

Abstract
Cleft lip is one of the most common human birth defects, yet our understanding of the mechanisms that regulate lip morphogenesis is limited. Here, we show in mice that sonic hedgehog (Shh)-induced proliferation of cranial neural crest cell (cNCC) mesenchyme is required for upper lip closure. Gene expression profiling revealed a subset of Forkhead box (Fox) genes that are regulated by Shh signaling during lip morphogenesis. During cleft pathogenesis, reduced proliferation in the medial nasal process mesenchyme paralleled the domain of reduced Foxf2 and Gli1 expression. SHH ligand induction of Foxf2 expression was dependent upon Shh pathway effectors in cNCCs, while a functional GLI-binding site was identified downstream of Foxf2. Consistent with the cellular mechanism demonstrated for cleft lip pathogenesis, we found that either SHH ligand addition or FOXF2 overexpression is sufficient to induce cNCC proliferation. Finally, analysis of a large multi-ethnic human population with cleft lip identified clusters of single-nucleotide polymorphisms in FOXF2. These data suggest that direct targeting of Foxf2 by Shh signaling drives cNCC mesenchyme proliferation during upper lip morphogenesis, and that disruption of this sequence results in cleft lip.
KEY WORDS: Sonic hedgehog, Orofacial morphogenesis, Cleft lip, Forkhead box, Cranial neural crest cell, Medial nasal process, Human, Mouse
Summary: The sonic hedgehog pathway controls expression of Foxf2 in mouse cranial neural crest mesenchyme, promoting proliferation and upper lip closure. Human FOXF2 polymorphisms are associated with cleft lip.
INTRODUCTION
Morphogenesis of the upper lip and palate requires orchestrated growth and fusion of embryonic facial growth centers (Jiang et al., 2006; Lan et al., 2015). Epithelial-mesenchymal cross-talk that drives cellular proliferation and tissue outgrowth is required for closure of both the upper lip and palate. Disruption of these processes results in orofacial clefts (OFCs), which are among the most common human birth defects (Rahimov et al., 2012; Murthy and Bhaskar, 2009). OFCs are genetically complex traits, but racial disparity in prevalence, heightened recurrence rates among siblings, and 50% concordance rates in monozygotic twins indicate an important hereditary component (Lidral et al., 2008). Genome-wide studies have identified multiple associated loci, but our understanding of the genetic underpinnings of OFCs is incomplete, particularly for cases considered non-syndromic (Lidral et al., 2008; Leslie et al., 2013).
Animal models have proven to be powerful for elucidating the mechanisms that drive human birth defects, but the vast majority of mouse OFC models exhibit clefts of the secondary palate with an intact upper lip, whereas most human OFC cases involve clefts of the upper lip (Watkins et al., 2014; Gritli-Linde, 2008, 2012). Consequently, our understanding of the molecular and cellular mechanisms of upper lip development is minimal relative to that of secondary palate morphogenesis.
Experiments in mouse and chick have shown that the sonic hedgehog (Shh) signaling pathway regulates morphogenesis of the tissues that form the midface. Specifically, secretion of SHH ligand from the ectoderm activates pathway activity in the adjacent cranial neural crest-derived mesenchyme (Marcucio et al., 2005; Hu and Marcucio, 2009; Hu et al., 2015; Xavier et al., 2016). Demonstrating clinical relevance, we have shown that transient in utero exposure to the Shh pathway inhibitor cyclopamine causes lateral clefts of the lip that typically extend into the primary and secondary palate, and that this model recapitulates human non-syndromic cleft lip with or without cleft palate (CL/P) (Lipinski et al., 2008b, 2010, 2014). Here, we utilized this pathway-specific mouse model of cleft lip to identify and investigate the mechanism of action of Shh target genes that mediate normal and abnormal upper lip morphogenesis. Along with providing new insight into the molecular and cellular mechanisms that regulate upper lip development, these studies identify novel candidate human CL/P genes that might contribute to this common human birth defect.
RESULTS
Transient Shh signaling inhibition attenuates outgrowth of the medial nasal process
We first investigated the morphogenesis of cleft lip resulting from transient Shh signaling inhibition by examining mouse embryos exposed in utero to cyclopamine or vehicle alone (Fig. 1). Although morphologically indistinguishable from vehicle-exposed embryos at gestational day (GD) 9.25, the medial nasal processes (MNPs) of cyclopamine-exposed embryos appeared deficient by GD10.25 (Fig. 1A,B,E,F). By GD11.0, the lower aspect of the MNP was markedly deficient, preventing contact and subsequent fusion with the maxillary process (Fig. 1C,G). By GD14.0, the majority of cyclopamine-exposed embryos exhibited unilateral or bilateral cleft lip in the absence of other gross craniofacial malformations (Fig. 1D,H).
Fig. 1.

Transient Shh signaling inhibition causes cleft lip due to a deficiency of the medial nasal process. Representative mouse embryos exposed to vehicle (A-D) or cyclopamine (E-H) are shown at the indicated stages of development (n=3 embryos from two independent litters per stage per treatment). The medial nasal process (MNP) is outlined. Note the deficient frontonasal prominence (FNP)-derived MNP of a cyclopamine-exposed embryo at GD10.25 that precludes fusion with the maxillary process (MxP) at GD11.0, leading to unilateral (shown in H) or bilateral cleft lip visible at GD14.0. LNP, lateral nasal process. Scale bars: 300 μm.
Identification of Shh-regulated genes in upper lip morphogenesis
To define the temporal dynamics of transient Shh signaling inhibition during CL/P pathogenesis, we assessed expression of the conserved Shh target gene Gli1 by in situ hybridization (ISH). Whereas no apparent difference was observed at GD9.0, Gli1 expression was markedly reduced in the ventral frontonasal prominence (FNP) of cyclopamine-exposed embryos by GD9.25 (Fig. 2A,B). Both the medial and lateral nasal processes develop from swellings of the FNP. To identify targets of Shh signaling during the initial pathogenesis of CL/P, FNP tissue was microdissected from stage-matched vehicle- and cyclopamine-exposed GD9.25 embryos (Fig. 2B, outlined) and subjected to cDNA microarray analysis. Of 210 significantly dysregulated genes that were identified, 120 were increased and 90 were decreased by cyclopamine exposure. Networking and gene annotation enrichment analyses confirmed Shh pathway modulation and revealed significant enrichment of the Forkhead box (Fox) family of transcription factors, with multiple members appearing dysregulated during the initial pathogenesis of cleft lip (Fig. 2C).
Fig. 2.
