

Summary
Malonyl-CoA is a central metabolite in mammalian fatty acid biochemistry generated and utilized in the cytoplasm; however, little is known about noncanonical organelle-specific malonyl-CoA metabolism. Intramitochondrial malonyl-CoA is generated by a malonyl-CoA synthetase, ACSF3, that produces malonyl-CoA from malonate, an endogenous competitive inhibitor of succinate dehydrogenase. To determine the metabolic requirement for mitochondrial malonyl-CoA, ACSF3 knockout (KO) cells were generated by CRISPR/Cas-mediated genome editing. ACSF3 KO cells exhibited elevated malonate and impaired mitochondrial metabolism. Unbiased and targeted metabolomics analysis of KO and control cells in the presence or absence of exogenous malonate revealed metabolic changes dependent on either malonate or malonyl-CoA. While ACSF3 was required for the metabolism and therefore detoxification of malonate, ACSF3-derived malonyl-CoA was specifically required for lysine malonylation of mitochondrial proteins. Together, these data describe an essential role for ACSF3 in dictating the metabolic fate of mitochondrial malonate and malonyl-CoA in mammalian metabolism.
Keywords: malonate, malonyl-CoA synthetase, ACSF3, acetyl-CoA, malonylation, mitochondrial metabolism, metabolomics, CRISPR/Cas, succinylation, SIRT5
Graphical abstract
Malonate is an endogenous metabolite that inhibits mitochondrial metabolism. Using genetically-modified human cells, Bowman et al. show that a mitochondrial malonyl-CoA synthetase, ACSF3, promotes malonate detoxification to enhance mitochondrial metabolic flux and is required for malonylation of mitochondrial proteins.
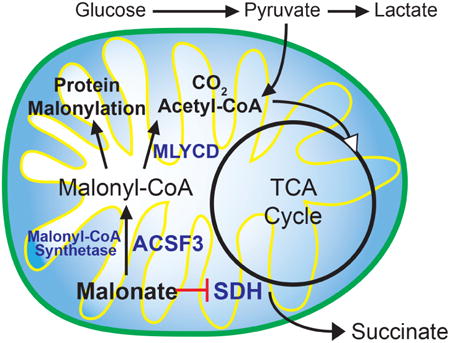
Introduction
Malonyl-CoA is positioned at a central regulatory node in mammalian metabolism to coordinate the synthesis and oxidation of fatty acids. Malonyl-CoA is generated in the cytoplasm and mitochondrial outer membrane by the biotin-dependent carboxylation of acetyl-CoA by the highly regulated Acetyl-CoA Carboxylase (Foster, 2012; Kerner et al., 2014; Ruderman et al., 2003; Saggerson, 2008; Wolfgang and Lane, 2006). Malonyl-CoA can then be used by Fatty Acid Synthase (FASN) to generate long-chain fatty acids, or be used for chain-elongation of fatty acids (Kerner et al., 2014). Therefore, malonyl-CoA represents the rate-determining and committed metabolite in de novo fatty acid synthesis. Concomitantly, malonyl-CoA acts as an allosteric inhibitor of Carnitine Palmitoyltransferase 1 (CPT1), the rate-setting step in the mitochondrial β-oxidation of long-chain fatty acids; therefore, malonyl-CoA is the metabolite that mediates the basic metabolic logic whereby fatty acid synthesis and oxidation do not occur simultaneously. Both acetyl-CoA and malonyl-CoA are membrane-impermeable and all of the canonical biosynthetic machinery for malonyl-CoA that has been described is localized exclusively to the cytoplasm. Even tissues with limited expression of FASN, such as mammalian muscle, generate cytoplasmic malonyl-CoA to regulate fatty acid oxidation (Funai et al., 2013; Pender et al., 2006). Muscle regulates malonyl-CoA largely by its decarboxylation via Malonyl-CoA Decarboxylase, MLYCD (Rodriguez et al., 2014; Saha et al., 2000). Inborn errors of MLYCD result in a combined malonic and methylmalonic aciduria (FitzPatrick et al., 1999; Gao et al., 1999; Wightman et al., 2003). Curiously, MLYCD contains putative peroxisomal and mitochondrial targeting sequences and can be readily found in mitochondria (FitzPatrick et al., 1999; Kerner and Hoppel, 2002; Laurent et al., 2013; Sambandam et al., 2004). How malonyl-CoA, which is membrane-impermeable, can be generated in the mitochondrial matrix has been a longstanding mystery. This has been at least partly resolved by the discovery of a eukaryotic mitochondrial malonyl-CoA synthetase, ACSF3 (Chen et al., 2011; Witkowski et al., 2011).
ACSF3 was originally described as an orphan member of the Acyl-CoA Synthetase (ACS) family of enzymes (Watkins et al., 2007). Recently, the Arabidopsis ACSF3 ortholog, Acyl Activating Enzyme 13, was described as a eukaryotic malonyl-CoA synthetase essential for plant growth and viability, especially in the presence of exogenous malonate (Chen et al., 2011; Guan and Nikolau, 2016). Like other acyl-CoA synthetases, ACSF3 ligates Coenzyme A to its co-substrate, malonate, in an ATP-dependent manner (Figure 1A). Human ACSF3 localizes to the mitochondrial matrix to produce malonyl-CoA from malonate within that organelle (Witkowski et al., 2011). Mitochondrial malonate is an endogenous metabolite that is a classic competitive inhibitor of succinate dehydrogenase, a component of the tricarboxylic acid (TCA) cycle and Complex II of the electron transport chain (Quastel and Wooldridge, 1928). As such, malonate is often used as a metabolic toxin to destroy striatal neurons in models of Parkinson's disease (Beal et al., 1993; Moy et al., 2000; Zeevalk et al., 1997). The severe malonic and methylmalonic aciduria characteristic of MLYCD deficiency is also accompanied by developmental delay, seizure disorders, hypoglycemia, and cardiomyopathy. Recently, patients that presented with combined malonic and methylmalonic aciduria without mutations in MLYCD were found to have nonsynonymous mutations in the ACSF3 gene (Alfares et al., 2011; Sloan et al., 2011). The similarities in the phenotypes of MLYCD and ACSF3 deficiencies suggest that they exist in the same biochemical pathway. This presents a metabolic rationale for why MLYCD is localized within mitochondria—toxic malonate may be metabolized within mitochondria through the subsequent activities of ACSF3 and MLYCD.
Figure 1. ACSF3 is a Mitochondrial Malonyl-CoA Synthetase.
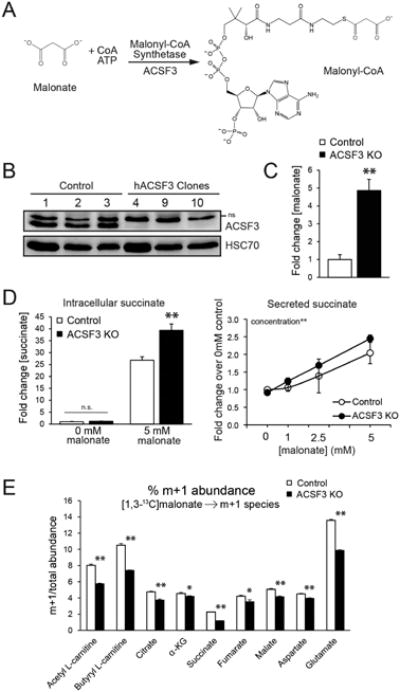
(A) The reaction catalyzed by acyl-CoA synthetase family member 3 (ACSF3), a mammalian mitochondrial malonyl-CoA synthetase.
(B) Immunoblot for ACSF3 in three control clones that were transfected with Cas9 only and three ACSF3 knockout (KO) clones. HSC70 is shown as a loading control. See Figure S1 for characterization of genomic mutations. (ns = non-specific band).
(C) Steady-state intracellular malonate and succinate (D) concentrations, in the presence or absence of 5 mM malonate for 24 hours (mean ± SEM, n=10).
(D) Succinate secretion into the culture medium upon increasing dose of malonate for 24h (mean ± SEM, n=4). Effect of concentration significant by 2-way ANOVA.
(E) [1,3-13C]malonate flux. % abundance of m+1-labeled metabolites determined by LC-MS/MS that are significantly down-regulated in ACSF3 KO cells labeled with 2.5 mM 1,3-13C-malonate for 4 hours (mean ± SEM, n=6, α-KG = α-ketoglutarate). *p<0.05, **p<0.001
Here we have taken advantage of CRISPR/Cas9-mediated genome editing to mutate ACSF3 in a human cell line that robustly expresses the enzyme. Using a combination of metabolic flux assays and measurement of steady-state metabolite concentrations, we demonstrate the requirement for ACSF3 in mitochondrial metabolism and reveal the metabolic fate of malonate in human cells. Furthermore, we demonstrate that ACSF3-derived malonyl-CoA is required for mitochondrial protein malonylation, a recently identified posttranslational lysine modification that can affect metabolic enzyme activity to alter cellular metabolism.
Results
Engineering ACSF3 Genomic Mutations in Human Cells
Human patient fibroblasts can be difficult to obtain and can have limited utility. Engineering mutations in well-characterized human cell lines is advantageous for elucidating the function of enzymes in cellular metabolism and bypasses the transient and often insufficient knockdown by RNA interference. Here we have taken advantage of CRISPR/Cas9-mediated genome engineering to induce mutations in the ACSF3 gene in human HEK293T cells (Bowman et al., 2016; Mali et al., 2013). ACSF3 is a 576 amino acid protein encoded by an 11-exon gene on human chromosome 16. We targeted a site in exon 3, the first protein-coding exon, by transfecting cells with a plasmid co-expressing human codon-optimized Cas9 nuclease and the designed guide RNA targeting ACSF3. Concomitantly, control cells were transfected with Cas9 without a guide RNA. Individual clones were selected by limited dilution plating and screened for loss of ACSF3 protein by immunoblotting (Figure 1B). Select clones were analyzed for genomic mutations in ACSF3 which are the result of non-homologous end-joining repair after Cas9-mediated endonuclease activity (Figure S1A). Consistent with its biochemical function, ACSF3 knockout (KO) cells have a 5-fold higher concentration of the enzyme's substrate, malonate, relative to control cells (Figure 1C). Total malonyl-CoA and acetyl-CoA abundance was unchanged in ACSF3 KO cells, demonstrating that compartment-specific defects in malonyl-CoA metabolism may not affect the cellular concentration of the enzyme's product (Figure S1B). Cytoplasmic routes of malonyl-CoA synthesis likely account for a greater proportion of cellular malonyl-CoA than ACSF3-derived mitochondrial malonyl-CoA. In this way, we have engineered cells with a similar biochemical phenotype to humans with inborn errors in ACSF3 in a cell line that can be easily cultured and manipulated in vitro.
