

Abstract
Background
In the past ten years there has been a growing interest in engineering Gram-positive bacteria for biotechnological applications, including vaccine delivery and production of recombinant proteins. Usually, bacteria are manipulated using plasmid expression vectors. The major limitation of this approach is due to the fact that recombinant plasmids are often lost from the bacterial culture upon removal of antibiotic selection. We have developed a genetic system based on suicide vectors on conjugative transposons allowing stable integration of recombinant DNA into the chromosome of transformable and non-transformable Gram-positive bacteria.
Results
The aim of this work was to select a strong chromosomal promoter from Streptococcus gordonii to improve this genetic system making it suitable for expression of single-copy recombinant genes. To achieve this task, a promoterless gene encoding a chloramphenicol acetyltransferase (cat), was randomly integrated into the S. gordonii chromosome and transformants were selected for chloramphenicol resistance. Three out of eighteen chloramphenicol resistant transformants selected exhibited 100% stability of the phenotype and only one of them, GP215, carried the cat gene integrated as a single copy. A DNA fragment of 600 base pairs exhibiting promoter activity was isolated from GP215 and sequenced. The 5' end of its corresponding mRNA was determined by primer extention analysis and the putative -10 and a -35 regions were identified. To study the possibility of using this promoter (PP) for single copy heterologous gene expression, we created transcriptional fusions of PP with genes encoding surface recombinant proteins in a vector capable of integrating into the conjugative transposon Tn916. Surface recombinant proteins whose expression was controlled by the PP promoter were detected in Tn916-containing strains of S. gordonii and Bacillus subtilis after single copy chromosomal integration of the recombinant insertion vectors into the resident Tn916. The surface recombinant protein synthesized under the control of PP was also detected in Enterococcus faecalis after conjugal transfer of a recombinant Tn916 containing the transcriptional fusion.
Conclusion
We isolated and characterized a S. gordonii chromosomal promoter. We demonstrated that this promoter can be used to direct expression of heterologous genes in different Gram-positive bacteria, when integrated in a single copy into the chromosome.
Background
In the past ten years there has been a growing interest in engineering Gram-positive bacteria for biotechnological applications, including vaccine delivery. [1-4], and in situ production of anti-infective protectants [5] and microbicides [6]. A common approach to genetic manipulation of bacteria is based on the use of plasmid expression vectors since these recombinant molecules can be introduced into bacterial cells by a variety of genetic techniques such as natural transformation, artificial transformation, transduction, conjugative mobilization, and electroporation [7-9]. However, the major limitation of this approach is due to the fact that recombinant plasmids are often lost from the bacterial culture upon removal of antibiotic selection. Certainly, this has consequences when using recombinant bacteria in vivo where their replication occurs in the absence of selection. An alternative approach is to integrate recombinant DNA molecules into the bacterial chromosome since this method allows increased in vivo stability of the genetic constructs. Therefore a lot of efforts have focused on the development of efficient expression systems based on chromosomal integration of expression cassettes [10,11]. Naturally transformable bacteria represent a convenient model, since heterologous DNA can be easily integrated into their chromosomes, whereas genetic manipulation of non-transformable bacteria is more difficult and relies mainly on electroporation and conjugative mobilization of foreign DNA molecules.
We have previously described a genetic system based on conjugative transposons allowing stable integration of recombinant DNA into the chromosome of transformable and non-transformable streptococci [12,13]. A series of transposon insertion vectors containing two regions of homology with Tn916 [14] have been created in order to manipulate both naturally transformable and non-transformable Gram-positive bacteria carrying Tn916 [12]. The aim of this work was to select a strong promoter to improve this genetic system making it suitable for expression of single-copy recombinant genes in a broad spectrum of Gram-positive bacteria.
