, sputa and bronchoalveolar lavages collected from patients suffering of bronchitis or pneumonia also supports the pathogenic potential of Parachlamydia [9-12]. The survival of these Chlamydia-like organisms within human macrophages [13] is an additional hint of parachlamydial pathogenicity.
Horn et al. [14], by sequencing and annotating the whole genome of the Parachlamydia-related UWE25 contributed much to the understanding of the evolution of chlamydiae. Indeed, they demonstrated that major virulence mechanisms of Chlamydiaceae such as the Type Three Secretion System (TTSS) and the Chlamydial Protease-like Activity Factor (CPAF) are also encoded by the chromosome of the evolutionary early-branching Parachlamydiaceae UWE25. Genome analysis of the parachlamydial endosymbiont also identified Open Reading Frames (ORFs) homologous to Type Four Secretion Systems (TFSS) and characterized by a high G+C content, suggesting that they result from an horizontal transfer. Based on their annotation revealing the apparent absence of genes necessary for DNA transfer, Horn et al. [14] proposed that this TFSS was involved in protein export but not in DNA transfer.
To date, numerous genomic islands (GIs) were already identified along whole chromosomal sequences of various bacterial species. For instance, 140 GIs are described in the Islander database, including GIs of proteobacteria, firmicutes, actinobacteria and cyanobacteria [15]. Thus, we wondered whether any GIs were located along the bacterial chromosome of the amoebal endosymbiont UWE25.
GIs are genetic elements which length vary from 10 to 200 kb and are inserted in a chromosome after a lateral transfer occurring, in some instances, between phylogenetically-distant microorganisms. Their heterologous origins are generally evidenced by a G+C content different from that of the remaining bacterial chromosome and by the presence of various mobility genes (i.e. involved in transposition, transduction or conjugative transfer), that are occasionally source of GI instability [16,17]. They are often flanked by particular DNA sequences, such as direct repeats or insertion sequences. Moreover, tRNA loci are generally used as insertion sites by GIs for their chromosomal integration [16-18]. Since no genetic tools are available for the study of this obligate intracellular bacteria, a bioinformatic approach was chosen to locate putative GIs.
Results
A genomic island is present in the genome of UWE25
Using standard G+C content analyses of Parachlamydia-related UWE25 chromosome, we observed a G+C-rich region (Figure 1A and 1B), similar to that shown by Horn et al. [14]. Using the residual cumulative G+C content analysis adapted from the GC profile of Zhang and Zhang [19], we were able to precisely define a 19-kb region (Figure 1C). The presence of 17-bp direct repeats flanking a 100-kb chromosome region (1648 to 1748 kb, Table 1) that encompasses the 19-kb DNA sequence enabled us to define a new region composed of 100 ORFs (See additional data file 1 for the description of these genes and their location on the chromosome of UWE25). Interestingly, this 100-kb region is characterized by a higher level of local gene coorientation (75/100) than that characterizing the remaining of the genome (1015/1931, 52.6%, p < 0.001) and by a particular signature in the cumulative GC skew analysis. Two identical gly-tRNA genes in tandem are located at the proximal end of this 100-kb genetic element (Figure 1A,1C and Table 1). Several mobility genes (eight putative transposases, one recombinase and seven bacteriophage related-proteins) are encoded within the 100-kb region (Figure 1C, Table 1). Thus, this region largely fulfills the accepted criteria of GIs [16-18]. We termed this newly described GI "Pam100G" (Protochlamydia amoebophila, 100-kb, Gly-tRNA) according to the nomenclature used in the Islander database [15].
Figure 1.
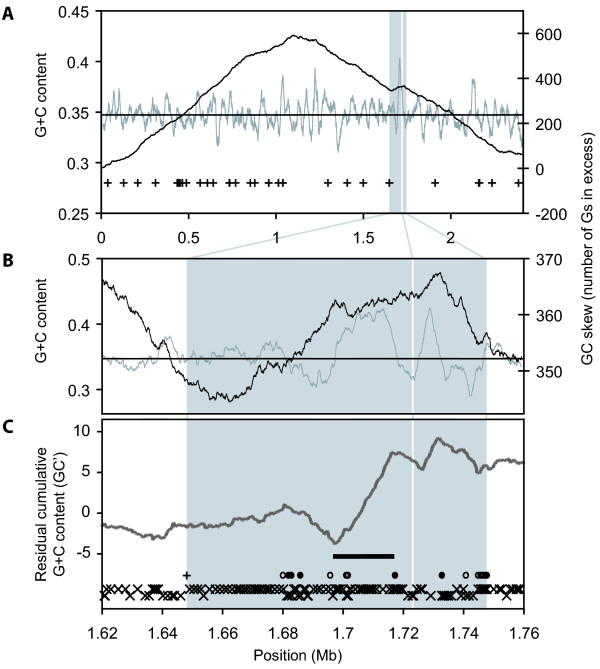
The genomic island (GI) present in the chromosome of the endosymbiont UWE25. (A) Position of the GI on the UWE25 genome, a 100-kb region (grey area) delimited by two direct repeats (Table 1) at both ends and by two gly-tRNAs genes in tandem (all tRNAs genes are represented by '+') at its proximal end. A third copy of the direct repeat (Table 1) is indicated by a white line disrupting the grey area. The region is characterized by a different slope in the cumulative GC skew analysis (black curve) and by a higher G+C content (grey curve, windows of 20 kb, 0.1-kb step). The horizontal line indicates the genomic G+C content average. (B) Closer view of the 100-kb region (black curve, cumulative GC skew; grey curve, G+C content windows of 5 kb, 0.1-kb step; horizontal line, average genomic G+C content). (C) Residual cumulative G+C content (GC') and genomic features of the 100-kb GI. This region encompasses the region with the highest G+C content in the 20-kb windows analysis of the UWE25 genome. The position of genes is represented by an 'X' on the upper line if encoded on the positive strand, otherwise by an 'X' on the bottom line (For details, see Table in Supplementary Material 1). A large majority of genes are co-oriented in the genome region flanked by the direct repeats. The tra operon (thick line), present on this GI, exhibits a G+C content (40.0%) clearly higher than that of the whole genome (34.7%). The positions of transposases (open circles) and of phage-related genes (full circles) are indicated.
