

Abstract
To what extent is the convergent evolution of protein function attributable to convergent or parallel changes at the amino acid level? The mutations that contribute to adaptive protein evolution may represent a biased subset of all possible beneficial mutations owing to mutation bias and/or variation in the magnitude of deleterious pleiotropy. A key finding is that the fitness effects of amino acid mutations are often conditional on genetic background. This context dependence (epistasis) can reduce the probability of convergence and parallelism because it reduces the number of possible mutations that are unconditionally acceptable in divergent genetic backgrounds. Here, I review factors that influence the probability of replicated evolution at the molecular level.
The convergent evolution of organismal phenotypes is often interpreted as a testament to the power of natural selection to craft uniquely favoured design solutions to common problems. Patterns of convergence may also reflect intrinsic biases in the production of variation, as propensities of development can increase the likelihood that similar traits will evolve in different lineages1,2. An important question in evolutionary genetics concerns the extent to which adaptive phenotypic convergence is caused by convergent or parallel changes in the underlying genes. Given the typical ‘many-to-one’ mapping of genotype to phenotype, a corollary question concerns the causes of convergence and parallelism at the molecular sequence level. If there are many possible solutions to a given problem, then it is all the more surprising when we discover that evolution has hit upon the same solution to the same problem time and time again. What properties distinguish the actualized solutions from those of the many non-actualized possibilities? These questions have important implications for understanding the repeatability (and, hence, predictability) of molecular adaptation3–8.
An especially powerful approach to examine the causes of molecular convergence and parallelism involves the examination of phylogenetically replicated changes in protein function that can be traced to specific amino acid substitutions. By focusing on genetically based changes in a well-defined biochemical phenotype, questions about the causes of molecular convergence and parallelism become experimentally tractable because it is possible to achieve a full account of causative mutations. Moreover, reverse genetics approaches such as site-directed mutagenesis make it possible to measure the functional effects of such mutations. Experimental studies of protein evolution therefore provide a manageable scale and level of focus for attempts to demarcate “…the boundary between predictability under invariant law and the multifarious possibilities of historical contingency” (REF. 9). Although I focus specifically on protein evolution in this Review, the same concepts are relevant to the evolution of RNA and DNA sequences.
In studies of organismal phenotypes, ‘convergence’ generally suffices as a term to describe the independent acquisition of similar traits in different species2,10. By contrast, the digital nature of molecular sequence data generally permits more refined inferences about the polarity of changes in character state, and it can be useful to make distinctions between different modes of replicated change. In comparisons between orthologous proteins from a given pair of species, convergent substitutions at a particular site refer to independent changes from different ancestral amino acids to the same derived amino acid in both species (FIG. 1a), whereas parallel substitutions refer to independent changes from the same ancestral amino acid11 (FIG. 1b). For brevity or for other principled reasons, some authors collectively refer to both types of replicated substitution as convergence12–14.
Figure 1. Convergent and parallel substitutions: phylogenetically replicated changes that involve different mutational paths.

a | In comparisons among orthologous proteins from a given set of species, convergent substitutions at a particular site refer to independent changes from different ancestral amino acids to the same derived amino acid. In this case, there was a change from G (the ancestral state) to T (the derived state) in one species, and a change from A to T in another species. The convergent substitutions are denoted by red bold lines. b | Parallel substitutions at a site refer to independent changes from the same ancestral amino acid to the same derived amino acid. In this case, changes from A to T occurred in two different species. The parallel substitutions are denoted by red bold lines. In sets of closely related species, parallelism is generally more common than convergence simply because — at any given site — close relatives will be more likely to share the same ancestral state prior to the occurrence of independent substitutions.
Strictly defined, convergent and parallel substitutions involve the fixation of identical-by-state alleles in different lineages; the alleles have independent mutational origins. Comparative studies of naturally evolved proteins have documented a number of striking cases of convergence and parallelism at the amino acid level, several of which involved experimental validation of presumably adaptive functional effects. These include studies of phylogenetically replicated substitutions in the visual opsins of various vertebrate species that mediate evolutionary transitions in spectral sensitivity15,16, substitutions in the haemoglobins of high-altitude Andean hummingbirds that mediate evolutionary transitions in blood-oxygen affinity, substitutions in Na+,K+-ATPase enzymes of herbivorous insects that mediate resistance to toxic, plant-derived cardenolides18,19, substitutions in Na+,K+-ATPase enzymes in squamate reptiles that mediate resistance to cardiac glycosides produced by toxic prey species20, and substitutions in voltage-gated sodium channel (Nav1) proteins in reptiles, amphibians and fish that mediate resistance to tetrodotoxin21,22. There are also numerous examples of molecular convergence and parallelism in proteins that mediate insecticide resistance23,24, herbicide resistance25 and antibiotic resistance26. In addition to comparative studies, experimental evolution studies involving microorganisms and viruses have documented remarkable cases of replicated amino acid substitutions during adaptation to novel environmental challenges27–32.
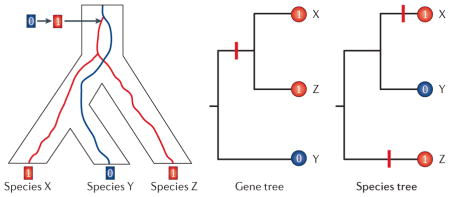
In addition to the replicated fixation of identical-by-state alleles that have independent mutational origins, the sharing of identical-by-descent alleles between species may be attributable to incomplete lineage sorting or introgressive hybridization; either way, the shared alleles do not have independent mutational origins, so the gene tree will not be congruent with the inferred species tree. As a result of this genealogical discordance, substitutions that are mapped onto the species tree (as opposed to the underlying gene tree) can make it look like single transitions in character state actually occurred multiple times independently. This phenomenon is termed ‘hemiplasy’33,34 to distinguish it from the true homoplasy produced by convergent and parallel substitutions. In the presence of hemiplasy, the phylogenetic replication of character-state transitions may be more apparent than real, as it simply results from forcing the discordant gene tree to conform to the branching topology of the species tree34 (BOX 1).
Box 1. Hemiplasy affects inferences about phenotypic convergence.
Genealogical discordance between gene trees and species trees affects inferences about the number and timing of character-state transitions, potentially resulting in the incorrect inference of convergence where none has occurred (termed hemiplasy)33,34. In the example shown in the figure (left panel), a set of three species (X, Y and Z) have retained an allelic polymorphism that was present in their last common ancestor (the ‘1’ allele is derived). As a result of incomplete lineage sorting, the identical-by-descent ‘1’ alleles do not coalesce in the common ancestor of the sister species X and Y, and instead coalesce in the last common ancestor of species X, Y and Z, thereby producing genealogical discordance between the gene tree ((X,Z),Y)) and the species tree ((X,Y),Z)). Mapping the 0→1 substitution onto the gene tree indicates a single character-state transition (middle panel). However, if the gene tree is assumed to be congruent with the topology of the species tree (right panel), then the phyletic distribution of character states produces the appearance that the 0→1 change occurred twice independently (once in the branch leading to species X and once in the branch leading to species Z, yielding so-called ‘collateral substitutions’7). Thus, the 0→1 substitution and any associated change in phenotype only appear to be phylogenetically replicated if we force the discordant gene tree to fit the species tree. This highlights the potentially illusory nature of ‘collateral substitutions’ and illustrates how incomplete lineage sorting can be mistaken for convergence when gene tree discordance is not taken into account34. Incomplete lineage sorting and the resultant hemiplasy in trait evolution is most likely to occur in sets of species that have undergone a relatively rapid radiation, such that time intervals between successive speciation events (that is, internodal times in the species tree) are short relative to effective population sizes124,125.
Here, I review factors that influence the probability of replicated substitutions in protein evolution. In addition to addressing questions of longstanding interest about the repeatability and predictability of evolutionary change, this topic is timely because there is currently a great deal of excitement about using whole-genome sequence data to test for evidence of adaptive molecular convergence and parallelism.
Causes of substitution bias
There are two main reasons why we might expect adaptive convergence in protein function to involve convergent or parallel substitutions at the amino acid level. First, it may be that there are simply a limited number of mutations capable of producing the requisite change in phenotype, reflecting intrinsic constraints imposed by the nature of structure–function relationships. This could be considered the ‘forced option’ hypothesis. Alternatively, it may be that there are many possible mutations that can produce the requisite change in phenotype, but particular mutations (or particular types of mutation) are preferentially fixed. This substitution bias may be attributable to variation in rates of origin; that is, sites vary in rates of mutation to alleles that produce the beneficial change in phenotype (mutation bias), or mutations with similar main effects on the selected phenotype vary in their probability of fixation once they arise (fixation bias) owing to variation in the magnitude of deleterious pleiotropy6.
