

Abstract
Autism spectrum disorder is a developmental condition of early childhood onset, which impacts socio-communicative functioning and is principally genetic in etiology. Currently, more than 50 genomic loci are deemed to be associated with susceptibility to autism spectrum disorder, showing de novo and inherited unbalanced copy number variants and smaller insertions and deletions (indels), more complex structural variants, as well as single-nucleotide variants deemed of pathological significance. However, the phenotypes associated with many of these genes are variable, and penetrance is largely unelaborated in clinical descriptions. This case report describes a family harboring two copy number variant microdeletions, which affect regions of NRXN1 and MBD5—each well-established in association with risk of autism spectrum disorder and other neurodevelopmental disorders. Although each copy number variant would likely be categorized as pathologically significant, both genomic alterations are transmitted in this family from an unaffected father to the proband, and shared by an unaffected sibling. This family case illustrates the importance of recognizing that phenotype can vary among exon overlapping variants of the same gene, and the need to evaluate penetrance of such variants in order to properly inform on risks.
Introduction
Autism spectrum disorder (ASD) is a developmental disorder of early childhood onset that impacts socio-communicative functioning, and which is principally genetic in etiology.1 It has a high rate of comorbidity with intellectual disability (ID) and other neurodevelopmental, neuropsychiatric, and medical disorders.1 ASD is relatively common, affecting ~1.5% of children,2 and is often associated with lifelong disability. Its core impairments and co-morbidities present a major challenge for caregivers and significant demands on health-care provision, and, by implication, health-care budget.3 Progress in elucidating its genetic etiopathogenesis will likely pave the way for new treatment options. To this end, significant progress has been made in the last decade with the advent of dense, high-throughput genotyping. More than 50 genes and loci harbouring de novo and inherited copy number variants (CNVs), structural variations, and single-nucleotide variants with diagnostic value (hereafter collectively referred to as ‘‘mutations’’ affecting the individuals discussed) have been implicated in ASD so far.4, 5 Functionally, many of these genes cluster in the post-synaptic density, whereas others are involved in neurite growth or histone modification.6
The term penetrance is used to describe the probability of a particular specified phenotype or set of phenotypes in those individuals harboring a particular mutation, whereas variable expression describes the range of phenotypic features observed among those with penetrant mutations.7 While some mutations are strongly associated with neurodevelopmental phenotypes such as intellectual disability and/or ASD,8 many of the mutations in the identified genes are characterized by incomplete penetrance, and variable expressivity and pleiotropy are seen in association with particular genotypes.9 For example, CNVs at 16p11–13 have been described in association with ASD in addition to a number of different neuropsychiatric disorders of variable severity,10 and the same is true of deletions in the SHANK 11 and NRXN genes.12–15 We are unaware of any susceptibility gene/locus that shows specificity for a single neurodevelopmental disorder.
Such variable expression and pleiotropy (multiple effects of a single gene) are not unusual, perhaps reflecting expression in different tissues, or shared pathophysiological mechanisms between disorders.7, 16 The ultimate phenotype may be influenced by the interplay of these with other factors:genetic (including sex), environmental (including maternal and hormonal influences) and epigenetic.7 A more striking observation is that some variants, classified as pathogenic, can be present without any apparent clinical consequence in some people—i.e., non-penetrant. In particular, some individuals with ASD or other neurodevelopmental disorders have been reported to share an ostensibly pathogenic variant with a phenotypically normal transmitting parent, and sometimes also one or more unaffected siblings.17–19 The underlying mechanism illustrated by such cases is poorly understood, although factors such as genomic context, impact on protein structure and function, and the effect of modifier genes may be important.7
Family based research does indicate that ASD itself is often expressed as a broader phenotype, beyond the bounds of the clinical spectrum, with family members often displaying mild, subclinical, traits.20 Indeed, the sibling recurrence for this broad autism phenotype is higher than for ASD.21 These milder traits do not often impact function, and, therefore, may not be immediately apparent or come to the attention of clinical services, but their importance lies in their implication for our understanding of the biology of ASD, and the penetrance and expression of the underlying genes.
We performed extensive genetic analyses including whole-genome sequencing in an individual with ASD and his family. We identified CNV deletions involving NRXN1 and MBD5 in the proband, but also in his father and sister, neither of whom had evidence of any clinically overt brain-related phenotype. NRXN1 and MBD5 are implicated in ASD, ID, and other neuropsychiatric disorders,12, 13, 22, 23 and functional mutations of either gene might be expected to have phenotypic consequences. We noted other variants of potential relevance to the ASD phenotype. This family illustrates that mutations anticipated to be highly penetrant may in fact be less so, and at times, apparently without phenotypic consequence.