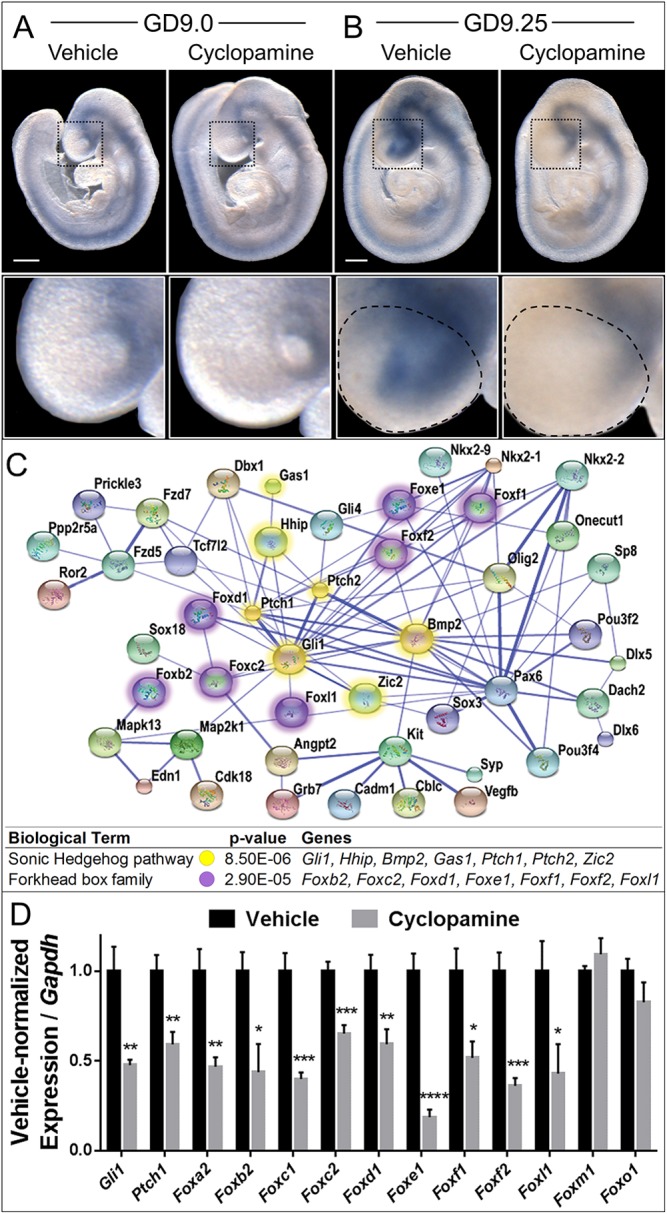
Shh regulates Fox genes in cleft lip pathogenesis. (A,B) Stage-matched GD9.0 (A) and GD9.25 (B) embryos exposed in utero to vehicle or cyclopamine and subjected to ISH for Gli1. The boxed region of the FNP is magnified beneath. Although Gli1 expression is grossly similar at GD9.0, downregulation of Gli1 is apparent in cyclopamine-exposed embryos by GD9.25. The microdissected FNP region used for the microarray and subsequent RT-PCR validation is outlined on the GD9.25 embryos. Scale bars: 300 μm. (C) A STRING network of select significantly differentially expressed genes identified by microarray. Colored nodes represent significant dysregulated genes; blue edges show functional and predicted interactions between gene products, with the thicker, darker lines representing interactions for which evidence is more abundant. Note the high degree of centrality of Shh signaling pathway genes (yellow) and Forkhead box (Fox) transcription factor genes (purple). P-value indicates significant enrichment for the Shh pathway and Fox family within the dysregulated gene set. (D) RT-PCR validation of the indicated genes in vehicle-exposed or cyclopamine-exposed FNP tissue. Values represent mean±s.e.m. expression of the cyclopamine-exposed group relative to Gapdh, normalized to the vehicle control group (n=6 pooled litters per group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (two-tailed t-test with Holm-Sidak correction).
RT-PCR analysis confirmed significant downregulation of the Shh target genes Gli1 and Ptch1, as well as nine Fox family members: Foxa2, Foxb2, Foxc1, Foxc2, Foxd1, Foxe1, Foxf1, Foxf2 and Foxl1 (Fig. 2D). Two additional Fox members (Foxm1 and Foxo1) expressed in the FNP were not differentially expressed in the microarray or as assessed by RT-PCR. These findings demonstrate that inhibition of Shh signaling results in downregulation of a specific subset of Fox genes during cleft lip pathogenesis.
Shh pathway activity and Foxf2 expression correspond with mesenchymal cell proliferation in the MNP
We next characterized the spatiotemporal expression domains of the identified Fox genes during early upper lip development. Because SHH ligand from the ectoderm signals to adjacent cranial neural crest cell (cNCC)-derived mesenchyme, we focused on Fox genes expressed in the mesenchyme of the FNP and MNP. Section ISH analysis revealed seven Fox genes expressed in the mesenchyme of the ventral FNP: Foxb2, Foxc1, Foxc2, Foxd1, Foxf1, Foxf2 and Foxl1 (Fig. 3). In most cases, expression persisted through GD10 and GD11 in the mesenchyme of the MNP tissue that forms the median aspect of the upper lip. The expression domains of Foxf1 and Foxf2 closely approximated with that of the canonical Shh target gene Gli1. As previously reported, Foxa2 expression was localized to the neuroectoderm, while Foxe1 expression was observed in the surface ectoderm of the facial primordia (data not shown) (Moreno et al., 2009; Ruiz i Altaba et al., 1993; Sasaki and Hogan, 1994).
Fig. 3.

Fox gene expression during upper lip development. (A) Oblique images of untreated GD9.25, GD10 and GD11 embryos illustrate the plane of section (blue trapezoids) in the panels beneath. (B) Representative images are shown for n=3 embryos from at least two independent litters per gene per stage. Gli1 and seven Fox genes are expressed in the mesenchyme of the tissues that form the upper lip, i.e. the FNP at GD9.25 and MNP at GD10 and GD11. (C) Schematic showing tissue compartments of the developing face and brain at GD9.25, GD10 and GD11. FNP, frontonasal prominence; MNP, medial nasal process; SE, surface ectoderm; M, mesenchyme; NE, neuroectoderm. Scale bars: 200 μm.
Single-nucleotide polymorphisms (SNPs) in FOXF2 were recently associated with non-syndromic CL/P in an Asian population (Bu et al., 2015). While Foxf2 has an intrinsic role in secondary palate development, its role in upper lip development and cleft lip pathogenesis had not been described (Wang et al., 2003; Xu et al., 2016; Nik et al., 2016). We therefore further examined the expression of Foxf2 during upper lip morphogenesis in our mouse model. Foxf2 colocalized with both Gli1 and Ptch1 in the mesenchyme of the MNP, where they are expressed in a medial-lateral gradient (Fig. 4A,C,E). Expression of these genes remained downregulated at GD10.25 following cyclopamine exposure (Fig. 4B,D,F).
Fig. 4.
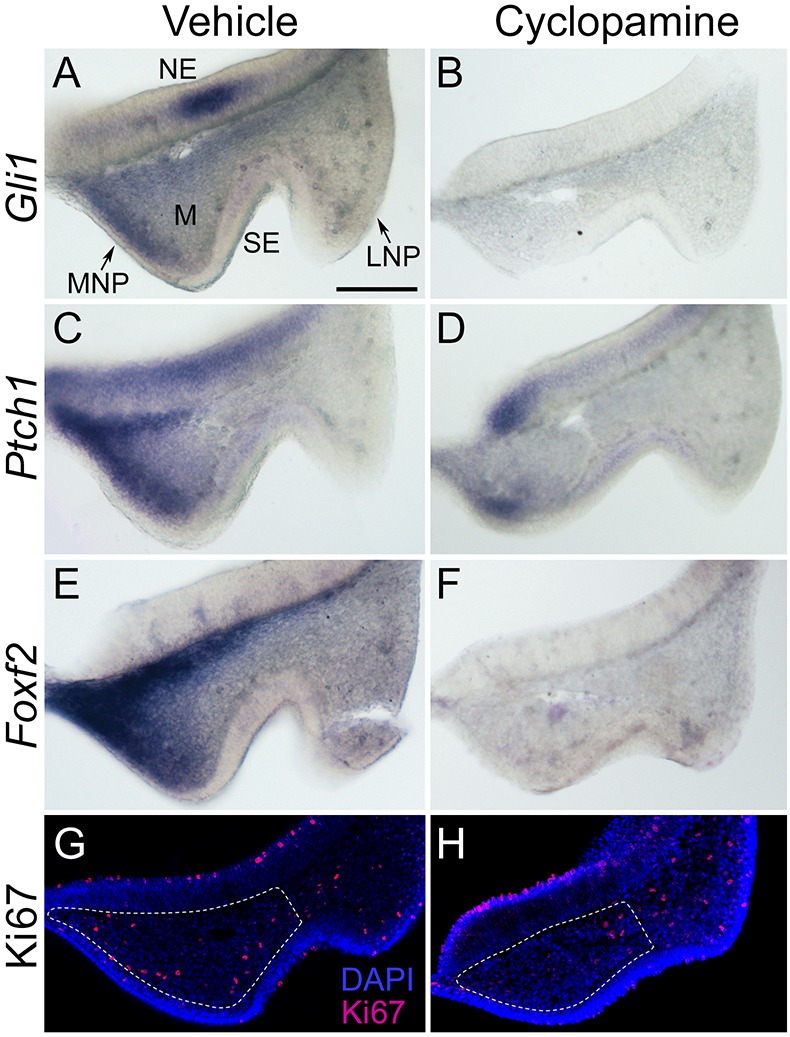
Concomitant downregulation of Gli1, Ptch1 and Foxf2 parallels reduced cell proliferation in the MNP during cleft lip pathogenesis. (A-F) Frontal sections through the MNP and LNP of GD10.25 embryos exposed in utero to vehicle or cyclopamine and subjected to ISH for Gli1 (A,B), Ptch1 (C,D) or Foxf2 (E,F). Gli1 and Ptch1 colocalize with Foxf2 in the MNP, and each is downregulated during cleft lip pathogenesis. (G,H) Immunostaining for Ki67 (magenta) and DAPI staining (blue) in the same plane of section shows that reduced proliferation is most apparent in the domain of Gli1, Ptch1 and Foxf2 expression following cyclopamine exposure. Dashed lines delineate mesenchyme of the MNP. MNP, medial nasal process; LNP, lateral nasal process; NE, neuroectoderm; M, mesenchyme; SE, surface ectoderm. Scale bar: 300 μm.