The probability of parallel substitution between two species, which both choose the next mutational step from the same distribution of n possible options (each with probability p(i)), is:
| (1) |
It follows that
| (2) |
where n is the number of possible options and V is the variance in the probabilities of those options. Equivalently,
| (3) |
where C is the coefficient of variation in the probabilities (see equation 8b in Chevin et al.5 for a similar formulation). In biological terms, n is the number of possible mutations, and C subsumes the effects of all mutational and selective factors that increase variability in the distribution of fixation probabilities of the n possible mutations. For a given n, the probability of parallel substitution is minimized if all n mutations occur at equal rates and if they all have identical selection coefficients; that is, Pr(//)=1/n when C = 0. The probability of parallel substitution increases linearly with decreasing n, and it increases monotonically with increasing C.
How might we go about estimating these key parameters that influence the probability of parallel substitution? Experimental insights into the effective number of mutations that are capable of producing a given change in protein function can be obtained via reverse-genetic screens of engineered mutational libraries, and insights into the causes of substitution bias can be obtained via directed evolution experiments and/or comparative studies of naturally evolved proteins. In comparative studies of orthologous proteins from multiple species, we can assess whether particular mutations or particular types of mutations have been fixed preferentially by testing whether average substitution rates are the same across sites6 (BOX 2). Below, I discuss potential causes of mutation bias and fixation bias.
Box 2. Are some mutations preferentially fixed during adaptive evolution?
Within a given gene, the mutations that contribute to adaptive modifications of protein function may represent a biased subset of all possible mutations that are capable of producing the same functional effect. For example, there may be numerous amino acid mutations that can produce the same adaptive improvement in the catalytic activity of an enzyme, but these mutations may be unequal in the eyes of natural selection if they vary in the magnitude of deleterious pleiotropy. Thus, mutations that have equivalent main effects on a selected phenotype may still have different selection coefficients (and, hence, different fixation probabilities). To assess whether particular types of amino acid mutation are preferentially fixed, we can test whether average substitution rates are the same for different mutation classes (for example, mutations in the active site versus mutations affecting protein allostery)6. In the simplest possible ‘origin-fixation’ model of molecular evolution, the substitution rate is given by:
| (4) |
where N is the size of a diploid population, μ is the per-copy rate of mutation, and is the probability that a new mutation becomes fixed once it has arisen. In this framework, we can specify the substitution rate as the product of the rate at which new alleles originate via mutation and the probability that they become fixed in the population once they arise39,126. This model is based on the ‘infinite sites’ assumption that each new mutation occurs at a site where variation is not currently segregating127. The origin-fixation formalism therefore describes a regime of mutation-limited evolution where the rate of evolution is directly proportional to the mutation rate39.
An important implication is that a bias in mutation rates can produce a bias in substitution rates, even when the substitutions are driven by positive selection35–37,39. In the absence of contributions from standing variation, a bias in rates of origin affects the joint probability of origin and fixation. As μ and λ can vary among different classes of mutations, site-specific substitution rates will vary accordingly126, so the expected rate of substitution for mutations in class i is Ki= 2Nμiλi. The class-specific mutation rate is μi = μθi, where μ is the overall rate of origin for mutations that produce a given change in phenotype and θi is the proportion of these mutations in class i, yielding Ki= 2Nμθiλi. Thus, the proportion of fixed mutations in class i is
| (5) |
We can assess evidence for substitution bias by testing whether each mutation class contributes equally to evolutionary changes in a particular protein function; that is, we can test the null hypothesis that ri= 1/n for all i. In a comparison between two discrete mutation classes, i and j, substitution bias is indicated if the different types of mutation have unequal rates of origin (θi ≠ θj) and/or unequal probabilities of fixation once they arise (λ i ≠ λj). The role of mutation bias can be assessed by testing the null hypothesis that θi= 1/n for all i. Using data from mutagenesis screens, the role of fixation bias can be assessed by testing the null hypothesis that the spectrum of spontaneous mutations that produce a given change in protein function is equal to the spectrum of substitutions that are responsible for evolutionary changes in protein function between species6.
For comparisons between different classes of site (or different classes of mutational change or mutational effect), this framework provides a means of assessing the extent to which an observed substitution bias is attributable to biased mutation rates and/or biased fixation probabilities6,35,37, and can therefore provide insights into possible causes of convergent and parallel substitutions during adaptive protein evolution.
Mutation bias as a cause of substitution bias
An under-explored question in evolutionary genetics concerns the extent to which mutation bias influences pathways of adaptive molecular evolution35–37. For example, in mammalian genomes the combination of transition:transversion bias and CpG bias results in especially high rates of C→T and G→A transition mutations. The genetic code then determines how these propensities of nucleotide change translate into propensities of amino acid change38. As explained in BOX 2, mutation bias can be an important orienting factor in both neutral and adaptive molecular evolution because an asymmetry in rates of origin can affect the joint probability of origin and fixation35–37,39.
Results of several experimental evolution studies have provided evidence that mutation bias can influence trajectories of adaptive protein evolution29,40–43; however, this has not been systematically investigated. In repeated single-step adaptive walks involving the same wild-type genotype of a single-strand DNA bacteriophage, ID11, the two amino acid mutations that fixed in the largest fraction of replicate lines did not have the largest selection coefficients but they were produced by mutationally favoured C→T transitions. By contrast, the fittest allele — which was fixed at a relatively lower rate — was produced by a mutationally unfavoured G→T transversion29. In this system, fixation probabilities could be accurately predicted by adjusting a model of origin-fixation dynamics (BOX 2) to incorporate the estimated transition:transversion bias.
Similar effects have been documented in studies of natural variation. For example, experiments on native haemoglobin variants and engineered, recombinant haemoglobin mutants demonstrated that a nonsynonymous mutation at a highly mutable CpG dinucleotide in the βA-globin gene of high-altitude Andean house wrens (Troglodytes aedon) contributed to an adaptive increase in haemoglobin-oxygen affinity, thereby enhancing blood-oxygen transport capacity under hypoxia44. There seems little reason to suppose that the causative amino acid mutation would have had a larger selection coefficient (and, hence, a higher fixation probability) than any number of other possible mutations that could have produced the same adaptive modification of protein function. However, if the rate of CpG mutation occurs at a higher rate than non-CpG mutations, then — in the absence of contributions from standing variation — the bias in mutation rate is expected to influence evolutionary outcomes in the same way as a commensurate bias in fixation probability35,37,39.
Fixation bias as a cause of substitution bias: the role of pleiotropy
Amino acid mutations commonly have pleiotropic effects on protein biochemistry as they can simultaneously affect multiple aspects of molecular function, structural stability, folding, solubility and propensity for aggregation45–52. Consequently, mutations that improve one aspect of protein function may simultaneously compromise other structural or functional properties. Within the set of mutations that have functionally equivalent effects on a selected biochemical phenotype, those that incur a lower magnitude of deleterious pleiotropy should have a higher fixation probability5,53, and may therefore contribute disproportionately to phenotypic evolution. Investigations into the genetic basis of evolutionary transitions in floral pigmentation in angiosperm plants provide evidence in support of this hypothesis6.
Patterns of replicated substitution in duplicated genes can also provide insight into the influence of pleiotropic constraints on fixation bias. Among the diverse insect taxa that have independently evolved resistance to toxic cardenolides via genetic changes in Na+,K+-ATPase, a preponderance of parallel substitutions occurred in those taxa that possess a single Na+,K+-ATPase gene (FIG. 2). In those taxa that possess two copies of the gene, a greater number of unique (non-shared) substitutions appeared to contribute to convergence in cardenolide resistance19. A possible explanation for this pattern is that the possession of two functionally redundant paralogues alleviates pleiotropic constraints, so a broader spectrum of function-altering mutations could be tolerated in one copy or the other and could therefore potentially contribute to adaptation.
Figure 2. Phylogenetic patterns of parallel and unique substitutions in ATPα1 that are associated with resistance to toxic cardenolides in herbivorous insects.

