

Abstract
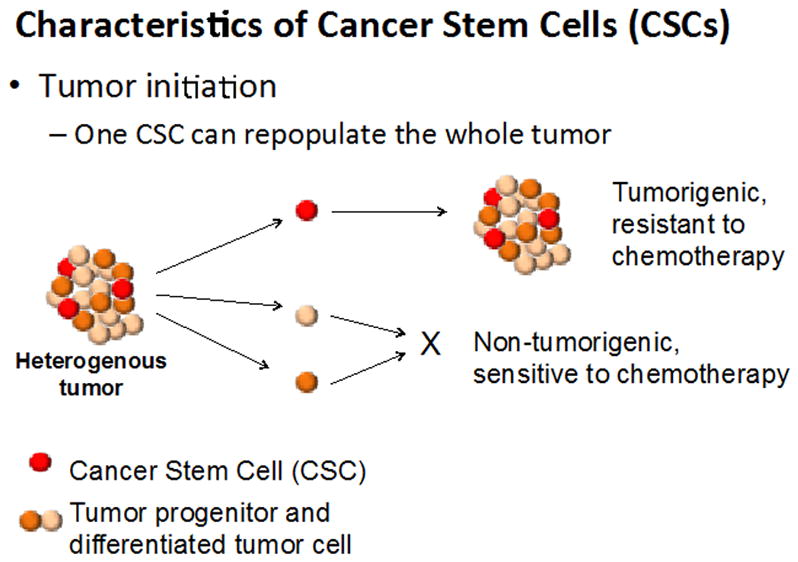
Cancer stem cells (CSCs), also called Tumor Initiating Cells (TICs), can be defined as cancer cells that are present within solid tumors or hematological cancers, which have characteristics associated with normal stem cells, but which can give rise to all cell types found in a particular cancer sample. CSCs, therefore, are transformed stem cells, which can self-renew, differentiate into diverse progenies, and drive continuous tumor growth. (Villani et al. 2015) (Zhou et al., 2009)(Kreso & Dick, 2014)(Schatton et al., 2008) (Figure 1).
Keywords: tumor initiating cells, phenotypic plasticity, humanized mice, innate and adaptive immunity
INTRODUCTION
The term Cancer Stem Cells (CSCs) was first introduced about 15 years ago (Villani et al., 2015)(Kreso & Dick, 2014)(Schatton et al., 2008)(Zhou et al., 2009). The concept quickly gained traction as a bone fide biological phenomenon in hematologic cancers (i.e. leukemias), followed shortly by solid evidence of CSCs in solid cancers, such as breast, brain, colon and pancreas. Soon, several reports suggested that melanoma could also be added to this list (Fang et al., 2005) (Quintana et al., 2008)( Quintana et al., 2012) (Schatton et al., 2008)(Frank, 2005)(Monzani et al., 2007) (Villani et al., 2015 and references therein), but other reports cast doubt on this conclusion (Monzani et al., 2007)(Cheli et al., 2014)(Perego et al., 2010)(Croteau et al., l. 2013)(Hoek & Goding, 2010). The conflicting data documented that melanomas did not consistently display markers associated with CSCs, but rather exhibited dramatic phenotypic plasticity as they readily adapted to different micro-environments, with continued tumor growth. The plethora of reports ascribing melanoma progression both to CSCs and to tumor heterogeneity and plasticity (or “phenotypic switching”(Hoek & Goding, 2010)), has left the issue of CSCs in melanoma unsettled. Here, we discuss arguments both “for” and “against” the concept of CSCs in melanoma and suggest a possible path forward to help resolve these conflicting data.