We next investigated the cellular basis of the MNP deficiency upon cyclopamine exposure by staining GD10.25 embryos for the proliferative marker Ki67. Following cyclopamine exposure, a reduction of proliferating cells was observed in the mesenchymal domain of the medial portion of the MNP that normally has the highest expression of Gli1, Ptch1 and Foxf2 (Fig. 4G,H). Quantification of Ki67-positive cells confirmed a tissue-specific reduction in cellular proliferation affecting the mesenchyme of the MNP but not the adjacent surface ectoderm or the neuroectoderm (Fig. S1). Multipotent cNCCs give rise to the mesenchyme of the MNPs as well as the neighboring lateral nasal processes that form the lateral aspects of the nostrils. However, reduced proliferation of cNCC-derived mesenchyme appeared specific to the MNP, consistent with the observation that cyclopamine exposure attenuates outgrowth of the MNPs without affecting morphogenesis of the lateral nasal processes (Fig. 1).
Foxf2 is a target of canonical Shh signaling in cNCCs
We then examined how Shh signaling regulates Foxf2 in the tissues that form the upper lip. Consistent with paracrine signaling activity and expression data shown in Fig. 4, we found that Shh, which encodes a secreted ligand, is expressed in the surface ectoderm of the MNP, whereas Gli1 and Foxf2 are predominantly expressed in the adjacent mesenchyme (Fig. 5A). To decipher how Shh signaling regulates Foxf2 at the cellular level, we utilized a mouse cNCC line (O9-1) that recapitulates the expression signature [AP-2α (Tfap2a), Twist1, Sox9, Cd44] and differentiation potential of the post-migrational neural crest-derived cranial mesenchyme (Ishii et al., 2012). Shh ligand (SHH) stimulation of cNCCs caused significant upregulation of both Gli1 and Foxf2 expression, which was blocked completely by the addition of vismodegib (Fig. 5B). Both vismodegib and cyclopamine act by binding to and inhibiting the Shh pathway transducing protein smoothened (SMO) (Robarge et al., 2009; Heyne et al., 2015). Because blocking SMO activity prevented induction of Foxf2, we next tested whether SMO overexpression could activate Foxf2 expression. Overexpression of a mutant form of human SMO (SMOM2) that generates constitutive downstream pathway activity (Xie et al., 1998) was sufficient to induce both Gli1 and Foxf2 expression to levels approximating SHH ligand stimulation (Fig. 5C). SMO acts upstream of three GLI proteins that regulate the transcription of canonical Shh target genes. To test whether GLI activity is required for transcriptional regulation of Foxf2, we expressed a dominant-negative form of GLI2, the primary transcriptional activator of Shh target genes (Roessler et al., 2005; Lipinski et al., 2006). In cNCCs expressing dominant-negative GLI2 (dnGLI2), Gli1 and Foxf2 expression was not significantly changed by the addition of SHH ligand (Fig. 5D).
Fig. 5.

Foxf2 is a target of Shh signaling in cranial neural crest cells (cNCCs). (A) Tissue-specific gene expression was determined for isolated GD11 MNP mesenchyme and surface ectoderm. Shh was more highly expressed in the MNP surface ectoderm, whereas Gli1 and Foxf2 were more highly expressed in the MNP mesenchyme (n=6 pooled litters per group). (B) Gene expression was determined for cNCCs cultured with or without SHH ligand and with or without the SMO antagonist vismodegib (Vis). SHH ligand caused an increase in Gli1 and Foxf2 expression, which was blocked by the addition of vismodegib (n=5). (C) Expression of Gli1 and Foxf2 are increased in cNCCs overexpressing a constitutively active form of human smoothened (SMOM2) relative to GFP only (control) (n=5). Inset shows SMO expression relative to Gapdh in GFP (control) and SMOM2-GFP-overexpressing cells. (D) Expression of Gli1 and Foxf2 is unchanged by SHH ligand treatment in cNCCs overexpressing a dominant-negative form of GLI2 (dnGLI2) (n=5). Inset shows mean±s.e.m. expression of dnGLI2 relative to Gapdh in vehicle-treated GFP-expressing (control) and dnGLI2-expressing cells. For all analyses, mean±s.e.m. expression relative to Gapdh is shown. **P<0.01, ****P<0.001 (two-tailed t-test with Holm-Sidak correction).
The zinc-finger proteins GLI1, GLI2 and GLI3 regulate the transcription of canonical Shh target genes by binding to GLI consensus binding sites in the DNA (Kinzler and Vogelstein, 1990; Yoon et al., 2002). Putative GLI-binding sites conserved between mouse and human were identified within a 20 kb region surrounding Foxf2. A conserved GLI-binding site containing two mismatches was identified 0.8 kb downstream from the Foxf2 coding sequence (Fig. 6B). We used electrophoretic mobility shift assays (EMSAs) to test whether this putative GLI-binding site can directly interact with GLI1 protein. Digoxigenin (DIG)-labeled oligonucleotide probes were synthesized for the identified sequence downstream of Foxf2 and control sequences. Purified GLI1 physically bound the sequence downstream of Foxf2, as well as a known GLI-binding site upstream of Foxl1 (Madison et al., 2009), causing each band to shift (Fig. 6A). The specificity of GLI1 binding to Foxf2 and Foxl1 was confirmed by competing away GLI1 from each DIG-labeled oligonucleotide probe using unlabeled probe. This competition was not observed with non-specific competitor probes with mutated GLI-binding sites. An additional putative GLI-binding site downstream of Foxb2 did not shift in the presence of GLI1, further supporting the specificity of the sequence identified downstream of Foxf2.
Fig. 6.

A novel GLI-binding site at the Foxf2 locus. (A) EMSAs were used to assess putative GLI-binding sites identified in silico. EMSAs of control and three unique putative GLI-binding sites near Foxf2, Foxl1 and Foxb2. Arrows indicate shifted bands marking DIG-labeled oligonucleotide probes bound by recombinant GLI1 protein. Lowest band (no arrow) marks free, unbound probe. An oligonucleotide probe containing a putative GLI-binding site 0.8 kb downstream of Foxf2 was bound by GLI1, causing a band shift. As a positive control, a probe was designed containing a known GLI-binding site near Foxl1, which was also bound by GLI1, causing a band shift. A probe containing a putative GLI-binding site near Foxb2 was not bound by GLI1. Consolidated gel images are shown. (B) A scale-drawn schematic of the mouse Foxf2 locus. Black boxes denote exons and a white box denotes the intron. The open arrowhead indicates the location of the novel GLI1 binding site 0.8 kb downstream of the Foxf2 coding sequence.