Results and discussion
Promoter selection by chromosomal integration
To select resident promoters from the genome of Streptococcus gordonii, we performed a random ligation of streptococcal DNA to a promoterless cat gene, conferring resistance to chloramphenicol (Cm). The ligation mixture was used to transform the naturally transformable S. gordonii «Challis» strain V288 and transformants were selected for Cm resistance. Chromosomal DNA flanking the promoterless cat gene provided the homology for the random integration of cat into the chromosome during transformation (Fig. 1). 71 Cm-resistant (CmR) transformants were isolated, presumably as a result of transcriptional fusions of streptococcal promoters to the promoterless cat gene. Eighteen CmR transformants were selected for further characterization. The strategy commonly used to select promoters is based on cloning random chromosomal DNA fragments in a promoter probe vector upstream of a promoterless reporter gene. However, integrating the promoterless reporter gene (cat) directly into the streptococcal chromosome, allowed us to select resident chromosomal promoters expressing cat after in vivo transcriptional fusion at a single locus on the chromosome. This is preferable when looking for promoters to express heterologous genes integrated into the chromosome in a single copy.
Figure 1.
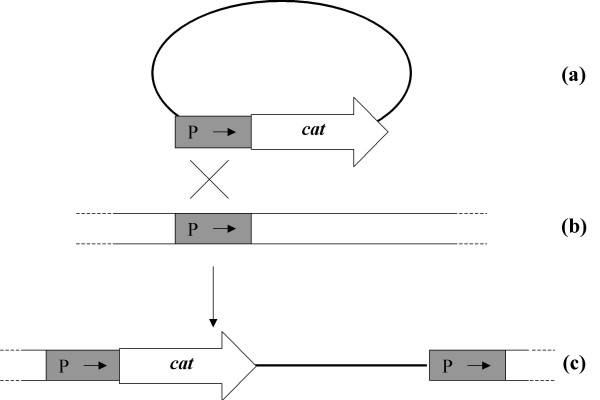
In vivo transcriptional fusion by integration of a promoterless cat gene into the chromosome of a naturally transformable streptococcus. A promoterless cat gene is ligated in vitro to random chromosomal fragments, using a restriction site a few base pairs upstream of its translation initiation codon (a). The ligation mixture is then used to transform the recipient strain (b), and the homologous sequences allow chromosomal integration of the promoterless cat gene (c). By this process, the cat gene is integrated into the chromosome between two direct repeats. Since some of the chromosomal fragments contain promoters (P), it is possible to obtain expression of the promoterless cat gene after in vivo transcriptional fusion with resident chromosomal promoters.
In vivo analysis of promoter strength was determined in the eighteen selected transformants by measuring the minimum inhibitory concentration (MIC) of Cm. Fifteen transformants exhibited a MIC of 16 μg/ml, whereas the remaining three transformants exhibited a MIC of 8 μg/ml. The fifteen strains with higher MIC were tested for the stability of the chloramphenicol-resistance phenotype: after 50 generations of growth without selection, bacterial cultures were plated on non-selective medium and at least 200 colonies were picked and tested for Cm resistance. The stability of the resistance phenotype varied considerably among the different transformants (data not shown). Three strains (GP214, GP215 and GP216) showed a 100% stability and were chosen for further analysis.
Cloning of a promoter from S. gordonii
The structure of the integrated cat gene in GP214, GP215 and GP216 was analyzed by Southern blot. Only GP215 showed to have a single cat copy integrated into the chromosome, whereas in GP214 and GP216 integration occurred at multiple sites (data not shown). In order to clone the regions flanking cat integration site in GP215, the chromosome of this strain was cut with TaqI whose recognition sequence is absent inside the cat gene. The derivative fragments were ligated to pBLUESCRIPT, and the ligation mixture was used to transform Escherichia coli cells; transformants were then selected for CmR. All transformants analyzed for plasmid content showed to carry a plasmid of the same size. One of these transformants (GP334) was selected for further analysis and the transforming plasmid was named pVMB5. By restriction analysis we showed that pVMB5 contains a 2.2 kb TaqI insert where a 600 base pairs (bp) streptococcal DNA fragment was cloned upstream of the cat gene. This DNA fragment was stably maintained in E. coli, where it retained its promoter activity, conferring CmR to E. coli. This is of considerable interest since it is known that very often cloning streptococcal promoters in a high copy number plasmid results in the failure of that plasmid to be established [15].