ACSF3 is Required for Malonate Oxidation
Loss of ACSF3 resulted in increased steady-state levels of malonate (Figure 1C), but the metabolic fate of malonate in human cells remained unclear. Malonate is an endogenous metabolite that acts as a classic competitive inhibitor of succinate dehydrogenase (Quastel and Wooldridge, 1928). As such, malonate is cytotoxic by blocking cellular respiration, and the effects of malonate are especially detrimental in cells that rely heavily on oxidative metabolism (Beal et al., 1993; Moy et al., 2000; Zeevalk et al., 1997). To determine if loss of ACSF3 would exacerbate malonate-induced toxicity, ACSF3 KO and control cells were treated with increasing concentrations of malonate and cell survival was assayed after 72 hours of exposure (Figure S1C). Malonate impairs proliferation of ACSF3 KO and control cells to the same extent, with only subtle differences in cell viability by genotype across the malonate concentrations tested. This suggests that, while ACSF3 participates in the clearance of mitochondrial malonate, loss of ACSF3 does not affect cell growth under normal culture conditions.
Consistent with the action of malonate as a potent inhibitor of succinate dehydrogenase, exposure of cells to 5 mM malonate for 24 hours dramatically increased steady-state succinate levels, and ACSF3 KO cells had 1.5-fold higher succinate accumulation than control cells upon malonate exposure (Figure 1D). In the absence of exogenous malonate, steady-state intracellular succinate concentrations were not different between ACSF3 KO and control cells, likely due to the ability of succinate to exit cells via the dicarboxylic acid transporter. Consistent with this, we observed that succinate secretion increased in a dose-dependent manner with increasing malonate exposure in both control and ACSF3 KO cells (Figure 1D). At baseline, ACSF3 KO cells had higher malonate levels, providing evidence that the endogenous malonate-clearance machinery may operate at low concentrations of substrate.
To determine the ACSF3-dependent metabolic fate of malonate, ACSF3 KO and control cells were labeled with [1,3-13C]malonate, and isotopomer distribution into short-chain acyl-carnitines, TCA cycle intermediates, and select amino acids was determined by LC-MS/MS (Figure 1E). ACSF3 KO cells had less m+1 labeled acetyl-L-carnitine, indicative of less acetyl-CoA formation from labeled malonate, which results from malonyl-CoA decarboxylase activity on labeled malonyl-CoA in either the cytoplasm or mitochondria. The abundances of m+1 mass isotopomers of TCA cycle intermediates were decreased in ACSF3 KO cells, suggestive of a decrease in malonyl-CoA-derived acetyl-CoA entering the TCA cycle in the absence of ACSF3. These data provide evidence that malonate can be metabolized by mammalian cells in a manner that depends on its activation to malonyl-CoA by ACSF3.
Loss of ACSF3 Perturbs Mitochondrial Metabolism
In addition to decreased labeling of TCA cycle intermediates from malonate in ACSF3 KO cells, the total abundances of α-ketoglutarate and fumarate were decreased while malate and citrate concentrations were unchanged by loss of ACSF3 (Figure S1D). Again, in the presence of exogenous malonate, cellular succinate increased in ACSF3 KO cells, consistent with the action of malonate as a succinate dehydrogenase inhibitor. To determine how impaired mitochondrial malonate metabolism would affect overall mitochondrial function, cellular oxygen consumption was measured in the presence of 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate. ACSF3 KO cells exhibited significantly lower basal respiration than control cells, as well as lower CCCP-stimulated maximal respiration, with no difference in non-mitochondrial respiration (Figure 2A). These data demonstrate an ACSF3-dependent defect in mitochondrial oxidative metabolism.
Figure 2. ACSF3 Regulates Mitochondrial Metabolic Efficiency.
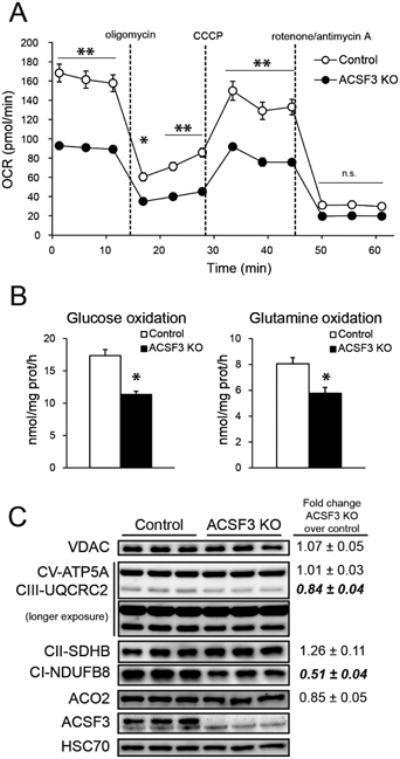
(A) Oxygen consumption rate of ACSF3 KO cells in the presence of 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate upon sequential administration of the specified mitochondrial inhibitors, normalized to cell number (mean ± SEM, n=5, representative of three independent experiments).
(B) Oxidation of [U-14C]D-glucose, [U-14C]L-glutamine, and [U-14C]L-alanine to 14CO2 in ACSF3 KO cells (mean ± SEM, n=6).
(C) Immunoblot of several mitochondrial proteins. VDAC (voltage-dependent anion channel 1) as outer mitochondrial membrane marker; ATP5A component of ATP synthase, Complex III subunit 2 (UQCRC2), SDHB subunit of Complex II, and NDUFB8 subunit of Complex I of the inner mitochondrial membrane; aconitase (ACO2) and ACSF3 of the mitochondrial matrix; and HSC70 as cytoplasmic loading control. Protein abundance was normalized to HSC70, and values shown are fold change proteinabundance in ACSF3 KO cells over control cells (mean ± SEM, n=5, p<0.05 are in bold).
*p<0.05, **p<0.001
To test for differences in substrate-specific oxidative metabolism, glucose, glutamine, and alanine oxidation rates in ACSF3 KO and control cells were determined with radiolabeled substrates (Figure 2B). Rates of oxidation of [U-14C]D-glucose to 14CO2 in ACSF3 KO cells were 65-75% of control cells, and the complete oxidation of [U-14C]L-glutamine and [U-14C]L-alanine was similarly impaired (Figure 2B). These defects in catabolic substrate utilization suggest a general impairment of mitochondrial metabolism in ACSF3 KO cells, consistent with a model of malonate-induced mitochondrial toxicity.
To determine whether the generalized impairment of mitochondrial metabolism in ACSF3 KO cells was a result of reduced mitochondrial content, immunoblotting for several mitochondrial proteins was conducted on ACSF3 KO and control cells (Figure 2C). While the abundance of the outer mitochondrial membrane voltage-dependent anion channel (VDAC) and the matrix enzyme aconitase (ACO2) was unchanged between ACSF3 KO and control cells, several subtle differences were observed among inner mitochondrial membrane proteins that are components of the oxidative phosphorylation machinery. Among these, the abundances of a Complex I subunit (NDUFB8) and a Complex III subunit (UQCRC2) were significantly lower in ACSF3 KO cells while ATP synthase subunit ATP5A expression was unchanged (Figure 2C). We next tested for complex-specific differences in respiratory chain function between ACSF3 KO and control cells in isolated mitochondria. No differences were observed in the individual activities of Complex I, Complex II, Complex IV, or Complex V (Figure S2A, S3B). These data suggest that the subtle differences in mitochondrial protein abundance were not contributing to the overall defects in mitochondrial metabolism observed in ACSF3 KO cells and the suppression in oxidative metabolism was due mainly to the competitive inhibition of SDH by malonate.
Impaired Mitochondrial Malonate Metabolism Affects Cellular Energy Status and Impairs De Novo Lipogenesis
To determine how impaired mitochondrial oxidative metabolism in ACSF3 KO cells affected cellular energy status, we tested the phosphorylation status of acetyl-CoA carboxylase (ACC). ACC is a substrate of the canonical energy-sensing AMP-activated protein kinase (AMPK), whose activity generates cytoplasmic malonyl-CoA. ACSF3 KO cells had more phosphorylated ACC (Ser79) than control cells (Figure 3A), with no difference in total ACC expression (Figure S2C). Since malonate inhibits succinate dehydrogenase, we reasoned that malonate treatment might also affect cellular energy status and therefore AMPK signaling. The degree of ACC phosphorylation in both ACSF3 KO and control cells was unchanged by malonate exposure (Figure 3A).
Figure 3. Metabolic Alterations in ACSF3-Deficient Cells.
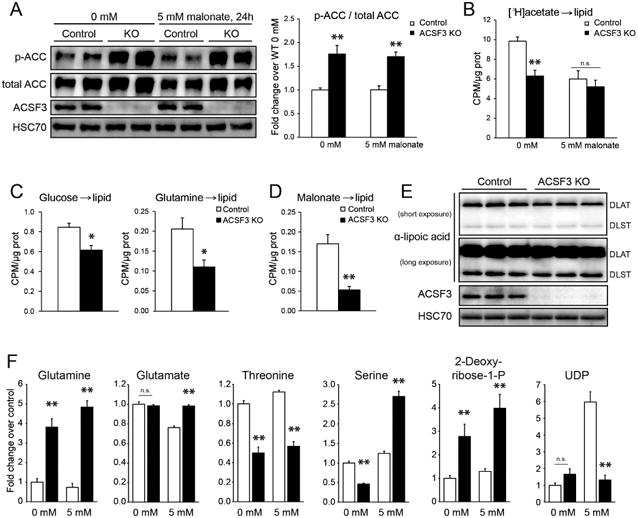
(A) Acetyl-CoA carboxylase (ACC) phosphorylation (Ser79) in ACSF3 KO and control cells in the presence or absence of 5 mM malonate for 24 hours (mean ± SEM, n=6 from two independent experiments, **p<0.01 for genotypic effect, with no effect of malonate treatment, determined by posttest after two-way ANOVA). See also Figure S2.