Shh-Foxf2 signaling promotes cNCC proliferation
Our in vivo data demonstrate that cleft lip resulting from transient Shh pathway inhibition follows concomitant decreases in cell proliferation and Foxf2 expression in the cNCC-derived MNP mesenchyme. We therefore tested whether Shh-Foxf2 signaling directly regulates cNCC proliferation. SHH ligand stimulation of wild-type cNCCs resulted in a 36% increase in cell number compared with the control. Consistent with the transcriptional response of Foxf2 to SHH ligand stimulation, the increase in cell proliferation was blocked by addition of the SMO antagonist vismodegib (Fig. 7A). To test the specific role of Foxf2 in regulating the proliferative response of cNCCs, we expressed either full-length FOXF2 or a truncated form that acts as a dominant negative (Ormestad et al., 2006) in wild-type cNCCs. Transient overexpression of full-length FOXF2 (pEVRF0-FOXF2) caused a 33% increase in cell number relative to cells transfected with empty pEVRF0 vector (Fig. 7B). Conversely, stable overexpression of dominant-negative FOXF2 dampened the proliferative response of cNCCs stimulated with SHH ligand as compared with control cells overexpressing empty pLenti vector only (Fig. 7C).
Fig. 7.

Foxf2 promotes cNCC proliferation. (A) cNCCs were cultured with or without SHH ligand and with or without vismodegib (Vis). SHH ligand caused cNCCs to proliferate, which was blocked by addition of vismodegib (n=4). (B) Transient overexpression of full-length FOXF2 resulted in a significant increase in cell number relative to control (transfected with empty pEVRF0) (n=4). Inset shows FOXF2 expression relative to Gapdh in pEVRF0 (control) and FOXF2-overexpressing cells (n=4). (C) In cNCCs with stable overexpression of an empty pLenti vector, SHH ligand stimulation caused a significant increase in cell number. In cNCCs with stable overexpression of a dominant-negative form of FOXF2 (dnFOXF2), the change in cell number following SHH ligand stimulation was not statistically significant (P=0.09, n=4). Inset shows dnFOXF2 expression relative to Gapdh in pLenti (control) and dnFOXF2-overexpressing cells (n=4). Mean±s.e.m. cell count is shown. *P<0.05 (two-tailed t-test).
FOXF2 SNPs are associated with human cleft lip
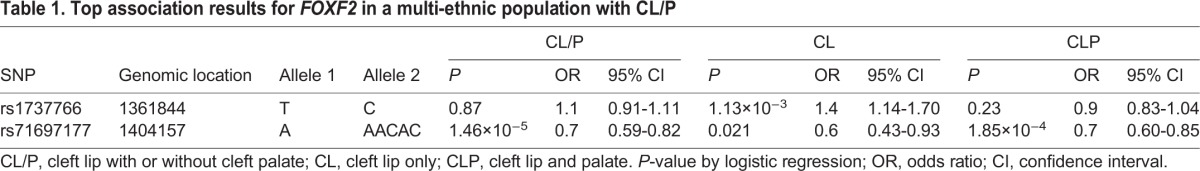
Mutations in FOXF2 have recently been linked to CL/P in humans, but existing studies are for a single Asian population (Bu et al., 2015). To assess the broader relevance of this gene in human clefting, we examined the FOXF2 locus within a large, multi-ethnic population of individuals with non-syndromic OFCs. We selected common SNPs within FOXF2 and a 30 kb flanking region. A cluster of SNPs located downstream of FOXF2 were associated with CL/P (lead SNP rs71697177, P=1.47×10−5) (Fig. S2). We then stratified the CL/P sample into those with a cleft of the upper lip only (CL) and those with cleft lip and cleft palate (CLP) (Table 1). The downstream peak associated with CL/P appeared more strongly associated with CLP (rs71697177, PCLP=1.85×10−4 and PCL=0.021). In the CL analysis, the most significant P-values were found for several SNPs upstream of FOXF2 (lead SNP rs1737766, P=1.13×10−3), although these were not formally significant after corrections for multiple testing. To account for discordant sample sizes between the CL and CLP analyses, we performed a formal test of the differences in effects of the top SNPs in each analysis. Whereas the rs1737766 association was specific to CL (ORCL=1.39, ORCLP=0.93; P=0.0003), there was no difference in effect for rs71697177 (ORCL=0.64, ORCLP=0.72; P=0.29).
Table 1.
Top association results for FOXF2 in a multi-ethnic population with CL/P
DISCUSSION
Shh signaling is a master regulator of the epithelial-mesenchymal interactions that drive orofacial development, but specific targets of the pathway in upper lip development had not been identified. In this study, we leveraged a mouse model of transient in utero inhibition of the Shh signaling pathway to investigate genetic and cellular drivers of lip morphogenesis and cleft lip pathogenesis. Whole-transcriptome expression profiling identified significant downregulation of canonical Shh pathway members, as well as multiple Fox family members during the initial pathogenesis of cleft lip. The defining feature of Fox transcription factors is a Forkhead, or winged-helix, DNA-binding domain. This family includes 44 genes in the mouse genome, with each having a close human ortholog (Jackson et al., 2010). Fox genes have been identified as targets of Shh signaling in diverse developmental contexts, including the gut and central nervous system, while GLI consensus binding sites have been identified in Foxa2, Foxe1, Foxf1 and Foxl1 (Madison et al., 2009; Eichberger et al., 2004; Sasaki et al., 1997).
Our study provides the first evidence that Shh signaling regulates multiple Fox genes during upper lip development and cleft lip pathogenesis. Transcriptional regulation of Fox genes by Shh signaling in facial morphogenesis was first reported by Jeong et al. (2004), who generated mice with conditional pathway inactivation in Wnt1-expressing cNCCs. However, in this previous study only five Fox family members were investigated, and affected embryos exhibited near-complete facial truncation, precluding examination of upper lip development. Here, we show that nine individual Fox genes are expressed in the ventral FNP and are downregulated during the initial pathogenesis of cleft lip resulting from temporally specific inhibition of the Shh pathway. Seven of these Fox genes exhibit dynamic expression domains in the cNCC-derived mesenchyme of the MNPs that form the median aspect of the upper lip. The expression domain of Foxf2 in particular parallels that of the canonical Shh target gene Gli1.
We show in mouse cNCCs (Ishii et al., 2012) that Shh pathway activation via addition of SHH ligand or overexpression of a constitutively active form of the pathway transducer SMO causes increased Foxf2 expression, and that this induction requires GLI2. In silico screening identified a novel GLI consensus binding site 0.8 kb downstream of Foxf2, and we show that purified GLI1 protein directly binds to this site in vitro. The EMSA utilized here has proven successful in identifying functional GLI-binding sites that regulate now well-recognized GLI targets such as Ccnd2 and Igfbp6 (Yoon et al., 2002; Lipinski et al., 2005; Xu et al., 2009; Lan and Jiang, 2009). Taken together, these data argue that Foxf2 is a direct target of canonical SHH-SMO-GLI signaling in post-migrational cNCCs.
Formation of the upper lip requires orchestrated growth and fusion of the bilaterally paired medial nasal and maxillary processes (Jiang et al., 2006). Therefore, clefts of the lip can arise from several distinct cellular and morphological mechanisms affecting the medial nasal and/or maxillary processes. We found that expression of several genes expressed in the surface ectoderm that are required for fusion of the medial nasal and maxillary processes [Pbx1-3, several Wnt genes, Tp63 (Trp63), Irf6] were not changed during cyclopamine-induced cleft lip (Fig. S3). This suggests that these genes are not regulated by Shh signaling, although it is possible that they act upstream of Shh in the surface ectoderm. Our observations support the alternative mechanism that epithelial SHH ligand induces pathway activity in the adjacent cNCC mesenchyme and that pathway inhibition reduces proliferation of the cNCC-derived mesenchymal cells of the MNP, causing a tissue deficiency that prevents contact with the opposing maxillary processes, resulting in cleft lip. Consistent with this in vivo observation, we show that direct addition of SHH ligand stimulates mouse cNCC proliferation in vitro. Moreover, transient overexpression of FOXF2 is sufficient to induce cNCC proliferation. Overexpression of a dominant-negative form of FOXF2 dampens the proliferative response of cNCCs to SHH ligand, such that the change is not statistically significant. Partial responsiveness might be explained by functional compensation by other Fox members, such as Foxf1, as has been described in other developmental contexts (Xu et al., 2016; Madison et al., 2009). Taken together, these data suggest that post-migrational cNCC proliferation in the MNP is dependent, at least in part, on SHH-mediated induction of Foxf2.