Substitutions at the indicated sites in Na+,K+-ATPase α1 subunit (ATPα1) are implicated in cardenolide-binding in insect taxa that feed on Apocynaceae. Names of insect orders are shown on the far right. Numbered columns correspond to sites that are implicated in cardenolide-binding based on experimental evidence. Grey sites correspond to sites that may have a role in cardenolide-binding based on structural considerations. Dots denote identity with the consensus sequence. Outgroup sequences of vertebrate Na+,K+-ATPase α1 subunit (ATP1A1; a homologue of ATPα1) from several taxa (sheep, pig and spiny dogfish) are shown for reference. Letters in red boxes represent amino acid replacements whose functional effects have been experimentally characterized. Underlined letters are substitutions that require at least two nonsynoymous substitutions relative to the ancestral sequence. Orange-shaded rows correspond to specialists that are known to sequester cardenolides. Grey-shaded rows represent species that either are not known to sequester cardenolides and/or are nonspecialists that are only occasionally found on Apocynaceae. Green circles represent inferred duplications of the gene encoding ATPα1. Numbers at the bottom of the figure correspond to the number of inferred substitutions associated with use of Apocynaceae. Red boxes at the bottom of the figure correspond to parallel substitutions (observed in more than one independent lineage). Blue boxes correspond to unique substitutions (observed in only one lineage). Note that a preponderance of parallel substitutions occurs only in taxa that possess a single copy of the gene encoding ATPα1. In those taxa that possess two duplicate copies, a greater number of unique (nonshared) substitutions have occurred. It may be that the possession of two functionally redundant paralogues alleviates pleiotropic constraints, so a broader spectrum of function-altering mutations can contribute to adaptation. A. nerii, Aphis nerii; A. pisum, Acyrthosiphon pisum; B. mori, Bombyx mori; B. tabaci, Bemisia tabaci; B. trivittata, Boisea trivittata; C. auratus, Chrysochus auratus; C. castaneus, Cyrtepistomus castaneus; C. lectularius, Cimex lectularius; C. tenera, Cycnia tenera; D. citri, Diaphorina citri; D. eresimus, Danaus eresimus; D. gilippus, Danaus gilippus; D. plexippus, Danaus plexippus; E. egle, Euchaetes egle; H. melpomene, Heliconius melpomene; L. archippus, Limenitis archippus; L. caryae, Lophocampa caryae; L. clivicollis, Labidomera clivicollis; L. halia, Lycorea halia; L. kalmii, Lygaeus kalmii; M. robiniae, Megacyllene robiniae; O. aries, Ovis aries; O. faciatus, Oncopeltus fasciatus; P. chalceus, Pogonus chalceus; P. glaucus, Papilio glaucus; P. versicolora, Plagiodera versicolora; R. lineaticollis, Rhyssomatus lineaticollis; S. acanthias, Squalus acanthias; S. scrofa, Sus scrofa; T. castaneum, Tribolium castaneum; T. legitima, Trichordestra legitima; T. tetrophthalmus, Tetraopes tetrophthalmus. Adapted from Zhen, Y., Aardema, M. L., Medina, E. M., Schumer, M. & Andolfatto, P. Parallel molecular evolution in an herbivore community. Science 337, 1634–1637 (2012). Reprinted with permission from the American Association for the Advancement of Science.
Pleiotropic constraints are often invoked to explain patterns of convergent and parallel molecular evolution3,4,7,19,54,55, but these inferences are often based on fairly indirect lines of evidence. Decisive tests of the deleterious pleiotropy hypothesis require direct, experimental measurements of the effects of individual mutations on a selected phenotype in conjunction with experimental measurements of mutational pleiotropy in the same genetic background45,52,56–58. This requires a decision about which particular molecular phenotypes to measure. Experimental assessments of mutational pleiotropy therefore require insight into the nature of possible trade-offs and an understanding of the biologically relevant dimensionality of phenotypic space for the protein in question.
Consideration of the role of pleiotropic constraints on molecular adaptation highlights a key difference between two alternative experimental approaches for investigating mechanisms of protein evolution. In directed evolution experiments, libraries of randomly mutagenized gene products are iteratively screened and selected for a desired biochemical property (for example, improved catalytic activity or novel substrate specificity)59–61. The advantage of this approach is that it permits the evaluation of a vast and unbiased set of mutant proteins, and can reveal large numbers of possible mutations that are capable of producing a particular change in function. The alternative strategy involves retrospective analysis of natural or laboratory evolution. This strategy explores a far more circumscribed region of sequence space, and is best suited to the goal of identifying the specific historical substitutions that caused changes in protein function in realized evolutionary pathways. In retrospective analyses, the protein under consideration will have evolved under pleiotropic constraints that are manifest in vivo, and adaptive modifications of particular functional properties may be more likely to require compromise-based solutions in the joint optimization of different structural and functional properties.
Intramolecular epistasis
Non-uniformity of mutational effects at orthologous sites
During the adaptive evolution of protein function, epistasis can affect the fixation probabilities of function-altering mutations in two ways: first, the effect of the mutation on a positively selected phenotype may be conditional on genetic background; and second, the pleiotropic effects of the mutation may be conditional on genetic background. In principle, both forms of epistatic interaction can influence which mutations contribute to molecular adaptation.
Protein engineering studies have documented pervasive epistasis between mutant sites in the same protein, both with respect to specific biochemical phenotypes58,62–71 and — in microbial and viral experimental evolution studies — with respect to fitness or fitness proxies8,40,48,56,72–81. An especially powerful experimental approach involves the synthesis of ‘combinatorially complete’ sets of recombinant mutants representing all possible mutational intermediates connecting a given pair of ancestral and derived alleles82. The chief merit of this approach is that additive and non-additive effects of amino acid changes can be quantified by testing each individual mutation in all possible multi-allelic combinations. Such protein engineering studies have demonstrated that the same mutations sometimes have opposite phenotypic effects on different genetic backgrounds — a phenomenon known as ‘sign epistasis’83. One consequence of sign epistasis is that the fitness effects of amino acid mutations will depend on the sequential order in which they occur during the course of an adaptive walk. A mutation that increases fitness on the ancestral genetic background (for example, when it occurs as the first step in an adaptive walk) may have neutral or deleterious effects on a background in which other substitutions have already occurred.
Sign epistasis can exert a deterministic effect on protein evolution by constraining the number of selectively accessible mutational pathways to high-fitness genotypes, thereby enhancing the repeatability of substitutions that represent intermediate steps in such pathways8,74,83. This notion of deterministic repeatability appears to be the basis for assertions that epistasis should generally increase the probability of molecular convergence and parallelism7,84. However, this form of repeatability applies specifically to replicated changes among independent iterations of the evolutionary process that are initiated with the same ancestral genotype. By contrast, questions about the causes of molecular convergence and parallelism in natural protein evolution generally pertain to replicated changes among lineages that evolved from disparate ancestral starting points. In comparisons of orthologous proteins in different species, sign epistasis for fitness should typically decrease the probability of convergence and parallelism, because a mutation that has a beneficial effect on the genetic background of one species will have a neutral or deleterious effect on the divergent genetic backgrounds of other species. Adaptive convergence and parallelism would only be expected for (non-sign-epistatic) mutations that retain their beneficial effects across all backgrounds.
Consider a pair of parallel substitutions that occur at the same site in orthologous proteins of two closely related species. If orthologues of the two species are identical or nearly identical in sequence, then the parallel substitutions are likely to have similar phenotypic effects (ignoring the possibility of intergenic epistasis). However, if the same substitution occurs in a more distantly related species, then it will be more likely to have a phenotypic effect that is different in magnitude or sign, simply because there would be more opportunity for divergence in sequence context (or, more specifically, there would be more opportunity for divergence at sites that epistatically interact with the focal residue than there would be in a closely related species). Convergent or parallel substitutions at orthologous sites in different species may have different fitness effects owing to lineage-specific changes in the fitness landscapes of individual residue positions (FIG. 3a). As a given site can be occupied by up to 20 amino acids, the set of fitness values conferred by each possible variant defines a vector of site-specific amino acid propensities for a given genetic background85–87. This single-position fitness landscape can be considered a cross-section of the complete fitness landscape (FIG. 3b), and can change through time owing to changes in the environment and/or changes in genetic background (FIG. 3c).
Figure 3. Schematic depiction of how single-position fitness landscapes change through time.

Horizontal rows correspond to each of the 20 possible amino acids at each site in a protein. a | At each site, the currently predominant amino acid (shown in bold outline) confers high fitness. b | The single-position fitness landscape of site 7, shown in isolation. c | Temporal change in the single-position fitness landscape. The relative fitness levels of different amino acids at the position change through time owing to changes in the genetic background (that is, substitutions at other sites) and/or changes in the environment. The depicted changes in fitness are modelled as a Poisson process. Adapted from Bazykin, G. A. Changing preferences: deformation of single position amino acid fitness landscapes and evolution of proteins. Biology Letters (2015) 11, 20150315, by permission of the Royal Society.
In addition to reducing the probability of convergent and parallel substitutions at orthologous sites, context-dependent fitness effects of mutations can make substitutions conditionally irreversible64,71,85,87–91. Even if a given substitution was beneficial, neutral or nearly neutral when it initially occurred, mutational reversions to the ancestral state may eventually become maladaptive owing to subsequent changes in sequence context — a phenomenon termed ‘entrenchment’87. Thus, epistasis can be expected to reduce levels of molecular homoplasy by decreasing the probability of convergent and parallel changes to a shared, derived state and by decreasing the probability of mutational reversions to the ancestral state.
