

Abstract
The phosphoinositide 3-kinase (PI3K) signaling pathway is one of the most frequently altered pathways in human cancer and has a critical role in driving tumor initiation and progression. Although PI3K and its lipid product phosphatidylinositol-3,4,5-trisphosphate (PIP3) have been shown to activate multiple downstream signaling proteins, the vast majority of studies have focused on the protein kinase AKT as the dominant effector of PI3K signaling. However, recent studies have demonstrated many contexts under which other PIP3-dependent signaling proteins critically contribute to cancer progression, illustrating the importance of understanding AKT-independent signaling downstream of PI3K. Here, we highlight three PI3K-dependent, but AKT-independent, signaling branches that have recently been shown to have important roles in promoting phenotypes associated with malignancy. First, the PDK1 -mTORC2-SGK axis can substitute for AKT in survival, migration, and growth signaling and has emerged as a major mechanism of resistance to PI3K and AKT inhibitors. Second, Rac signaling mediates the reorganization of the actin cytoskeleton to regulate cancer cell migration, invasion, and metabolism. Finally, the TEC family kinase BTK has a critical role in B cell function and malignancy and represents a recent example of an effective therapeutic target in cancer. These mechanisms highlight how understanding PI3K-dependent, but AKT-independent, signaling mechanisms that drive cancer progression will be crucial for the development of novel and more effective approaches for targeting the PI3K pathway for therapeutic benefit in cancer.
Introduction
Phosphoinositide 3-kinase (PI3K) signaling plays a central role in cellular physiology, coordinating insulin signaling during organismal growth and mediating critical cellular processes such as glucose homeostasis, protein synthesis, cell proliferation, and survival. This pathway has been an intense area of investigation, particularly in light of cancer genetics studies that have revealed it to be one of the most frequently altered pathways in human malignancies that controls most hallmarks of cancer, including cell proliferation, survival, genomic instability, and metabolism [1]. Consequently, PI3K signaling has emerged as an attractive target for cancer therapy, and many drugs that inhibit various pathway components are currently in clinical trials [2, 3].
Class I PI3K transduces upstream signals from receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) by phosphorylating the 3′-hydroxyl group of the inositol ring of phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3) [4, 5]. PIP3 serves as a critical lipid second messenger that recruits cytosolic proteins containing pleckstrin homology (PH) domains to the plasma membrane to promote either their activation or co-localization with other effector proteins [6–8]. It should be noted that only a small subset of PH domains in the human genome are thought to bind PIP3 with high affinity and specificity (10–20% out of ~290 PH domains have been shown to robustly bind phosphoinositides, with some of these robustly binding PI-3,4-P2 or PI-4,5-P2 but not PIP3) [9, 10]. Of the PH domain-containing proteins that do bind PIP3, the serine/threonine AGC-family protein kinase AKT has received the greatest attention, especially for its multi-faceted roles in promoting glucose metabolism and cancer [11, 12]. However, recent advances have demonstrated critical mechanisms by which other proteins with PIP3-binding PH domains contribute to cancer progression. Understanding the role of AKT-independent signaling downstream of PI3K is important because: a) AKT is not always hyperactivated in the context of mutations in PI3K pathway components such as PIK3CA and PTEN that elevate PIP3 levels in cancer; b) many critical cellular processes are driven by PI3K-dependent but AKT-independent signaling to promote malignant phenotypes, and; c) mechanisms of resistance to PI3K pathway inhibitors can involve the activation of PI3K-dependent signaling proteins that can substitute for AKT signaling. To illustrate this, in this review we highlight three AKT-independent signaling branches downstream of PI3K that have recently been shown to have critical roles in promoting cancer progression: the PDK1-mTORC2-SGK axis, Rac signaling, and the TEC family kinases.
Substituting for AKT signaling: The PDK1-mTORC2-SGK axis
PDK1 (3-phosphoinositide-dependent protein kinase 1) and the multi-protein complex mTORC2 (mechanistic target of rapamycin complex 2) are PI3K-dependent, PH domain-containing kinases that coordinately activate several growth factor-sensitive AGC kinases, including AKT (also known as protein kinase B), SGKs (serum and glucocorticoid-regulated kinase), and certain PKCs (protein kinase C), by phosphorylating their activation loops and hydrophobic motifs (HM), respectively [13]. PDK1 is a constitutively active kinase with two major regulatory domains: a C-terminal PH domain that binds PIP3, and a “PIF-binding pocket” within its catalytic domain that docks on the phosphorylated HM of AGC kinases, a region also known as the PDK1-interacting fragment (PIF) [14–17]. The PH domain allows PDK1 to co-localize with AKT at the plasma membrane and phosphorylate its activation loop upon PI3K activation. SGKs and PKCs, however, lack PH domains, and the PDK1 PH domain is not required for their phosphorylation; rather, PDK1 docks on the phosphorylated HM of these kinases through its PIF-binding pocket in order to phosphorylate their activation loops (atypical PKC isoforms contain acidic residues in their HMs that mimic constitutive phosphorylation) [18]. Two other growth factor-sensitive AGC kinases, S6K and RSK, are also activated by PDK1 in this manner but their HMs are not mTORC2 substrates and any dependence on PI3K is indirect. Therefore, at least for SGKs and some PKCs, activation loop phosphorylation by PDK1 is primed by phosphorylation of the HM by mTORC2, which is a multi-protein complex consisting of mTOR, Rictor, mSin1, mLST8, Protor1/2, and Deptor [19]. mTORC2 activity can be stimulated by PI3K activation and PIP3 [20], though the precise underlying mechanisms has long remained elusive. However, the mSin1 subunit of mTORC2 has been reported to contain a putative PH domain [21], and a recent study proposes that autoinhibition of mTOR kinase activity by the mSin1 PH domain is disrupted when mSin1-PH binds PIP3, resulting in mTORC2 activation downstream of PI3K [22••].