The studies described herein are the first to link Foxf2 to cleft lip pathogenesis in an animal model. Interestingly, Foxf2 null mice do not exhibit cleft lip but cleft palate only (CPO) (Wang et al., 2003). Functional compensation by other Fox genes might explain this dichotomy. At the same time, relative to humans, the mouse appears resistant to cleft lip, which may mask important developmental regulators of upper lip morphogenesis, such as Foxf2. In fact, the majority of mouse OFC models exhibit CPO, even those involving disruption of genes associated with both CPO and CL/P in humans, such as FOXE1 and IRF6 (Gritli-Linde, 2008; Juriloff and Harris, 2008; Leslie et al., 2016b; Ludwig et al., 2014). Although these OFC subtypes are traditionally considered distinct, development of the upper lip and secondary palate each involves outgrowth and fusion of facial growth centers that are largely composed of cNCC-derived mesenchyme (Jiang et al., 2006; Lan et al., 2015).
Comparison of our findings in upper lip development with recent evidence that Foxf2 plays an intrinsic role in secondary palate development suggests a divergence in the genetic regulation of these structures. Xu et al. (2016) found that SHH ligand from the palatal epithelium drives Foxf2 expression in the adjacent mesenchyme, which then suppresses mesenchymal Fgf18 expression. This results in loss of Shh expression in the palatal shelf epithelium and in CPO (Xu et al., 2016). Suggesting a likely parallel mechanism, Nik et al. (2016) found that Foxf2 expression regulates TGFβ signaling activity in the cNCC-derived mesenchyme of the secondary palate. Disruption of TGFβ signaling in these cells has been shown to cause defective proliferation, resulting in cleft palate (Iwata et al., 2012). However, these mechanisms do not appear to be conserved in upper lip morphogenesis. Neither Fgf18 nor markers of TGFβ signaling (Tgfbr3 and Itgb1) were altered during the initial pathogenesis of cyclopamine-induced cleft lip or following SHH ligand stimulation or downstream pathway activation in cNCCs in vitro (Fig. S4). Moreover, the proliferative response of cNCCs to SHH ligand or FOXF2 overexpression occurs without parallel changes in gene expression of Tfgbr3 or Itgb1. This suggests that although regulation of Foxf2 by Shh signaling is conserved between upper lip and secondary palate development, the downstream targets of FOXF2 that mediate its biological effects are context dependent. Identifying the unique transcriptional targets of FOXF2 in upper lip development is an important area of future investigation that could reveal additional mechanistic insights into cleft lip pathogenesis.
Increasing evidence from genetic association studies suggests that CL and CLP may have distinct risk factors in humans. This includes a locus near SPRY2 that is more strongly associated with CLP, a locus near GREM1 that appears to be associated with cleft lip and cleft soft palate, and the IRF6 enhancer variant that is more strongly associated with CL than CLP (Ludwig et al., 2012; Jia et al., 2015). Our results suggest that CL/P-associated SNPs downstream of FOXF2 are associated with both CL and CLP. However, we also identified evidence of association for several SNPs upstream of FOXF2 that appeared specific to CL. Although a biological mechanism behind stronger associations with specific cleft types remains generally elusive, our data suggest that the downstream targets of FOXF2 that mediate its biological effects are context dependent. The SNPs identified in our large multi-ethnic population have not been previously reported. These new associations, along with those previously reported in an Asian population (Bu et al., 2015), strongly link FOXF2 to CL/P in humans.
We demonstrate an integrated in vivo-in vitro-in silico approach to uncover and define the identity and mechanism of action of genetic regulators of upper lip development with relevance to human birth defects. In addition to Foxf2, the utility of this approach is underscored by our identification of Foxe1 as one of the genes most strongly downregulated by cyclopamine exposure. FOXE1 was recently identified as a major OFC locus linked to both CL/P and CPO in human populations (Moreno et al., 2009; Ludwig et al., 2014). In addition to Foxf2 and Foxe1, our study identifies seven other Fox genes that are downregulated during the initial pathogenesis of cleft lip. Inactivation of two of these genes (Foxc2 and Foxf1) is known to cause CPO in mouse, but examination of these genes in upper lip development has not been presented. For most of the other Fox genes that we have identified as Shh targets during cleft lip pathogenesis, knockout models have not been developed, or analysis of upper lip morphogenesis is precluded by early embryonic lethality. Further investigation of these genes, along with Foxf2, will improve our understanding of the mechanisms that regulate upper lip development and illuminate the complex etiology of CL/P in humans.
MATERIALS AND METHODS
Animals
This study was conducted in strict accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University of Wisconsin School of Veterinary Medicine Institutional Animal Care and Use Committee (protocol number 13–081.0). Eight-week-old wild-type C57BL/6J mice (Mus musculus, Linnaeus) were purchased from The Jackson Laboratory and housed under specific pathogen-free conditions in disposable, ventilated cages (Innovive, San Diego, CA, USA). Rooms were maintained at 22±2°C and 30-70% humidity on a 12-h light, 12-h dark cycle. Mice were fed 2920x Irradiated Harlan Teklad Global Soy Protein-Free Extruded Rodent Diet until day of plug, when dams received 2919 Irradiated Teklad Global 19% Protein Extruded Rodent Diet.
Timed pregnancies
One or two nulliparious female mice were placed with a single male for 1-2 h and then examined for copulation plugs. The beginning of the mating period was designated as GD0, and pregnancy was confirmed by assessing weight gain between GD7 and GD10, as previously described (Heyne et al., 2015). Pregnant dams were euthanized at the indicated gestational day ±1 h. Somite number was used as an indicator of embryo gestational stage. For microarray and RT-PCR validation, all analyzed embryos had 20-24 somite pairs, a subset of GD9.25 (Theiler, 1989). For ISH, embryos compared between treatment groups were stage matched ±1 somite pair.
Cyclopamine administration
Pregnant dams were administered 120 mg per kg per day cyclopamine (LC Laboratories, CAS #4449-51-8) or vehicle alone from GD8.25 to ∼GD9.375 using ALZET 2001D micro-osmotic pumps (Cupertino, CA, USA). This subcutaneous infusion exposure model yields steady-state serum concentrations within a few hours after pump implantation (GD8.25) that persist until dispensation is complete (∼GD9.375) (Lipinski et al., 2008b, 2010).
RNA extraction and purification
For in vivo assays, microdissected FNP tissues from six cyclopamine-exposed and six vehicle-exposed control litters were pooled by litter. RNA was isolated using GE Illustra RNAspin kits (GE Healthcare). This RNA was used for both microarray and RT-PCR validation assays. cDNA was synthesized from 1 µg total RNA using Promega GoScript reverse transcription reaction kits. Separation of mesenchyme and surface ectoderm of GD11 embryos was accomplished as previously described (Li and Williams, 2013). For separated tissues and in vitro cell cultures, cDNA was synthesized from 500 ng total RNA.
Microarray sample preparation, hybridization and scanning
Affymetrix Mouse Gene 2.0 ST GeneChip arrays were used for transcriptome analysis. Labeled cDNA was generated from 100-500 ng total RNA with Ambion Whole-Transcript Expression and Affymetrix GeneChip Whole-Transcript Terminal Labeling Reagents, according to manufacturers' specifications. GeneChips were scanned at 570 nm on an Affymetrix GC3000 G7 scanner, and signals were determined using Affymetrix GCOS and Affymetrix Expression Console software.
Microarray computational analysis
Significant differential gene expression was determined using the Affymetrix Transcriptome Analysis Console (TAC). Gene networking analysis was performed using STRING (Franceschini et al., 2013). Gene annotation enrichment analysis for pathways and biological processes was conducted using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009a,b).
Quantitative real-time RT-PCR
Singleplex RT-PCR was conducted as previously described (Heyne et al., 2016). Primer sequences are listed in Table S1. Gapdh was used as the housekeeping gene for expression normalization, and analyses were conducted with the 2−ΔΔCt method.