Results
Clinical characteristics of family
The proband (003) (Fig. 1) was diagnosed with attention deficit hyperactivity disorder (ADHD) at age 3 years and with ASD at age 5 years through a specialty ASD clinic. His mother and father were aged 33 and 34 years, respectively, at the time of his conception, and the mother reported a medically uneventful pregnancy. She had no history of miscarriage, and the male proband, her firstborn, was born by vaginal delivery following spontaneous labor at 39 weeks gestation. Birth weight was 4054 g. There were no neonatal complications, and no craniofacial dysmorphology noted. Development during the 1st year was normal, but by 36 months he began losing acquired language, and speech became echolalic and scripted. Although gross motor control was intact, fine motor was an additional area of early developmental difficulty. By 36 months, repetitive motor mannerisms and preoccupations become prominent. Assessment of intellectual ability and adaptive functioning were consistent with a diagnosis of intellectual disability, and in the specialty clinic an additional diagnosis of ADHD was made (Table 1). Further assessment in the clinic at the age of 5 years identified more significant socio-communicative vulnerabilities, and a diagnosis of ASD was given. At that time an Intelligence Quotient (IQ) of 53 was recorded (verbal IQ = 56, non-verbal IQ = 65), and his adaptive skills were largely consistent with function in the mildly impaired range (adaptive composite = 63). At age 12 years 10 months his head circumference was 57.9 cm, consistent with macrocephaly.
Fig. 1.
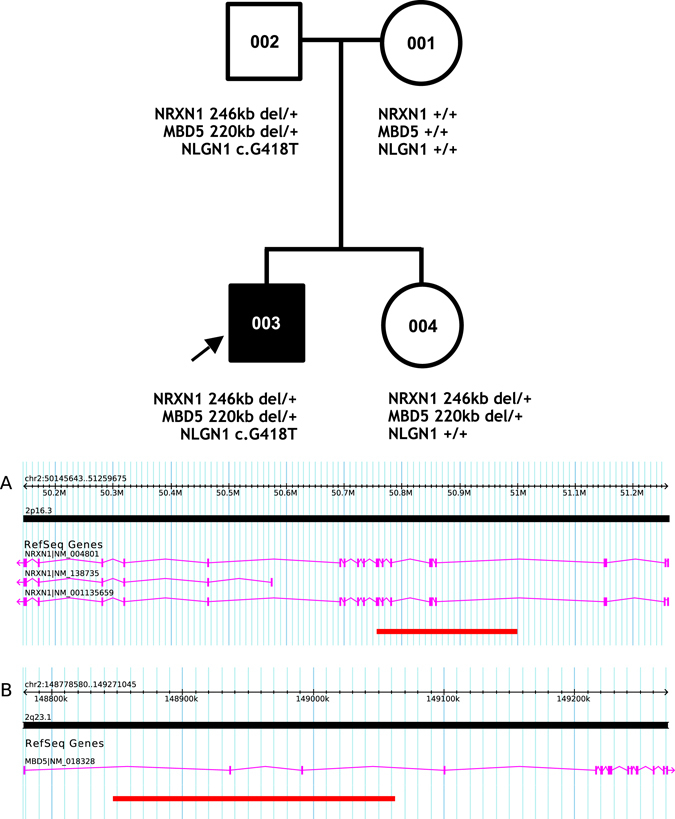
Pedigree with microarray results and annotated NRXN1 (a) and MBD5 (b) CNVs
Table 1.