Table 1.
Description of main features of the parachlamydial 100-kb genomic island. Chromosome location of direct repeats, tRNA genes, tra operon, transposases, bacteriophage-related proteins and proteins involved in DNA metabolism is listed below.
| Protein numbera | Positiona | |
| Direct repeat | - | 1648147–1648157 |
| gly-tRNAa,b | - | 1648172–1648243 |
| gly-tRNAa,b | - | 1648332–1648403 |
| Transposasec | pc1402 | 1679924–1680400 |
| Phage-related proteinc | pc1404 | 1681569–1682441 |
| Putative transcriptional regulatorc,d | pc1405 | 1679924–1680400 |
| Phage-related proteinc | pc1410 | 1685329–1686447 |
| Putative transposasec | pc1419 | 1695418–1696245 |
| tra operone | pc1420-1441 | 1696410–1716241 |
| Transposasese | pc1426-1427 | 1700887–1701896 |
| Putative DNA-binding proteinc | pc1443 | 1716648–1717004 |
| Phage-related proteinc | pc1444 | 1717137–1717400 |
| Direct repeat | - | 1723093–1723103 |
| Putative ATPase involved in DNA repairc | pc1451 | 1723169–1723504 |
| Probable Doc (death on cure) protein, bacteriophage P1a | pc1456 | 1732622–1732999 |
| Putative DNA-binding protein | pc1461 | 1735745–1736065 |
| Putative transposasec | pc1465 | 1740371–1741198 |
| Probable DNA double-strand break repair ATPase | pc1467 | 1742079–1744181 |
| Putative transposase | pc1468 | 1744634–1745023 |
| Probable resolvasea | pc1469 | 1745398–1745955 |
| Probable transposases, partial lengtha | pc1470-1471 | 1745807–1746692 |
| Probable Doc (death on cure) protein, bacteriophage P1a | pc1473 | 1747135–1747512 |
| Phage-related proteinc | pc1474 | 1747618–1747809 |
| Direct repeat | - | 1747915–1747925 |
a, according to Horn et al. [14];
b, positive strand (cooriented as the majority of the genes of the GI); gly-tRNAs are separated by 88 nt;
c, identified by BLAST [35; 36] and CLUSTALW [39] by ourselves;
d, phage-related protein based on additional BLAST hit;
e, partially annotated by Horn et al. [14] and further characterized by ourselves by BLAST [35, 36].
Mosaicism of the 100-kb genomic island
Interestingly, this GI can be divided into clearly distinct regions, according to their G+C content (Figure 1B, Table 2). The residual cumulative G+C content analysis highlights a modular structure with different slopes, each linear segment indicates that genes of this unit present a rather constant local G+C content (Figure 1C). A positive or a negative slope would indicate that each block of genes presents a G+C content higher or lower that of the UWE25 chromosome, respectively.
Table 2.
The genomic island of the UWE25 endosymbiont presents seven different modules. The limit of each module was determined by residual cumulative G+C content analysis.
| Modules | 1st mod. | 2nd mod. | 3rd mod. | 4th mod. | 5th mod. | 6th mod. | 7th mod. |
| Length | 32 kb | 16 kb | 19 kb | 10 kb | 6 kb | 12 kb | 2 kb |
| Mean G+C content (%) | 36.4%a | 34.1% | 40.9% | 33.4% | 41.8% | 33.3% | 38.7% |
| Number of ORFs | 28 | 18 | 21 | 13 | 1 | 13 | 6 |
| Number of genes having an homolog | 16 (57%) | 5 (28%) | 16 (85%) | 7 (54%) | 1 (100%) | 5 (38%) | 5 (83%) |
| Number of genes having no homolog | 12 | 13 | 5 | 6 | 0 | 8 | 1 |
| Best hits with homologs in: | |||||||
| - chlamydiae | 12 | 0 | 0 | 0 | 0 | 0 | 0 |
| - cyanobacteria | 2 | 2 | 0 | 3 | 0 | 2 | 0 |
| - plantsb | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| - α-proteobacteria | 0 | 0 | 6 | 0 | 0 | 0 | 0 |
| - β-proteobacteria | 1 | 0 | 3 | 1 | 0 | 0 | 0 |
| - γ-proteobacteria | 0 | 1 | 6 | 2 | 0 | 0 | 4c |
| - Bacteroidetes group | 0 | 0 | 1 | 0 | 0 | 2 | 1 |
| - others | 1 | 2 | 0 | 1 | 1 | 1 | 0 |
| Homologousd to: | |||||||
| - phage-related protein | 0 | 3 | 0 | 1 | 0 | 1 | 2 |
| - putative transposase | 1 | 1 | 2 | 0 | 0 | 2e | 2 |
| - resolvase | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| - protein involved in DNA metabolism | 0 | 1 | 0 | 2 | 0 | 2 | 0 |
a the G+C content is similar to that of 36.1% of the remaining genome (calculated on 1931 ORFs);
b 5% of the 2031 ORFs of the genome of UWE25 have products homologous to plant proteins, but no ORFs of the GI were homologous to plant counterparts;
c two ORFs which presented best homologs encoded by a plasmid of an uncultured bacteria present in activated sludge have a second best BLAST hit encoded by a gamma-proteobacterial ORFs (Pseudomonas sp.);
d not only the best BLAST hit is taking into consideration to determine the putative function encoded by the ORF;
e one of them has an e-value above 0.001.