In situ hybridization (ISH)
ISH analysis was performed as previously described (Heyne et al., 2016), using an established high-throughput technique (Abler et al., 2011). Embryos were processed whole or embedded in 4% agarose gel and cut in 50 μm sections using a vibrating microtome. Embryos were imaged using a MicroPublisher 5.0 camera connected to an Olympus SZX-10 stereomicroscope for whole mount or a Nikon Eclipse E600 microscope for sections. ISH riboprobe primers are listed in Table S2.
Immunohistochemistry
GD10.25 embryos were fixed in 4% paraformaldehyde in PBS overnight prior to graded dehydration into methanol and storage at −20°C. Embryos were subsequently rehydrated into PBS and 100 μm frontal sections were obtained using a vibrating microtome. Sections were blocked in 5% normalized goat serum (NGS) and 1% DMSO in PBSTx (PBS with 0.5% Triton X-100) for 1 h, incubated in monoclonal rabbit anti-Ki67 (1:250; Cell Signaling, #9027) (Gerdes et al., 1983), washed, and transferred to monoclonal goat anti-rabbit Alexa Fluor 594 secondary antibody (1:1000; Thermo Scientific, #R37117) in PBSTx containing 5% NGS for 1 h at room temperature. Finally, sections were incubated with DAPI (1:1000; Thermo Scientific) in PBS for 6 min, and imaged using a Leica SP8 confocal microscope. For Ki67 quantification, a blinded-rater processed each image to reduce background and optimize signal accuracy. Ki67-positive cells/area in each z-image were determined using the ImageJ (NIH) cell counter tool.
GLI transcription factor-binding site analysis
Evolutionarily conserved GLI-binding sites between mouse (mm10) and human (Hg19) were identified in silico using the VISTApoint tool (Loots et al., 2002). 10 kb regions upstream and downstream of each gene coding sequence were analyzed for GLI, GLI1, GLI2 and GLI3 consensus binding sites. Putative GLI-binding sites with fewer than three mismatches from the consensus GLI-binding sequence (TGGGTGGTC) were accepted for further analysis. GLI-binding sites were confirmed by EMSA using a DIG (digoxigenin) Gel Shift Kit (Roche), according to the manufacturer's specifications. Primers used for probe synthesis are listed in Table S3. Purified recombinant GLI1-protein or PinPoint control protein (Promega) was used for these assays. Samples were separated by electrophoresis on a 5% TBE gel (Bio-Rad), transferred onto a Zeta Probe GT membrane (Bio-Rad), and UV cross-linked. Probes were targeted using an anti-DIG, alkaline phosphatase-conjugated antibody (Roche). Specific protein-DNA complexes were visualized using ready-to-use CSPD chemiluminescent substrate (Roche).
In vitro cell culture
Immortalized O9-1 mouse cNCCs were cultured as described (Ishii et al., 2012). Cells were authenticated and tested for contamination. For treatment, O9-1 cells were plated at 5×105 cells/ml (0.4 ml per well in a 24-well plate) and allowed to attach in complete 15% FBS media for 24 h before media were replaced with DMEM containing 1% FBS with or without SHH-N ligand (R&D Systems) at 0.4 μg/ml and with or without 100 nM vismodegib (LC Laboratories, CAS No 879085-55-9) dissolved in DMSO.
Stable transfection experiments
O9-1 cells overexpressing GFP, SMOM2-GFP and dnGLI2-RFP (GLI2-mutB) were generated as described (Lipinski et al., 2008a). A pIRES shuttle vector carrying coding sequences for GFP, SMOM2-GFP and GLI2mutB-RFP was used to retrovirally infect wild-type O9-1 cells. Cells were plated at subconfluence in DMEM with 10% FBS. Cells were incubated with viral-conditioned medium at 37°C for 6 h. Following a 72 h propagation period, appropriate GFP+ or RFP+ populations were isolated using a BD FACSAria III fluorescence-activated cell sorter. Stably transfected cells were plated at 5×105 cells/ml (0.4 ml per well in a 24-well plate). After 24 h, cells were treated with DMEM containing 1% FBS with or without SHH-N ligand for 48 h. Cell lysates were collected for RNA extraction. Stable dnFOXF2-overexpressing O9-1 cells were created using pLenti CMV Blast empty vector (w263-1; Addgene plasmid #17486), provided by Dr Eric Campeau (Campeau et al., 2009). Dominant-negative FOXF2 (pGFP-C1-dnFOXF2) was provided by Dr Peter Carlsson (Hellqvist et al., 1996, 1998). The dnFOXF2 fragment was removed from the pGFP-C1 backbone with BamHI and XbaI sites and then inserted using the same sites into the pLenti CMV Blast empty transfer plasmid. A negative control cell line was generated in parallel by infecting with the empty pLenti CMV Blast vector. Infected cells were selected by blasticidin S. Cells were plated at 2×105 cells per well of a 24-well plate in duplicate. After 24 h, cells were treated with DMEM containing 1% FBS with or without SHH-N ligand for 48 h. Cell lysates were collected for RNA extraction or counted with a hemocytometer.
Transient transfection experiments
Full-length FOXF2 (pEVRF0-FOXF2) was provided by Dr Peter Carlsson (Hellqvist et al., 1996, 1998). Negative control (pEVRF0 empty vector) was made by removing FOXF2 with BamHI and SacI and religating with a short noncoding sequence. O9-1 cells were plated at 1.25×105 cells/ml (0.4 ml per well in a 24-well plate) in duplicate. After 24 h, media were changed to DMEM with 1% FBS and transiently transfected with 0.75 µl Lipofectamine 3000 (Thermo Fisher Scientific), 1 µl P3000 reagent (Thermo Fisher Scientific) and 500 ng pEVRF0-FOXF2 or pEVRF0 empty vector in Opti-MEM (Gibco) following manufacturer's instructions. After 48 h, cell lysates were collected for RNA extraction or counted by hemocytometer.
Statistics
Affymetrix TAC was used for determination of significant differential expression in microarray experiments, utilizing an FDR P-value of 0.5 (Benjamini-Hochberg) based on independent RT-PCR validation. DAVID was used for determination of significant enrichment of differentially expressed genes (Huang et al., 2009a,b). Two-tailed t-tests with Holm-Sidak correction were used to determine whether gene expression was changed by cyclopamine exposure in vivo or with SHH ligand stimulation with or without vismodegib in O9-1 cNCCs. Two-tailed t-tests were used for analysis of proliferation assays and tissue-specific gene expression. GraphPad Prism 6 was used for all non-microarray statistical analyses. An alpha value of 0.05 was maintained for determination of significance for all non-microarray experiments.
Human genetics analysis
The cohort for this association analysis comes from a world-wide, multi-ethnic recruitment in the Pittsburgh Orofacial Cleft (POFC) study. University of Pittsburg Institutional Review Board approval and informed consent was obtained for all subjects. Participants comprising 823 CL/P cases, 1700 controls and 1319 case-parent trios were recruited from 13 countries worldwide. Full details of the recruitment, genotyping and quality control procedures have been described previously (Leslie et al., 2016a). Briefly, samples were genotyped on the Illumina HumanCore+Exome array, phased with SHAPEIT2, and imputed to 1000 Genomes Phase 3 reference panel. Genotype probabilities were converted to most-likely genotypes using Plink v1.9 if the genotype with the highest probability was >0.9. SNPs with minor allele frequencies <1%, INFO score <0.5, or deviating from Hardy–Weinberg equilibrium in European controls (P<0.0001) were removed from the analysis. Analysis was further limited to 550 SNPs located within a 66 kb interval (FOXF2 and 30 kb flanking sequence). Separate analyses for cases and controls (logistic regression assuming an additive genetic model and including 18 principal components of ancestry) and trios (transmission disequilibrium test) were combined in an inverse variance-weighted fixed-effects meta analysis. Analysis was performed for CL/P and stratified into CL and CLP. A Bonferroni P-value threshold of 4.3×10−4 was considered statistically significant based on 116 independent tests (Gao et al., 2010). Differences in effects between CL and CLP were formally tested using a Q statistic (Schenker and Gentleman, 2001).