Summary of family’s genotypes and phenotypes
| −001 | −002 | −003 | −004 | |
|---|---|---|---|---|
| Sex | Female | Male | Male | Female |
| Microarray | NRXN1 +/+ MBD5 +/+ | NRXN1 del/+ MBD5 del/+ | NRXN1 del[pat]/+ MBD5 del[pat]/+ | NRXN1 del[pat]/+ MBD5 del[pat]/+ |
| WGS | – | NLGN1, c.G418T [p.D140Y] | ASB14, c.461 462del [p.L154fs] NLGN1, c.G418T [p.D140Y] | Targeted Sanger: NLGN1 +/+ |
| Clinical diagnosis | None | None | ASD, ID, ADHD | None |
| IQ | NA | NA | WPPSI-IIIF | WASI |
| – | – | FSIQ = 53, | FSIQ = 121, | |
| – | – | VIQ = 56, | VIQ = 124, | |
| – | – | NVIQ = 65 | NVIQ = 112 | |
| Vineland adaptive behavior scales (comm = communication, daily = daily living) | NA | NA | Comm 3%ile, daily 1%ile, social <1%ile | NA |
| Morphology (HC = head circumference) | HC = 56 cm, height = 166.4 cm, weight = 60.3 kg (age 45 years) | HC = 58 cm, height = 182.9 cm, weight = 83.9 kg (age 44 years) | HC = 57.9 cm, height = 166.5 cm, weight = 58.1 kg (age 12:10) | HC = 51 cm, height = 106.7 cm, weight = 17.7 kg (age 6:6) |
| Medical | Nil | Nil | Nil | Nil |
| Language | CC-A: | CC-A: | OWLS: | CCC: |
| Language 42%ile, pragmatics 7%ile, social engagement 7%ile | Language 7%ile, pragmatics 9%ile, social engagement 19%ile | Standard score = 50 (<0.1%ile) CCC: GCC = 30 (1%ile) | GCC = 71 (32%ile) | |
| SRS | T-score = 61 (normal) | T-score = 43 (normal) | NA | T score = 45 (normal) |
| PPVT | 109 (73%ile) | 101 (%ile) | 86 (9%ile, aged 7:4) | 73 (86%ile, aged 3:7) |
| RMET | 31 (86% correct) | 19 (53% correct) | NA | NA |
NA not available, CC-A communication checklist–adult, CCC children's communication checklist, OWLS oral and written language scales, SRS social responsiveness scale, PPVT Peabody Picture Vocabulary Test, RMET Reading the Mind in the Eyes Test, FSIQ fullscale IQ, VIQ verbal IQ, NVIQ nonverbal IQ
The proband’s younger sister (004) was born following an uneventful pregnancy by normal vaginal delivery. Birth weight was 3856 g. She required incubation and monitoring for transient tachypnea, which resolved spontaneously, but otherwise there were no perinatal complications. Her early language and motor milestones were attained without delay. She did experience stuttering at 36 months, but otherwise exhibited no social or communicative vulnerabilities. Her cognitive function was in the superior range on the Wechsler Abbreviated Scale of Intelligence (WASI-II) age 6 years, and she had above average expressive and receptive language skills. Her scores on the Autism Diagnostic Interview-Revised (ADI-R) and the Autism Diagnostic Observation Schedule (ADOS, module 3), both completed when she was aged 9 years, did not indicate ASD symptoms. Similarly, neither children’s communication checklist (CCC)24 nor the child version of the social responsiveness scale (SRS)25 revealed any such developmental vulnerabilities.
We evaluated both parents (001 and 002) for the presence of neurocognitive vulnerabilities and neuropsychiatric diagnoses (Table 1). Both graduated high school and attained professional level employment. The SRS was not consistent with any ASD traits in either parent, although the communication checklist—adult (CC-A)26 indicated some maternal and paternal communication vulnerabilities, and father’s score on the Reading the Mind in the Eyes Test (RMET)27 was below average, suggesting some impairment in theory of mind abilities. No additional social or communication vulnerabilities were apparent, and neither parent had findings consistent with ASD. Moreover, besides maternal post-natal depression, both parents denied any neuropsychiatric history.
Genetic characterization of family
All four family members provided blood for genotyping. We initially identified hemizygous microdeletions in the chromosomal regions 2p16.3 (50,754,487–50,996,179 [hg19]) and 2q23.1 (148,851,175–149,059,335 [hg19])] in both offspring and their father. By microarray, we estimated the deletion at 2p16.3 to be ~242 kb eliminating exons 6 to 16 of NRXN1 (Fig. 1). The 2q23.1 deletion was ~215 kb in size, eliminating non-coding exons 2 and 3 in the 5’ untranslated region (UTR) of MBD5 (Fig. 1). We validated both deletions using SYBR-Green based real-time quantitative PCR. We found no other CNVs deemed clinically significant or of uncertain clinical significance according to the American College of Medical Genetics’ guidelines,28 in any family member.
We undertook whole-genome sequencing of the proband and both parents using the BGI platform as previously described.29 This validated the NRXN1 and MBD5 deletions. The breakpoints of the NRXN1 deletion were further mapped to 50,754,222–51,000,379 [hg19] by Sanger sequencing, and visual inspection of BAM files mapped the MBD5 breakpoints to 148,843,025- 149,062,962 [hg19], thereby adjusting the size of the NRXN1 and MBD5 deletions to ~246 and ~220 kb, respectively. There were no additional structural alterations at the breakpoints. In the proband and his father we found a missense variant (c.G418T [p.D140Y]) in NLGN1 (an ASD risk gene), which was predicted by in silico algorithms to be damaging. Targeted Sanger sequencing of the unaffected sibling’s DNA did not identify the variant. Finally, the proband had a de novo 2 bp deletion (c.461_462del [p.L154fs]) involving ASB14. Although the mutation was predicted to lead to a frameshift of the protein, this gene has not been associated with ASD or other neurodevelopmental disorders, to date. We identified no other rare loss-of-function or de novo missense SNVs this family’s genomic sequences.