(B) [3H]acetate incorporation into total cellular lipids in the presence or absence of malonate (5mM) (mean ± SEM, n=6, representative of two independent experiments, **p<0.01 for genotypic effect, malonate treatment effect p=0.001 with significant interaction (p<0.05), determined by two-way ANOVA). (CPM = counts per minute).
(C) [U-14C]glucose and [U-14C]glutamine incorporation into the total lipid fraction (mean ± SEM, n=6, *p<0.05).
(D) [2-14C]malonate into total cellular lipids (n=12, pooled from two independent experiments).
(E) Immunoblotting for lipoic acid-modified proteins, DLAT (E2 of pyruvate dehydrogenase) and DLST (E2 of α-ketoglutarate dehydrogenase), with HSC70 as loading control.
(F) Select metabolite levels in the presence or absence of 5 mM malonate for 24 hours (mean ± SEM, n=10, **p<0.01 by posttest after two-way ANOVA). More metabolites and two-way ANOVA results in Table S1.
Because phosphorylation and inactivation of ACC affects cytoplasmic production of malonyl-CoA and, therefore, de novo lipogenesis, we next measured the flux of radiolabeled substrates into total cellular lipids. Consistent with the phosphorylation status of ACC, [3H]acetate incorporation into lipid was decreased in ACSF3 KO cells relative to controls (Figure 3B). Interestingly, the presence of malonate significantly decreased lipid synthesis from acetate in control cells while ACSF3 KO cells were unaffected by malonate exposure. This observation supports a role for ACSF3 in promoting malonate clearance to prevent a mitochondrial bioenergetic stress. While acetate incorporation into lipid does not require mitochondrial metabolism to generate acetyl-CoA, the synthesis of cellular lipids from the oxidative metabolism of glucose, glutamine, and alanine requires mitochondrial production of citrate. Consistent with impaired oxidation of these substrates in ACSF3 KO cells (Figure 2B), the flux into total cellular lipids was decreased to a similar degree (Figure 3C, Figure S2D). These deficits provide further evidence of suppressed mitochondrial metabolic capacity upon loss of ACSF3.
Protein Lipoylation is Unaffected by ACSF3 Deficiency
Mitochondrial malonyl-CoA derived from ACSF3 has been implicated as a significant source of carbon for type II fatty acid synthesis in the mitochondrial matrix (Guan and Nikolau, 2016; Witkowski et al., 2011). The octanoyl-ACP (acyl-carrier protein) produced by de novo fatty acid synthesis in the mitochondrial matrix is a substrate for the synthesis of lipoic acid, an essential cofactor for α-ketoacid dehydrogenase complexes (reviewed in (Hiltunen et al., 2010; Hiltunen et al., 2009)). To test the contribution of ACSF3-derived malonyl-CoA to total cellular lipids, cells were labeled with [2-14C]malonate and contribution to the total lipid fraction was determined (Figure 3D). ACSF3 KO cells had significantly lower malonate incorporation into lipid than control cells, consistent with a requirement for ACSF3 in malonate metabolism. In order to more specifically evaluate protein lipoylation status, immunoblotting of ACSF3 KO and control cell lysates was performed with an antibody that recognizes lipoylated residues (Figure 3E). No defects in lipoylation of DLAT (E2 of pyruvate dehydrogenase) or DLST (E2 of α-ketoglutarate dehydrogenase) were detected in ACSF3 KO cells, suggesting that lipoic acid deficiency is not contributing to the impaired mitochondrial metabolic phenotypes in ACSF3 KO cells.
Unbiased Metabolomics of ACSF3 KO Cells Reveal Malonate- and Malonyl-CoA-Dependent Metabolic Alterations
To gain a more global understanding of how ACSF3 regulates cellular metabolism, we performed unbiased global steady-state metabolomics on ACSF3 KO and control cells in the presence or absence of 5 mM malonate for 24 hours. No signs of toxicity were observed at this concentration of malonate (Figure S1C). There were 355 unique metabolites detected, and 186 were changed greater than two-fold in either direction by genotype or malonate treatment or both. Metabolic alterations that were dependent upon malonate inhibition of SDH were clearly present (Figure 3F; Table S1). However, these steady-state data demonstrate broad metabolic changes that cannot be easily explained by SDH inhibition alone and likely reflect both malonate- and malonyl-CoA-dependence. The differences observed by the loss of ACSF3 and exogenous malonate reveal intriguing and unique metabolic effects of the reaction's substrate and product.
ACSF3 Is Required for Mitochondrial Protein Malonylation
The complexity of metabolic changes observed in ACSF3 KO cells suggests a more complex role for malonate than inhibition of SDH alone. Malonate and other structurally similar dicarboxylic acids traverse membranes via dedicated transporters in a concentration-dependent manner. ACSF3-derived mitochondrial malonyl-CoA, however, is trapped within that organelle. To further delineate the fate of malonate and mitochondrial malonyl-CoA, cells were labeled with [2-14C]malonate and a biphasic extraction protocol was used to separate and recover polar, non-polar, protein, and nucleic acid fractions (Hosios et al., 2016). ACSF3 KO cells had higher [2-14C]malonate accumulation than control cells (total counts) and higher 14C signal in the polar metabolite fraction (Figure 4A), consistent with the elevated steady-state malonate concentrations observed in ACSF3 KO cells (Figure 1C). Interestingly, although malonate incorporation into protein only accounted for ∼5% of total label incorporation in control cells (Figure S2E), ACSF3 KO cells had significantly lower malonate incorporation into protein (∼1% of total label incorporation). These results suggest that the ACSF3-dependent incorporation of malonate into the protein fraction may be part of an adaptive response to cellular malonate exposure. Furthermore, an inability to direct malonate toward this fate may account for some of the metabolic derangements observed in ACSF3 KO cells, particularly in the presence of exogenous malonate.
Figure 4. ACSF3 Is Required for Mitochondrial Protein Malonylation.
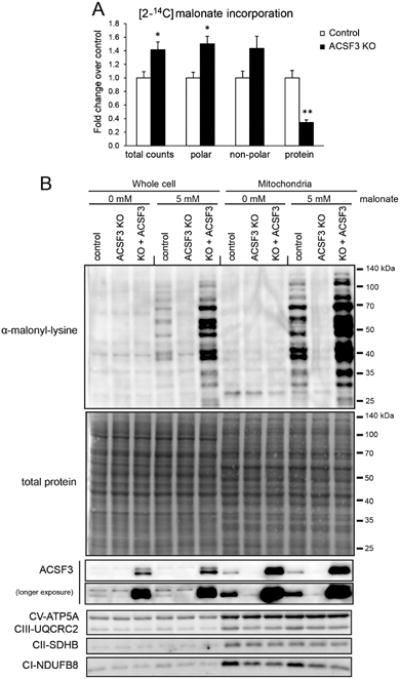
(A) [2-14C]malonate incorporation into different classes of biomolecules by biphasic extraction after 4h incubation with 0.2 μCi at 0.1 mM malonate (mean ± SEM, n=3, representative of two independent experiments, *p<0.05, **p<0.01). See also Figure S2C.
(B) Immunoblotting for malonylated lysine residues in whole cell and mitochondrial extracts of cells in the presence or absence of 5 mM malonate for 24h. ACSF3 KO cells were transiently transfected with a human ACSF3 expression vector, and control cells were transfected with a mitochondrially-targeted YFP. Total protein staining by amido black shown as loading control. See also Figure S3.
Protein malonylation has been recognized as a dynamically regulated posttranslational lysine modification that can affect mitochondrial and nucleocytoplasmic proteins (Du et al., 2011; Nishida et al., 2015; Peng et al., 2011), and malonyl-CoA is the likely substrate for protein malonylation (Colak et al., 2015; Hirschey and Zhao, 2015; Kulkarni et al., 2017). Using an antibody that specifically recognizes malonylated lysine residues, we immunoblotted for this modification in whole cell lysates and after enriching for mitochondria. Exogenous malonate treatment robustly increased mitochondrial protein malonylation and the majority of protein malonylation signal was localized to mitochondria (Figure 4B). Importantly, ACSF3 was required for mitochondrial protein malonylation, and transient re-expression of ACSF3 was sufficient to rescue the impaired mitochondrial malonylation in ACSF3 KO cells (Figure 4B). Transient re-expression of ACSF3 in KO cells did not alter the observed differences in protein abundance of particular inner mitochondrial membrane components of the oxidative phosphorylation machinery (Figure 4B). Interestingly, immunoprecipitation of a mitochondrially-targeted YFP and immunoblotting for malonylated lysine residues demonstrated that a heterologous protein can be malonylated in the mitochondrial matrix of control cells but not in ACSF3 KO cells (Figure S3). This suggests that protein malonylation may be proportional to malonyl-CoA concentrations, as others have suggested (Pougovkina et al., 2014), and that ACSF3 is the principal source of mitochondrial malonyl-CoA in cells treated with malonate.
Mitochondrial Protein Malonylation Is a Stable Posttranslational Modification that Alters Cellular Metabolism
We next sought to define the metabolic effects of mitochondrial protein malonylation through a malonate washout paradigm where cells were cultured in the presence of 5 mM malonate for 24 hours before a washout with malonate-free growth media for up to 24 hours. We observed that mitochondrial protein malonylation is a stable modification that persists after removing malonate from the media (Figure 5A). To understand potential roles of mitochondrial malonylation, we performed a malonate washout experiment with steady-state metabolite concentrations determined by 1H-NMR over the time course. Cellular succinate concentrations decreased rapidly (within 3h) after removal of exogenous malonate, consistent with the acute and reversible inhibition of SDH by malonate (Figure 5B). Across the same time course, cellular lactate and pyruvate increased, while threonine concentrations remained consistently lower in ACSF3 KO cells, independent of time from malonate washout (Figure 5B). These data further demonstrate malonate-dependent and malonyl-CoA-dependent effects of ACSF3 deficiency.
Figure 5. Mitochondrial Protein Malonylation Persists Following Malonate Washout.