The roles of PDK1 and mTORC2 in cancer are frequently linked to their activation of AKT, but recent evidence has demonstrated that other downstream AGC kinases also contribute to cancer malignancy. This is highlighted by the finding that reduced anchorage-independent growth and increased apoptosis caused by PDK1 silencing cannot necessarily be rescued by the expression of constitutively active AKT [23]. Recent studies have identified the SGKs, which consist of three isoforms SGK1, SGK2, and SGK3, as critical mediators of AKT-independent signaling downstream of PDK1 and mTORC2 in cancer (Figure 1). The SGK isoforms share high sequence similarity, and isoform-specific functions remain an open question. SGK knockout mice have revealed little about their physiological functions: SGK1-null mice have sodium balance intolerances, SGK3 knockout mice have defective hair follicle development, and SGK2 knockout mice have not been studied [24, 25]. Since SGKs and AKT share highly similar substrate specificities and have overlapping substrates [26–28], SGKs can substitute for AKT signaling in survival, migration, and growth signaling (Figure 1). For instance, a subset of PIK3CA mutant breast cancer cell lines that exhibit minimal AKT activation were found to rely on the PDK1-mTORC2-mediated activation of SGK3, and not AKT, for anchorage-independent growth [29]. Interestingly, although the SGKs lack PH domains, SGK3 possesses a phox homology (PX) domain that binds phosphatidylinositol-3-phosphate (PI-3-P), which is predominantly produced at the endosome by the Class III PI3K hVps34 [30]. In addition to activation at the endosome, SGK3 can also be activated at the plasma membrane by PI3K-dependent PI-3-P production through the sequential dephosphorylation of PIP3 to PI-3,4-P2 and PI-3-P by the lipid phosphatases SHIP1/2 (SH2 domain-containing inositol phosphatase) and INPP4B (Inositol polyphosphate 4-phosphatase type II), respectively (Figure 1) [31–34••]. In PIK3CA mutant breast cancer cells, SGK3 activation correlates with elevated expression of INPP4B, which positively regulates SGK3 activity to promote cell proliferation, anchorage-independent growth, cell migration, and tumor growth in vivo [35••].
Figure 1. The PDK1-mTORC2-SGK signaling axis.
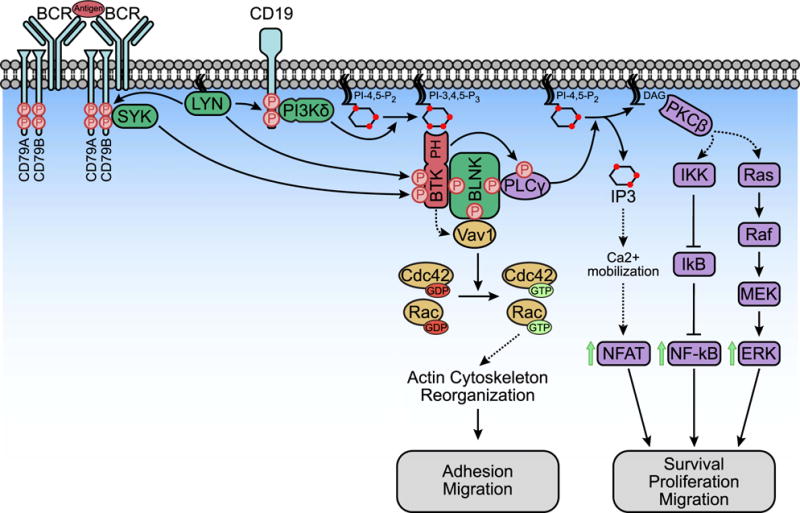
SGK3 is activated at either the plasma membrane or the endosome, where its PX domain binds to PI-3-P. PI-3-P is produced both at the plasma membrane through the sequential dephosphorylation of PIP3 by SHIP1/2 and INPP4B and at the endosome by the Class III PI3K hVps34. After PI-3-P binding, SGK3 is first phosphorylated on the hydrophobic motif (HM) by mTORC2. PDK1 then binds to the phosphorylated HM through its PIF-binding pocket and phosphorylates SGK3 on its activation loop. SGK1 is not regulated by PI-3-P because it lacks a PX domain, but it is phosphorylated by mTORC2 and PDK1 in the same manner as SGK3. Upon full activation, SGK1/3 phosphorylates its substrates with an optimal consensus motif of R-X-R-X-X-S/T to regulate cellular processes including cell growth, proliferation, migration, invasion, and resistance to PI3K and AKT inhibitors.
Activation of SGK signaling has also recently emerged as an important mechanism of resistance to PI3K and AKT inhibitors. For example, treatment of breast cancer cells with PI3K or AKT inhibitors results in increased expression and activation of SGK3, which under these conditions depends on hVps34 for activation by PDK1 and mTORC2. SGK3 subsequently substitutes for AKT by phosphorylating TSC2 and inhibiting the TSC complex to activate the kinase complex mTORC1 [36••, 37]. Similarly, elevated SGK1 expression and activation by PDK1/mTORC2 is also observed in breast cancer cell lines resistant to PI3K/AKT inhibitors. In this setting, SGK1 maintains signaling downstream of AKT by phosphorylating TSC2 to activate mTORC1 while also phosphorylating and inhibiting the transcription factor FOXO3 [38, 39••]. As mTORC1 and FOXO are major coordinators of anabolic metabolism and growth, their regulation by SGK can allow cells to overcome PI3K/AKT inhibition [40]. These results suggest that inhibitors of PDK1 or SGKs, listed in Table 1, may synergize with PI3K/AKT inhibitors to induce more robust antitumoral responses.
Table 1.