The first module begins with a direct repeat and two identical gly-tRNAs in tandem. Composed of 28 ORFs, this unit exhibits a G+C content (36.4%) similar to that of the remaining of the genome of UWE25 (1931 ORFs, 36.1%). Sixteen homologs to these 28 genes (57%) were found in databases, 12 of them (75%) exhibiting a best score in BLAST analyses with a Chlamydiaceae ORF (See additional data file 1). Interestingly, no gene of the other modules of the 100-kb GI exhibited a best hit in similarity analyses with any Chlamydiaceae counterpart. Some of the genes present in the first module, such as sctN and sctQ, are part of a TTSS also present in Chlamydiaceae. The other TTSS genes are disseminated along the chromosome of UWE25. The presence of some TTSS genes in the first module and of a gene encoding a putative transposase at the distal end of the first module of this 100-kb GI suggests that this first unit was acquired by chromosomal rearrangements. (See additional data file 1 for the results of BLAST analyses).
Characterized by a low G+C content (34.1%), the 2nd module encodes 18 ORFs. Only five are similar to known protein sequences (28%), four of them being identified as mobility genes (three bacteriophage-related genes and one putative transposase encoding gene).
The 3rd module (19 kb), exhibiting the second highest G+C content of the UWE25 genome (40.9%), comprises 21 ORFs. Some of these genes were identified as tra genes by Horn et al. [14]. Using BLAST analyses and alignment tools, we re-annotated the whole module (see below) and, if we except two transposase genes and one ORF of unknown function, we unveiled that all ORFs of this module belong to a genetic unit similar to the tra operons encoding the TFSS previously described in proteobacterial genomes (Figure 2, See additional data file 1 for the re-annotations of this module).
Figure 2.
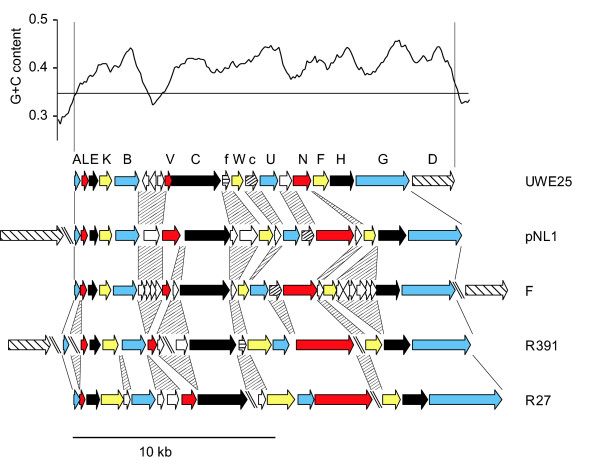
Comparison of the tra unit of the endosymbiont UWE25 with similar operons of pNL1 (Novosphingobium aromaticivorans), F (Escherichia coli), R391 (Providencia rettgeri) and R27 (Salmonella Typhi) plasmids. In the upper part of the figure: the G+C content of the UWE25 chromosome, around the 1.71 Mb location (1-kb sliding window average, 0.1-kb step). The horizontal line represents the genomic G+C content average. Only the ORFs composed of more than hundred amino-acids are presented on genetic maps of tra units/operons by arrows according to their transcription direction (adapted from Lawley et al. [20]). Colors or patterns are used to indicate tra gene homologs. White genes represent non-conserved transfer genes. Upper case letters refer to the corresponding tra genes, whereas lower case letters f and c stand for trsF and trbC, respectively. Double slashes indicate non-contiguous regions. Interestingly, the G+C-rich genes encoded by the UWE25 chromosome correspond to the ORFs presenting tra homologs.
Presenting a low G+C content (33.4%), the 4th module (10 kb) is composed of 13 ORFs. All these ORFs were previously annotated by Horn et al. [14] as encoding hypothetical proteins or without homolog. Our BLAST analysis identified one ORF homologous to genes encoding bacteriophage-related proteins and two genes of proteins involved in DNA metabolism (Table 1 and 2, see also the table in the additional data file 1). Interestingly, a direct repeat is located between the 9th and 10th genes of the module. This 17-bp direct repeat, that presents 3 mismatches is similar to those present at the proximal and distal ends of the GI, exhibiting the same 14 conserved nucleotides. It may reflect a complex evolutionary history of the GI, possibly enabling it to be mobile as 25-kb, 75-kb or 100-kb DNA segments.
A single large protein is encoded along the 5th module (6 kb). Its G+C content is one of the highest of the UWE25 chromosome (41.8%). By BLAST analysis, this protein exhibits the strongest similarity with the human Nod3 protein.
The 6th module (12 kb) is characterized by a low G+C content (33.3%). This unit is composed of 13 ORFs, the first ORF encoding a product similar to the Death on cure (Doc) protein of P1 bacteriophage. Two ORFs code for proteins involved in DNA metabolism and an additional ORF encodes a putative transposase.
The 7th module is short (2 kb) and present a G+C-rich unit (38.7%). Five of the six ORFs of this unit encode a probable resolvase, three putative transposases and a phage-related Doc protein. The final direct repeat is located at the end of this module. With the only exception of the phage-related protein, all other ORFs of the 7th module appear to be similar to gamma-proteobacterial proteins, possibly explaining the observed different signal in the G+C content analysis.
Role of the type IV secretion system encoded by the 100-kb genomic island
The functions of genes encoded by GIs may be related, among others, to pathogenicity such as the ability to exploit the host intracellular environment. Since no genetic system has been described for any obligate intracellular chlamydiae, we investigated the putative functions of this GI by bioinformatics. We focused our analyses on the TFSS, for which a previous annotation of the tra genes showed a genetic unit unable to transfer DNA [14]. Using different protein comparison methods described in the additional data file 1, we identified supplementary tra genes, and compared the general organization of this tra unit with other genetic elements encoding TFSS genes [20]. The UWE25 tra unit displays a striking colinearity with tra operons encoding F-like conjugative DNA transfer system, especially to those of the F and pNL1 plasmids of Escherichia coli and Novosphingobium aromaticivorans, respectively (Figure 2). All homologous genes essential for DNA transfer in plasmid F (traF, traG, traH, traN, traU, traW, and trbC) and involved in sex pilus retraction and mating pair stabilization [20] are present, strongly suggesting that, similarly to the other F-like TFSSs, the gene products encoded by the UWE25 tra unit are devoted to DNA transfer. With the only exception of traG, these genes are not present on P-like and I-like plasmids, reinforcing the close relationship prevailing between the UWE25 tra unit and their F-like plasmids counterparts.