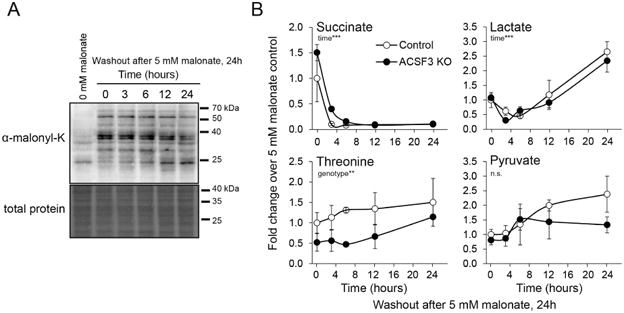
(A) Immunoblotting for malonylated lysine residues in mitochondrial extracts of cells treated with 5 mM malonate for 24h before switching to malonate-free media for the times indicated (0-24h after malonate washout). See also Figure S4.
(B) Steady-state concentrations of cellular succinate, lactate, pyruvate, and threonine determined by 1H-NMR (mean ± SEM, n=3) after 24h 5 mM malonate followed by incubation in malonate-free media for the times indicated. Statistical significance for time and genotype effects determined by two-way ANOVA, **p<0.01, ***p<0.001.
ACSF3 Enables Metabolite-Specific Mitochondrial Protein Acylation
We reasoned that global changes in protein malonylation may affect other lysine acyl modifications that are altered by cellular metabolism. Despite the defects in mitochondrial metabolism in ACSF3 KO cells related to SDH inhibition by malonate, neither malonate treatment nor loss of ACSF3 dramatically affected mitochondrial lysine acetylation (Figure 6A). The removal of the malonyl-lysine modification is catalyzed by the mitochondrial sirtuin SIRT5 (Du et al., 2011; Peng et al., 2011), which also removes the structurally similar succinyl-lysine modification (Du et al., 2011; Rardin et al., 2013). The loss of Sirt5 enhances both malonylation and succinylation and has clear metabolic effects (Nishida et al., 2015; Rardin et al., 2013). We hypothesized that the increase in cellular succinate upon SDH inhibition by malonate may concurrently increase lysine succinylation and malonylation. Surprisingly, exogenous malonate did not affect mitochondrial protein succinylation, and ACSF3 KO and control cells exhibited no differences in mitochondrial protein succinylation (Figure 6B, 6C). In order to increase succinate and succinyl-CoA concentrations by an orthogonal method, cells were treated with the SDH suicide inhibitor, 3-nitropropanoate (3-NP). Cellular succinate concentrations were dramatically increased by 3-NP (Figure S5) and corresponded with increased mitochondrial protein succinylation independent of ACSF3 (Figure 6C). Lysine succinylation and malonylation may occur on the same residues, so we also treated cells with both 3-NP and malonate simultaneously to test whether there is a propensity for one modification over the other. The presence of both malonate and 3-NP decreased the intensity of the malonyl-lysine and succinyl-lysine signals relative to their respective controls with only one inhibitor (Figure 6B, 6C). These results show that ACSF3, by being required for protein malonylation, participates in the metabolite-specific acylation of mitochondrial proteins.
Figure 6. ACSF3 Does Not Affect Mitochondrial Protein Acetylation or Succinylation.
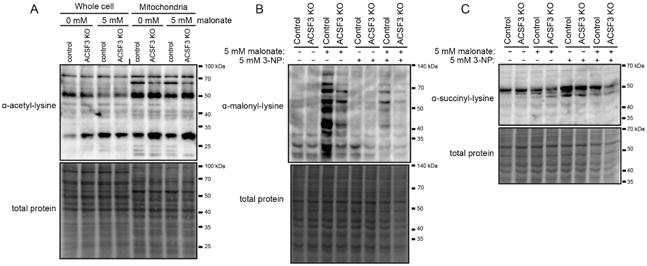
(A) Immunoblotting for acetylated lysine residues in whole cell and mitochondrial extracts of cells in the presence or absence of 5 mM malonate for 24h. Total protein staining by amido black shown as loading control.
(B-C) Immunoblotting for malonylated (B) and succinylated (C) lysine residues in mitochondrial extracts of ACSF3 KO and control cells treated with 5 mM malonate, 5 mM 3-nitropropanoate (3-NP, SDH suicide inhibitor), or both for 24h. See also Figure S5.
Tissue-specific Differences in Mitochondrial Protein Malonylation Reflect Acsf3 Abundance
ACSF3 expression across tissues and under different physiological conditions may provide insight into the regulation of mitochondrial malonate metabolism in vivo. ACSF3 mRNA is highly expressed in mitochondria-rich tissues in mouse such as brown adipose tissue (BAT), soleus muscle, kidney, and liver (Ellis et al., 2015). Others have shown that protein malonylation is higher in fed liver than fasted liver (Nishida et al., 2015), and obese leptin-deficient ob/ob mice exhibit a dramatic increase in hepatic protein malonylation (Du et al., 2015). Both fed liver and ob/ob liver are characterized by high rates of de novo fatty acid synthesis and, therefore, elevated cytoplasmic malonyl-CoA. We posit that hydrolysis of cytoplasmic malonyl-CoA and transport into the mitochondrial matrix could represent a significant source of malonate for ACSF3-derived malonyl-CoA that can be metabolized or modify mitochondrial proteins.
In four mitochondria-rich tissues (liver, kidney, heart, and BAT), we observed different levels of mitochondrial protein malonylation that positively correlated with Acsf3 expression (Figure 7A). Malonylation of mitochondrial proteins was most robust in BAT, kidney, and liver. We confirm the findings of Du et al. that showed increased total protein malonylation in livers of ob/ob mice; and furthermore, we demonstrate that mitochondrial proteins from ob/ob mice have higher lysine malonylation than wild-type (WT) mitochondrial extracts (Figure 7B) (Du et al., 2015). Interestingly, mitochondrial protein succinylation was the same in WT and ob/ob liver (Figure 7B), suggesting that the regulation observed in ob/ob mice is metabolite-specific. In BAT, both malonylation and succinylation of mitochondrial proteins was reduced in ob/ob mice relative to WT consistent with diminished nonshivering thermogenesis and BAT function in this model. Here we demonstrate that mitochondrial protein malonylation is positively correlated with Acsf3 expression and that alterations in lysine acylation in ob/ob mice may reflect tissue- and metabolite-specific differences relevant to the physiology of these tissues.
Figure 7. Tissue-specific Differences in Mitochondrial Protein Malonylation Reflect Acsf3 Abundance.
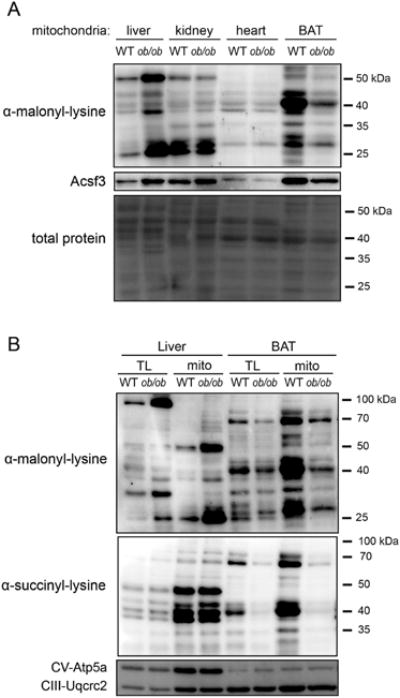
(A) Immunoblot of malonylated lysine residues in mitochondrial extracts from liver, kidney, heart, and brown adipose tissue (BAT) of wild-type (WT) and ob/ob mice. Total protein staining by amido black shown as loading control.
(B) Immunoblot of malonylated and succinylated lysine residues in mitochondrial extracts (mito) and total lysates (TL) of liver and BAT in WT and ob/ob mice. Complex V Atp5a subunit and Complex III component Uqcrc2 shown as loading control. Representative of n=3.
Discussion
Cytoplasmic malonyl-CoA is a well-characterized metabolite that simultaneously drives de novo fatty acid synthesis while inhibiting the rate-setting step in mitochondrial β-oxidation of long-chain fatty acids (reviewed in (Foster, 2012; Ruderman et al., 2003; Saggerson, 2008; Wolfgang and Lane, 2006)). The role and requirement for malonyl-CoA in the mitochondrial matrix is poorly understood, yet two enzymes have (so far) been identified as participants in the metabolism of this metabolite. Acyl-CoA synthetase family member 3 (ACSF3) uses ATP and Coenzyme A to activate malonate to malonyl-CoA within the mitochondrial matrix (Chen et al., 2011; Watkins et al., 2007; Witkowski et al., 2011), and mitochondrially-localized malonyl-CoA decarboxylase (MLYCD) can decarboxylate malonyl-CoA to generate acetyl-CoA and CO2. Malonate is an endogenous metabolic intermediate and a well-characterized competitive inhibitor of succinate dehydrogenase (SDH) (Quastel and Wooldridge, 1928). As such, malonate is cytotoxic by blocking the TCA cycle and cellular respiration. We propose that ACSF3 and MLYCD work in concert to clear malonate from mammalian mitochondria. In addition, another fate of mitochondrial malonyl-CoA is modification of lysine residues of mitochondrial proteins. To this end, we have shown that mammalian cells are capable of metabolizing labeled malonate in an ACSF3-dependent manner and that ACSF3 is required for mitochondrial protein malonylation.