Prevalence and penetrance of overlapping mutations
We next examined clinical and population data sets to investigate the penetrance of the putative mutations identified. For NRXN1 and MBD5, we specifically focused on CNV deletions with at least a 50% reciprocal overlap with that of the proband (hereafter termed ‘‘overlapping CNVs’’). First, we examined clinical data sets comprising individuals (N = 19,237, comprising N = 5273 cases and their family members) ascertained by way of one or more different neurodevelopmental diagnoses, including ASD,6, 30 developmental delay,31 OCD32 and cerebral palsy (CP).19 These individuals had been genotyped on a variety of platforms, each allowing a CNV detection threshold of 10 kb using five or more probes. Only CNVs called with two or more algorithms were considered. Thus, we identified one clinical case with an overlapping NRXN1 CNV (chr2: 50,761,808–51,037,134 [hg19]), and two clinical cases with overlapping MBD5 CNVs (chr2: 148,787,060- 149,106,568 and 148,842,503–149,059,335 [hg19]) from a total of 6 and 7 exon-impacting NRXN1 and MBD5 CNVs, respectively. All three individuals have developmental delay but no further phenotype information was available. We next examined population data sets, comprising samples genotyped on the Illumina 2.5M platform (KORA and COGEND)33, 34 and Illumina 1M platform (WTCCC, SAGE, ONC, and HABC),35–37 giving rise to a total sample size of 13,871. A CNV detection threshold of 30 kb was employed using a minimum of five probes. We considered all CNVs called by two or more algorithms, identifying from this pooled data set one NRXN1 CNV and one MBD5 CNV from a total of 61 (six exonic) and 19 (seven exonic) NRXN1 and MBD5 CNV deletions, respectively. Consequently, there was no statistical evidence for a greater prevalence of overlapping NRXN1 or MBD5 CNVs among cases than among controls.
We also considered overlapping CNVs recorded in DECIPHER—a clinician-submitted sample of 21,688 individuals with identified phenotypes and validated CNVs.8 Of this sample, eight individuals had overlapping NRXN1 CNVs, and one an overlapping MBD5 CNV (Fig. 2). Similarly, the International Standards for Cytogenetic Arrays (ISCA) clinical database38 included 11 overlapping NRXN1 CNVs and 8 overlapping MBD5 CNVs. The phenotypes described among these 19 individuals all included ID with an additional diagnosis of ASD in five. Among these CNVs, DECIPHER inheritance pattern is described as de novo for the one MBD5 deletion, and variable for the NRXN1 deletions (de novo for 3, inherited for 3, and unknown for 2).
Fig. 2.
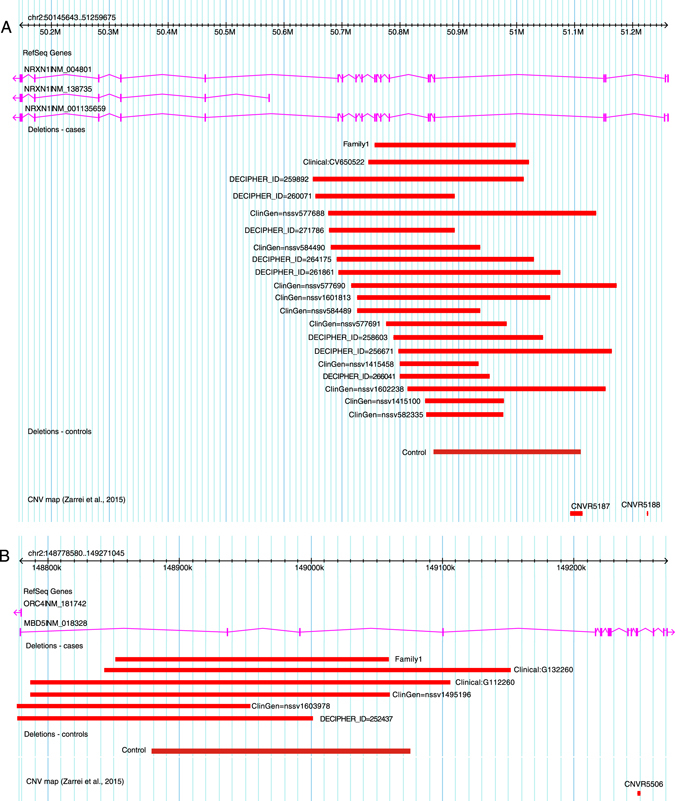
CNVs overlapping the family’s NRXN1 (a) and MBD5 (b) CNV deletion from clinical (‘‘clinical’’) and population (‘‘controls’’) data sets, and DECIPHER and ClinGen
We also examined our whole-genome sequenced ASD families (comprising N = 5205 probands, siblings and parents, see methods) for any additional individuals with damaging NLGN1 variants. We found overlapping variants in three individuals, two of with ASD, and one an unaffected father. Exome Aggregation Consortium (ExAC),39 a data set which spans 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies, lists two individuals with an overlapping mutation.