Figure 3 shows that the UWE25 tra unit clusters within F-like TFSSs, confirming that it may function as a F-like conjugative system. Drawn as an UPGMA tree (Figure 3A), the comparison of the genetic organization of all tra units was performed as a gene order breakpoint analysis developed for the study of the mitochondrial genome evolution [21]. This analysis clearly shows that the closest relatives of the UWE25 tra units are the tra operons of the F-like conjugative plasmids. The Fitch-Margoliash- and the minimum evolution comparisons performed on the same dataset presented the same tree topologies, confirming the former UPGMA results (data not shown). An omit test performed on this tree confirms that the results are robust: with one exception (involving the deep branching of one cluster on one tree), all 11 trees were congruent in all their nodes. Figure 3B shows an UPGMA tree comparing the Kimura corrected p-distances (the proportion p of nucleotide sites at which two sequences are different, taking into account the proportion of transversion- and transition-substitution rates) of nucleotide sequences of the concatenated traA, traK, traB, traV, and traC genes. A similar topology is observed with (i) neighbor-joining- and minimum evolution trees inferred using the Kimura-corrected p-distances and (ii) UPGMA, neighbor-joining- and minimum evolution trees performed on p-distance of the whole coding sequences of the concatenated tra genes (See additional data file 2 for these trees). Neighbor-joining- and minimum evolution methods comparing Kimura-corrected p-distances of the complete coding sequences confirmed that the tra unit of UWE25 is phylogenetically closely related to the tra operons of the F-like plasmids: the bootstrap values of 94% and 91% respectively, support the node separating the concatenated tra genes of the chromosomal UWE25 and the R27 plasmid, a gamma-proteobacterial F-like conjugative plasmid, from those of all other plasmids (See the additional data file 2 for these trees). In neighbor-joining and minimum evolution analyses of p-distances, the tra unit of UWE25 also clusters with the tra operons of gamma-proteobacterial F-like plasmids: the bootstrap of 96% and 92%, respectively, support the node separating the concatenated tra genes of UWE25 and RTS1, SXT, R391, three gamma-proteobacterial F-like conjugative plasmid, from their closest relative, R27 plasmid (See additional data file 2 for these trees). Taken together, all these data strongly suggest that the UWE25 tra unit is closely related to F-like conjugative tra operons.
Figure 3.
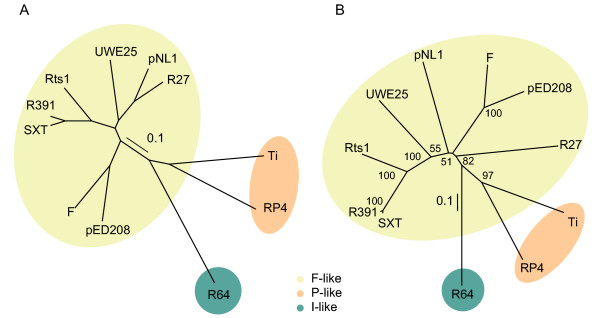
Similarity and phylogenetic analyses of tra units showing the close relatedness of the UWE25 tra unit with the operons involved in the F-like conjugative systems: (A) UPGMA tree of gene order analysis and (B) UPGMA tree comparing the Kimura corrected p-distances of the concatenated traA, traK, traB, traV, and traC gene present along the UWE25 tra unit and the F-like, I-like and P-like plasmids [20]. The bar represents estimated evolutionary distance scale. The numbers at each node are the results of a bootstrap analysis; each value is derived from 100 samples.
Origin of the genomic island and of its type four secretion system
Our BLAST analyses [22] reveal that a majority (24/43) of genes not presenting a best hit for chlamydial genes but having homologs in other taxa are more related to proteobacterial genes (see Table 2 and the additional data file 1 for similarity analyses indicating for each parachlamydial tra gene the most similar gene and its taxonomical background). Moreover, the BLAST analyses of the 21 ORFs of the third module, encoding the tra genes, show that most ORFs of this unit (15/21) are of proteobacterial origin. However, since six of them present the highest similarity to alpha-proteobacterial genes and six others to gamma-proteobacterial genes, a more precise origin of the parachlamydial tra unit could not be precisely defined by this first approach.
The presence of gly-tRNA at the proximal end of the GI of UWE25 is consistent with a close relatedness between this GI and proteobacteria: out of 14 GIs described in the Islander database of Mantri et al. [15,22] inserted along a chromosome by a gly-tRNA (14/140), 12 of them (86%) were sequenced in a proteobacterial genome. No GI of Gram-positives described in the Islander database are inserted in a chromosome within a gly-tRNA gene. Again, a precise proteobacterial origin could not be proposed, because the distribution of gly-tRNA genes in alpha- (4/22) and gamma-proteobacterial (8/72) GIs is not significantly different: by including only the non-redundant GIs, the distribution of gly-tRNA genes in alpha and gamma-proteobacterial GIs is 2/20 and 7/71, respectively.
Comparison of gene order between all tra units also failed in assigning a precise origin to the UWE25 tra unit since it branched near the alpha- and gamma-proteobacterial tra operons (Figure 3A). The only first hint of a possible gamma-proteobacterial origin for the UWE25 tra unit was brought by the phylogenetic analyses (Figure 3B and additional files 1 &2). Thus, bootstraps values of 94, 91, 96 and 92% supported the node separating the concatenated tra genes of UWE25 and several tra operons of gamma-proteobacterial F-like plasmids from the F-plasmids of an alpha-proteobacteria and of other gamma-proteobacteria. (See above, and additional data file 2 for these trees).