Acetyl-CoA derived from MLYCD-catalyzed decarboxylation of mitochondrial malonyl-CoA can enter the TCA cycle and be fully oxidized, analogous to some bacterial species (An and Kim, 1998; Lee et al., 2000). Indeed, in legume plants, rhizobacteria are significant utilizers of malonate; however, the exact molecular source of malonate in eukaryotic cells is not well defined. We suggest that much of the mitochondrial malonate may derive from hydrolysis of cytoplasmic malonyl-CoA and subsequent transport into the mitochondrial matrix via dicarboxylic acid transporters of the inner mitochondrial membrane. In this way, ACSF3 may play a critical role in metabolic proofreading of a toxic side reaction (Van Schaftingen et al., 2013). In plants and in pig heart, malonate was shown to be generated from the decarboxylation of oxaloacetate (de Vellis et al., 1963; Vennesland and Evans, 1944). Additionally, some bacterial species can generate malonate from pyrimidines (Hayaishi and Kornberg, 1952). Interestingly, several pyrimidine-related metabolites were up-regulated in ACSF3 KO cells (Table S1). It has also been proposed that malonate can be formed from the oxidation of malondialdehyde, a product of lipid peroxidation, that is often used as an indicator of oxidative stress (Chen et al., 2011; Kim and Yu, 1989). Alternatively, prior to the identification of ACSF3 as the eukaryotic malonyl-CoA synthetase, it was proposed that malonyl-CoA could be non-specifically synthesized by propionyl-CoA carboxylase from acetyl-CoA in the mitochondrial matrix, albeit at a much lower rate than propionyl-CoA carboxylation (Hegre et al., 1959; Kim and Kolattukudy, 1978). Although the relative contributions of each of these potential sources of mitochondrial malonate remains to be determined, our model describes the roles for ACSF3 and MLYCD in regulating mitochondrial malonate concentrations. Consistent with our model, mutations in human ACSF3 or MLYCD result in toxic accumulation of malonate and systemic metabolic acidosis (Alfares et al., 2011; Sloan et al., 2011).
To further determine the metabolic role of ACSF3 in mammalian cells, we generated an ACSF3 loss-of-function genetic model in cultured cells by CRISPR/Cas9-mediated genome editing. Prior to recent advances in genome editing in mammalian cells, the use of patient primary cells has been the preferred model to study inborn errors of metabolism and to determine the molecular mechanisms behind human disease. However, these cultured patient fibroblasts or lymphoblasts often do not sufficiently express the genes of interest to determine the consequence of the mutations (Asadollahi et al., 2014). Here we demonstrate the utility of generating stable mutations in well-defined cultured cells to accomplish this same purpose. ACSF3 knockout (KO) cells have elevated steady-state malonate and succinate concentrations (Figure 1C and Table S1), consistent with the biochemical function of ACSF3 and the inhibitory role of its substrate malonate. Although methods are currently lacking to measure concentrations of malonate and malonyl-CoA with subcellular resolution (Ellis and Wolfgang, 2012), mitochondrial protein malonylation may be a reasonable proxy for mitochondrial matrix malonyl-CoA concentrations. Indeed, fibroblasts from patients with MLYCD deficiency had higher protein malonylation, consistent with an inability to catabolize malonyl-CoA to acetyl-CoA (Colak et al., 2015; Pougovkina et al., 2014). Elevated protein malonylation in MLYCD-deficient cells also corresponded with impaired mitochondrial function, and here we demonstrate that a lack of mitochondrial protein malonylation in ACSF3-deficient cells also correlated with impaired mitochondrial function. The similar metabolic phenotypes observed in MLYCD- and ACSF3-deficient cells corroborate the similar clinical presentation of ACSF3 and MLYCD mutations in combined malonic and methylmalonic aciduria (CMAMMA).
Elevated levels of malonate in plasma (2-10 μM) and urine (15-70 μM) have been described in patients with mutations in ACSF3 (Sloan et al., 2011). Loss of the plant homolog of ACSF3 also resulted in dramatically elevated malonate concentrations in plant shoots as well as a dose-dependent increase in both malonate and succinate upon treatment with exogenous malonate (Chen et al., 2011). In a similar manner, we expected cells deficient in ACSF3 to be more sensitive to malonate-induced cytotoxicity. Surprisingly, ACSF3-deficient cells were as sensitive to increasing concentrations of malonate as control cells. The doses of malonate tested are comparable to the concentrations used to treat plants (Chen et al., 2011) and lower than the concentrations used to induce cytotoxicity in neuronal cultures (10-75 mM) (Moy et al., 2000; Zeevalk et al., 1995); however, it is possible that these doses simply overwhelm the endogenous malonate-clearance machinery. Like other metabolite proofreading systems, we suggest that mitochondrial malonate metabolism operates at low levels of substrate (Van Schaftingen et al., 2013), and, consistent with this, malonyl-CoA concentrations were lower than other short-chain acyl-CoAs in vivo (Sadhukhan et al., 2016). Additionally, in cell culture models, the culture media represents a significant metabolic sink, and toxic intermediates may be secreted into the media instead of being accumulated to the same extent as might occur in vivo. Specifically, we propose that malonate-induced inhibition of SDH does not result in dramatic accumulation of succinate because this metabolite is free to exit the cell via dicarboxylic acid transporters. Nevertheless, the observation that there is no difference in malonate-induced cytotoxicity between ACSF3 KO and control cells facilitates interpretation of differences in metabolic flux data and steady-state metabolite concentrations, as these are not complicated by differences in cell viability.
ACSF3 KO cells demonstrate impaired mitochondrial metabolism as evidenced by decreased oxygen consumption and impaired flux of glucose and glutamine to CO2 and to total cellular lipids. Additionally, steady-state concentrations of several TCA cycle intermediates were slightly decreased in ACSF3 KO cells relative to controls. This generalized impairment of mitochondrial flux is not the result of defective protein lipoylation, as ACSF3 KO cells have normal levels of lipoylated proteins. Instead, we hypothesize that malonate-induced toxicity accounts for the impaired mitochondrial metabolism observed in ACSF3 KO cells, and impaired mitochondrial protein malonylation may also contribute to the observed impairments. Widespread changes in malonylation as a result of ACSF3 deficiency could have the capacity to broadly affect several metabolic pathways, and metabolites identified from our steady-state metabolomic analysis as being regulated in a malonyl-CoA-dependent manner could represent candidate pathways that are regulated by malonylation.
ACSF3 is a recently identified mitochondrial malonyl-CoA synthetase. Patients with mutations in ACSF3 or MLYCD display genetic concordance with systemic malonic aciduria and clinical features such as developmental delay, seizure disorders, hypoglycemia, and cardiomyopathy (Alfares et al., 2011; Sloan et al., 2011), although the tissue-autonomous role for mitochondrial malonate metabolism is unknown. While there is much to be learned about the role and requirement for ACSF3 in vivo, we have generated ACSF3-deficient human cells using CRISPR/Cas9-mediated genome engineering to begin deciphering the cell-autonomous requirements for mitochondrial malonate metabolism. Using a combination of metabolic flux assays and measurement of steady-state metabolite concentrations, we have shown that malonate can be oxidized by mammalian cells and that ACSF3 plays an important role in preventing malonate-induced impairment of mitochondrial metabolism. Our approach is broadly applicable to target other metabolic enzymes implicated in human disease and may yield mechanistic insight into metabolic regulation in health and disease.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Wolfgang (mwolfga1@jhmi.edu).
Experimental Model and Subject Details
Cell Lines
HEK293T cells were grown in DMEM supplemented with 10% bovine calf serum and 1% pen/strep antibiotic (Invitrogen) at 37°C in a humidity-controlled incubator at 10% CO2. As described in Method Details, ACSF3 KO cells were generated by transfecting hCas9 and the hACSF3 gRNA or hCas9 alone and deriving individual clones that were screened by immunoblotting and sequencing the ACSF3 target site.
Mice
All procedures were performed in accordance with the NIH's Guide for the Care and Use of Laboratory Animals and under the approval of the Johns Hopkins Medical School Animal Care and Use Committee. Male, 8-week-old, chow-fed, wild-type and ob/ob mice (C57BL/6J) were housed in a facility with ventilated racks on a 14h light/10h dark cycle with ad libitum access to a standard rodent chow (2018SX;Teklad Global; 18% protein).
Method Details
CRISPR/Cas9 Cloning Strategy
Human codon-optimized Cas9 expression vector was obtained from Addgene, plasmid #41815 (Mali et al., 2013), and hCas9 was cloned into a pEF6 expression vector, downstream and in-frame with a nuclear-localized YFP, linked by a viral 2A bicistronic peptide, such that nls-YFP and Cas9 are expressed in approximate equimolar quantities. A guide RNA (gRNA) targeting human ACSF3 (Figure S1A) was designed according to the recommendations of Mali et al. (Mali et al., 2013) and was computed as a candidate unique gRNA target. The gRNA, including the U6 promoter, was synthesized as a 500bp gBlocks fragment (Integrated DNA Technologies) and was cloned into the pEF6-nls-YFP-2A-Cas9 vector by In-Fusion Cloning (Clontech), as previously described (Bowman et al., 2016).
Derivation of ACSF3 KO clones
HEK293T cells were grown in DMEM supplemented with 10% bovine calf serum and 1% pen/strep antibiotic (Invitrogen) at 37°C in a humidity-controlled incubator at 10% CO2. Cells were transfected with plasmid expressing both hCas9 and the hACSF3 gRNA or hCas9 alone using Fugene HD (Promega) transfection reagent according to manufacturer's instructions. Selection with 7.5 μg/mL blasticidin was started 48 hours post-transfection and continued for two weeks. Individual clones were selected by limited dilution plating and screened for loss of ACSF3 protein by immunoblotting. For transient addback experiments, ACSF3 KO cells were transfected with a human ACSF3 expression vector and control cells were transfected with a plasmid encoding mitochondrially-targeted YFP (Clontech). Assays were conducted 48-72 h post-transfection. For cell viability assay, cells were counted, plated in 96-well format, and treated with 0, 2.5, 5, or 10 mM malonate (Sigma-Aldrich) for 72 hours, and cell viability was determined by CellTiter 96 Aqueous One Solution tetrazolium-based cell proliferation assay (Promega).
Characterization of ACSF3 Mutations
Genotyping primers were designed to span the gRNA target in exon 3 of ACSF3 (F: 5′ACACAGAGGAAGTGGTCTTCT3′/R:5′CGTTAGCGCATAGGAAGGAGA3′) and PCR was conducted on genomic DNA from ACSF3 KO clones by standard methods. PCR products were separated on agarose gels, and bands were excised, gel-extracted, and cloned into pCRII vector (Invitrogen) for Sanger sequencing.
Measurement of Cellular Oxygen Consumption
Oxygen consumption was measured using the Seahorse XFe96 extracellular flux analyzer (Seahorse Bioscience). 24,000 cells were plated in poly-L-lysine-coated 96-well microplates 18h before assaying cellular respiration. Cells were incubated with Seahorse base medium supplemented with 10mM glucose, 2 mM glutamine, 1 mM pyruvate for 1h before starting the assay. Oxygen consumption was measured repeatedly over the course of the experiment as the following inhibitors were added sequentially at the specified concentrations: 2 μM oligomycin A, 250 nM CCCP, 0.5 μM rotenone/0.5 μM antimycin A. Cell number was determined by CyQUANT (Invitrogen) staining of cellular nucleic acids and was used to normalize oxygen consumption rate in pmol/min.