Selected list of inhibitors of AKT-independent PI3K signaling
| Inhibitor | Target | Refs |
|---|---|---|
|
PDK1/mTORC2-SGK signaling
| ||
| GSK2334470 | PDK1 | [82, 83] |
| Torin 1 | mTOR kinase | [84] |
| PP242 | mTOR kinase | [85] |
| AZD8055 | mTOR kinase | [86] |
| AZD2014 | mTOR kinase | [87] |
| MLN0128 (formerly INK128) | mTOR kinase | [88] |
| VPS34-IN1 | hVps34 | [34] |
| SAR405 | hVps34 | [89•] |
| GSK650394 | SGK1/2/3 | [90] |
| 14h | SGK1/3 | [36, 91•] |
| SI113 | SGK1 | [92–95•] |
| EMD638683 | SGK1/2/3 | [96] |
|
PI3K/Rac signaling | ||
| NSC23766 | Rac | [97, 98] |
| EHop-16 | Rac | [99, 100•] |
| EHT 1864 | Rac | [101–103] |
|
BTK and PI3Kδ inhibitors | ||
| Ibrutinib (PCI-32765) | BTK (1st gen) | [75, 76, 104, 105] |
| Acalabrutinib (ACP-196) | BTK (2nd gen) | [77, 78] |
| ONO/GS-4059 | BTK (2nd gen) | [78, 106] |
| BGB-3111 | BTK (2nd gen) | [78, 107] |
| CC-292 | BTK (2nd gen) | [108, 109] |
| Idelalisib (GS-1101; CAL-101) | PI3Kδ | [79, 80] |
| AMG-319 | PI3Kδ | [80, 110] |
| TGR-1202 | PI3Kδ | [80, 111] |
| INCB050465 | PI3Kδ | [80] |
Taken together, these studies demonstrate that AKT-independent signaling initiated by PDK1 and mTORC2 downstream of PI3K contributes significantly to cancer progression. Understanding the specific contexts under which the PDK1-mTORC2-SGK axis provides an alternative mechanism for PI3K signaling to drive tumorigenesis will be critical for the development of more effective strategies for targeting the PI3K pathway for cancer treatment.
Actin cytoskeleton remodeling: Rac signaling
One of the most important PI3K-dependent cellular processes that primarily occurs independently of AKT is remodeling of the actin cytoskeleton. A major effector of this process is the Rac proteins (RAC1, 2, and 3), which are a subfamily of the Rho family of small GTPases (Figure 2) [41]. Like all G proteins, Rac alternates between its inactive GDP-bound and active GTP-bound states, which are promoted by GTPase-activating proteins (GAPs) and guanine-nucleotide exchange factors (GEFs), respectively. PI3K functions as an upstream activator of Rac by stimulating PIP3-sensitive Rac-GEFs, which include PREX1, PREX2, Vav1, Sos1, and SWAP-70 (Figure 2). These Rac-GEFs each contain a PH domain that interacts with PIP3, which subsequently relieves the inhibition of the GEF catalytic guanine-nucleotide exchange domain [42]. Upon activation, Rac-GTP then regulates cytoskeleton reorganization by binding and activating relevant target proteins, most notably p21-activated kinase (PAK) and WAVE [43–46].
Figure 2. PI3K-Rac signaling regulates actin cytoskeleton reorganization.
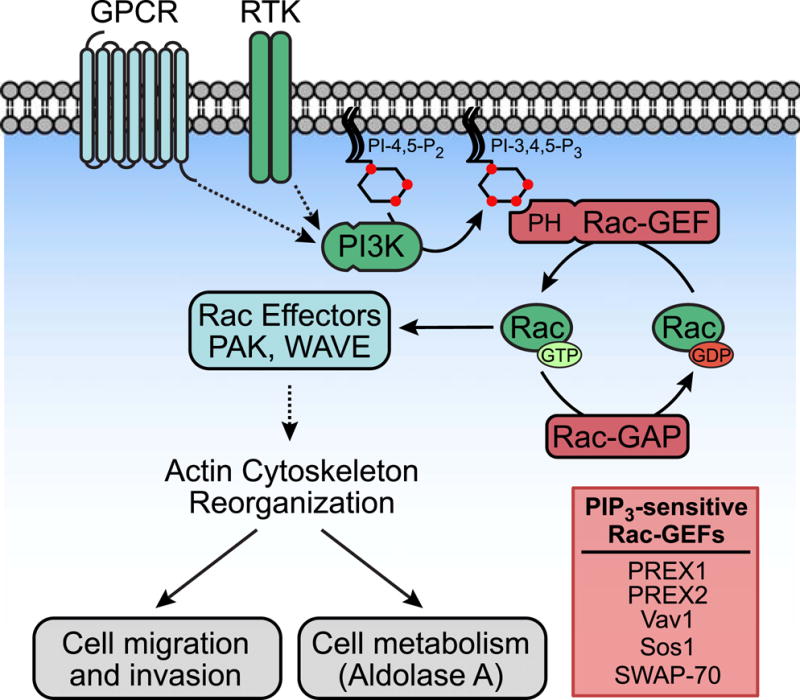
Upon binding to PIP3 via their PH domains, PIP3-sensitive Rac-GEFs catalyze the exchange of GDP for GTP on Rac. Rac activation is negatively regulated by Rac-GAPs, which facilitate GTP hydrolysis by Rac. When Rac-GTP is activated, it regulates cytoskeleton reorganization by binding and activating relevant target proteins, most notably PAK and WAVE. Remodeling of the actin cytoskeleton modulates cellular processes such as cell migration, invasion, and metabolism.
While many studies have demonstrated that AKT regulates cancer cell invasion and migration [47, 48], Rac signaling also promotes this phenotype independently of AKT by remodeling the actin cytoskeleton [49, 50]. Recent studies have focused on the Rac-GEFs PREX1 and PREX2, which have been implicated in multiple cancers. For example, PREX1 is up-regulated in breast cancer, metastatic prostate cancer, and melanoma, and it activates RAC1 to promote cell migration and invasion through the formation of lamellipodia. PREX1 overexpression is sufficient to increase the metastatic potential of non-metastatic prostate cancer cells in vivo, and Prex1−/− mice are resistant to metastasis in an Nras/INK4a-driven mouse melanoma model [51–53]. Likewise, PREX2 is up-regulated in many cancers and is also frequently mutated, especially in melanoma [54–56]. These oncogenic PREX2 mutations increase its Rac-GEF activity, which contributes to NRAS-driven melanoma development and promotes breast cancer cell migration [57••, 58••]. Interestingly, PREX2 is not only activated by PIP3, but also directly binds to PTEN and inhibits its lipid phosphatase activity to amplify PI3K signaling [59]. As a potential mechanism to prevent a feed-forward circuit in which PREX2-induced PI3K signaling results in unrestrained PIP3-mediated PREX2 activation, PREX2 is reciprocally inhibited by PTEN independently of its lipid phosphatase activity and PIP3 [58••]. Remarkably, oncogenic PREX2 mutants are able to escape PTEN-mediated inhibition, allowing them to both amplify PI3K signaling and activate Rac to promote tumor growth and cell invasion to drive cancer progression [57••, 58••].