Discussion
We showed that the Parachlamydia-related endosymbiont UWE25 presents a 100-kb region largely fullfilling the criteria of GIs [16-18]. Indeed, this DNA region characterized by a high level of gene co-orientation presents a G+C content different from that of the remainder of the genome. The presence of direct repeats flanking this chromosome region enabled us to focus on 100 ORFs. Two identical gly-tRNA genes in tandem are present at the proximal end of this genetic element. Moreover, several mobility genes encoding transposases and bacteriophage related-proteins are located within this chromosome region.
The cumulative residual G+C content analysis shows that this GI is composed of seven modules. Such a chimeric organization was already described in other GIs [23,24]. The first module contains chlamydiae genes probably brought by chromosome rearrangements. Some of these genes, homologous to TTSS genes of Chlamydiaceae, might provide selective advantages to strains that retained the GI. The 2nd, 4th and 6th modules are mainly composed of bacteriophage-related protein genes, that could reflect a putative phage implication in GI formation.
The 3rd module codes for a TFSS similar to tra operons. We propose that this tra unit is devoted to DNA transfer, based (i) on similarity analyses demonstrating the presence of all genes encoding proteins used during a DNA transfer, (ii) on phylogenetic analyses of tra unit genes and, (iii) on comparison of gene order. These analyses clearly demonstrate that the UWE25 tra unit is strongly more related to F-like conjugative system than to P-like and I-like secretion systems. The significant bootstraps of all trees obtained by standard gene phylogeny and their congruent topologies with others obtained by the gene order breakpoint analysis not biased by codon usage homing, strongly support the validity of these analyses confirming the F-like conjugative nature of the parachlamydial tra unit. Thus, our model significantly differs from the other proposed by Horn et al. [14], who did not identify traA, traL, traK, traV, and concluded that the UWE25 tra unit is involved in protein export, and not in DNA transfer.
The 5th module presents a nucleotide composition similar to the tra unit and is composed of a single high G+C 6-kb gene, whose product is similar to the human Nod3 protein. The Nod (Nucleotide-binding oligomerization domain) proteins are members of a family that also includes the apoptosis regulator Apaf1 (Apoptotic protease activating factor 1) and plant disease-resistance gene products [25]. The function of the human Nod3 is still unknown. Like Nod1 and Nod2, Nod3 might be involved in the recognition of conserved motifs present at the surface of bacteria, such as peptidoglycan.
The nucleotide G+C composition of the 2nd, 4th, and 6th modules are similar, explaining the observed similar negative slope of the residual G+C curves. Moreover, these three modules encode phage-related proteins and proteins involved in DNA metabolism. These modules probably involved in mobility might have a common origin, the ancestral single phage module being currently separated in three pieces by the presence of the tra unit and of the Nod3-like protein encoding gene.
The positive slope in the G+C analysis of the 7th module echoes those of the tra unit (3rd module) and of the Nod3-like protein (5th module). The 7th module encodes a transposition resolvase and three transposases similar to gamma-proteobacterial homologs. With the only exception of the phage-related Doc protein, that has an homolog at the beginning of the sixth module, and that might be located there after transposition, the 7th module appears thus to have a different origin than the 2nd, 4th and 6th modules, though also encoding mobility genes.
The presence of a F-like tra unit along the sequences of UWE25 is the first evidence of a putative conjugative system in chlamydiae. If conjugation occurs, it most probably takes place within free-living amoebae, that may contain several hundreds of Parachlamydia bacteria tightly packed in their vacuoles [4]. Such a conjugation system would be a mechanism to transfer DNA between internalized bacteria sharing an amoebal vacuole. Moreover, it may provide molecular genetic tools for obligate intracellular bacteria.
The presence of tra units/operons in the parachlamydial UWE25 and in proteobacteria could be explained by an emergence of this unit in a common ancestor of both clades, and by its subsequent loss in Chlamydiaceae. Another evolutionary scenario is that the tra unit was acquired from a proteobacteria by a Parachlamydiaceae in a common amoebal vacuole. Since the tra unit is absent from all sequenced Chlamydiaceae genomes, this transfer would have occurred after the divergence of Parachlamydiaceae and Chlamydiaceae, at a time when Parachlamydia was already an intracellular bacteria. An intra-amoebal transfer of this GI is supported by the permissivity of free-living amoebae to proteobacteria [26], and by several hints suggesting its proteobacterial origin. Though phylogenetic analyses suggested a gamma-proteobacterial origin of the F-like parachlamydial tra, further analyses have to confirm whether this GI module was acquired from an alpha-, beta-, or gamma-proteobacteria unit. We hypothesize that the F-like parachlamydial tra unit has been brought by a lateral transfer from a proteobacterial genome. This hypothesis is strongly supported by the cumulative GC skew analysis [27-30] producing a signal of the GI differing from that of the remaining of the genome (Figure 1A and 1B). The value of nucleotide skew analyses as good taxonomical markers is supported by (i) routine analyses on prokaryotic genome by cumulative TA-skews [30] and (ii) comparison of intragenic nucleotide skews of small subunit ribosomal RNA of the whole living world [31]. The genometric approach appeared to be able to identify GIs of Chlamydiales. Sequencing additional genomes of environmental chlamydiae, that present a large biodiversity [3], will provide major insights on bacterial evolution and hopefully a better comprehension of the emergence of this parachlamydial GI.
Conclusions
We showed that a GI present on the UWE25 chromosome encodes a potentially functional F-like DNA conjugative system. This is the first hint of a putative conjugative system in chlamydiae. Conjugation most probably occurs within free-living amoebae, that may contain hundreds of Parachlamydiaceae bacteria tightly packed in vacuoles. Such a conjugative system might be involved in DNA transfer between internalized bacteria. Since this system is absent from the sequenced genomes of Chlamydiaceae, we hypothesize that it was acquired after the divergence between Parachlamydiaceae and Chlamydiaceae, when the Parachlamydia-related symbiont was an intracellular bacteria. It suggests that this heterologous DNA was acquired by a Parachlamydiaceae from phylogenetically-distant bacteria sharing an amoebal vacuole. Since Parachlamydiaceae are emerging agents of pneumonia [2] and since many GIs are also considered as pathogenicity islands [17], the Pam100G GI might be involved in pathogenicity. In future, conjugative systems might be developed as genetic tools for studying Chlamydiales.