Acknowledgements
We thank Drs Mamoru Ishii and Robert Maxson for providing O9-1 cells; Drs Peter Carlsson and Eric Campeau for FOXF2 and lentiviral plasmid constructs; Galen Heyne for experimental support; Dr John Svaren for help on binding site analysis; Jenna Carlson, Drs John Shaffer and Eleanor Feingold for association analyses; the UW-Madison Biotechnology Center; the UW-Madison Carbone Cancer Center Flow Cytometry Laboratory; the UW-Madison Biotron Laboratory; and the UW-Madison Research Animals Resource Center.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.L.E., R.J.L.; Methodology: J.L.E., D.M.F., J.W.Y., E.J.L., H.M.C.; Formal analysis: J.L.E., D.M.F., J.W.Y., E.J.L., H.W.K.; Investigation: J.L.E., D.M.F., J.W.Y., E.J.L., H.W.K., L.J.A.-W., M.L.M.; Writing - original draft: J.L.E., R.J.L.; Writing - review & editing: J.L.E., D.M.F., H.M.C., R.J.L., L.J.A-W., H.W.K., J.W.Y., D.O.W., E.J.L., M.L.M.; Funding acquisition: M.L.M., R.J.L.
Funding
This work was supported by the National Institutes of Health [R00DE022010-02 to R.J.L., T32ES007015-37 to J.L.E., T35OD011078 to L.J.A.-W., K99DE025060 to E.J.L., X01HG007485 and R01DE016148 to M.L.M.]. Deposited in PMC for release after 12 months.
Data availability
Microarray data generated in this study are available at Gene Expression Omnibus under accession number GSE98336 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse98336).
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.149930.supplemental
References
- Abler L. L., Mehta V., Keil K. P., Joshi P. S., Flucus C.-L., Hardin H. A., Schmitz C. T. and Vezina C. M. (2011). A high throughput in situ hybridization method to characterize mRNA expression patterns in the fetal mouse lower urogenital tract. J. Vis. Exp. 54, e2912 10.3791/2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu L., Chen Q., Wang H., Zhang T., Hetmanski J. B., Schwender H., Parker M., Chou Y.-H. W., Yeow V., Chong S. S. et al. (2015). Novel evidence of association with nonsyndromic cleft lip with or without cleft palate was shown for single nucleotide polymorphisms in FOXF2 gene in an Asian population. Birth Defects Res. A Clin. Mol. Teratol 103, 857-862. 10.1002/bdra.23413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K. and Kaufman P. D. (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529 10.1371/journal.pone.0006529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichberger T., Regl G., Ikram M. S., Neill G. W., Philpott M. P., Aberger F. and Frischauf A.-M. (2004). FOXE1, a new transcriptional target of GLI2 is expressed in human epidermis and basal cell carcinoma. J. Invest. Dermatol. 122, 1180-1187. 10.1111/j.0022-202X.2004.22505.x [DOI] [PubMed] [Google Scholar]
- Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C. et al. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808-D815. 10.1093/nar/gks1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Becker L. C., Becker D. M., Starmer J. D. and Province M. A. (2010). Avoiding the high Bonferroni penalty in genome-wide association studies. Genet. Epidemiol. 34, 100-105. 10.1002/gepi.20430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes J., Schwab U., Lemke H. and Stein H. (1983). Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 31, 13-20. 10.1002/ijc.2910310104 [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. (2008). The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 84, 37-138. 10.1016/S0070-2153(08)00602-9 [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. (2012). The mouse as a developmental model for cleft lip and palate research. Front. Oral Biol. 16, 32-51. 10.1159/000337523 [DOI] [PubMed] [Google Scholar]
- Hellqvist M., Mahlapuu M., Samuelsson L., Enerbäck S. and Carlsson P. (1996). Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J. Biol. Chem. 271, 4482-4490. 10.1074/jbc.271.8.4482 [DOI] [PubMed] [Google Scholar]
- Hellqvist M., Mahlapuu M., Blixt A., Enerbäck S. and Carlsson P. (1998). The human forkhead protein FREAC-2 contains two functionally redundant activation domains and interacts with TBP and TFIIB. J. Biol. Chem. 273, 23335-23343. 10.1074/jbc.273.36.23335 [DOI] [PubMed] [Google Scholar]
- Heyne G. W., Plisch E. H., Melberg C. G., Sandgren E. P., Peter J. A. and Lipinski R. J. (2015). A simple and reliable method for early pregnancy detection in inbred mice. J. Am. Assoc. Lab. Anim. Sci. 54, 368-371. [PMC free article] [PubMed] [Google Scholar]
- Heyne G. W., Everson J. L., Ansen-Wilson L. J., Melberg C. G., Fink D. M., Parins K. F., Doroodchi P., Ulschmid C. M. and Lipinski R. J. (2016). Gli2 gene-environment interactions contribute to the etiological complexity of holoprosencephaly: evidence from a mouse model. Dis. Model. Mech. 9, 1307-1315. 10.1242/dmm.026328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D. and Marcucio R. S. (2009). A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development 136, 107-116. 10.1242/dev.026583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Young N. M., Li X., Xu Y., Hallgrímsson B. and Marcucio R. S. (2015). A dynamic Shh expression pattern, regulated by SHH and BMP signaling, coordinates fusion of primordia in the amniote face. Development 142, 567-574. 10.1242/dev.114835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1-13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ishii M., Arias A. C., Liu L., Chen Y.-B., Bronner M. E. and Maxson R. E. (2012). A stable cranial neural crest cell line from mouse. Stem Cells Dev. 21, 3069-3080. 10.1089/scd.2012.0155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Hacia J. G., Suzuki A., Sanchez-Lara P. A., Urata M. and Chai Y. (2012). Modulation of noncanonical TGF-β signaling prevents cleft palate in Tgfbr2 mutant mice. J. Clin. Invest. 122, 873-885. 10.1172/JCI61498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B. C., Carpenter C., Nebert D. W. and Vasiliou V. (2010). Update of human and mouse forkhead box (FOX) gene families. Hum. Genomics 4, 345-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J., Mao J., Tenzen T., Kottmann A. H. and Mcmahon A. P. (2004). Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18, 937-951. 10.1101/gad.1190304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z., Leslie E. J., Cooper M. E., Butali A., Standley J., Rigdon J., Suzuki S., Gongorjav A., Shonkhuuz T. E., Natsume N. et al. (2015). Replication of 13q31.1 association in nonsyndromic cleft lip with cleft palate in Europeans. Am. J. Med. Genet. A 167, 1054-1060. 10.1002/ajmg.a.36912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R., Bush J. O. and Lidral A. C. (2006). Development of the upper lip: morphogenetic and molecular mechanisms. Dev. Dyn. 235, 1152-1166. 10.1002/dvdy.20646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff D. M. and Harris M. J. (2008). Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 82, 63-77. 10.1002/bdra.20430 [DOI] [PubMed] [Google Scholar]
- Kinzler K. W. and Vogelstein B. (1990). The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 10, 634-642. 10.1128/MCB.10.2.634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y. and Jiang R. (2009). Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 136, 1387-1396. 10.1242/dev.028167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., Xu J. and Jiang R. (2015). Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 115, 59-84. 10.1016/bs.ctdb.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. J., Mancuso J. L., Schutte B. C., Cooper M. E., Durda K. M., L'Heureux J., Zucchero T. M., Marazita M. L. and Murray J. C. (2013). Search for genetic modifiers of IRF6 and genotype-phenotype correlations in Van der Woude and popliteal pterygium syndromes. Am. J. Med. Genet. A 161, 2535-2544. 10.1002/ajmg.a.36133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. J., Carlson J. C., Shaffer J. R., Feingold E., Wehby G., Laurie C. A., Jain D., Laurie C. C., Doheny K. F., Mchenry T. et al. (2016a). A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum. Mol. Genet. 25, 2862-2872. 10.1093/hmg/ddw104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. J., Koboldt D. C., Kang C. J., Ma L., Hecht J. T., Wehby G. L., Christensen K., Czeizel A. E., Deleyiannis F. W.-B., Fulton R. S. et al. (2016b). IRF6 mutation screening in non-syndromic orofacial clefting: analysis of 1521 families. Clin. Genet. 90, 28-34. 10.1111/cge.12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Williams T. (2013). Separation of mouse embryonic facial ectoderm and mesenchyme. J. Vis. Exp. 74, e50248 10.3791/50248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidral A. C., Moreno L. M. and Bullard S. A. (2008). Genetic factors and orofacial clefting. Semin. Orthod. 14, 103-114. 10.1053/j.sodo.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R. J., Cook C. H., Barnett D. H., Gipp J. J., Peterson R. E. and Bushman W. (2005). Sonic hedgehog signaling regulates the expression of insulin-like growth factor binding protein-6 during fetal prostate development. Dev. Dyn. 233, 829-836. 10.1002/dvdy.20414 [DOI] [PubMed] [Google Scholar]