Rac signaling has also recently been shown to contribute to PI3K-dependent regulation of cellular metabolism, which has typically been thought to be predominantly controlled by AKT [60–62]. A recent study reported that PI3K acutely stimulates glycolysis independently of AKT by regulating aldolase A in a Rac1-dependent manner. In quiescent cells, aldolase A is bound to the actin cytoskeleton, which inhibits its enzymatic activity. Upon growth factor-induced PI3K activation, activation of Rac and its effectors PAK and WAVE leads to actin cytoskeleton reorganization that releases aldolase A, and thus increases its activity [63••]. Interestingly, another study demonstrated that inhibition of PI3K, but not AKT, selectively inhibits the non-oxidative branch of the pentose phosphate pathway (non-oxPPP), resulting in decreased ribose 5-phosphate and nucleotide production [64•]. Since glyceraldehyde 3-phosphate, a product of aldolase A, serves as an entry point into the non-oxPPP, PI3K/Rac-mediated aldolase A activation may promote the PI3K-dependent, but AKT-independent, stimulation of the non-oxPPP to contribute to nucleotide synthesis. Given recent interest in therapeutically exploiting cancer-associated metabolic vulnerabilities, a deeper understanding of how PI3K-Rac signaling regulates glycolysis and other metabolic pathways may reveal novel strategies for targeting the PI3K pathway for cancer therapy.
These studies highlight the importance of Rac signaling in modulating cellular processes associated with actin cytoskeleton remodeling to promote cancer malignancy. Importantly, Rac also controls other processes independently of cytoskeleton reorganization such as proliferation and survival, which are not discussed in this review [41]. Therefore, developing inhibitors of Rac (Table 1) and exploring their potential as therapeutic options for cancer treatment is an active area of investigation [65].
Activation of non-receptor tyrosine kinase signaling: TEC family kinases
Of the 90 tyrosine kinases in the human genome, 58 are membrane-spanning receptor tyrosine kinases (RTKs), while the remaining 32 lack transmembrane domains and are classified as nonreceptor tyrosine kinases (NRTKs). NRTKs function within the nucleocytoplasm to trigger signaling cascades downstream of cell surface receptors and tend to play an especially important role in propagating signals downstream of receptors that do not contain intrinsic enzymatic domains [66]. For instance, in immune cells members of the SRC and TEC subfamilies of NRTKs mediate critical signaling events downstream of the T cell receptor (TCR), B cell receptor (BCR), Toll-like receptors (TLRs), and integrins [67]. Like RTKs, NRTKs can activate PI3K by phosphorylating its regulatory subunit or creating phospho-tyrosine docking sites on adaptors and receptors that recruit and activate PI3K. However, members of one NRTK subgroup, the TEC family, conversely rely on PI3K for their activation, distinguishing them from other NRTKs [68].
The five-member TEC kinase family includes: TEC (tyrosine kinase expressed in hepatocellular carcinoma), BMX (bone marrow tyrosine kinase gene on chromosome X; also known as ETK), BTK (bruton’s tyrosine kinase), ITK (interleukin-2-inducible T-cell kinase; also known as EMT or TSK), and TXK (also known as RLK). These share a common domain architecture including a C-terminal tyrosine kinase domain, SH2 and SH3 protein-binding domains, and, with the exception of TXK, an N-terminal PH domain that binds PIP3 [68]. The binding of PI3K-produced PIP3 results in the accumulation of TEC, BMX, BTK, and ITK at the plasma membrane where they are phosphorylated and activated by other NRTKs (as described for BTK in more detail below). Although PI3K may simultaneously activate AKT in the same cell, TEC kinase activation is AKT-independent. Hence, PI3K activity can dynamically promote membrane localization and activation of TEC kinases in contrast to, for instance, SRC family members which constitutively associate with membranes via their myristoyl and palmitoyl lipid modifications. Although the TEC kinases are expressed in multiple organ systems and preclinical studies have implicated them in multiple cancers including breast, colorectal, prostate, and glioblastoma, their role in the immune system has received the greatest attention [67, 68]. Here we highlight the role of the TEC family member BTK and its regulation by the delta isoform of PI3K in B lymphocyte (B cell) function and malignancy.
The B cell receptor (BCR) plays a pivotal role in both the development and activation of B cells. In naïve B cells, antigen binding to the BCR induces clustering of BCRs and associated CD79A/CD79B heterodimers that triggers tyrosine kinase signaling by NRTKs (Figure 3). BCR-associated LYN, a SRC family member that tethers to the membrane via dual lipid modifications, phosphorylates tyrosine motifs on the CD79A/CD79B heterodimer. The NRTK SYK then binds tyrosine-phosphorylated CD79A/CD79B, is activated, and subsequently phosphorylates a tyrosine motif on the co-receptor CD19 to which Class I PI3K binds via its SH2 domain-containing regulatory subunit. Production of PIP3 from PI-4,5-P2 by PI3K recruits BTK (via its PH domain) to the plasma membrane, where it can undergo activating phosphorylation by LYN and SYK. PI3K-dependent AKT activation also occurs in this context and regulates certain B-cell functions, but does not directly regulate BTK and is not discussed further here [69]. Through its SH2 domain, BTK participates in a signaling complex nucleated by a scaffolding protein BLNK (B cell linker; also known as SLP65) that is phosphorylated on multiple tyrosines by other NRTKs. PLCγ and the Rho family GEF Vav1 also dock on tyrosine-phosphorylated BLNK via their SH2 domains. Phosphorylation and activation of PLCγ by BTK triggers calcium mobilization and PKC signaling that ultimately turns on Ras signaling along with NF-κB and NFAT transcriptional programs. BTK-dependent activation of Vav1 turns on the Rho family small GTPases Cdc42 and Rac, which control actin remodeling. Together, these and other signals cooperate to govern the survival, differentiation, proliferation, and migration responses that underlie B cell development and activation [70].
Figure 3. PI3K and BTK Signaling in Response to BCR activation in B cells.