Discussion
In the family described here, CNV deletions in two ASD-implicated genes, NRXN1 and MBD5, are shared by an ASD proband, his typically developing sibling, and their unaffected father. Based on extensive literature,40 our clinical diagnostic laboratory would have assigned either one of these CNVs as ‘‘likely pathogenic’’ for ASD. In addition, WGS identified a missense variant in the putative ASD gene NLGN1 that was paternally transmitted to only the proband. This is of interest for the known interaction between the NLGN1 and NRXN1 proteins,41 and in this family the missense mutation was predicted to be damaging. A de novo frameshift variant in ASB14 was also identified in the proband, and although not brain expressed, we cannot rule out an etiological role for this mutation.
NRXN1 is one of three neurexin scaffolding proteins; aberrations in its gene are strongly associated with cognitive, neurodevelopmental and neuropsychiatric phenotypes.12, 13 Deletions in NRXN1 are relatively common in ASD (0.45%) and ID (0.12%) cohorts,12 but much less frequently seen in population based surveys (~0.02%) (ref. 12). Other studies have also highlighted the fact that deletions can occur in apparently healthy individuals (ref. 12 and references therein). An estimate of penetrance for all CNVs for schizophrenia and neurodevelopmental phenotypes for NRXN1 is 33% (ref. 42). The ExAC constraint metric for this gene is −0.13 (ref. 39), which is consistent with tolerance to copy number variation. However, its low burden of mutations in the healthy population coupled with high expression levels (messenger RNA and protein) results in its categorization as critical to brain development.43 MBD5 encodes a methylated-DNA binding protein, which has previously been described in the literature as highly penetrant, characterized in all cases by intellectual disability, ASD and, more variably, craniofacial abnormalities.22, 23 Most of the cases described so far have been de novo, although transmission is sometimes unknown. In the present family, the proband’s phenotype is largely consistent with previous descriptions connected with mutations of NRXN1 and MBD5,12, 13, 22, 23 characterized by moderate ID and ASD in the absence of dysmorphism. The ExAC constraint measure for MBD5 is 0.69 (ref. 39) indicating some degree of intolerance to variation, although it is not classified critical to brain development.43
Beyond the gene per se, the exact genomic location of a CNV may be important in the determination of functional aberration and phenotypic consequence. In this family, the 2p16.3 deletion impacted exons 6–16, and our analysis indicated that overlapping CNVs were rare among clinical cases or population controls. Crucially, overlapping CNVs were not enriched among the cases compared with controls. We speculate that this hemizygous deletion impacting only exons 6–16 may be less penetrant than others reported for this gene. Indeed, most clinical cases seem to cluster around exons 1–4 at the 5’ end of the gene, with deletions that impact the subsequent exons showing evidence of lower penetrance.13 This may be due to influence of the lncRNA AK127244 adjacent to the promotor of alpha-NRXN1.13 Confounding the argument of lower penetrance, however, is the 20 individuals with overlapping CNVs in ISCA, DECIPHER, and the other clinical data sets we examined with variable but largely overlapping phenotypes.
Similarly, overlapping MBD5 deletions were not enriched among clinical cases, although mutations described largely overlap with that of our patient, impacting one or more exons in the 5’-UTR. Two additional individuals in our clinical data set, with developmental delay, had identical MBD5 CNVs. Although these exons are not translated, all 5’-UTR deletions result in haploinsufficiency, with peripheral expression of MBD5 approximately halved.23 Many cases described, including the one in DECIPHER, have de novo mutations.
Finally, NLGN1, is of potential interest, forming complexes with NRXN1, and implicated in both structural integrity and function of synapses.44 While the function of NLGN1 has been well described, particularly in the context of its interaction with NRXN1, the phenotype associated with gene mutations has not been elaborated. One genome-wide CNV analysis of ASD cases identified enrichment for CNVs in NLGN1 compared with population controls,45 and another provided evidence of association between common variants in NLGN1 and schizophrenia in the Han Chinese population.46 However, the penetrance of the mutation described in our family is unclear in light of the identification of a similar number of cases in our clinical data set and ExAC.