Methods
Sequence
The genome sequence of UWE25 [14] (Accession number: NC_005861) is available at the NCBI website [32,33]. In this contribution, the acronym UWE25 refers only to the Parachlamydia-related endosymbiont UWE25, and thus not to the Acanthamoeba sp. strain UWE25 from which the parachlamydial endosymbiont UWE25 was recovered [3]. Horn et al. recently proposed UWE25 as the type strain of a new bacterial species: Protochlamydia amoebophila [34].
BLAST analyses
BLAST analyses were performed with BLASTP 2.2.9 [35] available on the NCBI website [36] using the BLOSUM62 matrix, and gap penalties of 11 and 1. Each ORF was compared against all genes of non-redundant databases available at the NCBI website. An e-value of 0.001 was selected as a standard cut-off. To further identify possible homologous ORFs, we also BLASTed each tra gene of F plasmid versus all genes of the full genome of Parachlamydia and conversely, each ORF of the putative parachlamydial tra unit versus counterparts of the different F-like plasmids. CLUSTALW was used to detect the best relatedness of a given parachlamydial Tra protein with its possible homologs encoded by the F and pNL1 plasmids.
Residual cumulative GC content
The residual cumulative G+C content, a slightly modified version of the cumulative GC profile defined by Zhang and Zhang [19], was used to reveal local variations of G+C content of a genome, without using sliding windows of arbitrary size. First, a G+C content analysis was performed on 100-bp windows of the selected chromosome sequence, as for a cumulative GC skew analysis. The cumulative G+C content GCn of the nth window is obtained by cumulating the G+C contents from the first to the nth window:
![]()
where, in the window i, Gi and Ci are the numbers of Gs and Cs, respectively, and Ni is the total number of nucleotides. To visualize genomic regions differing from the average G+C content, a linear regression y defined by a slope k is performed on the cumulative curve using the least square methods:
y(n) = kn
where n is the position of the center of the nth window. The residual cumulative G+C content curve GC' can then be drawn as a function of the position of each window center:
GC'n = GCn - kn
Zhang and Zhang [19] recently demonstrated that, in some instances, abrupt changes in the residual cumulative G+C content curve correspond to genomic islands.
Repeats identification
The perfect tandem repeats identification was first performed using the EQUICKTANDEM software (Richard Durbin, Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SA, UK) [37] on a 200-kb DNA sequence (UWE25 genome position: 1.6 to 1.8 Mb) encompassing the tra genes previously identified by Horn et al. [14]. The duplicated genes and the ORFs containing internal repeats were removed. For each pair of direct repeats, potential unperfect matches of flanking nucleotides were scanned using DNA strider 1.2.1 [38], with the following settings: a minimal size of 11 bp and 3 mismatches. Furthermore, sequences similar to direct repeats were searched along the whole chromosome, and sequences also found outside the selected 200-kb region were discarded from our analysis. Finally, the direct repeats positions were compared to the G+C content analysis, the cumulative GC skew curve, and to tRNA genes locations.
Phylogenetic analyses
Since Horn et al. [14] did not identified traA, traL, traK, traV, re-annotation of the UWE25 tra unit was necessary for phylogenetic analyses. We used i) the genes of F-like plasmids encoding the following tra genes, i.e. traA, traK, traB, traV, traC, and ii) the corresponding ORFs of P- and I-like plasmids [20], i.e. trbC/VirB2, trbG/VirB9, trbI/VirB10, trbH/VirB7, trbE/VirB4 of P-plasmids and traX, traN, traO, traI, traU of I-plasmids, respectively. The genes were concatenated to obtain a single nucleotide sequence and aligned with CLUSTALW ([39] as it was already performed for genes of ribosomal proteins [40]. Using this alignment and the MEGA 2.1 software [41], we inferred phylogenetic relationships by drawing trees using p-distances (the proportion p of nucleotide sites at which two sequences compared are different) and Kimura corrected p-distance (correction for the rates of transition and transversion) with Unweighted Pair Group Method with Arithmetic Mean (UPGMA), neighbor-joining, and minimum evolution methods. To prevent alignment biases, trees were drawn using the complete deletion option implemented on MEGA 2.1.
Gene order breakpoint analyses
To quantify the inversion and transposition events leading to the current organization of tra operons, the gene order breakpoint analysis developed for small genomes (mitochondria) by Blanchette et al. [21] was used to estimate the similarity of gene order existing between the tra unit of UWE25 and the tra operons reviewed by Lawley et al. [20]. The distance calculated for two given operons Oi and Oj containing homologous genes proposed by Blanchette et al. [21] was slightly modified to take into account the variation of gene numbers of tra operons: instead of counting the number of minimal breakpoints existing between two tra operons, a distance was estimated by measuring the proportion of conserved gene pairs between both genomic entities. Next, a comparison matrix is established by calculating the distance for each pairwise comparison. Finally, a dissimilarity matrix is obtained by subtracting each distance from 1.
For instance, if the operon Oi encodes sequentially four genes (a, b, c, and d) and the operon Oj, six genes (a, b, e, -d, -c, and -f; genes labeled by a minus sign are encoded by the complementary strand), the gene order breakpoint analysis reveals that two gene pairs are conserved: ab and cd. The dissimilarity distances existing between the operons i) Oi and Oj, and ii) Oj and Oi would be: 1-(2/3) = 1/3 and 1-(2/5) = 3/5, respectively.
From the square dissimilarity matrix, phylogenetic trees were drawn. Three different distance-matrix analyses were used: the UPGMA, the Fitch-Margoliash- and the minimum evolution methods. To assess the robustness of the tree, an omit test [42] was performed on 11 UPGMA trees, in each one organism is missing.