CSCs IN MELANOMA
One of the earliest reports on CSCs in melanoma seems to conclusively demonstrate the presence of CSCs (Fang et al., 2005). The authors investigated whether stem cell–like populations existed in human melanomas. They found that about 20% of the metastatic melanomas cultured in medium that supported the growth of human embryonic stem cells also supported a subpopulation of cells, which could be propagated as non-adherent spheres. However, if the cells were grown in standard medium, cells grew only as only adherent monolayers. Thus, sphere formation, originally defined as a characteristic central to stem cells, was definitively linked to growth of a subpopulation of melanoma cells. Further, single cells from these melanoma spheres could differentiate, under appropriate culture conditions, into several cell lineages, such as melanocytic, adipocytic, osteocytic, and chondrocytic lineages, recapitulating the plasticity of neural crest stem cells. Since these are the cells from which melanomas arise, the findings supported the concept of “stem-ness,” a term that has been used to describe the integration of the various molecular programs that control and maintain a cell as a stem cell (Villani et al., 2015)(Tang, 2012)(Rycaj & Tang, 2015)(Zhou et al., 2009). Thus, the authors concluded that melanomas can contain a subpopulation of stem cells that contribute to heterogeneity and tumorigenesis (Fang et al., 2005).
Along with these biochemical data, which, indeed, suggested that CSCs were present in melanoma, there were also data indicating that melanomas were very heterogeneous tumors, containing different sub-populations. However, the biological significance of these subpopulations was unclear, although it was reported that one particular sub-population might be responsible for stem-ness (Roesch et al., 2010). This subpopulation of melanoma cells expressed the biomarker, the H3K4 demethylase JARID1B (KDM5B/PLU-1/RBP2-H1). It was also slow-cycling, with a doubling time of more than 4 weeks, and was found within the larger population of rapidly proliferating cells. These JARID1B-positive melanoma cells gave rise to a highly proliferative progeny, and knockdown of JARID1B led to accelerated tumor growth, which was followed by exhaustion. Perhaps then, this small population of JARID1B-positive was required for continuous tumor growth. However, expression of JARID1B was not consistent, and did not follow a hierarchical cancer stem cell model: even JARID1B-negative cells could become positive and even single melanoma cells were tumorigenic in xenografts (Roesch et al., 2010). Were they true CSCs or a small population of melanoma cells that could display dramatic plasticity, changing their pattern of proliferation and varying expression of JARID1B?
Another confounding variable is the apparent inherent chemo-resistance of a subpopulation of melanomas (Frank, 2005)(Murphy et al. 2014). This resistance has, at least in part, been attributed to expression of, ABCB5 (ATP-binding cassette, subfamily B, member 5), a drug transporter and a mediator of chemo-resistance. Conceivably, it could also be a molecular marker for a subpopulation of chemo-resistant tumor cells with a stem cell phenotypic marker found within the bulk population of melanoma cells. In support of this concept, blockade of ABCB5 in melanoma cells reversed resistance to the chemical agent doxorubicin. In addition, along with the expression of ABCB5, this slow-cycling population could be resistant to traditional chemotherapies because they are not rapidly dividing and the cell-cycle is a traditional target for many chemotherapeutic agents (Roesch et al., 2010).
TUMOR HETEROGENEITY AND PHENOTYPIC PLASTICITY
Concomitant with reports of CSC markers and stem-ness, expression of many different cell surface markers on putative melanoma CSCs was reported. While this may suggest a cell of origin with multilineage differentiation abilities, it also supports the concept of phenotypic plasticity, i.e., the ability a cell to change its phenotype in response to changes in the environment. There is a remarkably exhaustive list of reported markers: CD20, CD24, CD44 and CD117, CD133, CD166, and nestin and CD133 as well as ABCB5 and aldehyde dehydrogenase (ALDH) activity. Other genes associated with CSCs include MITF, SOX2, NANOG, OCT4 and KLF4 (Fang et al., 2005) (Perego et al., 2010)(Cheli et al., 2014)(Croteau et al., 2013)(Seftor et al., 2014) (Monzani et al., 2007) (Hoek & Goding, 2010)(Villani et al., 2015). There has, therefore, been no consensus about which markers on melanomas were consistently linked to stem-ness, or whether any marker actually identified a bone fide CSC. Perhaps there are no CSCs in melanoma. Rather, instead of representing a true CSC, which could differentiate within an appropriate tumor microenvironment (TME), growth of melanoma tumors is mediated by clonal heterogeneity within the tumor. Indeed, the concepts of heterogeneity and plasticity are supported by extensive reports of clonal variation within a given population of melanoma cells (Cheli et al., 2014)(Monzani et al., 2007)(Perego et al., 2010)(Croteau et al., 2013)(Hoek & Goding, 2010)). Convincing data document plasticity in the clonal expression of putative markers and on the ability of these clones to readily form spheres (melanospheres) when cultured under the appropriate conditions. Thus, there is striking intra-tumor heterogeneity in melanomas, which has lead to the suggestion that tumorigenicity among different clones of melanoma cells may be driven by different molecular pathways. If, in fact, multiple mechanisms are responsible for melanoma progression within a tumor, successful therapies may require different targeted approaches, thereby complicating the development of successful (targeted) therapies (Croteau et al., 2013).