Complexity of the etiology underlying ASD is well demonstrated in families like that presented here, where the most advanced genomic technologies have provided a comprehensive genetic profile, and the variants detected are shared among family members with and without ASD. We are reminded to acknowledge what remains unknown (e.g., the role of environmental factors and epigenetic regulation) and not to overstate the causal impact of variants. We are spurred to investigate the mechanisms whereby genotype can lead to phenotype in some, but not others.47 Variously, this may be a function of the type of variant (i.e., loss-of-function, missense, deletion), its location (i.e., exonic, intronic, regulatory region, intergenic), or the resultant transcript/isoform.9 We must, however, move from a genetic to a genomic perspective, recognizing that no gene or gene product functions in isolation. Indeed, each of the three genetic aberrations in this family might have been deemed sufficient to explain ASD in the proband, but all were non-penetrant in other family members. For future investigations related to penetrance, we recommend the approach of comparing only highly overlapping CNVs, rather than all CNVs involving the same gene. A true estimate of penetrance will require a more robust approach than ours, with access to comprehensive control data from pedigrees48 and large data sets.49 For example, although many variants may be very rare in the population, those that are inherited can be tracked through family members and their segregation with disease phenotype examined. This allows a quantification of their pathogenicity to be determined,48 as well as a Bayesian Factor to be estimated, which can be used as a test of the hypothesis of causality by examining its distribution under the hypothesis of neutrality.50 Although not a direct measurement of penetrance per se, this approach does go some way to quantifying the probability of disease in association with particular mutations.
Context is crucial. The impact of the wider genomic landscape, including the epigenome, along with factors such as age, sex and the early environmental milieu, will undoubtedly contribute to a person’s evolving phenotypes. The rich tapestry of protein interactions at the cellular level translate more proximally into endophenotypes, which, rather than global diagnostic fields, are the more internal phenotypic elements or markers revealed by specific measures. In the family presented here, the father’s vulnerabilities decoding emotions from facial stimuli may, for example, represent such an endophenotype. The ‘‘vulnerable brain’’ may be impacted by another factor to result in the full expression of a clinical phenotype. These mechanisms will become untangled as a result of large, epidemiological studies, but also the accumulation of evidence from case studies such as this one.
Methods
The family described was recruited as part of ongoing studies of the genetics of ASD (www.mss.ng).51 This data set currently comprises ~2500 probands with ASD and, in most cases, both parents. ASD diagnoses are made by expert clinicians using the ADI-R and the ADOS combined with clinical judgment. Probands and their available first degree relatives have all undergone phenotyping as described below, and have provided DNA for the identification of CNVs and SNVs (see below). All data were collected following informed consent from participants or substitute decision makers, and the study is conducted with approval from respective local research ethics boards. The family described in detail in this paper has provided specific written consent for their data to be shared in the scientific literature in the form of this case report.
Phenotypes
In addition to the ADI-R and ADOS-G, the proband underwent a cognitive assessment using the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) and a language assessment with the oral and written language scales (OWLS-II). Additionally, measures of the proband’s adaptive functioning (Vineland Adaptive Behavior Scales-II) was completed with his parents. Both parents were assessed using the Peabody Picture Vocabulary Test and the RMET (a measure of theory of mind).27 In addition, both parents completed the CC-A26 and the SRS.25 The proband’s sibling underwent assessment with the WASI-II, the ADI and the ADOS. Her parents completed a measure of her social communication (CCC).24 Height, weight, and head circumference were measured for each family member.
Genotypes
We called CNVs as previously described.31 Briefly, four different CNV calling algorithms were used to annotate high-confidence CNVs. These included the Affymetrix Chromosome Analysis Suite (ChAS), iPattern,52 Nexus,53 and Partek.54 A stringent set of variants was defined for further analyses. This set included variants detected by one or both of ChAS or iPattern, and if detected by only one of these, then also by one of Nexus or Partek. For stringent calls on the X chromosome, we required calling by both ChAS and iPattern. Only CNVs with five probes or more on the array were called, with a minimum length cutoff of 30 kb. CNVs were filtered to prioritize rare variants that occurred with a frequency of<0.1% in control samples (N = 9611). For the purpose of filtering, CNVs with >50% reciprocal overlap were deemed overlapping. We also removed all CNVs that had >70% overlap with a known segmental duplication. We further restricted our list to those with more than 75% overlap with copy number stable regions, according the stringent CNV map of the human genome.55 All CNVs described in the index family have been validated using the SYBR green based quantitative PCR method. The genomic coordinates presented in this paper are based on the February 2009 Human Genome Build (GRCh37/hg19).