Authors' contributions
GG and CAR initiated the project. GG and FC reannotated the parachlamydial tra unit and performed all BLAST analyses. FC and LG delimited the GI by direct repeat analyses. GG drew the phylogenetic trees of the concatenated tra genes. After developing the residual cumulative G+C content analysis used for a software development, LG performed the G+C content analyses and the gene order comparison of tra units by the gene order breakpoint analysis. CAR established the correlation existing between the tRNA genes and the GIs according to taxonomy and coordinated the team work. GG wrote the first draft of the paper. All authors improved the manuscript and approved its final version.
Supplementary Material
Supplementary table. Results of BLAST [35,36] analyses of 100 ORFs present in the 100-kb region. BLAST analyses were performed using BLOSUM62 matrix and gap penalties of 11 and 1. Chromosome location of each ORF, its G+C content, coding strand, and the presence of at least one homolog in Chlamydiaceae are presented. Direct repeats (DR), gly-tRNA genes and limits of each modules of the GI are highlighted.
Supplementary figure. Phylogenetic analyses suggest that the UWE25 tra unit is phylogenetically closely related to F-like DNA conjugative tra operons: (A) UPGMA-, (B) Neighbor-joining-, and (C) minimum evolution-trees comparing p-distances and Kimura corrected p-distances of nucleotide sequences of the concatenated traA, traK, traB, traV, and traC genes (the UPGMA tree comparing the Kimura corrected p-distances of tra genes, shown in Figure 3B, is presented here to facilitate comparison with the other trees). Interestingly, in neighbor-joining and minimum evolution analyses of the p-distances, the tra unit of UWE25 is clustered with tra operons of gamma-proteobacterial F-like plasmids: bootstrap values of 96% and 92%, respectively, support the node separating the concatenated tra genes of UWE25 and RTS1, SXT, R391, three gamma-proteobacterial F-like conjugative plasmids, from their closest relative R27 plasmid. Similarly, in neighbor-joining and minimum evolution analyses of the Kimura corrected p-distances, bootstrap values of 94% and 91%, respectively, support the node separating the concatenated tra genes of the chromosomal UWE25 and the R27 plasmid, another gamma-proteobacterial F-like conjugative plasmid, from those of all other plasmids.
Contributor Information
Gilbert Greub, Email: gilbert.greub@hospvd.ch.
François Collyn, Email: francois.collyn@unil.ch.
Lionel Guy, Email: lionel.guy@unil.ch.
Claude-Alain Roten, Email: claude-alain.roten@unil.ch.
References
- Amann R, Springer N, Schonhuber W, Ludwig W, Schmid EN, Muller KD, Michel R. Obligate intracellular bacterial parasites of Acanthamoebae related to Chlamydia spp. Appl Environ Microbiol. 1997;63:115–21. doi: 10.1128/aem.63.1.115-121.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Raoult D. Parachlamydiaceae: potential emerging pathogens. Emerg Infect Dis. 2002;8:625–30. doi: 10.3201/eid0806.010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche TR, Horn M, Wagner M, Herwig RP, Schleifer KH, Gautom RK. Phylogenetic diversity among geographically dispersed Chlamydiales endosymbionts recovered from clinical and environmental isolates of Acanthamoeba spp. Appl Environ Microbiol. 2000;66:2613–19. doi: 10.1128/AEM.66.6.2613-2619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Raoult D. Crescent bodies of Parachlamydia acanthamoeba and its life cycle within Acanthamoeba polyphaga: an electron micrograph study. Appl Environ Microbiol. 2002;68:3076–84. doi: 10.1128/AEM.68.6.3076-3084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtles RJ, Rowbotham TJ, Storey C, Marrie TJ, Raoult D. Chlamydia-like obligate parasite of free-living amoebae. Lancet. 1997;349:925–26. doi: 10.1016/s0140-6736(05)62701-8. [DOI] [PubMed] [Google Scholar]
- Marrie TJ, Raoult D, La Scola B, Birtles RJ, de Carolis E. Legionella-like and other amoebal pathogens as agents of community-acquired pneumonia. Emerg Infect Dis. 2001;7:1026–29. doi: 10.3201/eid0706.010619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Boyadjiev I, La Scola B, Raoult D, Martin C. Serological hint suggesting that Parachlamydiaceae are agents of pneumonia in polytraumatized intensive care patients. Ann N Y Acad Sci. 2003;990:311–19. doi: 10.1111/j.1749-6632.2003.tb07381.x. [DOI] [PubMed] [Google Scholar]
- Greub G, La Scola B, Raoult D. Parachlamydia acanthamoeba is endosymbiotic or lytic for Acanthamoeba polyphaga depending on the incubation temperature. Ann N Y Acad Sci. 2003;990:628–34. doi: 10.1111/j.1749-6632.2003.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Ossewaarde JM, Meijer A. Molecular evidence for the existence of additional members of the order Chlamydiales. Microbiology. 1999;145:411–17. doi: 10.1099/13500872-145-2-411. [DOI] [PubMed] [Google Scholar]
- Corsaro D, Venditti D, Le Faou A, Guglielmetti P, Valassina M. A new Chlamydia-like 16S rDNA sequence from a clinical sample. Microbiology. 2001;147:515–16. doi: 10.1099/00221287-147-3-515. [DOI] [PubMed] [Google Scholar]
- Corsaro D, Venditti D, Valassina M. New parachlamydial 16S rDNA phylotypes detected in human clinical samples. Res Microbiol. 2002;153:563–67. doi: 10.1016/S0923-2508(02)01369-4. [DOI] [PubMed] [Google Scholar]
- Greub G, Berger P, Papazian L, Raoult D. Parachlamydiaceae as rare agents of pneumonia. Emerg Infect Dis. 2003;9:755–56. doi: 10.3201/eid0906.020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Mege JL, Raoult D. Parachlamydia acanthamoeba enters and multiplies within human macrophages and induces their apoptosis. Infect Immun. 2003;71:5979–85. doi: 10.1128/IAI.71.10.5979-5985.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes HW, Wagner M. Illuminating the evolutionary history of Chlamydiae. Science. 2004;304:728–30. doi: 10.1126/science.1096330. [DOI] [PubMed] [Google Scholar]