Mitochondrial Isolation and Functional Assays
Mitochondria were isolated as previously described (Frezza et al., 2007). Briefly, cells were scraped into cold isolation buffer (10 mM Tris, 10 mM MOPS, 1 mM EGTA, 200 mM sucrose, supplemented with 20 mM nicotinamide, pH 7.4) and homogenized on ice with a glass homogenizer for 45 strokes before centrifuging at 600g, resuspending the pellet, and centrifuging again at 600g. All centrifugation steps were for 10 min at 4°C. The supernatant was transferred to new tubes and centrifuged at 7,000g. The supernatant was discarded and the pellet was resuspended, and centrifuged at 7,000g followed by a resuspension of the pellet and centrifugation at 10,000g. The mitochondrial pellet was resuspended in lysis buffer, sonicated, and protein concentration determined by BCA assay prior to SDS-PAGE and immunoblotting.
For in-gel assays of respiratory complex function, mitochondria were solubilized in 1.25% digitonin in basic lysis buffer (20mM HEPE-KOH, pH 7.4, 100mM NaCl, 20mM imidazole, 1mM CaCl2, 10% glycerol) with protease inhibitors (1mM PMSF, 10μM leupeptin, 2μM pepstatin A) on ice for 30 min. Insoluble material was pelleted at 21,000g for 30min at 4°C and supernatant was combined with 10× native sample buffer (5% Coomassie Brilliant Blue G-250, 0.5M 6-aminocaproic acid, 100mM BisTris-HCl, pH 7.0). 150μg of solubilized mitochondria were loaded per well and separated on 5-12% or 6-16% BisTris acrylamide gels. For Complex I NADH Nitrotetrazolium blue (NTB) reductase activity assay, gels were incubated in 2.5mg/mL NTB, 0.1mg/mL NADH, and 5mM Tris-HCl, pH 7.4 for 2h at room temperature, then fixed in 50% (v/v) methanol, 10% (v/v) acetic acid and destained. For Complex IV cytochrome c oxidase activity, native gels were incubated in 0.5mg/mL diaminobenzidine, 0.025mM equine heart cytochrome c, 50mM potassium phosphate, pH 7.4 for 5h at room temperature, protected from light, then fixed in 50% (v/v) methanol, 10% (v/v) acetic acid and destained. For Complex V ATP hydrolysis activity, native gels were equilibrated in 270mM glycine, 35mM Tris-HCl for 1h at room temperature, then switched to assay buffer containing 270mM glycine, 35mM Tris-HCl with 14mM MgSO4, 0.2% Pb(NO3)2, and 8mM ATP, and incubated overnight. Gels were fixed in 50% (v/v) methanol and destained before imaging. Complex II activity was assayed by immunoaffinity microplate-based colorimetric assay (Abcam ab109908). Mitochondria were solubilized as per manufacturer's recommendations and 50μg of mitochondria were used per well. Activity is quantified as the rate of decrease in absorbance at 600nm as the production of ubiquinol reduces the blue dye DCPIP to a colorless product.
Immunoblotting
Cells were harvested in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100) with protease inhibitor cocktail (Roche) followed by brief sonication and pelleting of cellular debris at 12,000g (5 min at 4°C). 20 mM nicotinamide was included in the lysis buffer for immunoblotting for lysine acylation. Protein concentration of lysate was determined by bicinchoninic acid (BCA) assay (Thermo Scientific), and 30μg of cell lysate was separated by Tris-Glycine SDS-PAGE (12% polyacrylamide). Proteins were transferred to PVDF membranes (Immobilon), blocked in 5% nonfat-milk TBST, and incubated with primary antibodies overnight. Primary antibodies used include: ACSF3 rabbit polyclonal at 1:1000 (ThermoFisher PA5-25803), heat shock chaperone 70 (HSC70) mouse monoclonal at 1:1000 (Santa Cruz sc-7298), MitoProfile total OXPHOS mouse monoclonal antibody cocktail at 1:500 (Abcam ab110413), voltage-dependent anion channel 1 (VDAC1) rabbit polyclonal at 1:1000 (Calbiochem AB10527), aconitase 2 (ACO2) rabbit polyclonal at 1:1000 (Cell Signaling 6922), α-lipoic acid rabbit polyclonal at 1:1000 (Calbiochem #437695), phospho-ACC (Ser79) rabbit polyclonal at 1:1000 (Cell Signaling 11818), total ACC rabbit polyclonal at 1:1000 (Cell Signaling 3676), malonyl-lysine rabbit monoclonal mix at 1:1000 (Cell Signaling 14942), acetyl-lysine rabbit polyclonal at 1:1000 (Cell Signaling 9441), succinyl-lysine rabbit polyclonal at 1:1000 (PTM Biolabs 401), GFP mouse monoclonal at 1:1000 (Santa Cruz sc-9996). Cy3-conjugated anti-mouse secondary antibodies (Invitrogen M30010) at 1:1500 or horseradish peroxidase (HRP)-conjugated anti-mouse (GE Healthcare NA931V) or anti-rabbit secondary antibodies (GE Healthcare NA934V) at 1:2000 were incubated with washed membranes, and proteins were visualized with Amersham Prime enhanced chemiluminescent substrate (GE Healthcare) or epifluorescence on an Alpha Innotech MultiImage III instrument. Protein abundance was quantified using Alpha Innotech FluorChem Q software (Santa Clara, CA) and was normalized to HSC70 expression. For amido black total protein staining, PVDF membranes were incubated in 0.1% naphthol blue black (w/v), 10% methanol (v/v), 2% acetic acid (v/v) for 5 min and destained in 10% acetic acid (v/v), 25% isopropanol (v/v) for 1 min before imaging. For GFP immunoprecipitation (IP), cells were lysed in lysis buffer with 20 mM nicotinamide, and 800 μg of protein was incubated with 10 μL of GFP nanobody magnetic beads (Allele Biotechnology) for 4h at 4°C in 400 μL final volume binding buffer (10 mM Tris HCl, 150 mM NaCl, 20 mM nicotinamide). Beads were washed once in binding buffer and twice in wash buffer (10 mM Tris HCl, 500 mM NaCl, 20 mM nicotinamide) before eluting with 40 μL elution buffer (200 mM glycine, pH 2.5) and neutralizing with 4 μL 1M Tris. IP samples from an equivalent of 200 μg cell lysate were resolved by SDS-PAGE as described above.
Radiolabeled Substrate Metabolic Flux Assays
For glucose oxidation assays, cells were incubated in stoppered T-25 flasks with 0.1μCi/flask [U-14C]D-glucose (Moravek Biochemicals) in serum-free DMEM containing 2.5 mM glucose, 2 mM glutamine, 0.5 mM pyruvate for 4 h. 14CO2 was trapped on a filter paper suspended in the headspace of the flask by addition of 200μL 70% perchloric acid to the flask and 150μL 1M NaOH to the filter paper and incubating the samples at 55°C for 1 h. The filter paper was placed in scintillation fluid and counted, and counts were normalized to total protein. Glutamine and alanine oxidation assays were performed as described for glucose except 0.2μCi/flask [U-14C]L-glutamine (New England Nuclear Radiochemicals/PerkinElmer) with 0.5 mM cold glutamine or 0.2μCi/flask [U-14C]L-alanine (Moravek Biochemicals) with 0.1 mM cold alanine were used, respectively.
To determine substrate incorporation into the total lipid fraction, cells were plated in 24-well dishes and labeled with 14C-labeled substrate for 4h. Total lipids were extracted with 2:1 chloroform:methanol via the Folch method (Folch et al., 1957), and radioactivity was counted by liquid scintillation. For [3H]acetate incorporation, cells were labeled for 4h with 0.2μCi/well in DMEM supplemented with 10% bovine calf serum. For glucose, glutamine, and alanine incorporation, 0.2μCi/mL of radioactivity was used with the same media compositions as used for oxidation assays. For [2-14C]malonate incorporation, cells were labeled for 4h with 0.2μC/well in 0.1mM unlabeled malonate. All counts were normalized to total protein as determined by BCA assay (Thermo Scientific). In order to separate and recover polar, non-polar, protein, and nucleic acid fractions from cells labeled with 0.2μCi/well [2-14C]malonate (0.1mM unlabeled malonate) for 4h, a biphasic extraction protocol was used (Hosios et al., 2016).
Metabolite Extraction and Analysis
For LC-QTOF, control and ACSF3 KO cells were exposed to 0 or 5 mM malonate for 24h prior to collection of cell extracts in ice-cold HPLC-grade 80:20 methanol: water and rapid quenching in liquid nitrogen. Cellular debris was pelleted by centrifugation, and the supernatant was dried by vacuum centrifugation prior to resuspension and unbiased metabolomics analysis by LC-QTOF (Bowman et al., 2016; Ellis et al., 2013).
For stable isotope labeling experiments, cells (5E6 cells plated 24h before labeling) were incubated with 2.5 mM [1,3-13C]malonate (Sigma-Aldrich) for 4 hours and washed twice in PBS before cell extracts were collected in ice-cold 80:20 methanol:water with rapid quenching in liquid nitrogen. The abundance and isotopic enrichment of select water-soluble metabolites (including short-chain acyl-carnitines, TCA cycle intermediates, and select amino acids) were quantified by LC-MS/MS on an Agilent 6490 triple-quadropole system (Agilent Technologies, Santa Clara, CA), as previously described (Bowman et al., 2016). Briefly, the MS/MS experiment was performed in positive/negative switching electrospray mode, as described previously (Alves et al., 2015), and all data processing was performed with the Mass Hunter Quantitative Analysis software package. Raw metabolite abundances were normalized to protein content.
For capillary electrophoresis-MS (CE-MS), cells were washed with 5% mannitol and extracted with methanol spiked with an internal standard solution as specified by Human Metabolome Technologies. Insoluble material was pelleted and supernatant was filtered through centrifugal filters. Filtrate was dried under vacuum and resuspended for analysis by CE-TOF MS for cationic metabolites and CE-QqQ MS for anionic metabolites. Metabolite abundances were normalized to protein content.