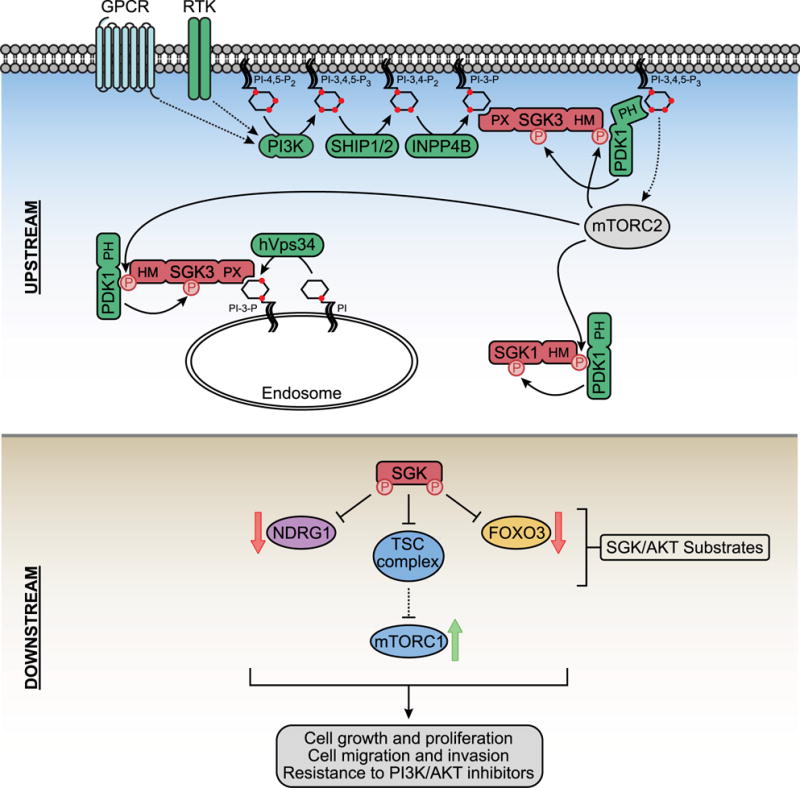
Antigen engagement of the BCR receptor induces clustering of the BCR, associated CD79A/CD79B heterodimers, and the non-receptor tyrosine kinase (NRTK) LYN which is tethered to the membrane by dual lipid modifications. LYN phosphorylates tyrosine motifs on CD79A/CD79B and the co-receptor CD19 to which the NRTK SYK and PI3K dock respectively and become activated. PIP3 produced by PI3K recruits BTK to the membrane where it is phosphorylated and activated by SYK and LYN. BTK also forms a complex with the adaptor protein BLNK, the Rho family GEF Vav, and the phospholipase PLCγ. Following its BTK-dependent activation, Vav promotes GTP loading and activation of Cdc42 and Rac which then trigger actin cytoskeleton remodeling through a variety of effectors. Simultaneously, BTK phosphorylates and activates PLCγ which cleaves PI-4,5-P2 to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG is bound by PKCβ which triggers IKK signaling and NFkB-dependent transcription plus activation of Ras-Raf-MEK-ERK signaling. IP3 stimulates calcium (Ca2+) mobilization which results in NFAT-dependent transcription. These signaling events cooperate to control survival, proliferation, and adhesion signals that underlie B cell development and activation.
In humans, inactivating mutations in BTK are the molecular basis for the immunodeficiency disorder X-linked agammaglobulinemia which is characterized by severe defects in B cell development and function [71, 72]. BTK knockout mice or mice treated with small molecule inhibitors of BTK have similar B cell deficiencies, illustrating the critical role played by BTK in B cell biology [73–75]. Aberrant BCR signaling can promote B cell transformation, and due to the tissue-specific importance of BTK and PI3Kδ in B cells, there has been an intense effort to therapeutically target these for the treatment of B cell malignancies [70]. It is worth noting that the role of BTK is more specific to B cells compared with PI3Kδ, which is broadly important in leukocytes. A first generation irreversible BTK inhibitor Imbruvica (Ibrutinib) is FDA-approved for treatment of chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and Waldenström macroglobulinemia (Table 1) [75, 76]. As Imbruvica inhibits multiple Tec kinases that are important in other immune cell types, more potent and selective second generation BTK inhibitors, such as Acalabrutinib, have been developed and are now showing promise in Phase I and II clinical trials (Table 1) [77••, 78]. A PI3Kδ inhibitor, Zydelig (Idelalisib), has also been FDA-approved for the treatment of relapsed CLL, follicular B cell non-Hodgkin lymphoma (FL) and small lymphocytic lymphoma (SLL), again underscoring the critical role played by PI3K in B cells [79, 80]. Interestingly, there is some evidence that these inhibitors may also prove useful in treating solid tumors. In a mouse model of pancreas ductal adenocarcinoma (PDAC), inhibitors of BTK and, in this case, PI3Kγ, were shown to alter lymphocyte-macrophage cross-talk that ultimately promoted immunosuppression of the tumor [81••]. Therefore, PI3K-dependent BTK signaling is a key example of an AKT-independent branch of the PI3K network that has emerged as an effective therapeutic target in cancer. The clinical results achieved with BTK and PI3Kδ inhibitors represent one of the recent success stories for rationally developed, precision medicine.