The above considerations suggest that epistasis may often reduce probabilities of molecular convergence and parallelism because it reduces the number of possible mutations that have unconditionally acceptable effects in divergent genetic backgrounds. As explained below, research on compensatory mutations has provided strong evidence for the pervasiveness of such context-dependent fitness effects.
Compensatory substitutions
Pleiotropic trade-offs can give rise to a context dependence of mutational effects as evidenced by cases where the fitness impact of a given mutation is determined by compensatory (conditionally beneficial) mutations at other sites in the same protein. For example, the selective fixation of function-altering mutations that confer a net fitness benefit may select for compensatory mutations to mitigate deleterious pleiotropic effects of the functional change46,92,93. Alternatively, function-altering mutations may be selectively permissible only on a background in which the requisite compensatory (or ‘permissive’) mutations have already occurred47,52,63,68,94–96. These compensatory mutations are neutral or deleterious on their own; they are only beneficial in combination with the function-altering mutation.
In addition to experimental evidence for sign-epistatic interactions between mutant sites in the same protein, sign epistasis for fitness is indirectly implicated in cases where pathogenic amino acid variants in one species are observed as wild type in orthologous proteins of other species97–105. The inference is that the deleterious effect of the mutation in the focal species is mitigated by one or more compensatory substitutions in the orthologue of the other species. More direct evidence for sign-epistatic effects comes from cases where a given substitution has an experimentally well-documented effect on protein function in one species, but it has a different phenotypic effect when it occurs at the orthologous site in another species. For example, two lineages of foregut-fermenting mammals, ruminant artiodactyls and colobine monkeys, have convergently evolved digestive RNases with reduced ribonuclease activity against double-stranded RNA. However, these convergent changes in ribonuclease activity were caused by different amino acid substitutions, and one substitution that produced a significant increase in ribonuclease activity in the ruminant RNase produced the opposite effect in the monkey RNase106. Investigations into the evolution of spectral sensitivity in vertebrate opsins have revealed numerous cases where identical substitutions in the homologues of different species shift the wavelength of maximal absorbance in opposite directions16. Similarly, spontaneous mutations at highly conserved carboxy-terminal residue positions in the β-chain of human haemoglobin are known to completely abolish the Bohr effect (the pH sensitivity of haemoglobin-oxygen affinity), a deleterious reduction of allosteric regulatory control that compromises tissue-oxygen delivery. Surprisingly, however, identical substitutions at homologous sites in the haemoglobins of crocodilians107, birds108 and ground squirrels109 do not compromise the Bohr effect.
Sign epistasis and nonlinearity of the phenotype-fitness map
In principle, sign epistasis for fitness can stem directly from the non-additivity of mutational effects on a selected phenotype, in which case nonlinearity in the mapping of genotype to phenotype gives rise to nonlinearity in the mapping of phenotype to fitness. However, even when mutations have additive effects on phenotype, sign epistasis for fitness can result from a nonlinear relationship between phenotype and fitness46,72,78,80,87,110,111. Sign epistasis for fitness is therefore an inherent property of stabilizing selection even if causative mutations have strictly additive effects on the selected phenotype (FIG. 4). This has important implications for understanding how epistasis influences probabilities of molecular convergence and parallelism. Consider a pair of species that are adapting to a shared selection pressure. Even if a given mutation has identical phenotypic effects in the two species, it will have different effects on fitness if the two species start out at different distances from the phenotypic optimum (FIG. 4). This effect of sign epistasis illustrates how stochastic vagaries of history can reduce the probability of molecular convergence and parallelism111. The mapping function that relates phenotype to fitness will inevitably vary from one species to the next owing to idiosyncratic differences in population size and past histories of selection, so the same mutations may often have different effects on fitness (thereby reducing the likelihood that they would contribute to adaptive convergence) even when they have identical effects on the selected phenotype.
Figure 4. Mutations with additive effects on phenotype can have epistatic effects on fitness.

As a result of nonlinearity in the relationship between phenotype and fitness, mutations with additive effects on phenotype can have epistatic effects on fitness. In this hypothetical example, the same mutation is introduced into three genotypes (labelled a, b and c on the figure) that have different phenotypes. The mutation has the same phenotypic effect (Δp = 1) on each of the three backgrounds, but the change in fitness (Δw) is different in each case. The mutation increases fitness in genotypes a and b (although the magnitude of the increment is different) and it reduces fitness in genotype c. Adapted from REF. 111.
Tests of repeated molecular adaptation
Molecular homoplasy can be produced by chance as each amino acid site in a protein has only 20 possible character states. In practice, a far more restricted number of amino acids can generally be tolerated at any given residue position, so the effective number of possible character states will typically be far less than 20 (REFS 11,14,85,112–116). Many parallel sequence changes in protein evolution involve neutral or nearly neutral back-and-forth exchanges between physicochemically similar amino acids that can be interconverted by single mutational steps11,38,112,113. As nonrandom patterns of molecular homoplasy can be produced by mutation bias and/or purifying selection that constrains substitutions to a restricted number of exchangeable amino acids, clear evidence is required to invoke positive selection as a cause of molecular convergence and parallelism.
Zhang117 listed four requirements for establishing that convergent or parallel substitutions are responsible for adaptive convergence in protein function: replicated substitutions are observed in independent lineages; the proteins under investigation have independently evolved derived changes in function; replicated substitutions are responsible for the convergent changes in protein function; and the number of replicated substitutions is greater than expected by chance alone. Most claims of adaptive molecular convergence and parallelism satisfy one or two of these requirements, and the third requirement on the list (establishing a causal connection between change in sequence and change in phenotype) is the one that most often remains unfulfilled because it requires experimental data on the functional effects of individual substitutions.
Experimental approaches
Surveys of sequence variation in the haemoglobin genes of Andean waterfowl revealed numerous amino acid substitutions that occurred in parallel in different high-altitude lineages118. The authors concluded that these parallel substitutions must have contributed to adaptive increases in haemoglobin-oxygen affinity (which would enhance arterial oxygen-loading under hypoxia), although the postulated functional effects were not experimentally validated. The parallel substitutions in the waterfowl globin genes were highlighted as ‘hotspots’ for molecular adaptation and were cited in support of the claim that available adaptive solutions were constrained owing to the ‘limited number of effective mutations’55. However, a subsequent study by Natarajan et al.108 experimentally measured the phenotypic effects of each of the putatively adaptive substitutions and demonstrated that only one of the parallel changes actually contributed to a convergent increase in haemoglobin-oxygen affinity in separate highland taxa. Most convergent increases in haemoglobin-oxygen affinity in highland taxa were attributable to unique substitutions, and most parallel substitutions were functionally inconsequential with respect to the oxygenation properties of haemoglobin108. This highlights the importance of experimentally validating claims about molecular adaptation, and demonstrates why convergence and parallelism at the amino acid level should not be interpreted as prima facie evidence for positive selection on protein function. It seems likely that many published claims about adaptive molecular convergence and parallelism would not hold up under experimental scrutiny, as few comparative studies of naturally evolved proteins have rigorously tested the functional effects of putatively causative substitutions. Upon closer inspection, many convergent and parallel substitutions at sites that are assumed to be ‘hotspots’ of molecular adaptation55 may turn out to be nothing more than hotspots of neutral homoplasy.
Null models for testing adaptive molecular convergence and parallelism
Rigorous assessments of the prevalence of adaptive molecular convergence and parallelism require a properly formulated null model. This is highlighted by a recent study that claimed to have documented genome-wide convergence in protein-coding genes in two mammalian lineages that have independently evolved the capacity for echolocation: microchiropteran bats and toothed whales119. Re-analysis of the data using appropriate null models revealed no preponderance of convergent substitutions in the two lineages120,121, refuting the conclusions of the original study. In fact, the re-analyses actually revealed a lower level of genome-wide convergence in the comparison between microbats and toothed whales than in comparisons between microbats and equally divergent non-echolocating mammals120,121.