- Mantri Y, Williams KP. Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res. 2004;32:D55–D58. doi: 10.1093/nar/gkh059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–79. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- Dobrindt U, Hochhut B, Hentschel U, Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. 2004;2:414–24. doi: 10.1038/nrmicro884. [DOI] [PubMed] [Google Scholar]
- Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997;23:1089–97. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang CT. A systematic method to identify genomic islands and its applications in analyzing the genomes of Corynebacterium glutamicum and Vibrio vulnificus CMCP6 chromosome I. Bioinformatics. 2004;20:612–22. doi: 10.1093/bioinformatics/btg453. [DOI] [PubMed] [Google Scholar]
- Lawley TD, Klimke WA, Gubbins MJ, Frost LS. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett. 2003;224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kunisawa T, Sankoff D. Gene order breakpoint evidence in animal mitochondrial phylogeny. J Mol Evol. 1999;49:193–203. doi: 10.1007/pl00006542. [DOI] [PubMed] [Google Scholar]
- Islander Database http://129.79.232.60/cgi-bin/islander/islander.cgi
- Pickard D, Wain J, Baker S, Line A, Chohan S, Fookes M, Barron A, Gaora PO, Chabalgoity JA, Thanky N, Scholes C, Thomson N, Quail M, Parkhill J, Dougan G. Composition, acquisition, and distribution of the Vi exopolysaccharide-encoding Salmonella enterica pathogenicity island SPI-7. J Bacteriol. 2003;185:5055–65. doi: 10.1128/JB.185.17.5055-5065.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collyn F, Billault A, Mullet C, Simonet M, Marceau M. YAPI, a New Yersinia pseudotuberculosis Pathogenicity Island. Infect Immun. 2004;72:4784–90. doi: 10.1128/IAI.72.8.4784-4790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–82. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Greub G, Raoult D. Microorganisms resistant to free-living amoebae. Clin Microbiol Rev. 2004;17:413–33. doi: 10.1128/CMR.17.2.413-433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26:2286–90. doi: 10.1093/nar/26.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank AC, Lobry JR. Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene. 1999;238:65–77. doi: 10.1016/S0378-1119(99)00297-8. [DOI] [PubMed] [Google Scholar]
- Frank AC, Lobry JR. Oriloc: prediction of replication boundaries in unannotated bacterial chromosomes. Bioinformatics. 2000;16:560–561. doi: 10.1093/bioinformatics/16.6.560. [DOI] [PubMed] [Google Scholar]
- Roten CA, Gamba P, Barblan JL, Karamata D. Comparative Genometrics (CG): a database dedicated to biometric comparisons of whole genomes. Nucleic Acids Res. 2002;30:142–44. doi: 10.1093/nar/30.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L, Roten C-AH. Genometric analyses of the organization of circular chromosomes: a universal pressure determines the direction of ribosomal RNA genes transcription relative to chromosome replication. Gene. 2004;340:45–52. doi: 10.1016/j.gene.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Church DM, Edgar R, Federhen S, Helmberg W, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Suzek TO, Tatusova TA, Wagner L. Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res. 2004;32:D35–D40. doi: 10.1093/nar/gkh073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI web site http://www.ncbi.nlm.nih.gov/
- Protochlamydia amoebophila Genome Database http://webclu.bio.wzw.tum.de/genre/proj/uwe25/
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI BLAST Tools http://www.ncbi.nlm.nih.gov/BLAST/
- EQUICKTANDEM Software http://bioweb.pasteur.fr/seqanal/interfaces/equicktandem.html
- Marck C. 'DNA Strider': a 'C' program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic Acids Res. 1988;16:1829–36. doi: 10.1093/nar/16.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV. Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol. 2001;1:8. doi: 10.1186/1471-2148-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: Molecular Evolutionary Genetics Analysis software. Arizona State University ed. Tempe, Arizona, USA; 2001. [DOI] [PubMed] [Google Scholar]
- Greub G, Raoult D. History of the ADP/ATP-translocase-encoding gene, a parasitism gene transfered from a Chlamydiales ancestor to Plants 1 billion years ago. Appl Environ Microbiol. 2003:5530–5. doi: 10.1128/AEM.69.9.5530-5535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table. Results of BLAST [35,36] analyses of 100 ORFs present in the 100-kb region. BLAST analyses were performed using BLOSUM62 matrix and gap penalties of 11 and 1. Chromosome location of each ORF, its G+C content, coding strand, and the presence of at least one homolog in Chlamydiaceae are presented. Direct repeats (DR), gly-tRNA genes and limits of each modules of the GI are highlighted.
Supplementary figure. Phylogenetic analyses suggest that the UWE25 tra unit is phylogenetically closely related to F-like DNA conjugative tra operons: (A) UPGMA-, (B) Neighbor-joining-, and (C) minimum evolution-trees comparing p-distances and Kimura corrected p-distances of nucleotide sequences of the concatenated traA, traK, traB, traV, and traC genes (the UPGMA tree comparing the Kimura corrected p-distances of tra genes, shown in Figure 3B, is presented here to facilitate comparison with the other trees). Interestingly, in neighbor-joining and minimum evolution analyses of the p-distances, the tra unit of UWE25 is clustered with tra operons of gamma-proteobacterial F-like plasmids: bootstrap values of 96% and 92%, respectively, support the node separating the concatenated tra genes of UWE25 and RTS1, SXT, R391, three gamma-proteobacterial F-like conjugative plasmids, from their closest relative R27 plasmid. Similarly, in neighbor-joining and minimum evolution analyses of the Kimura corrected p-distances, bootstrap values of 94% and 91%, respectively, support the node separating the concatenated tra genes of the chromosomal UWE25 and the R27 plasmid, another gamma-proteobacterial F-like conjugative plasmid, from those of all other plasmids.