Summary and outlook
As one of the most frequently altered pathways in human cancer, PI3K signaling has clearly emerged as a highly attractive drug target for the treatment of human cancers. Given the predominant focus on studying AKT as the primary effector downstream of PI3K, much of the effort in targeting this pathway has centered around the development of drugs that effectively inhibit PI3K/AKT signaling. However, as this review has illustrated, it is becoming increasingly clear that other PI3K-dependent but AKT-independent signaling branches are just as important, if not more important, in promoting cancer initiation and progression. Indeed, a growing recognition of this idea has led to the development of drugs that target these other pathways downstream of PI3K (Table 1). Given that PI3K and AKT inhibitors currently in the clinic have shown limited efficacy with dose-limiting toxicities, these drugs may provide opportunities to rationalize new therapeutic combinations that may target the PI3K pathway more effectively. It is also important to note that the three pathways highlighted in this review constitute only a subset of the PH domain-containing proteins that are regulated by PIP3 (or other phosphoinositides derived from PI3K-produced PIP3). Therefore, we believe that there is still much to learn about how these various PI3K-dependent signaling proteins contribute to cancer malignancy, and that this knowledge will greatly strengthen our ability to develop novel and more effective strategies for targeting the PI3K pathway for the treatment of human cancers.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health and the Ludwig Center at Harvard. E.C.L. is a pre-doctoral fellow of the NSF graduate research fellowship program (NSF DGE1144152). C.C.D is supported by a grant from the National Cancer Institute (K99-CA194314).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 3.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 5.Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 6.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 7.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 8.Toker A. Phosphoinositide 3-kinases-a historical perspective. Subcell Biochem. 2012;58:95–110. doi: 10.1007/978-94-007-3012-0_4. [DOI] [PubMed] [Google Scholar]
- 9.Park WS, Heo WD, Whalen JH, O’Rourke NA, Bryan HM, Meyer T, Teruel MN. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell. 2008;30:381–392. doi: 10.1016/j.molcel.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 12.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 15.Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J. 1999;337:575–583. [PMC free article] [PubMed] [Google Scholar]
- 16.Balendran A, Biondi RM, Cheung PC, Casamayor A, Deak M, Alessi DR. A 3-phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Czeta (PKCzeta) and PKC-related kinase 2 by PDK1. J Biol Chem. 2000;275:20806–20813. doi: 10.1074/jbc.M000421200. [DOI] [PubMed] [Google Scholar]
- 17.Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000;19:979–988. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 2011;286:10998–11002. doi: 10.1074/jbc.M110.195016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan D, Matsuura Y. Structures of the pleckstrin homology domain of Saccharomyces cerevisiae Avo1 and its human orthologue Sin1, an essential subunit of TOR complex 2. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:386–392. doi: 10.1107/S1744309112007178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22••.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, Wang B, Blenis J, Cantley LC, Toker A, et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015;5:1194–1209. doi: 10.1158/2159-8290.CD-15-0460. Suggests that PIP3 activates mTORC2 by binding to the PH domain of mSin1, which subsequently releases its inhibition of the mTOR kinase domain to induce mTORC2 activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gagliardi PA, di Blasio L, Orso F, Seano G, Sessa R, Taverna D, Bussolino F, Primo L. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia. 2012;14:719–731. doi: 10.1593/neo.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 25.McCormick JA, Feng Y, Dawson K, Behne MJ, Yu B, Wang J, Wyatt AW, Henke G, Grahammer F, Mauro TM, et al. Targeted disruption of the protein kinase SGK3/CISK impairs postnatal hair follicle development. Mol Biol Cell. 2004;15:4278–4288. doi: 10.1091/mbc.E04-01-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339:319–328. [PMC free article] [PubMed] [Google Scholar]
- 28.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tessier M, Woodgett JR. Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J Biol Chem. 2006;281:23978–23989. doi: 10.1074/jbc.M604333200. [DOI] [PubMed] [Google Scholar]
- 31.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 32.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wisniewski D, Strife A, Swendeman S, Erdjument-Bromage H, Geromanos S, Kavanaugh WM, Tempst P, Clarkson B. A novel SH2-containing phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase (SHIP2) is constitutively tyrosine phosphorylated and associated with src homologous and collagen gene (SHC) in chronic myelogenous leukemia progenitor cells. Blood. 1999;93:2707–2720. [PubMed] [Google Scholar]
- 34••.Bago R, Malik N, Munson MJ, Prescott AR, Davies P, Sommer E, Shpiro N, Ward R, Cross D, Ganley IG, Alessi DR. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J. 2014;463:413–427. doi: 10.1042/BJ20140889. Identifies a selective inhibitor of hVps34 that reduces PI(3)P at the endosome and partially suppresses SGK3 activity. Inhibition of hVps34 in combination with a Class I PI3K inhibitor completely inhibits SGK3, indicating that SGK3 is activated by two pools of PI(3)P: one at the endosome, and one at the plasma membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35••.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 Mediates INPP4B-Dependent PI3K Signaling in Breast Cancer. Mol Cell. 2014;56:595–607. doi: 10.1016/j.molcel.2014.09.023. Shows that in oncogenic PIK3CA mutant breast cancer cells with elevated expression of INPP4B, amplification of SGK3 and activation of SGK3 signaling functions independently of AKT to drive cell proliferation, invasive migration, and tumorigenesis in vivo. The effects of SGK3 on cell migration and invasion are due in part to its phosphorylation of NDRG1, which is then directed towards degradation by Fbw7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.Bago R, Sommer E, Castel P, Crafter C, Bailey FP, Shpiro N, Baselga J, Cross D, Eyers PA, Alessi DR. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016;35:1902–1922. doi: 10.15252/embj.201693929. Demonstrates that SGK3 expression and activation increases in response to prolonged treatment of breast cancer cells with PI3K or AKT inhibitors. SGK3 mediates resistance to these inhibitors by substituting for AKT to phosphorylate TSC2 in order to activate mTORC1, and SGK3 inhibition synergizes with an AKT inhibitor to induce tumor regression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J. 2013;452:499–508. doi: 10.1042/BJ20130342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39••.Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell. 2016;30:229–242. doi: 10.1016/j.ccell.2016.06.004. Shows that PDK1 inhibition restores the sensitivity of cancer cells resistant to PI3Kα inhibition towards PI3Kα inhibitors. Resistance to PI3Kα inhibitors is mediated by the PDK1-dependent activation of SGK1, which represses the FOXO-mediated transcriptional signature and maintains mTORC1 activity by phosphorylating TSC2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ilagan E, Manning BD. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer. 2016;2:241–251. doi: 10.1016/j.trecan.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell Signal. 2012;24:353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–97. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 43.Whale A, Hashim FN, Fram S, Jones GE, Wells CM. Signalling to cancer cell invasion through PAK family kinases. Front Biosci (Landmark Ed) 2011;16:849–864. doi: 10.2741/3724. [DOI] [PubMed] [Google Scholar]