While these findings support the concepts of heterogeneity and plasticity, an alternative argument is that some stem-like cells from solid tumors are dynamic: they may transiently acquire properties of stem-ness, depending on the tumor context (Roesch et al., 2010). In this model of dynamic stem-ness, at any point in time, and within the bulk of a tumor, there exists a sub-population of slow-cycling, self-renewing tumor cells, which may continuously appear or disappear and, therefore, is a “moving target”(Kreso & Dick, 2014). Consequently, it has been proposed that the sine qua non for assessing the existence of a CSCs is whether a particular clone(s) of cells exhibits the potential for unlimited growth, a central characteristic of the most primitive cells, which possess stem-ness property of self-renewal (Kreso & Dick, 2014). To test this paradigm, it has been suggested that potential CSCs be studied by repeated cloning of individual tumor cells, followed by genetic and biochemical analyses and subsequent transplantation into mice (Figure 2) (Kreso & Dick, 2014). Once tumors appear, they need to be resected and re-analyzed. Prospective purification of CSCs should be carried out to determine if hierarchical organization is present, by enumerating CSC frequency in the mice that develop tumors, the response of the tumors to therapy and the ability of the isolated cells to self-renew. By understanding the properties of stem-ness within tumors, the cells that drive sequential rounds of tumor growth could be identified. As a result, genetically true CSCs could be distinguished from heterogeneous phenotypically plastic cells that are responsive to non-genetic influences (Kreso & Dick, 2014) (Tang, 2012)(Rycaj & Tang, 2015)
Figure 2.

Experimental Approach to Investigate CSC Properties in the Context of Genetic Sub-clones. Studying CSCs will require separation of distinct genetic sub-clones, because CSCs cannot be reliably identified in genetically heterogeneous tumors. One method by which sub-clones can be separated is by transplanting cancer cells at clonal cell doses over multiple recipients. Following engraftment of the human cancer cells, the hierarchical composition of a particular sub-clone can be studied using prospective purification of cells. Sequential transplantations of cancer cells allows for the tracking of further clonal evolution. For solid tumors, sampling different geographical regions from the primary tumor will be important for capturing distinct sub-clones. (From Kreso and Dick, 2014)
IN VIVO MODEL SYSTEMS: HELPING TO FIND THE FIRE AMID THE SMOKE
In vivo studies designed to identify CSCs are an essential complement to cellular and molecular studies carried out in vitro. However, of necessity, these in vivo studies have relied on xenografts in immune-compromised mice. Even though these mouse models have provided an enormous trove of information on mechanisms mediating tumor growth and metastasis in an in vivo environment, there are inherent limitations (Villani et al., 2015). These include: (1) the melanoma cells, themselves; (2) the site of implantation; and (3) the strain of mouse. Some investigations (Fang et al., 2005) (Seftor et al., 2014) (Cheli et al., 2014) have used melanomas from primary tumors, which have not previously been adapted to culture, and thus may more closely represent the biology of human melanomas. Other studies (Frank, 2005)(Monzani et al., 2007)( Quintana et al., 2008) (Croteau et al., 2013) have relied on established melanoma cell lines. Reasons for using these established cell lines are logical and pragmatic: the lines can be readily cloned, the cells grow well in culture, and they can be manipulated genetically so as allow well-controlled studies. But these cell lines, long separated from their original human host, may well have undergone “genetic drift” where clonal variations among the original cells have had survival advantages, depending on culture conditions.