The proband and both parents also underwent whole-genome sequencing (WGS) by BGI as described previously.29 All identified variants were subsequently validated by Sanger sequencing. In addition, the unaffected sibling underwent targeted Sanger sequencing. We annotated the Identified SNVs, and prioritized those likely to be damaging using a filtering algorithm. This captured all those SNVs that were rare (≤1% minor allele frequency), and involved loss of function (nonsense, splice site, and frameshift), and damaging de novo missense mutations (damaging as evidenced by two of the following criteria: SIFT ≤ 0.05, Polyphen2 ≥ 0.95, CADD ≥ 15, Mutation Assessor score ≥ 2, placental mammal PhyloP ≥ 2.4 and vertebrate PhyloP ≥ 4).29
Data availability
Sequence data has been deposited at the European Genome–phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under accession number EGAS00001001023. The data, as part of a larger autism whole-genome sequencing project, are also available in the MSSNG database on Google Genomics (for access see http://www.mss.ng/researchers).
Acknowledgements
We thank the family described in this paper for supporting this publication. We also thank all other families described herein for their contribution to our ongoing studies of the genetics of ASD. We thank The Centre for Applied Genomics (TCAG), which is funded by Genome Canada and the Ontario Genomics Institute, Canada Foundation for Innovation (CFI), and the Ontario Research Fund of the Government of Ontario. M.W.S. is supported by a Clinical Investigatorship Award from the Canadian Institutes of Health Research (CIHR) Institute of Genetics; S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children. This study makes use of data generated by the DECIPHER community and the MSSNG-Autism Speaks Whole Genome Autism Sequencing project. A full list of centers whom contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was also provided by the Wellcome Trust, Autism Speaks and CIHR.
Author contributions
M.W.S. conceived the case report, reviewed, and compiled the background literature and wrote the first draft of the manuscript. R.N., N.H., and C.C. were involved in the collection and interpretation of clinical data, and A.T. in managing the database of clinical information. M.U. and M.Z. were directly involved in interpretation and analysis of CNV data. R.K.C.Y. and S.W. analyzed and interpreted the sequence data. J.H. assisted in data analysis and interpretation, and P.S. and J.B. with the conceptual ideas. All authors directly contributed to manuscript revisions. S.W.S. assisted in conceiving the case report, funding the research project and staff, and was involved in writing and revising the manuscript.
Competing interests
The authors declare that they have no competing financial interests.
References
- 1.Anagnostou E, et al. Autism spectrum disorder: advances in evidence-based practice. Can. Med. Assoc. J. 2014;186:509–519. doi: 10.1503/cmaj.121756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014;63:1–21. [PubMed] [Google Scholar]
- 3.Buescher AV, Cidav Z, Knapp M, Mandell DS. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 2014;168:721–728. doi: 10.1001/jamapediatrics.2014.210. [DOI] [PubMed] [Google Scholar]
- 4.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012;22(3):229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Geshwind D, State M. Gene-hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015;14:1109–1120. doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013;132:1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Firth HV, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeRubeis, S. & Buxbaum, J. D. Genetics and genomics of autism spectrum disorder: embracing complexity Hum. Mol. Genet. 24, 1–8 (2015). [DOI] [PMC free article] [PubMed]
- 10.Hanson E, et al. The cognitive and behavioral phenotype of the 16p11. 2 deletion in a clinically ascertained population. Biol. Psychiatry. 2015;77:785–793. doi: 10.1016/j.biopsych.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 2014;74:113–122. doi: 10.1002/dneu.22128. [DOI] [PubMed] [Google Scholar]
- 12.Dabell MP, et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am. J. Med. Genet. A. 2013;161A:717–731. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 13.Lowther, C. et al. Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet. Med. (2016). doi:10.1038/gim.2016.54. [DOI] [PMC free article] [PubMed]
- 14.Szatmari P, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39(3):319–327. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall C, et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 17.Lee C, Scherer SW. The clinical context of copy number variation in the human genome. Expert Rev. Mol. Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 18.Salyakina D, et al. Copy number variants in extended autism spectrum disorder families reveal candidates potentially involved in autism risk. PLoS ONE. 2011;6:e26049. doi: 10.1371/journal.pone.0026049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oskoui M, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat. Commun. 2015;6:7949. doi: 10.1038/ncomms8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piven J. The broad autism phenotype: a complementary strategy for molecular genetic studies of autism. Am. J. Hum. Genet. 2001;105:34–35. doi: 10.1002/1096-8628(20010108)105:1<34::AID-AJMG1052>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Bolton P, et al. A case-control family history study of autism. J. Child. Psychol. Psychiatry. 1994;35(5):877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 22.Hodge JC, et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol. Psychiatry. 2014;19:368–379. doi: 10.1038/mp.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talkowski ME, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop, D. V. M. The Children’s Communication Checklist Version 2 (CCC-2) (Psychological Corporation, 2003).