Sequence analysis
The streptococcal DNA upstream of the cat gene in pVMB5 was sequenced (GenBank accession number: U74080). Analysis of the sequence revealed the presence of an open reading frame (ORF1), preceded by a typical ribosome binding site (RBS) (Fig. 2). A BLAST search with the partial sequenced genome of S. gordonii http://www.tigr.org/index.shtml showed that ORF1 represents the 5'-end of a gene encoding the first 48 amino acids (aa) of an uncharacterized protein conserved in bacteria and whose function is unknown. As indicated in figure 2, this truncated streptococcal protein is translationally fused to the N-terminus of CAT. Sequence analysis also revealed that the first 122 bp of the cloned streptococcal fragment belong to the 3'-end of a gene encoding a putative acetyltransferase (ORF2). To obtain information about the chromosomal region containing the 600 bp streptococcal DNA fragment cloned in pVMB5, we looked throughout the whole contig sequence and we found out that upstream ORF2, and partially overlapping with it, there is an ORF encoding a DltD horthologue (dltD), a protein involved in D-alanine incorporation into lipoteichoic acid (LTA) [16], whereas downstream ORF1, there is a gene encoding a Tmp7 transmembrane protein (Fig. 2).
Figure 2.
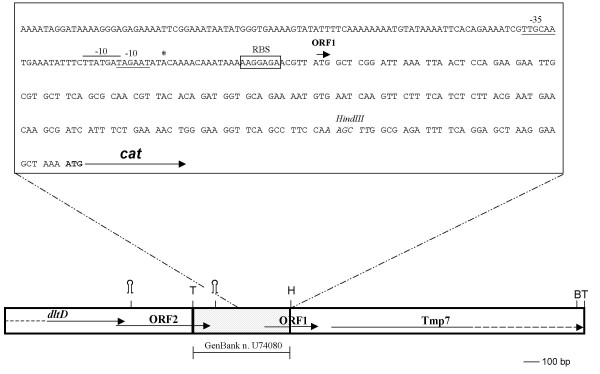
Schematic representation of the S. gordonii locus containing PP promoter. DltD orthologue, an ORF encoding a putative acetyltransferase (ORF2), ORF1, the gene encoding a Tmp7 transmembrane protein, and the cat gene are indicated by arrows. Dashed arrows designate that the ORF is only partially represented in the scheme. The 600 bp TaqI-HindIII fragment including PP promoter and cloned upstream of the cat gene in pVMB5 is indicated by a dotted box. Nucleotide sequence of the PP promoter region (332 bp) is reported inside the box. The transcriptional start site determined by primer extention analysis is marked with an asterisk. Proposed -35 and -10 regions are underlined. A second putative -10 sequence is overlined. ORF1 putative ribosome binding site (RBS) is boxed. ATG initiation codon of cat is in bold characters and the sequence of HindIII site is in italic letters. The complete sequence of the 600 bp cloned fragment is available on GenBank (Accession number: U74080). T, TaqI site; B, BamHI site; H, HindIII site ; loop, putative transcriptional terminator.
Identification of the transcriptional start site
To identify the sequences responsible for the observed promoter activity, the 5'-end of the cat-specific mRNA was mapped by primer extension with a specific primer. Total RNA was isolated from E. coli GP334 (containing pVMB5) and S. gordonii GP215. In both strains, the position of the 5'-end of the mRNA was located at the same purine residue at position 423 of the 600 bp region of streptococcal DNA, 26 nucleotides upstream of the ORF1 translational start site (Fig. 2 and 3). Putative -35 and -10 sequences closely resembling the consensus E. coli σ70 and Bacillus subtilis σ43 [17] recognition sequences TTGACA (-35) and TATAAT (-10) could be identified. The -35 region was TTGCAA, and the -10 region was TAGAAT. The spacing between the -35 and -10 region was 17 bp and was thus similar to the spacing in B. subtilis (17 to 19 bp) and E. coli (16 to 18 bp) promoters. Moreover, a TG nucleotide pair was found 1 bp upstream of the -10 region; such a structure is typical of Gram-positive bacteria promoters [18]. Based on this information, we concluded that indeed we isolated a streptococcal promoter which was designated PP. The spacing between the 5'-end of the mRNA and the -10 hexanucleotide was only 2 bp, which is unusually short. A second putative -10 region could be identified in the TATGAT hexanucleotide (Fig. 2), whose distance from the 5' end of the mRNA is 7 bp. However, since this sequence is not preceded by the TG nucleotide pair typical of Gram-positive promoters, and is separated from the -35 region only by 12 bp, we suppose that this -10 region of PP is probably not active.