Second, the site of implantation may also contribute to experimental outcome. Primary melanomas arise within the dermal/epidermal junction where they establish the appropriate niche and TME for optimal growth and survival, and perhaps metastatic potential (Villani et al., 2015). However, investigators may opt for sub-cutaneous implantation, which does not mimic the orthotopic site within the dermis. Although abundant vascularization and a local milieu rich in adipose tissue may enhance tumor cell survival with subcutaneous implantation, the TME is artificial for a melanoma cell. Further, IV injection has been used to mimic metastatic behavior, but it does not in any way recapitulate the processes of local tissue invasion from the primary site and the mechanisms needed to facilitate either hematologic or lymphatic spread. Still other studies may incorporate the melanoma cells in Matrigel® (Villani et al., 2015) (Quintana et al., 2012) with the hope of facilitating tumor take, either by helping to sequester growth factors/cytokines produced by the tumor cells, themselves, and/or by providing factors that may be present in the Matrigel®.
Third, these in vivo experiments have depended on xenografts of human tumors in mice with varying degrees of a compromised immune system (Figure 3)(Villani et al., 2015). Even with these differences in the extent of immune-deficiency, there are there are intrinsic constraints on the data since an autologous system with an intact immune response is not possible. Indeed, under conditions where the mouse is severely immune-compromised (nonobese diabetic (NOD)-scid IL2r null; NSG) (Figure 3)(Villani et al., 2015), even injection of a single melanoma cell isolated from a primary melanoma has resulted in the successful outgrowth of a tumor (Quintana et al., 2008). In this experimental system, melanoma cells are given “free reign” with the complete absence of any constraints imposed by immune surveillance. Does this mean that CSCs in melanoma do, in fact, exist but that they are held in check by an immune system that preferentially targets them? Or does it mean that a heterogeneous and phenotypically plastic (sub)population can adapt to this permissive TME, with resultant tumor growth?
Figure 3.
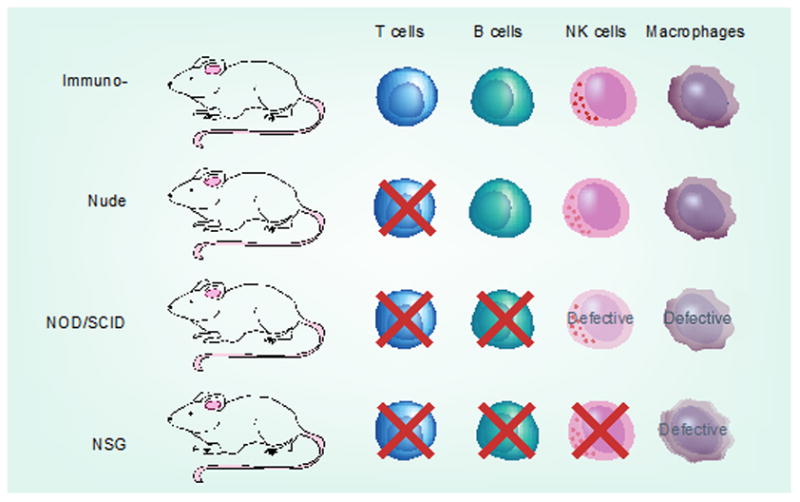
Immunologic characteristics of animal model systems commonly used in melanoma research. NK: Natural killer; NOD: Nonobese diabetic; NSG: NOD-scid IL2rγnull ; SCID: Severe combined immunodeficiency. Melanoma. (From Villani et al. 2015).