For 1H-NMR, cells were washed with ice-cold PBS and scraped into ice-cold methanol and snap frozen in liquid nitrogen before two additional freeze-thaw rounds with methanol extraction. For 1H-NMR of culture medium for succinate measurements, 600μL of media was extracted with twice the volume ice-cold methanol, snap frozen in liquid nitrogen and thawed. Cell extracts and media samples were subjected to a final spin at 15,000g for 1 min at 4°C, then the supernatant was dried under vacuum and the extract resuspended in 20 mM phosphate buffer, pH 7.4 ± 0.1, with 0.1 mM TMSP as an internal reference and 0.1 mM sodium azide. 1H spectra were recorded on a Bruker Avance III 500MHz (Bruker Instruments, Germany) NMR spectrometer, operating at 499.9MHz and equipped with room temperature quadruple nuclei probe. Typical 1H spectra were acquired using presaturation solvent suppression pulse sequence (noesyprld). Acquisition parameters were set as follows; spectral width of 8012.820 with a 64K data points, 512 scans, with a relaxation delay of 7s for a total collection time of 1.14h. Samples were automatically tuned and match, and shimmed to TMSP signal. Spectra were exported into Bruker format and were processed with Chenomx NMR Suit 8.2 Professional (Chenomx Inc, Edmonton, Alberta, Canada). TMSP signal (0.0 ppm) was used as a reference peak, spectra manually phase corrected and spline function was applied for the baseline correction. Metabolites were profiled and quantified using built-in Chenomx 500MHz library. Metabolite concentrations were normalized to protein content.
Protein Malonylation in Mouse Tissue Lysates
Mitochondria were isolated based on Frezza et al. 2007, and tissue lysates and mitochondrial extracts were prepared in RIPA buffer (50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 0.25% deoxycholate) with protease inhibitor, supplemented with 20mM nicotinamide, followed by pelleting of insoluble material at 13,000g for 15 min at 4°C.
Quantification and Statistical Analysis
Student's t-test was used to evaluate statistical significance for quantitation of metabolic flux assays and quantitation of immunoblots between control and ACSF3 KO cells. Repeated measures ANOVA with Bonferroni post-tests were used to test statistical significance in cellular oxygen consumption experiments and 1H-NMR time course. For steady-state metabolomics analysis, two-way ANOVA was conducted using the freely available, web-based MetATT metabolomics tool for two-factor datasets (Xia et al., 2011). Statistical details of individual experiments can be found in Figure Legends.
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Acyl-CoA synthetase family member 3 (ACSF3), rabbit polyclonal | ThermoFisher | Cat#PA5-25803 |
| Heat shock chaperone 70 (HSC70), mouse monoclonal | Santa Cruz | Cat#sc-7298 |
| MitoProfile total OXPHOS, mouse monoclonal antibody mix | Abcam | Cat#ab110413 |
| Voltage-dependent anion channel 1 (VDAC1), rabbit polyclonal | Calbiochem | Cat#AB10527 |
| Aconitase 2 (ACO2), rabbit polyclonal | Cell Signaling | Cat#6922 |
| α-lipoic acid, rabbit polyclonal | Calbiochem | Cat#437695 |
| Phospho-acetyl-CoA carboxylase (ACC) (Ser79), rabbit polyclonal | Cell Signaling | Cat#11818 |
| Total ACC, rabbit polyclonal | Cell Signaling | Cat#3676 |
| α-malonyl-lysine, rabbit monoclonal antibody mix | Cell Signaling | Cat#14942 |
| α-acetyl-lysine, rabbit polyclonal | Cell Signaling | Cat#9441 |
| α-succinyl-lysine, rabbit polyclonal | PTM Biolabs | Cat#401 |
| GFP, mouse monoclonal | Santa Cruz | Cat#sc-9996 |
| Cy3-conjugated anti-mouse secondary antibody | Invitrogen | Cat#M30010 |
| Horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody | GE Healthcare | Cat#NA931V |
| HRP-conjugated anti-rabbit secondary antibody | GE Healthcare | Cat#NA934V |
| GFP nanobody magnetic beads | Allele Biotechnology | Cat#ABP-NAB-GFPM050 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Malonate | Sigma-Aldrich | M1296; CAS: 141-82-2 |
| [U-14C]D-glucose | Moravek Biochemicals | MC144W |
| [U-14C]L-glutamine | New England Nuclear/PerkinElmer | NEC451050UC |
| [U-14C]L-alanine | Moravek Biochemicals | MC132 |
| [3H]acetate | New England Nuclear/PerkinElmer | NET003025MC |
| [2-14C]malonate | Moravek Biochemicals | MC312 |
| [1,3-13C]malonate | Sigma-Aldrich | 490199; CAS: 99524-14-8 |
| 3-Nitropropionic acid | Sigma-Aldrich | N5636; CAS: 504-88-1 |
| Critical Commercial Assays | ||
| CellTiter 96 Aqueous One Cell Proliferation Assay | Promega | Cat#G3582 |
| Amersham Prime Enhanced Chemiluminescent Substrate | GE Healthcare | Cat#RPN2232 |
| XFe96 Extracellular Flux Assay Kit | Seahorse Bioscience | Cat#102601-100 |
| CyQUANT Cell Proliferation Assay Kit | Invitrogen | Cat#C7026 |
| Complex II Enzyme Activity Microplate Assay | Abcam | Cat#ab109908 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat#CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wild-type C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: ob/ob obese, leptin-deficient: B6.Cg-Lepob/J | The Jackson Laboratory | JAX: 000632 |
| Oligonucleotides | ||
| ACSF3 Forward: 5′-ACACAGAGGAAGTGGTCTTCT-3′ | This paper | N/A |
| ACSF3 Reverse: 5′-CGTTAGCGCATAGGAAGGAGA-3′ | This paper | N/A |
| Recombinant DNA | ||
| Human codon-optimized Cas9 expression vector | Mali et al., 2013 | Addgene Plasmid #41815 |
| pEF6-nls-YFP-2A-Cas9 for bicistronic expression of nuclear YFP and Cas9 | Bowman et al., 2016 | N/A |
| Mitochondrially-targeted YFP expression vector (pEYFP-Mito) | Clontech | Cat#6115 |
| Cytoplasmic YFP expression vector (pEYFP-C1) | Clontech | Cat#6341 |
| Other | ||
| FuGENE HD Transfection Reagent | Promega | Cat#E2311 |
| In-Fusion HD Cloning Enzyme | Clontech | Cat#639636 |
Significance.
The regulation of cytoplasmic malonyl-CoA to promote fatty acid synthesis and inhibit mitochondrial β-oxidation of long-chain fatty acids is well described, but the source and metabolic fate of mitochondrial malonyl-CoA has remained a mystery until the recent identification of a mitochondrial malonyl-CoA synthetase, ACSF3. By engineering loss-of-function mutations in ACSF3 in cultured human cells, we defined the requirement for mitochondrial malonate metabolism and gained mechanistic insight into disease-causing genetic deficiencies in malonyl-CoA metabolism. Malonate is an endogenous metabolite that potently inhibits succinate dehydrogenase, and we demonstrated that ACSF3 is required for malonate detoxification to promote mitochondrial metabolic flux. Malonate can be oxidized by human cells via the subsequent activities of ACSF3 and mitochondrial malonyl-CoA decarboxylase. ACSF3-deficient cells exhibited elevated malonate concentrations and impaired mitochondrial metabolism. A combination of steady-state metabolite measurements and metabolic flux studies was used to describe malonate- and malonyl-CoA-dependent metabolic alterations. Lysine malonylation of mitochondrial proteins was identified as another significant fate of ACSF3-derived malonyl-CoA. ACSF3 was specifically required for malonylation but did not affect lysine acetylation or succinylation. ACSF3-deficient cells represent an excellent model system to define the regulation and metabolic consequences of mitochondrial protein malonylation to delineate the metabolic underpinnings of human metabolic disease.
Highlights.
Malonate, a toxic metabolic by-product, is metabolized by conversion to malonyl-CoA
A mitochondrial malonyl-CoA synthetase, ACSF3, generates malonyl-CoA from malonate
ACSF3 detoxifies mitochondrial malonate to promote mitochondrial metabolism
Human cells lacking ACSF3 have impaired mitochondrial protein malonylation
Acknowledgments
We would like to thank Natasha Zachara for essential reagents. This work was supported in part by a National Institutes of Health grant R01NS072241 to M.J.W. and R01HL108882 to S.M.C. and a pilot grant from the National Organization of Rare Disease to M.J.W. C.E.B. was supported in part by a fellowship from the American Heart Association 15PRE25090309. M.G.A. was supported in part by a fellowship from the American Heart Association 16PRE31140006.
Footnotes
Author contributions: C.E.B., S.R., E.S.A. and M.G.A. conducted the experiments and analyzed the results. E.S.A. conducted the 1H-NMR experiments and analyzed the results. L.Z. and T.H. conducted the 13C-malonate labeling experiment and the unbiased metabolomics analysis of ACSF3 KO and control cells and analyzed the results. P.A.W., S.M.C. and M.J.W. conceived the idea for the project and provided essential reagents. C.E.B. and M.J.W. wrote the paper with input and approval of all authors.