Figure 3.
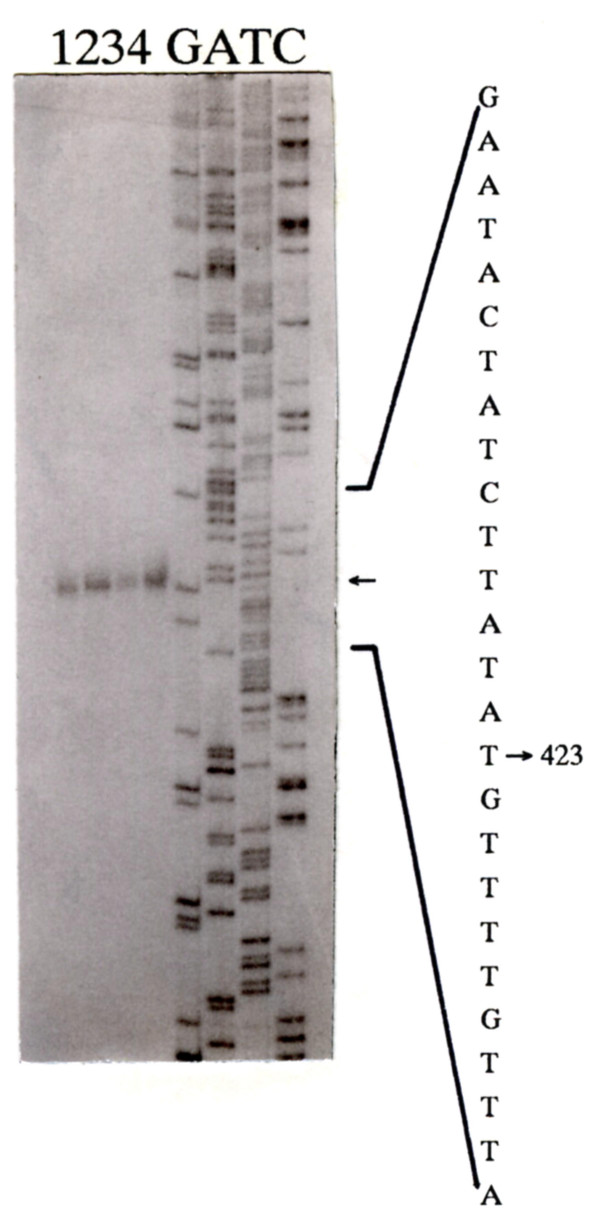
Primer extension analysis of the PP promoter. Localization by primer extension of the transcriptional start site of the cat mRNA specified by the PP promoter in E. coli GP334 (lanes 1–2), and S. gordonii GP215 (lanes 3–4). The sequence of the region upstream of the cat gene in pVMB5 was used as standard. The A residue, complementary to the T at position 423, indicated by an arrow, represents the transcriptional start site used in both strains.
Insertion vectors to express heterologous proteins
We have previously developed a genetic system based on the use of Streptococcus pyogenes surface fibrillar M6 protein, as a partner for the construction of translational fusions to deliver foreign proteins on the surface of S. gordonii [10]. To determine the possibility of using PP for chromosomal single copy heterologous gene expression in Gram-positive bacteria, we first generated a transcriptional fusion of PP with a promoterless gene encoding the M6 protein (emm6) in pSMB47, a suicide vector capable of integrating heterologous DNA into the conjugative transposon Tn916 via homologous recombinantion [12]. The recombinant plasmid was named pSMB139 (see Methods) (Fig. 4). To construct a Tn916 insertion vector that could be used to express translational fusions between M6 and any heterologous proteins under the PP promoter control, a 900 bp AvrII-HindIII fragment internal to emm6 in pSMB139 was replaced by a 390 bp AvrII-HindIII fragment of pSMB55 containing a multiple cloning site [10] (Fig. 4). The resulting vector, named pSMB148 (Fig. 4), allows to create translational fusions of heterologous proteins between the first 122 N-terminal aa and the last 140 aa of the M6 protein, which provides sequences necessary for cell wall anchoring.