Thus, the importance of immune-editing on a (sub)population of melanoma cells can not be overstated. Increasingly, evidence points to roles for innate immunity, particularly NK (Natural Killer) cells in eliminating melanoma cells, particularly those expressing markers co-incident with CSCs (Pietra et al., 2009)(Ames et al., 2015)(Iannello & Raulet, 2014) (O’Sullivan et al., 2012)(Villani et al., 2015)(Tallerico et al., 2016). Other components of the immune system – both innate (e.g. macrophages) and adaptive (T and B cells) surely cooperate to enhance killing of tumor cells, whether or not they are CSCs (Iannello & Raulet, 2014)(O’Sullivan et al., 2012). Importantly, the emergence of humanized mice harboring the full complement of human cells mediating immunity could be enormously beneficial in dissecting out how components of the immune system help or hinder the growth of a (sub)population of heterogeneous melanomas cells and/or melanoma CSCs (Pattabiraman & Weinberg, 2014)(Theocharides, et al., 2015)(Kozlowska et al., 2016)(Hasgur et al., 2016). Specifically, studies with humanized mice with an intact (human) immune system, together with stringent in vitro biochemical and molecular analyses (Figure 2)(Kreso & Dick, 2014) could help to determine whether immune surveillance is targeting bone fide CSCs or a sub-population of melanoma cells that can modify their behavior in response to their TME.
Lastly, it is possible that the targeted manipulation of the genome by CRISPR (clustered regularly interspaced short palindromic repeats) technology (Komor et al., 2017), can be used to generate strains of mice that can be bred to give an intact immune system with the desired genetic make-up, compatible with the melanoma to be studied. CRISPR technology is already being used to knock-in genes (https://www.jax.org/contact-jax) and this technology may well provide a critical tool for carefully controlled molecular analysis of the role(s) of the immune system in the survival (or not) of CSCs and/or phenotypically plastic heterogeneous populations of melanoma cells. This advanced technology may allow us to finally distinguish the smoke sent up by heterogeneous melanoma cells from the actual fire provided by CSCs.
SUMMARY AND CONCLUSIONS
Substantial data convincingly implicate a role for CSCs in melanoma progression. However, data also indicate enormous diversity and heterogeneity among different melanomas, and even within a single melanoma tumor. Further, the inability of these tumors to consistently display a marker associated with stem-ness has led to the concept of extensive phenotypic plasticity in melanoma, so much so that the concept of the existence of CSCs has been severely questioned. In contrast, evidence that CSCs do exist is provided by studies in severely immune-compromised mice, where injection of only a single melanoma cell results in outgrowth of a tumor. However, the use of severely immune-compromised mice as an in vivo vesicle neglects the potential and essential roles of innate and adaptive immunity in either favoring or inhibiting the emergence of melanomas driven by CSCs. Rigorous in vitro studies are needed to clone and re-clone individual melanomas to demonstrate whether they can retain stem-ness. Coupled with in vivo experiments that directly compare tumorigenicity of these clones in immune-compromised and in immune-competent (i.e., humanized mice) murine hosts may help discriminate between the existence of true melanoma CSCs and a highly adaptable melanoma cell that can respond to changes in its TME with extreme phenotypic plasticity, so that it remains tumorigenic without displaying hallmark characteristics of CSCs.
Figure 1.
Acknowledgments
Supported by NIH grants to CEB: AR-26599 and CA-77267
LITERATURE CITED
- Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, Murphy WJ. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. Journal of Immunology. 2015;195:4010–4019. doi: 10.4049/jimmunol.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Bonnazi VF, Jacquel A, Allegra M, De Donatis GM, Bahadoran P, Ballotti R. CD271 is an imperfect marker for melanoma initiating cells. Oncotarget. 2014;5:5272–83. doi: 10.18632/oncotarget.1967. http://doi.org/10.18632/oncotarget.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau W, Jenkins MH, Ye S, Mullins DW, Brinckerhoff CE. Differential mechanisms of tumor progression in clones from a single heterogeneous human melanoma. Journal of Cellular Physiology. 2013;228:773–780. doi: 10.1002/jcp.24225. http://doi.org/10.1002/jcp.24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Research. 2005;65:20, 9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. http://doi.org/10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Frank NY. ABCB5-Mediated Doxorubicin Transport and Chemoresistance in Human Malignant Melanoma. Cancer Research. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. Retrieved from http://cancerres.aacrjournals.org/cgi/doi/10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- Hasgur S, Aryee KE, Shultz DL, Greiner DL, Brehm MA. Mouse Models for Drug Discovery. Methods in Molecular Biology. 2016;1438:67–78. doi: 10.1007/978-1-4939-3661-8_4. http://doi.org/10.1007/978-1-4939-3661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell and Melanoma Research. 2010;23:746–759. doi: 10.1111/j.1755-148X.2010.00757.x. http://doi.org/10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- Iannello A, Raulet DH. Immunosurveillance of senescent cancer cells by natural killer cells. Oncoimmunology. 2014;3:e27616. doi: 10.4161/onci.27616. http://doi.org/10.4161/onci.27616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, Liu DR. Review CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell. 2017:1–17. doi: 10.1016/j.cell.2016.10.044. in press http://doi.org/10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed]
- Kozlowska AK, Kaur K, Topchyan P, Jewett A. Adoptive transfer of osteoclast-expanded natural killer cells for immunotherapy targeting cancer stem-like cells in humanized mice. Cancer Immunology, Immunotherapy. 2016;65:835–845. doi: 10.1007/s00262-016-1822-9. http://doi.org/10.1007/s00262-016-1822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. http://doi.org/10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, La Porta CAM. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. European Journal of Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. http://doi.org/10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Murphy GF, Wilson BJ, Girouard SD, Frank NY, Frank MH. Stem cells and targeted approaches to melanoma cure. Molecular Aspects of Medicine. 2014;39:33–49. doi: 10.1016/j.mam.2013.10.003. http://doi.org/10.1016/j.mam.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Bui JD. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. The Journal of Experimental Medicine. 2012;209:1869–1882. doi: 10.1084/jem.20112738. http://doi.org/10.1158/1538-7445.AM2012-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. http://doi.org/10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M, Tortoreto M, Tragni G, Mariani L, Deho P, Carbone A, Castelli C. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. Journal of Investigative Dermatology. 2010;130:1877–1886. doi: 10.1038/jid.2010.69. http://doi.org/10.1038/jid.2010.69. [DOI] [PubMed] [Google Scholar]
- Pietra G, Manzini C, Vitale M, Balsamo M, Ognio E, Boitano M, Mingari MC. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. International Immunology. 2009;21:793–801. doi: 10.1093/intimm/dxp047. http://doi.org/10.1093/intimm/dxp047. [DOI] [PubMed] [Google Scholar]
- Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, Morrison SJ. Human Melanoma Metastasis in NSG Mice Correlates with Clinical Outcome in Patients. Sci Transl Med. 2012;4:159. doi: 10.1126/scitranslmed.3004599. http://doi.org/10.1126/scitranslmed.3004599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. http://doi.org/nature07567 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Herlyn M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. http://doi.org/10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycaj K, Tang DG. Cell-of-origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Research. 2015;75:4003–4011. doi: 10.1158/0008-5472.CAN-15-0798. http://doi.org/10.1158/0008-5472.CAN-15-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Frank MH. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. http://doi.org/10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seftor EA, Seftor REB, Weldon DS, Kirsammer GT, Margaryan NV, Gilgur A, Hendrix MJC. Melanoma tumor cell heterogeneity: A molecular approach to study subpopulations expressing the embryonic morphogen nodal. Seminars in Oncology. 2014;41:259–266. doi: 10.1053/j.seminoncol.2014.02.001. http://doi.org/10.1053/j.seminoncol.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallerico R, Garofalo C, Carbon E. A new biological feature of natural killer cells: The recognition of solid tumor-derived cancer stem cells. Frontiers in Immunology. 2016;7:5–10. doi: 10.3389/fimmu.2016.00179. http://doi.org/10.3389/fimmu.2016.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Research. 2012;22:457–472. doi: 10.1038/cr.2012.13. http://doi.org/10.1038/cr.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharides APA, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica. 2015;101:5–19. doi: 10.3324/haematol.2014.115212. http://doi.org/10.3324/haematol.2014.115212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani V, Sabbatino F, Ferrone CR, Ferrone S. Melanoma initiating cells: where do we stand? Melanoma Management. 2015;2:109–114. doi: 10.2217/mmt.15.2. http://doi.org/10.2217/mmt.15.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B-BS, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nature Reviews Drug Discovery. 2009;8:806–823. doi: 10.1038/nrd2137. http://doi.org/10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]