Zou and Zhang116 analysed genome-wide alignments of protein-coding genes from a number of eukaryotic taxa to assess whether observed levels of molecular convergence and parallelism could generally be explained without invoking positive selection. For the alignment of each set of orthologous sequences, they estimated branch lengths of the tree (based on known phylogenetic relationships of the species under consideration), the substitution rate of each site relative to the average across the entire protein, and ancestral sequences for each internal node of the phylogeny. For each pair of 1:1 orthologues between a given pair of species, they then compared the inferred number of replicated substitutions with neutral expectations derived from several different substitution models (BOX 3). The most simple substitution model was based on average substitution patterns across many proteins, where expected equilibrium frequencies of the 20 amino acids were set equal to the observed frequencies in the protein under consideration. Under this simple model, the observed numbers of convergent and parallel substitutions were significantly higher than the null expectation. At face value, this result could be uncritically interpreted as evidence for pervasive positive selection that favoured the same sequence changes in different lineages. However, the authors obtained a different result when they used a more sophisticated model that accounted for the fact that different sites in a protein are subject to different physicochemical constraints, such that the particular amino acids that are acceptable at one site may be different from those that are acceptable at another site (the equilibrium frequencies of the 20 amino acids were set equal to the observed frequencies at each site across all sequences in the alignment, rather than averaging across sites). Under this model, the observed number of convergent and parallel substitutions did not exceed the neutral expectation116.
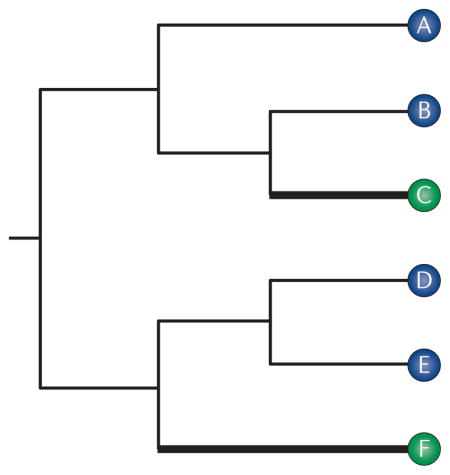
Box 3. Null models for testing adaptive molecular convergence and parallelism.
How do we test whether observed levels of molecular convergence and parallelism are attributable to positive selection using comparative sequence data? That is, how do we formulate a rigorous neutral expectation?
Consider an alignment of orthologous sequences of a given enzyme from a set of six species, the phylogenetic relationships of which are depicted in the figure. Suppose that species C and species F have convergently evolved derived increases in the catalytic activity of the enzyme. We want to assess whether the convergent functional changes in the enzyme were caused by convergent or parallel substitutions, and we want to assess whether it is necessary to invoke positive selection to explain the observed patterns of replicated substitution. That is, we want to test whether the numbers of convergent or parallel substitutions along the two thick branches in the figure exceed the neutral expectation11,116. The first step is to infer the ancestral amino acids at all internal nodes of the phylogeny for each site in the amino acid alignment. The total number of sites that have undergone replicated substitutions on the thick branches is tallied up as the ‘observed’ number of such substitutions.
Assuming that amino acid substitutions at a site follow a Markov process, we can compute the probabilities of convergent and parallel substitutions along the thick branches given the following: a rate matrix of amino acid substitutions; the rate of substitution at the focal site relative to the average rate across all sites in the alignment; the equilibrium frequencies of each amino acid; and estimates of branch lengths based on the expected number of substitutions per site. The expected number of sites with convergent or parallel substitutions is the sum of these probabilities across all sites. Using this framework, the observed and expected numbers of convergent and parallel substitutions can be computed using a probabilistic model of amino acid substitution. The choice of model is important for formulating an appropriate neutral expectation. In particular, recent empirical and simulation studies have demonstrated the importance of using substitution models that account for variation in functional constraint among sites and site-specific changes in functional constraint through time14,116.
These results demonstrate the importance of using substitution models that account for site-specific variation in functional constraints, reflected by among-site heterogeneities in equilibrium amino acid frequencies, and changes in the nature of those constraints through time14,85,116. Analyses that are based on simple time-averaged and site-averaged substitution models underestimate the expected number of replicated substitutions under neutrality, and can therefore lead to spurious inferences about the prevalence of adaptive molecular convergence and parallelism.
Rates of parallelism decrease with time
Another key finding to emerge from model-based studies of protein evolution is that the number of parallel substitutions tends to decrease as a function of sequence divergence14,86,88,89,112,115,116. This pattern may be attributable to pervasive epistatic interactions among sites in the same protein, as changes in site-specific functional constraints occur as a result of changes in sequence context52,85–87,89,90,114–116. In the presence of intramolecular epistasis, the set of amino acids that is acceptable at a given site depends on which amino acids are present at interacting sites. Orthologous sites in the proteins of different species will gradually diverge in the sets of mutually acceptable amino acids owing to divergence in sequence context. Thus, epistasis reduces the probability that the same substitution will be acceptable at orthologous sites in different species, and this probability is expected to decrease as a function of sequence divergence.
Although intramolecular epistasis is a plausible cause of the apparent decline in molecular parallelism as a function of divergence time, it will be important to confirm that this explanation is robust to the effects of genealogical discordance caused by incomplete lineage sorting. Recent preliminary work122 demonstrates that analysing substitutions on discordant gene trees in the context of a single fixed species tree can produce the appearance that single substitutions have occurred twice independently — an artefactual result that could lead to misleading inferences about the prevalence of molecular parallelism.
Summary and future outlook
The number of possible mutations that are capable of producing an adaptive change in protein function depends on the particular protein under consideration and the nature of the selected biochemical phenotype. If adaptive modifications of protein function require fine-tuned changes in catalytic activity or substrate specificity, then the number of potentially adaptive mutations may be limited to a fairly restricted set of active-site residues (with regard to equation 3, n would be small). If the adaptive change involves a more generalized property, such as thermostability or net charge, then numerous possible mutations at numerous possible sites may be capable of producing the requisite change (n would be larger). The former scenario involves fewer degrees of freedom and is therefore expected to involve a higher frequency of convergent or parallel substitutions. The probability of replicated substitution may be further enhanced by variation among sites in the rate at which beneficial alleles originate, and by variation in their probabilities of fixation once they arise, owing to variation in dominance coefficients or the magnitude of deleterious pleiotropy. These factors would increase the coefficient of variation in fixation probabilities (increasing C in equation 3).
Results of protein-engineering experiments demonstrate that the particular mutations that could potentially contribute to adaptive improvements in a given protein function may often depend on the particular amino acids that are present at other sites in the same protein. As the phenotypic effects of function-altering changes are often context-dependent — and as orthologous proteins of different species provide different sequence contexts to the extent that they have diverged owing to drift and/or lineage-specific selection pressures — the number and identity of potentially acceptable mutations can be expected to vary from one lineage to the next. Thus, epistasis may generally reduce the probability of molecular convergence and parallelism in orthologues of different species.
Whole-genome sequencing will continue to provide a starting point for many comparative and experimental studies of molecular convergence and parallelism. For comparative studies of naturally evolved proteins, increasingly sophisticated modelling approaches are needed to assess whether it is necessary to invoke positive selection to explain observed patterns of molecular convergence and parallelism. Although computational analyses of sequence variation will continue to play a key part in suggesting hypotheses, experimental appraisals of putatively causative mutations are needed to provide definitive tests of those hypotheses. Functional experiments provide the most decisive means of adjudicating claims about the adaptive significance of replicated substitutions, and can provide direct, mechanistic insights into the causes of genetic constraints on adaptation. Moreover, as answers to fundamental questions about the genetics of adaptation require data that speak in terms of “...individual mutations that have individual effects” (REF. 123), experimental approaches that involve direct measurements of mutational effects are essential.
Acknowledgments
The author thanks M. W. Hahn, M. J. Harms, D. McCandlish, M. D. Rausher, A. Stoltzfus and anonymous reviewers for helpful suggestions. This work was supported by grants from the US National Institutes of Health (HL087216) and the US National Science Foundation (MCB-1517636 and IOS-0949931).
Glossary
- Orthologous
A form of homologous relationship between genes from different species, indicating that they trace their common ancestry back to speciation events (represented by internal nodes of the species tree) rather than gene duplication events.
- Fixation
The process by which the frequency of a mutant allele increases to 100% in a population, thereby supplanting the ancestral allele.
- Identical-by-state
The identity of allelic gene copies that is attributable to independent mutational changes to a shared character state. The alleles in question have independent mutational origins.
- Cardenolides
Plant-derived steroidal toxins that have an important role in defence against insect herbivores by inhibiting the Na+/K+-ATPase enzyme.
- Identical-by-descent
The identity of allelic gene copies that is attributable to direct descent from a single-copy ancestral allele. The alleles in question have a single mutational origin.
- Incomplete lineage sorting
The retention of ancestral polymorphism from one population-splitting event to the next, followed by stochastic sorting of allelic lineages among descendant species. A common cause of genealogical discordance between gene trees and species trees.
- Introgressive hybridization
The incorporation of allelic variants from one species into the gene pool of another species via hybridization and repeated backcrossing.