- 44.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suetsugu S, Kurisu S, Oikawa T, Yamazaki D, Oda A, Takenawa T. Optimization of WAVE2 complex-induced actin polymerization by membrane-bound IRSp53, PIP(3), and Rac. J Cell Biol. 2006;173:571–585. doi: 10.1083/jcb.200509067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 47.Chin YR, Toker A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol Cell. 2010;38:333–344. doi: 10.1016/j.molcel.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009;21:470–476. doi: 10.1016/j.cellsig.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 50.Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–112. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40:877–892. doi: 10.1016/j.molcel.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin J, Xie Y, Wang B, Hoshino M, Wolff DW, Zhao J, Scofield MA, Dowd FJ, Lin MF, Tu Y. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene. 2009;28:1853–1863. doi: 10.1038/onc.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindsay CR, Lawn S, Campbell AD, Faller WJ, Rambow F, Mort RL, Timpson P, Li A, Cammareri P, Ridgway RA, et al. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat Commun. 2011;2:555. doi: 10.1038/ncomms1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57••.Lissanu Deribe Y, Shi Y, Rai K, Nezi L, Amin SB, Wu CC, Akdemir KC, Mahdavi M, Peng Q, Chang QE, et al. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc Natl Acad Sci U S A. 2016;113:E1296–305. doi: 10.1073/pnas.1513801113. Shows that PREX2 mutations frequently found in human cancers increase its Rac-GEF activity, abolish PREX2 binding to PTEN to activate PI3K/AKT signaling, and accelerate NRAS-driven melanoma in a genetically engineered mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58••.Mense SM, Barrows D, Hodakoski C, Steinbach N, Schoenfeld D, Su W, Hopkins BD, Su T, Fine B, Hibshoosh H, Parsons R. PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci Signal. 2015;8:ra32. doi: 10.1126/scisignal.2005840. Demonstrates that PTEN binds to PREX2 and inhibits its Rac-GEF activity independently of its lipid phosphatase activity, resulting in decreased PREX2-induced cell migration and invasion. PREX2 mutants found in human cancers are able to escape PTEN-mediated inhibition while maintaining their ability to inhibit PTEN lipid phosphatase activity to activate PI3K/AKT signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325:1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robey RB, Hay N. Is Akt the “Warburg kiπase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lien EC, Lyssiotis CA, Cantley LC. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016;207:39–72. doi: 10.1007/978-3-319-42118-6_3. [DOI] [PubMed] [Google Scholar]
- 62.Lien EC, Lyssiotis CA, Juvekar A, Hu H, Asara JM, Cantley LC, Toker A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat Cell Biol. 2016;18:572–578. doi: 10.1038/ncb3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63••.Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. Shows that PI3K stimulates glycolysis independently of AKT by activating Rac, which leads to the reorganization of the actin cytoskeleton and the release of actin-bound aldolase A, which increases aldolase activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64•.Juvekar A, Hu H, Yadegarynia S, Lyssiotis CA, Ullas S, Lien EC, Bellinger G, Son J, Hok RC, Seth P, et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci U S A. 2016;113:E4338–47. doi: 10.1073/pnas.1522223113. Shows that inhibition of PI3K, but not AKT, induces DNA damage in BRCA1-mutant breast cancers by selectively inhibiting the non-oxidative branch of the pentose phosphate pathway to reduce the production of ribose 5-phosphate and nucleotides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013;12:1925–1934. doi: 10.1158/1535-7163.MCT-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gocek E, Moulas AN, Studzinski GP. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit Rev Clin Lab Sci. 2014;51:125–137. doi: 10.3109/10408363.2013.874403. [DOI] [PubMed] [Google Scholar]
- 67.Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22:1175–1184. doi: 10.1016/j.cellsig.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 68.Horwood NJ, Urbaniak AM, Danks L. Tec family kinases in inflammation and disease. Int Rev Immunol. 2012;31:87–103. doi: 10.3109/08830185.2012.670334. [DOI] [PubMed] [Google Scholar]
- 69.Szydłowski M, Jabłońska E, Juszczyński P. FOXO1 transcription factor: a critical effector of the PI3K-AKT axis in B-cell development. Int Rev Immunol. 2014;33:146–157. doi: 10.3109/08830185.2014.885022. [DOI] [PubMed] [Google Scholar]
- 70.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–232. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 71.Vetrie D, Vorechovský I, Sideras P, Holland J, Davies A, Flinter F, Hammarström L, Kinnon C, Levinsky R, Bobrow M. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 72.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 73.Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, Witte ON, Perlmutter RM. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 74.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Müller S, Kantor AB, Herzenberg LA. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 75.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77••.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. This article reports on the first clinical trial in which patients with a B cell malignancy were treated with the second generation BTK inhibitor acalabrutinib. Phase 1–2 trial results showed promising safety and efficacy for acalabrutinib in patients with relapsed chronic lymphocytic leukemia (CLL). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016;9:80. doi: 10.1186/s13045-016-0313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016;6:1090–1105. doi: 10.1158/2159-8290.CD-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81••.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–285. doi: 10.1158/2159-8290.CD-15-0827. This study demonstrates that BTK inhibitors may be useful for treating solid tumors in some settings. Ibrutinib treatments improved the response of a PDAC mouse model to chemotherapy by increasing the anti-tumor immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Medina JR, Becker CJ, Blackledge CW, Duquenne C, Feng Y, Grant SW, Heerding D, Li WH, Miller WH, Romeril SP, et al. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J Med Chem. 2011;54:1871–1895. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]