- Lipinski R. J., Gipp J. J., Zhang J., Doles J. D. and Bushman W. (2006). Unique and complimentary activities of the Gli transcription factors in Hedgehog signaling. Exp. Cell Res. 312, 1925-1938. 10.1016/j.yexcr.2006.02.019 [DOI] [PubMed] [Google Scholar]
- Lipinski R. J., Bijlsma M. F., Gipp J. J., Podhaizer D. J. and Bushman W. (2008a). Establishment and characterization of immortalized Gli-null mouse embryonic fibroblast cell lines. BMC Cell Biol. 9, 49 10.1186/1471-2121-9-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R. J., Hutson P. R., Hannam P. W., Nydza R. J., Washington I. M., Moore R. W., Girdaukas G. G., Peterson R. E. and Bushman W. (2008b). Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol. Sci. 104, 189-197. 10.1093/toxsci/kfn076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R. J., Song C., Sulik K. K., Everson J. L., Gipp J. J., Yan D., Bushman W. and Rowland I. J. (2010). Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: Phenotypic characterization and clinical implications. Birth Defects Res. A Clin. Mol. Teratol. 88, 232-240. 10.1002/bdra.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R. J., Holloway H. T., O'leary-Moore S. K., Ament J. J., Pecevich S. J., Cofer G. P., Budin F., Everson J. L., Johnson G. A. and Sulik K. K. (2014). Characterization of subtle brain abnormalities in a mouse model of hedgehog pathway antagonist-induced cleft lip and palate. PLoS ONE 9, e102603 10.1371/journal.pone.0102603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots G. G., Ovcharenko I., Pachter L., Dubchak I. and Rubin E. M. (2002). rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 12, 832-839. 10.1101/gr.225502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig K. U., Mangold E., Herms S., Nowak S., Reutter H., Paul A., Becker J., Herberz R., Alchawa T., Nasser E. et al. (2012). Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 44, 968-971. 10.1038/ng.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig K. U., Böhmer A. C., Rubini M., Mossey P. A., Herms S., Nowak S., Reutter H., Alblas M. A., Lippke B., Barth S. et al. (2014). Strong association of variants around FOXE1 and orofacial clefting. J. Dent. Res. 93, 376-381. 10.1177/0022034514523987 [DOI] [PubMed] [Google Scholar]
- Madison B. B., Mckenna L. B., Dolson D., Epstein D. J. and Kaestner K. H. (2009). FoxF1 and FoxL1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J. Biol. Chem. 284, 5936-5944. 10.1074/jbc.M808103200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucio R. S., Cordero D. R., Hu D. and Helms J. A. (2005). Molecular interactions coordinating the development of the forebrain and face. Dev. Biol. 284, 48-61. 10.1016/j.ydbio.2005.04.030 [DOI] [PubMed] [Google Scholar]
- Moreno L. M., Mansilla M. A., Bullard S. A., Cooper M. E., Busch T. D., Machida J., Johnson M. K., Brauer D., Krahn K., Daack-Hirsch S. et al. (2009). FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum. Mol. Genet. 18, 4879-4896. 10.1093/hmg/ddp444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy J. and Bhaskar L. V. K. S. (2009). Current concepts in genetics of nonsyndromic clefts. Indian J. Plast. Surg. 42, 68-81. 10.4103/0970-0358.53004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik A. M., Johansson J. A., Ghiami M., Reyahi A. and Carlsson P. (2016). Foxf2 is required for secondary palate development and Tgfβ signaling in palatal shelf mesenchyme. Dev. Biol. 415, 14-23. 10.1016/j.ydbio.2016.05.013 [DOI] [PubMed] [Google Scholar]
- Ormestad M., Astorga J., Landgren H., Wang T., Johansson B. R., Miura N. and Carlsson P. (2006). Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development 133, 833-843. 10.1242/dev.02252 [DOI] [PubMed] [Google Scholar]
- Rahimov F., Jugessur A. and Murray J. C. (2012). Genetics of nonsyndromic orofacial clefts. Cleft Palate Craniofac. J. 49, 73-91. 10.1597/10-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robarge K. D., Brunton S. A., Castanedo G. M., Cui Y., Dina M. S., Goldsmith R., Gould S. E., Guichert O., Gunzner J. L., Halladay J. et al. (2009). GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 19, 5576-5581. 10.1016/j.bmcl.2009.08.049 [DOI] [PubMed] [Google Scholar]
- Roessler E., Ermilov A. N., Grange D. K., Wang A., Grachtchouk M., Dlugosz A. A. and Muenke M. (2005). A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 14, 2181-2188. 10.1093/hmg/ddi222 [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A., Prezioso V. R., Darnell J. E. and Jessell T. M. (1993). Sequential expression of HNF-3 beta and HNF-3 alpha by embryonic organizing centers: the dorsal lip/node, notochord and floor plate. Mech. Dev. 44, 91-108. 10.1016/0925-4773(93)90060-B [DOI] [PubMed] [Google Scholar]
- Sasaki H. and Hogan B. L. M. (1994). HNF-3 beta as a regulator of floor plate development. Cell 76, 103-115. 10.1016/0092-8674(94)90176-7 [DOI] [PubMed] [Google Scholar]
- Sasaki H., Hui C., Nakafuku M. and Kondoh H. (1997). A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313-1322. [DOI] [PubMed] [Google Scholar]
- Schenker N. and Gentleman J. F. (2001). On judging the significance of differences by examining the overlap between confidence intervals. Am. Stat. 55, 182-186. 10.1198/000313001317097960 [DOI] [Google Scholar]
- Theiler K. (1989). The House Mouse: Atlas of Embryonic Development. New York: Springer. [Google Scholar]
- Wang T., Tamakoshi T., Uezato T., Shu F., Kanzaki-Kato N., Fu Y., Koseki H., Yoshida N., Sugiyama T. and Miura N. (2003). Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev. Biol. 259, 83-94. 10.1016/S0012-1606(03)00176-3 [DOI] [PubMed] [Google Scholar]
- Watkins S. E., Meyer R. E., Strauss R. P. and Aylsworth A. S. (2014). Classification, epidemiology, and genetics of orofacial clefts. Clin. Plast. Surg. 41, 149-163. 10.1016/j.cps.2013.12.003 [DOI] [PubMed] [Google Scholar]
- Xavier G. M., Seppala M., Barrell W., Birjandi A. A., Geoghegan F. and Cobourne M. T. (2016). Hedgehog receptor function during craniofacial development. Dev. Biol. 415, 198-215. 10.1016/j.ydbio.2016.02.009 [DOI] [PubMed] [Google Scholar]
- Xie J., Murone M., Luoh S.-M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C.-W., Hynes M., Goddard A. et al. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90-92. 10.1038/34201 [DOI] [PubMed] [Google Scholar]
- Xu X.-F., Guo C.-Y., Liu J., Yang W.-J., Xia Y.-J., Xu L., Yu Y.-C. and Wang X.-P. (2009). Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells. J. Carcinog. 8, 13 10.4103/1477-3163.55429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Liu H., Lan Y., Aronow B. J., Kalinichenko V. V. and Jiang R. (2016). A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet. 12, e1005769 10.1371/journal.pgen.1005769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. W., Kita Y., Frank D. J., Majewski R. R., Konicek B. A., Nobrega M. A., Jacob H., Walterhouse D. and Iannaccone P. (2002). Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 277, 5548-5555. 10.1074/jbc.M105708200 [DOI] [PubMed] [Google Scholar]