- Genealogical discordance
Topological discrepancies among the allelic genealogies (gene trees) of different loci in the same organismal phylogeny.
- Homoplasy
Sharing of character states between species that is attributable to convergence, parallelism, or evolutionary reversal rather than direct inheritance from a common ancestor.
- Pleiotropy
The phenomenon where the same mutation (or genetic locus) affects multiple phenotypes.
- Selection coefficients
Measures of the relative fitnesses of particular genotypes in comparison with a reference genotype in a defined environment.
- Transition:transversion bias
The commonly observed excess of transition mutations (exchanges between purine DNA bases [A↔G] or between pyrimidine bases [C↔T]) relative to transversion mutations (exchanges between purines and pyrimidines).
- CpG bias
If the DNA nucleotide cytosine (C) is immediately 5′ to guanine (G) on the same coding strand (a so-called ‘CpG’ dinucleotide), and if the C is methylated to form 5′-methylcytosine, then C→T and G→A transition mutations occur at an elevated rate relative to mutations at non-CpG sites.
- Adaptive walks
Adaptive evolution that occurs via the sequential fixation of beneficial mutations. The process can be conceptualized as the movement of a population through genotype space via discrete mutational steps, following a trajectory of progressively increasing fitness.
- Nonsynonymous mutation
A point mutation in coding sequence that causes an amino acid replacement in the encoded protein.
- Standing variation
Allelic variation that is currently segregating in a population, as opposed to allelic variants produced by de novo mutation.
- Epistasis
Non-additive interactions between alleles at two or more loci, such that the phenotypic effect of the different alleles in combination cannot be predicted by the sum of the individual effects of each allele by itself.
- Stabilizing selection
Selection on phenotypic variation that favours intermediate trait values.
- Purifying selection
Selection that removes deleterious allelic variants.
- Equilibrium frequencies
Expected frequencies of amino acids in a given sequence or at each site within a sequence. In most models of amino acid sequence evolution, the frequencies are assumed to remain constant over the time period under consideration.
- Markov process
A ‘memoryless’ process of stochastic change with the property that future states depend only on the present state, not on the sequence of antecedent states.
Footnotes
Competing interests statement
The author declares no competing interests.
References
- 1.Wake DB. Homoplasy: the result of natural selection, or evidence of design limitations? Am Naturalist. 1991;138:543–567. [Google Scholar]
- 2.Losos JB. Covergence, adaptation, and constraint. Evolution. 2011;65:1827–1840. doi: 10.1111/j.1558-5646.2011.01289.x. [DOI] [PubMed] [Google Scholar]
- 3.Stern DL, Orgogozo V. The loci of evolution: how predictable is genetic evolution? Evolution. 2008;62:2155–2177. doi: 10.1111/j.1558-5646.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern DL, Orgogozo V. Is genetic evolution predictable? Science. 2009;323:746–751. doi: 10.1126/science.1158997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevin LM, Martin G, Lenormand T. Fisher’s model and the genomics of adaptation: restricted pleiotropy, heterogeneous mutation, and parallel evolution. Evolution. 2010;64:3213–3231. doi: 10.1111/j.1558-5646.2010.01058.x. [DOI] [PubMed] [Google Scholar]
- 6.Streisfeld MA, Rausher MD. Population genetics, pleiotropy, and the preferential fixation of mutations during adaptive evolution. Evolution. 2011;65:629–642. doi: 10.1111/j.1558-5646.2010.01165.x. This paper describes a statistical framework for testing whether different classes of mutations have made disproportionate contributions to adaptive phenotypic evolution. [DOI] [PubMed] [Google Scholar]
- 7.Stern DL. The genetic causes of convergent evolution. Nat Rev Genet. 2013;14:751–764. doi: 10.1038/nrg3483. [DOI] [PubMed] [Google Scholar]
- 8.de Visser JAGM, Krug J. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet. 2014;15:480–490. doi: 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- 9.Gould SJ. Wonderful Life: The Burgess Shale and the Nature of History. W. W. Norton and Company; 1989. [Google Scholar]
- 10.Arendt J, Reznick D. Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol Evol. 2008;23:26–32. doi: 10.1016/j.tree.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JZ, Kumar S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol. 1997;14:527–536. doi: 10.1093/oxfordjournals.molbev.a025789. [DOI] [PubMed] [Google Scholar]
- 12.Castoe TA, et al. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc Natl Acad Sci USA. 2009;106:8986–8991. doi: 10.1073/pnas.0900233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christin PA, Weinreich DM, Besnard G. Causes and evolutionary significance of genetic convergence. Trends Genet. 2010;26:400–405. doi: 10.1016/j.tig.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein RA, Pollard ST, Shah SD, Pollock DD. Nonadaptive amino acid convergence rates decrease over time. Mol Biol Evol. 2015;32:1373–1381. doi: 10.1093/molbev/msv041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama S, Tada T, Zhang H, Britt L. Elucidation of phenotypic adaptations: molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci USA. 2008;105:13480–13485. doi: 10.1073/pnas.0802426105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoyama S, Yang H, Starmert WT. Molecular basis of spectral tuning in the red- and green-sensitive (M/LWS) pigments in vertebrates. Genetics. 2008;179:2037–2043. doi: 10.1534/genetics.108.090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Projecto-Garcia J, et al. Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds. Proc Natl Acad Sci USA. 2013;110:20669–20674. doi: 10.1073/pnas.1315456110. This integrative study documents a striking example of adaptive convergence in protein function, where parallel amino acid substitutions at the same site produced repeated increases in haemoglobin-oxygen affinity in multiple highland lineages of Andean hummingbirds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobler S, Dalla S, Wagschal V, Agrawal AA. Community-wide convergent evolution in insect adaptation to toxic cardenolides by substitutions in the Na, K-ATPase. Proc Natl Acad Sci USA. 2012;109:13040–13045. doi: 10.1073/pnas.1202111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhen Y, Aardema ML, Medina EM, Schumer M, Andolfatto P. Parallel molecular evolution in an herbivore community. Science. 2012;337:1634–1637. doi: 10.1126/science.1226630. References 18 and 19 document a striking pattern of parallel molecular evolution in the ATPase enzymes of herbivorous insects that have convergently evolved resistance to plant-derived toxins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ujvari B, et al. Widespread convergence in toxin resistance by predictable molecular evolution. Proc Natl Acad Sci USA. 2015;112:11911–11916. doi: 10.1073/pnas.1511706112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldman CR, et al. Constraint shapes convergence in tetrodotoxin-resistant sodium channels of snakes. Proc Natl Acad Sci USA. 2012;109:4556–4561. doi: 10.1073/pnas.1113468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodie ED, 3rd, Brodie ED., Jr Predictably convergent evolution of sodium channels in the arms race between predators and prey. Brain Behav Evol. 2015;86:48–57. doi: 10.1159/000435905. [DOI] [PubMed] [Google Scholar]
- 23.ffrench-Constant RH. The molecular and population-genetics of cyclodiene insecticide resistance. Insect Biochem Mol Biol. 1994;24:335–345. doi: 10.1016/0965-1748(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 24.ffrench-Constant RH, Pittendrigh B, Vaughan A, Anthony N. Why are there so few resistance-associated mutations in insecticide target genes? Phil Trans R Soc Series B-Biol Sci. 1998;353:1685–1693. doi: 10.1098/rstb.1998.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broser M, et al. Structural basis of cyanobacterial photosystem II inhibition by the herbicide terbutryn. J Biol Chem. 2011;286:15964–15972. doi: 10.1074/jbc.M110.215970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toprak E, et al. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2012;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bull JJ, et al. Exceptional convergent evolution in a virus. Genetics. 1997;147:1497–1507. doi: 10.1093/genetics/147.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. Different trajectories of parallel evolution during viral adaptation. Science. 1999;285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]
- 29.Rokyta DR, Joyce P, Caudle SB, Wichman HA. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat Genet. 2005;37:441–444. doi: 10.1038/ng1535. [DOI] [PubMed] [Google Scholar]
- 30.Rokyta DR, Abdo Z, Wichman HA. The genetics of adaptation for eight microvirid bacteriophages. J Mol Evol. 2009;69:229–239. doi: 10.1007/s00239-009-9267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer JR, et al. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science. 2012;335:428–432. doi: 10.1126/science.1214449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Ditmarsch D, et al. Convergent evolution of hyperswarming leads to impaired biofilm formation in pathogenic bacteria. Cell Rep. 2013;4:697–708. doi: 10.1016/j.celrep.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avise JC, Robinson TJ. Hemiplasy: a new term in the lexicon of phylogenetics. Syst Biol. 2008;57:503–507. doi: 10.1080/10635150802164587. [DOI] [PubMed] [Google Scholar]