- 83.Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J. 2011;433:357–369. doi: 10.1042/BJ20101732. [DOI] [PubMed] [Google Scholar]
- 84.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 87.Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, Martin NM, Ruston L, Pass SL, Pass M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212–1216. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 88.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89•.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–1019. doi: 10.1038/nchembio.1681. Identifies a novel inhibitor of hVps34, SAR405, that alters vesicle trafficking, autophagy, and displays anti-tumor activity in combination with the mTORC1 inhibitor everolimus. [DOI] [PubMed] [Google Scholar]
- 90.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68:7475–7483. doi: 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91•.Halland N, Schmidt F, Weiss T, Saas J, Li Z, Czech J, Dreyer M, Hofmeister A, Mertsch K, Dietz U, et al. Discovery of N-[4-(1H-Pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as Highly Active and Selective SGK1 Inhibitors. ACS Med Chem Lett. 2015;6:73–78. doi: 10.1021/ml5003376. Identifies a set of candidate small molecules that selectively inhibit SGK1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ortuso F, Amato R, Artese A, D’antona L, Costa G, Talarico C, Gigliotti F, Bianco C, Trapasso F, Schenone S, et al. In silico identification and biological evaluation of novel selective serum/glucocorticoid-inducible kinase 1 inhibitors based on the pyrazolo-pyrimidine scaffold. J Chem Inf Model. 2014;54:1828–1832. doi: 10.1021/ci500235f. [DOI] [PubMed] [Google Scholar]
- 93•.D’Antona L, Amato R, Talarico C, Ortuso F, Menniti M, Dattilo V, Iuliano R, Gigliotti F, Artese A, Costa G, et al. SI113, a specific inhibitor of the Sgk1 kinase activity that counteracts cancer cell proliferation. Cell Physiol Biochem. 2015;35:2006–2018. doi: 10.1159/000374008. Characterizes SI113 as a specific inhibitor of SGK1 that inhibits the growth and induces cell death in various cancer cell lines. SI113 also increases the sensitivity of cancer cells towards paclitaxel. [DOI] [PubMed] [Google Scholar]
- 94•.Talarico C, D’Antona L, Scumaci D, Barone A, Gigliotti F, Fiumara CV, Dattilo V, Gallo E, Visca P, Ortuso F, et al. Preclinical model in HCC: the SGK1 kinase inhibitor SI113 blocks tumor progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget. 2015;6:37511–37525. doi: 10.18632/oncotarget.5527. Shows that the SGK1 inhibitor SI113 inhibits tumor growth and induces apoptosis in hepatocellular carcinoma, both in vitro and in vivo. SI133 also synergizes with ionizing radiation to induce cell death. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95•.Talarico C, Dattilo V, D’Antona L, Barone A, Amodio N, Belviso S, Musumeci F, Abbruzzese C, Bianco C, Trapasso F, et al. SI113, a SGK1 inhibitor, potentiates the effects of radiotherapy, modulates the response to oxidative stress and induces cytotoxic autophagy in human glioblastoma multiforme cells. Oncotarget. 2016;7:15868–15884. doi: 10.18632/oncotarget.7520. Shows that SGK1 is over expressed in malignant gliomas and that the SGK1 inhibitor SI113 inhibits the growth of and induces apoptosis in glioblastoma cell lines. SGK1 overexpression protects cells against oxidative stress, and SI113 sensitizes cells towards oxidative stress and ionizing radiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ackermann TF, Boini KM, Beier N, Scholz W, Fuchss T, Lang F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem. 2011;28:137–146. doi: 10.1159/000331722. [DOI] [PubMed] [Google Scholar]
- 97.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Akbar H, Cancelas J, Williams DA, Zheng J, Zheng Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. Methods Enzymol. 2006;406:554–565. doi: 10.1016/S0076-6879(06)06043-5. [DOI] [PubMed] [Google Scholar]
- 99.Montalvo-Ortiz BL, Castillo-Pichardo L, Hernández E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 2012;287:13228–13238. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100•.Castillo-Pichardo L, Humphries-Bickley T, De La Parra C, Forestier-Roman I, Martinez-Ferrer M, Hernandez E, Vlaar C, Ferrer-Acosta Y, Washington AV, Cubano LA, et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl Oncol. 2014;7:546–555. doi: 10.1016/j.tranon.2014.07.004. Demonstrates that the Rac inhibitor EHop-016 inhibits tumor growth, metastasis, and angiogenesis in a breast cancer xenograft model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Désiré L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, Bachelot F, Leblond B, Taverne T, Beausoleil E, et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005;280:37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 102.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 103.Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 2008;439:111–129. doi: 10.1016/S0076-6879(07)00409-0. [DOI] [PubMed] [Google Scholar]
- 104.Lee CS, Rattu MA, Kim SS. A review of a novel, Bruton’s tyrosine kinase inhibitor, ibrutinib. J Oncol Pharm Pract. 2016;22:92–104. doi: 10.1177/1078155214561281. [DOI] [PubMed] [Google Scholar]
- 105.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med. 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, Quittet P, Shah N, Hutchinson CV, Honda H, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127:411–419. doi: 10.1182/blood-2015-08-664086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tam C, Grigg AP, Opat S, Ku M, Gilbertson M, Anderson MA, Seymour JF, Ritchie DS, Dicorleto C, Dimovski B, et al. The BTK inhibitor, Bgb-3111, is safe, tolerable, and highly active in patients with relapsed/refractory B-cell malignancies: initial report of a phase 1 first-in-human trial. Blood. 2015;126:832. [Google Scholar]
- 108.Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT, Barr PM, Foran JM, Miller TP, Burger JA, et al. Phase I study of single-agent CC-292, a highly selective Bruton’s tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2016;101:e295–8. doi: 10.3324/haematol.2015.140806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arnason JE, Brown JR. B cell receptor pathway in chronic lymphocytic leukemia: specific role of CC-292. Immunotargets Ther. 2014;3:29–38. doi: 10.2147/ITT.S37419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cushing TD, Hao X, Shin Y, Andrews K, Brown M, Cardozo M, Chen Y, Duquette J, Fisher B, Gonzalez-Lopez de Turiso F, et al. Discovery and in vivo evaluation of (S)-N-(1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and related PI3Kδ inhibitors for inflammation and autoimmune disease. J Med Chem. 2015;58:480–511. doi: 10.1021/jm501624r. [DOI] [PubMed] [Google Scholar]
- 111.Locatelli SL, Careddu G, Inghirami G, Castagna L, Sportelli P, Santoro A, Carlo-Stella C. The novel PI3K-δ inhibitor TGR-1202 enhances Brentuximab Vedotin-induced Hodgkin lymphoma cell death via mitotic arrest. Leukemia. 2016 doi: 10.1038/leu.2016.224. [DOI] [PubMed] [Google Scholar]