- 25.Constantino, J. N. & Gruber, C. P. The Social Responsiveness Scale Manual (Western Psychological Services, 2005).
- 26.Whitehouse, A. J. O. & Bishop, D. V. M. Communication Checklist Adult. (The Psychological Corporation, 2009).
- 27.Baron-Cohen S, Jolliffe T, Mortimore C, Robertson M. Another advanced test of theory of mind: evidence from very high functioning adults with autism or Asperger syndrome. J. Child. Psychol. Psychiatry. 1997;38:813–822. doi: 10.1111/j.1469-7610.1997.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 28.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuen RK, et al. Genome-wide characteristics of de novo mutation in autism. NPJ Genom. Med. 2016;1:16027. doi: 10.1038/npjgenmed.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uddin M, et al. A high-resolution copy-number variation resource for clinical and population genetics. Genet. Med. 2014;17:747–752. doi: 10.1038/gim.2014.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazzellone, M. J. et al. Uncovering obsessive-compulsive disorder risk genes using a high-resolution genome-wide CNV approach. J. Neurodev. Dis8, 36 (2016) [DOI] [PMC free article] [PubMed]
- 33.Verhoeven VJ, et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 2013;45:314–318. doi: 10.1038/ng.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bierut LJ, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum. Mol. Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bierut LJ, et al. A genome-wide association study of alcohol dependence. Proc. Natl Acad. Sci. U.S.A. 2010;107:5082–5087. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cotterchio M, et al. Red meat intake, doneness, polymorphisms in genes that encode carcinogen-metabolizing enzymes, and colorectal cancer risk. Cancer Epidemiol. Biomarkers Prev. 2008;17:3098–3107. doi: 10.1158/1055-9965.EPI-08-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coviello AD, et al. A genome-wide association meta-analysis of circulating sex hormone-binding globulin reveals multiple Loci implicated in sex steroid hormone regulation. PLoS Genet. 2012;8:e1002805. doi: 10.1371/journal.pgen.1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riggs ER, et al. Towards an evidence based process for the clinical interpretation of copy number variation. Clin. Genet. 2012;81:403–412. doi: 10.1111/j.1399-0004.2011.01818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tammimies K, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314:895–903. doi: 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 41.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirov G, et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry. 2014;75:378–385. doi: 10.1016/j.biopsych.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uddin M, et al. Indexing effects of copy number variation on genes involved in developmental delay. Sci. Rep. 2016;6:28663. doi: 10.1038/srep28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reissner C, Klose M, Fairless R, Missler M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc. Natl Acad. Sci. U.S.A. 2008;105:15124–15129. doi: 10.1073/pnas.0801639105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, et al. Evidence for association of cell adhesion molecules pathway and NLGN1 polymorphisms with schizophrenia in Chinese Han population. PLoS ONE. 2015;10:e0144719. doi: 10.1371/journal.pone.0144719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen R, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotech. 2016;34(5):531–538. doi: 10.1038/nbt.3514. [DOI] [PubMed] [Google Scholar]
- 48.Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 2016;98:1077–1081. doi: 10.1016/j.ajhg.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minikel EV, et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016;8:322ra9. doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson DT, Easton DF, Goldgar DE. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am. J. Hum. Genet. 2003;73:652–655. doi: 10.1086/378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuen RKC, et al. Whole-genome sequence based resource for autism research. Nat. Neurosci. 2017;20:602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinto D, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011;29:512–520. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darvishi, K. Application of Nexus copy number software for CNV detection and analysis. Curr. Protoc. Hum. Genet. 65, 4.14.1–4.14.28 (2010). [DOI] [PubMed]
- 54.Downey T. Analysis of a multifactor microarray study using partek genomics solution. Methods Enzymol. 2006;411:256–270. doi: 10.1016/S0076-6879(06)11013-7. [DOI] [PubMed] [Google Scholar]
- 55.Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat. Rev. Genet. 2015;16:172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequence data has been deposited at the European Genome–phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under accession number EGAS00001001023. The data, as part of a larger autism whole-genome sequencing project, are also available in the MSSNG database on Google Genomics (for access see http://www.mss.ng/researchers).