- 34.Hahn MW, Nakhleh L. Irrational exuberance for resolved species trees. Evolution. 2016;70:7–17. doi: 10.1111/evo.12832. This paper explains how genealogical discordance between gene trees and species trees (due to incomplete lineage sorting or introgressive hybridization) can lead to misleading inferences about trait evolution. [DOI] [PubMed] [Google Scholar]
- 35.Yampolsky LY, Stoltzfus A. Bias in the introduction of variation as an orienting factor in evolution. Evol Dev. 2001;3:73–83. doi: 10.1046/j.1525-142x.2001.003002073.x. [DOI] [PubMed] [Google Scholar]
- 36.Stoltzfus A. Mutationism and the dual causation of evolutionary change. Evol Dev. 2006;8:304–317. doi: 10.1111/j.1525-142X.2006.00101.x. [DOI] [PubMed] [Google Scholar]
- 37.Stoltzfus A, Yampolsky LY. Climbing Mount Probable: mutation as a cause of nonrandomness in evolution. J Hered. 2009;100:637–647. doi: 10.1093/jhered/esp048. [DOI] [PubMed] [Google Scholar]
- 38.Yampolsky LY, Stoltzfus A. The exchangeability of amino acids in proteins. Genetics. 2005;170:1459–1472. doi: 10.1534/genetics.104.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCandlish DM, Stoltzfus A. Modeling evolution using the probability of fixation: history and implications. Quarterly Rev Biol. 2014;89:225–252. doi: 10.1086/677571. [DOI] [PubMed] [Google Scholar]
- 40.Lozovsky ER, et al. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA. 2009;106:12025–12030. doi: 10.1073/pnas.0905922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weigand MR, Sundin GW. General and inducible hypermutation facilitate parallel adaptation in Pseudomonas aeruginosa despite divergent mutation spectra. Proc Natl Acad Sci USA. 2012;109:13680–13685. doi: 10.1073/pnas.1205357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong A, Rodrigue N, Kassen R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PloS Genet. 2012;8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Couce A, Rodriguez-Rojas A, Blazquez J. Bypass of genetic constraints during mutator evolution to antibiotic resistance. Proc R Soc B. 2015;282:20142698. doi: 10.1098/rspb.2014.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galen SC, et al. Contribution of a mutational hotspot to adaptive changes in hemoglobin function in high-altitude Andean house wrens. Proc Natl Acad Sci USA. 2015;112:13958–13963. doi: 10.1073/pnas.1507300112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang XJ, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 46.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat Rev Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 47.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 49.Tokuriki N, Stricher F, Serrano L, Tawfik DS. How protein stability and new functions trade off. PLoS Comput Biol. 2008;4:e1000002. doi: 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Harms MJ, Thornton JW. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet. 2013;14:559–571. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631. doi: 10.7554/eLife.00631. This elegant experimental study documents the context-dependent fitness effects of stabilizing and destabilizing amino acid substitutions during the long-term evolution of an influenza nucleoprotein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto SP. Two steps forward, one step back: the pleiotropic effects of favoured alleles. Proc R Soc Lond B. 2004;271:705–714. doi: 10.1098/rspb.2003.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gompel N, Prud’homme B. The causes of repeated genetic evolution. Dev Biol. 2009;332:36–47. doi: 10.1016/j.ydbio.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 55.Martin A, Orgogozo V. The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution. 2013;67:1235–1250. doi: 10.1111/evo.12081. [DOI] [PubMed] [Google Scholar]
- 56.Brown KM, et al. Compensatory mutations restore fitness during the evolution of dihydrofolate reductase. Mol Biol Evol. 2010;27:2682–2690. doi: 10.1093/molbev/msq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schenk MF, et al. Role of pleiotropy during adaptation of TEM-1 beta-lactamase to two novel antibiotics. Evol Appl. 2015;8:248–260. doi: 10.1111/eva.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tufts DM, et al. Epistasis constrains mutational pathways of hemoglobin adaptation in high-altitude pikas. Mol Biol Evol. 2015;32:287–298. doi: 10.1093/molbev/msu311. This experimental study shows that the phenotypic effects of amino acid mutations are conditional on the sequential order in which they occur during the course of an adaptive walk; some mutations had opposite phenotypic effects depending on the genetic background in which they occurred. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bloom JD, Arnold FH. In the light of directed evolution: pathways of adaptive protein evolution. Proc Natl Acad Sci USA. 2009;106:9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet. 2015;16:379–394. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 62.Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312:97–101. doi: 10.1681/01.asn.0000926836.46869.e5. [DOI] [PubMed] [Google Scholar]
- 63.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bridgham JT, Ortlund EA, Thornton JW. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci USA. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Natarajan C, et al. Epistasis among adaptive mutations in deer mouse hemoglobin. Science. 2013;340:1324–1327. doi: 10.1126/science.1236862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reetz MT. The importance of additive and non-additive mutational effects in protein engineering. Angew Chem Int Ed. 2013;52:2658–2666. doi: 10.1002/anie.201207842. [DOI] [PubMed] [Google Scholar]
- 68.Harms MJ, Thornton JW. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature. 2014;512:203–207. doi: 10.1038/nature13410. This innovative study integrates a directed evolution approach with reconstructive inference to demonstrate that rare permissive mutations may represent an important source of contingency in the evolution of novel protein functions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yokoyama S, et al. Epistatic adaptive evolution of human color vision. PloS Genet. 2014;10:e1004884. doi: 10.1371/journal.pgen.1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bank C, Hietpas RT, Jensen JD, Bolon DNA. A systematic survey of an intragenic epistatic landscape. Mol Biol Evol. 2015;32:229–238. doi: 10.1093/molbev/msu301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaltenbach M, Jackson CJ, Campbell EC, Hollfelder F, Tokuriki N. Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. eLife. 2015;4:e06492. doi: 10.7554/eLife.06492. This study dissects the mechanistic basis of intramolecular epistatic interactions in an experimentally evolved enzyme, and demonstrates how such interactions can prevent site-specific mutational reversions to ancestral amino acids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lunzer M, Milter SP, Felsheim R, Dean AM. The biochemical architecture of an ancient adaptive landscape. Science. 2005;310:499–501. doi: 10.1126/science.1115649. [DOI] [PubMed] [Google Scholar]
- 73.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 74.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. Empirical fitness landscapes reveal accessible evolutionary paths. Nature. 2007;445:383–386. doi: 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- 75.da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics. 2010;185:293–303. doi: 10.1534/genetics.109.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lunzer M, Golding GB, Dean AM. Pervasive cryptic epistasis in molecular evolution. Plos Genet. 2010;6:e1001162. doi: 10.1371/journal.pgen.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kvitek DJ, Sherlock G. Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet. 2011;7:e1002056. doi: 10.1371/journal.pgen.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rokyta DR, et al. Epistasis between beneficial mutations and the phenotype-to-fitness map for a ssDNA virus. Plos Genet. 2011;7:e1002075. doi: 10.1371/journal.pgen.1002075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salverda MLM, et al. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 2011;7:e1001321. doi: 10.1371/journal.pgen.1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schenk MF, Szendro IG, Salverda MLM, Krug J, de Visser JAGM. Patterns of epistasis between beneficial mutations in an antibiotic resistance gene. Mol Biol Evol. 2013;30:1779–1787. doi: 10.1093/molbev/mst096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parera M, Angel Martinez M. Strong epistatic interactions within a single protein. Mol Biol Evol. 2014;31:1546–1553. doi: 10.1093/molbev/msu113. [DOI] [PubMed] [Google Scholar]
- 82.Weinreich DM, Lan Y, Wylie CS, Heckendorn RB. Should evolutionary geneticists worry about higher-order epistasis? Curr Opin Genet Dev. 2013;23:700–707. doi: 10.1016/j.gde.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinreich DM, Watson RA, Chao L. Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–1174. [PubMed] [Google Scholar]
- 84.Rosenblum EB, Parent CE, Brandt EE. The molecular basis of phenotypic convergence. Annu Rev Ecol Evol Syst. 2014;45:203–226. [Google Scholar]