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfares A, Nunez LD, Al-Thihli K, Mitchell J, Melancon S, Anastasio N, Ha KC, Majewski J, Rosenblatt DS, Braverman N. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet. 2011;48:602–605. doi: 10.1136/jmedgenet-2011-100230. [DOI] [PubMed] [Google Scholar]
- Alves TC, Pongratz RL, Zhao X, Yarborough O, Sereda S, Shirihai O, Cline GW, Mason G, Kibbey RG. Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell metabolism. 2015;22:936–947. doi: 10.1016/j.cmet.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Kim YS. A gene cluster encoding malonyl-CoA decarboxylase (MatA), malonyl-CoA synthetase (MatB) and a putative dicarboxylate carrier protein (MatC) in Rhizobium trifolii--cloning, sequencing, and expression of the enzymes in Escherichia coli. European journal of biochemistry / FEBS. 1998;257:395–402. doi: 10.1046/j.1432-1327.1998.2570395.x. [DOI] [PubMed] [Google Scholar]
- Asadollahi R, Oneda B, Joset P, Azzarello-Burri S, Bartholdi D, Steindl K, Vincent M, Cobilanschi J, Sticht H, Baldinger R, et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J Med Genet. 2014;51:677–688. doi: 10.1136/jmedgenet-2014-102588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- Bowman CE, Zhao L, Hartung T, Wolfgang MJ. Requirement for the Mitochondrial Pyruvate Carrier in Mammalian Development Revealed by a Hypomorphic Allelic Series. Mol Cell Biol. 2016;36:2089–2104. doi: 10.1128/MCB.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kim HU, Weng H, Browse J. Malonyl-CoA synthetase, encoded by ACYL ACTIVATING ENZYME13, is essential for growth and development of Arabidopsis. Plant Cell. 2011;23:2247–2262. doi: 10.1105/tpc.111.086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, Cheng Z, Park J, Wan X, Liu X, et al. Proteomic and Biochemical Studies of Lysine Malonylation Suggest Its Malonic Aciduria-associated Regulatory Role in Mitochondrial Function and Fatty Acid Oxidation. Mol Cell Proteomics. 2015;14:3056–3071. doi: 10.1074/mcp.M115.048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vellis J, Shannon LM, Lew JY. Malonic Acid Biosynthesis in Bush Bean Roots. I. Evidence for Oxaloacetate as Immediate Precursor. Plant Physiol. 1963;38:686–690. doi: 10.1104/pp.38.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Cai T, Li T, Xue P, Zhou B, He X, Wei P, Liu P, Yang F, Wei T. Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins. Mol Cell Proteomics. 2015;14:227–236. doi: 10.1074/mcp.M114.041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Bowman CE, Wolfgang MJ. Metabolic and Tissue-Specific Regulation of Acyl-CoA Metabolism. PLoS One. 2015;10:e0116587. doi: 10.1371/journal.pone.0116587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Wolfgang MJ. A genetically encoded metabolite sensor for malonyl-CoA. Chem Biol. 2012;19:1333–1339. doi: 10.1016/j.chembiol.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Wong GW, Wolfgang MJ. Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol. 2013;33:1869–1882. doi: 10.1128/MCB.01548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzPatrick DR, Hill A, Tolmie JL, Thorburn DR, Christodoulou J. The molecular basis of malonyl-CoA decarboxylase deficiency. Am J Hum Genet. 1999;65:318–326. doi: 10.1086/302492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest. 2012;122:1958–1959. doi: 10.1172/JCI63967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Funai K, Song H, Yin L, Lodhi IJ, Wei X, Yoshino J, Coleman T, Semenkovich CF. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J Clin Invest. 2013;123:1229–1240. doi: 10.1172/JCI65726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Waber L, Bennett MJ, Gibson KM, Cohen JC. Cloning and mutational analysis of human malonyl-coenzyme A decarboxylase. J Lipid Res. 1999;40:178–182. [PubMed] [Google Scholar]
- Guan X, Nikolau BJ. AAE13 encodes a dual-localized malonyl-CoA synthetase that is crucial for mitochondrial fatty acid biosynthesis. Plant J. 2016;85:581–593. doi: 10.1111/tpj.13130. [DOI] [PubMed] [Google Scholar]
- Hayaishi O, Kornberg A. Metabolism of cytosine, thymine, uracil, and barbituric acid by bacterial enzymes. J Biol Chem. 1952;197:717–732. [PubMed] [Google Scholar]
- Hegre CS, Halenz DR, Lane MD. The Enzymatic Carboxylation of Butyryl Coenzyme-A. J Am Chem Soc. 1959;81:6526–6527. [Google Scholar]
- Hiltunen JK, Autio KJ, Schonauer MS, Kursu VA, Dieckmann CL, Kastaniotis AJ. Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta. 2010;1797:1195–1202. doi: 10.1016/j.bbabio.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. Mitochondrial fatty acid synthesis type II: more than just fatty acids. J Biol Chem. 2009;284:9011–9015. doi: 10.1074/jbc.R800068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Zhao Y. Metabolic regulation by lysine malonylation, succinylation and glutarylation. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, Vander Heiden MG. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell. 2016;36:540–549. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner J, Hoppel CL. Radiochemical malonyl-CoA decarboxylase assay: activity and subcellular distribution in heart and skeletal muscle. Anal Biochem. 2002;306:283–289. doi: 10.1006/abio.2002.5696. [DOI] [PubMed] [Google Scholar]
- Kerner J, Minkler PE, Lesnefsky EJ, Hoppel CL. Fatty acid chain elongation in palmitate-perfused working rat heart: mitochondrial acetyl-CoA is the source of two-carbon units for chain elongation. J Biol Chem. 2014;289:10223–10234. doi: 10.1074/jbc.M113.524314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Yu BP. Characterization of age-related malondialdehyde oxidation: the effect of modulation by food restriction. Mech Ageing Dev. 1989;50:277–287. doi: 10.1016/0047-6374(89)90105-x. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kolattukudy PE. Purification and properties of malonyl-CoA decarboxylase from rat liver mitochondria and its immunological comparison with the enzymes from rat brain, heart, and mammary gland. Arch Biochem Biophys. 1978;190:234–246. doi: 10.1016/0003-9861(78)90273-4. [DOI] [PubMed] [Google Scholar]
- Kulkarni RA, Worth AJ, Zengeya TT, Shrimp JH, Garlick JM, Roberts AM, Montgomery DC, Sourbier C, Gibbs BK, Mesaros C, et al. Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem Biol. 2017;24:231–242. doi: 10.1016/j.chembiol.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. 2013;50:686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, An JH, Kim YS. Identification and characterization of a novel transcriptional regulator, MatR, for malonate metabolism in Rhizobium leguminosarum bv. trifolii. European journal of biochemistry / FEBS. 2000;267:7224–7230. doi: 10.1046/j.1432-1327.2000.01834.x. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy LY, Zeevalk GD, Sonsalla PK. Role for dopamine in malonate-induced damage in vivo in striatum and in vitro in mesencephalic cultures. J Neurochem. 2000;74:1656–1665. doi: 10.1046/j.1471-4159.2000.0741656.x. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol Cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender C, Trentadue AR, Pories WJ, Dohm GL, Houmard JA, Youngren JF. Expression of genes regulating malonyl-CoA in human skeletal muscle. J Cell Biochem. 2006;99:860–867. doi: 10.1002/jcb.20944. [DOI] [PubMed] [Google Scholar]
- Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougovkina O, Te Brinke H, Wanders RJ, Houten SM, de Boer VC. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. J Inherit Metab Dis. 2014;37:709–714. doi: 10.1007/s10545-014-9684-9. [DOI] [PubMed] [Google Scholar]
- Quastel JH, Wooldridge WR. Some properties of the dehydrogenating enzymes of bacteria. Biochem J. 1928;22:689–702. doi: 10.1042/bj0220689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell metabolism. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Ellis JM, Wolfgang MJ. Chemical-genetic induction of Malonyl-CoA decarboxylase in skeletal muscle. BMC Biochem. 2014;15:20. doi: 10.1186/1471-2091-15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A. 2016;113:4320–4325. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253–272. doi: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- Saha AK, Schwarsin AJ, Roduit R, Masse F, Kaushik V, Tornheim K, Prentki M, Ruderman NB. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside. J Biol Chem. 2000;275:24279–24283. doi: 10.1074/jbc.C000291200. [DOI] [PubMed] [Google Scholar]
- Sambandam N, Steinmetz M, Chu A, Altarejos JY, Dyck JR, Lopaschuk GD. Malonyl-CoA decarboxylase (MCD) is differentially regulated in subcellular compartments by 5′AMP-activated protein kinase (AMPK). Studies using H9c2 cells overexpressing MCD and AMPK by adenoviral gene transfer technique. Eur J Biochem. 2004;271:2831–2840. doi: 10.1111/j.1432-1033.2004.04218.x. [DOI] [PubMed] [Google Scholar]
- Sloan JL, Johnston JJ, Manoli I, Chandler RJ, Krause C, Carrillo-Carrasco N, Chandrasekaran SD, Sysol JR, O'Brien K, Hauser NS, et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet. 2011;43:883–886. doi: 10.1038/ng.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schaftingen E, Rzem R, Marbaix A, Collard F, Veiga-da-Cunha M, Linster CL. Metabolite proofreading, a neglected aspect of intermediary metabolism. J Inherit Metab Dis. 2013;36:427–434. doi: 10.1007/s10545-012-9571-1. [DOI] [PubMed] [Google Scholar]
- Vennesland B, Evans EA. The formation of malonic acid from oxalacetic acid by pig heart preparations. J Biol Chem. 1944;156:783–784. [Google Scholar]
- Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48:2736–2750. doi: 10.1194/jlr.M700378-JLR200. [DOI] [PubMed] [Google Scholar]
- Wightman PJ, Santer R, Ribes A, Dougherty F, McGill N, Thorburn DR, FitzPatrick DR. MLYCD mutation analysis: evidence for protein mistargeting as a cause of MLYCD deficiency. Hum Mutat. 2003;22:288–300. doi: 10.1002/humu.10264. [DOI] [PubMed] [Google Scholar]
- Witkowski A, Thweatt J, Smith S. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J Biol Chem. 2011;286:33729–33736. doi: 10.1074/jbc.M111.291591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang MJ, Lane MD. The role of hypothalamic malonyl-CoA in energy homeostasis. J Biol Chem. 2006;281:37265–37269. doi: 10.1074/jbc.R600016200. [DOI] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Wishart DS. MetATT: a web-based metabolomics tool for analyzing time-series and two-factor datasets. Bioinformatics. 2011;27:2455–2456. doi: 10.1093/bioinformatics/btr392. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Derr-Yellin E, Nicklas WJ. Relative vulnerability of dopamine and GABA neurons in mesencephalic culture to inhibition of succinate dehydrogenase by malonate and 3-nitropropionic acid and protection by NMDA receptor blockade. J Pharmacol Exp Ther. 1995;275:1124–1130. [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Hoppe J, Sonsalla P. In vivo vulnerability of dopamine neurons to inhibition of energy metabolism. Eur J Pharmacol. 1997;320:111–119. doi: 10.1016/s0014-2999(96)00892-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.