- 85.Pollock DD, Thiltgen G, Goldstein RA. Amino acid coevolution induces an evolutionary Stokes shift. Proc Natl Acad Sci USA. 2012;109:E1352–E1359. doi: 10.1073/pnas.1120084109. Similar to findings reported in reference 87, this computational study demonstrates that individual amino acid substitutions at a given site can alter the amino acid propensities of other sites in the same protein; consequently, once a given substitution has occurred, the protein will tend to equilibrate to the newly altered structural context via substitutions at other sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bazykin GA. Changing preferences: deformation of single position amino acid fitness landscapes and evolution of proteins. Biol Lett. 2015;11:20150315. doi: 10.1098/rsbl.2015.0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shah P, McCandlish DM, Plotkin JB. Contingency and entrenchment in protein evolution under purifying selection. Proc Natl Acad Sci USA. 2015;112:E3226–E3235. doi: 10.1073/pnas.1412933112. This study demonstrates that the set of acceptable amino acid substitutions at a given site is highly contingent on prior substitutions and — likewise — once a substitution has occurred at a site, mutational reversions to the ancestral state become increasingly deleterious owing to changes in structural context caused by substitutions at other sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rogozin IB, Thomson K, Csueroes M, Carmel L, Koonin EV. Homoplasy in genome-wide analysis of rare amino acid replacements: the molecular-evolutionary basis for Vavilov’s law of homologous series. Biol Direct. 2008;3:7. doi: 10.1186/1745-6150-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Povolotskaya IS, Kondrashov FA. Sequence space and the ongoing expansion of the protein universe. Nature. 2010;465:922–926. doi: 10.1038/nature09105. [DOI] [PubMed] [Google Scholar]
- 90.Naumenko SA, Kondrashov AS, Bazykin GA. Fitness conferred by replaced amino acids declines with time. Biol Lett. 2012;8:825–828. doi: 10.1098/rsbl.2012.0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soylemez O, Kondrashov FA. Estimating the rate of irreversibility in protein evolution. Genome Biol Evol. 2012;4:1213–1222. doi: 10.1093/gbe/evs096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Poon A, Chao L. The rate of compensatory mutation in the DNA bacteriophage phi X174. Genetics. 2005;170:989–999. doi: 10.1534/genetics.104.039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poon AFY, Chao L. Functional origins of fitness effect-sizes of compensatory mutations in the DNA bacteriophage phi X174. Evolution. 2006;60:2032–2043. [PubMed] [Google Scholar]
- 94.Bloom JD, et al. Thermodynamic prediction of protein neutrality. Proc Natl Acad Sci USA. 2005;102:606–611. doi: 10.1073/pnas.0406744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bloom JD, Romero PA, Lu Z, Arnold FH. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci USA. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. This influential study documents cases where pathogenic amino acid variants in human proteins are present as wild-type residues in the orthologues of other mammals, suggesting that the fitness effects of mutations are highly dependent on the genetic background in which they occur. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gao LZ, Zhang JZ. Why are some human disease-associated mutations fixed in mice? Trends Genet. 2003;19:678–681. doi: 10.1016/j.tig.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 99.Kulathinal RJ, Bettencourt BR, Hartl DL. Compensated deleterious mutations in insect genomes. Science. 2004;306:1553–1554. doi: 10.1126/science.1100522. [DOI] [PubMed] [Google Scholar]
- 100.Azevedo L, Suriano G, van Asch B, Harding RM, Amorim A. Epistatic interactions: how strong in disease and evolution? Trends Genet. 2006;22:581–585. doi: 10.1016/j.tig.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of compensated mutations in terms of structural and physico-chemical properties. J Mol Biol. 2007;365:249–256. doi: 10.1016/j.jmb.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 102.Baresic A, Hopcroft LEM, Rogers HH, Hurst JM, Martin ACR. Compensated pathogenic deviations: analysis of structural effects. J Mol Biol. 2010;396:19–30. doi: 10.1016/j.jmb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 103.Ivankov DN, Finkelstein AV, Kondrashov FA. A structural perspective of compensatory evolution. Curr Opin Struct Biol. 2014;26:104–112. doi: 10.1016/j.sbi.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu J, Zhang J. Why human disease-associated residues appear as the wild-type in other species: genome-scale structural evidence for the compensation hypothesis. Mol Biol Evol. 2014;31:1787–1792. doi: 10.1093/molbev/msu130. Results of this bioinformatic study indicate that conditionally deleterious amino acid mutations are often compensated by second-site mutations in close proximity in the same protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jordan DM, et al. Identification of cis-suppression of human disease mutations by comparative genomics. Nature. 2015;524:225–229. doi: 10.1038/nature14497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang JZ. Parallel adaptive origins of digestive RNases in Asian and African leaf monkeys. Nat Genet. 2006;38:819–823. doi: 10.1038/ng1812. This study integrates statistical analyses of sequence divergence with manipulative in vitro experiments to make inferences about the adaptive significance of parallel amino acid substitutions in the RNase isozymes of leaf-eating monkeys. [DOI] [PubMed] [Google Scholar]
- 107.Weber RE, Fago A, Malte H, Storz JF, Gorr TA. Lack of conventional oxygen-linked proton and anion binding sites does not impair allosteric regulation of oxygen binding in dwarf caiman hemoglobin. Am J Physiol Regul Integ Comp Physiol. 2013;305:R300–R312. doi: 10.1152/ajpregu.00014.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Natarajan C, et al. Convergent evolution of hemoglobin function in high-altitude Andean waterfowl involves limited parallelism at the molecular sequence level. PloS Genet. 2015;11:e1005681. doi: 10.1371/journal.pgen.1005681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Revsbech IG, et al. Hemoglobin function and allosteric regulation in semi-fossorial rodents (family Sciuridae) with different altitudinal ranges. J Exp Biol. 2013;216:4264–4271. doi: 10.1242/jeb.091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin G, Elena SF, Lenormand T. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nat Genet. 2007;39:555–560. doi: 10.1038/ng1998. [DOI] [PubMed] [Google Scholar]
- 111.Pearson VM, Miller CR, Rokyta DR. The consistency of beneficial fitness effects of mutations across diverse genetic backgrounds. PLoS ONE. 2012;7:e43864. doi: 10.1371/journal.pone.0043864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bazykin GA, et al. Extensive parallelism in protein evolution. Biol Direct. 2007;2:20. doi: 10.1186/1745-6150-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rokas A, Carroll SB. Frequent and widespread parallel evolution of protein sequences. Mol Biol Evol. 2008;25:1943–1953. doi: 10.1093/molbev/msn143. Results of this comparative sequence analysis suggest that a large fraction of parallel amino acid substitutions may be attributable to purifying selection that constrains substitutions to a restricted set of physicochemically similar amino acids. [DOI] [PubMed] [Google Scholar]
- 114.Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490:535–538. doi: 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- 115.Usmanova DR, Ferretti L, Povolotskaya IS, Vlasov PK, Kondrashov FA. A model of substitution trajectories in sequence space and long-term protein evolution. Mol Biol Evol. 2015;32:542–554. doi: 10.1093/molbev/msu318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zou Z, Zhang J. Are convergent and parallel amino acid substitutions in protein evolution more prevalent than neutral expectations? Mol Biol Evol. 2015;32:2085–2096. doi: 10.1093/molbev/msv091. This comparative genomic analysis demonstrates that inferred levels of molecular convergence and parallelism in eukaryotic proteins are largely consistent with neutral expectations, provided that among-site variation in functional constraint is taken into account. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang J. Parallel functional changes in the digestive RNases of ruminants and colobines by divergent amino acid substitutions. Mol Biol Evol. 2003;20:1310–1317. doi: 10.1093/molbev/msg143. [DOI] [PubMed] [Google Scholar]
- 118.McCracken KG, et al. Parallel evolution in the major haemoglobin genes of eight species of Andean waterfowl. Mol Ecol. 2009;18:3992–4005. doi: 10.1111/j.1365-294X.2009.04352.x. [DOI] [PubMed] [Google Scholar]
- 119.Parker J, et al. Genome-wide signatures of convergent evolution in echolocating mammals. Nature. 2013;502:228–231. doi: 10.1038/nature12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thomas GWC, Hahn MW. Determining the null model for detecting adaptive convergence from genomic data: a case study using echolocating mammals. Mol Biol Evol. 2015;32:1232–1236. doi: 10.1093/molbev/msv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zou Z, Zhang J. No genome-wide protein sequence convergence for echolocation. Mol Biol Evol. 2015;32:1237–1241. doi: 10.1093/molbev/msv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mendes FK, Hahn MW. Gene tree discordance causes apparent substitution rate variation. bioRxiv. 2016 doi: 10.1093/sysbio/syw018. http://dx.doi.org/10.1101/029371. [DOI] [PubMed]
- 123.Orr HA. The genetic theory of adaptation: a brief history. Nat Rev Genet. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- 124.Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- 125.Maddison WP. Gene trees in species trees. Syst Biol. 1997;46:523–536. [Google Scholar]
- 126.Orr HA. The population genetics of adaptation: The distribution of factors fixed during adaptive evolution. Evolution. 1998;52:935–949. doi: 10.1111/j.1558-5646.1998.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 127.Kimura M. Number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 1969;61:893–903. doi: 10.1093/genetics/61.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]