Figure 4.
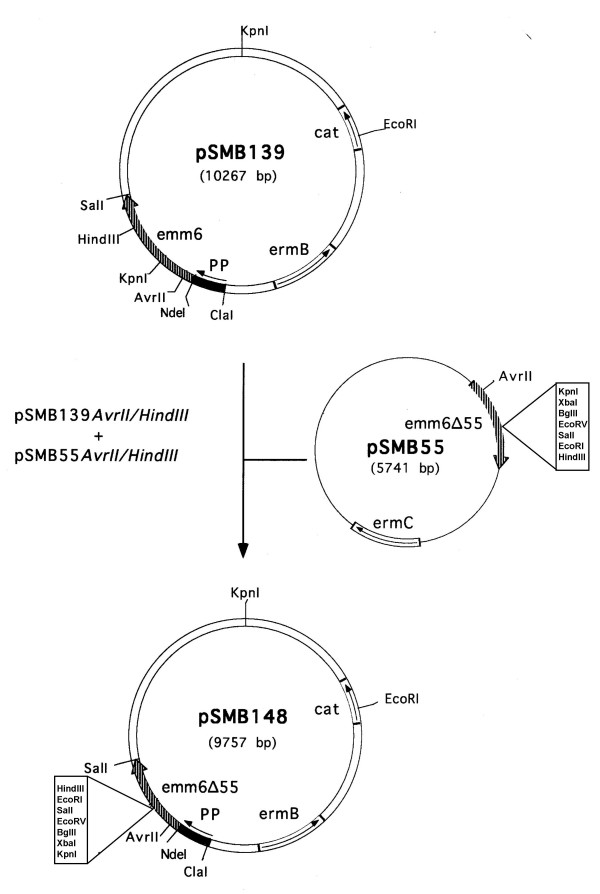
Construction of the insertion plasmid pSMB148. pSMB139 was constructed by introducing a 2.0 kb fragment, containing a transcriptional fusion of PP promoter with emm6, in the Tn916 insertion vector pSMB47 (see Methods). pSMB148 is a derivative of pSMB139 in which a 900 bp AvrII-HindIII fragment was replaced with a 390 bp AvrII-HindIII fragment from pSMB55 containing a multiple cloning site [10].
pSMB148 was used to create two derivative vectors in which the emm6 gene was fused respectively with a DNA sequence encoding 339 aa of the chicken ovalbumin (OVA) (pSMB156), and a DNA sequence encoding 458 aa of the tetanus toxin fragment C (TTFC) (pSMB288) (see Methods). A schematic representation of the recombinant proteins is shown in Fig. 5A.
Figure 5.
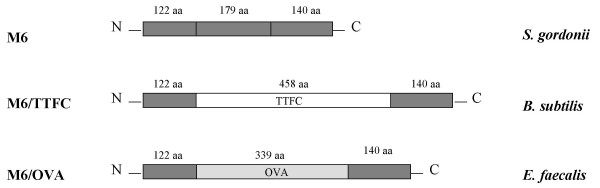
Schematic representation of recombinant M6, M6/TTFC and M6/OVA expressed on the surface of S. gordonii, B. subtilis and E. faecalis. The 458 aa protein TTFC (white bar) and the 339 aa protein OVA (light gray bar) were fused with the first 122 N-terminal aminoacids and the last 140 C-terminal aminoacids of M6 (dark gray bar). The predicted molecular weight of M6, M6/TTFC and M6/OVA is 49 kDa, 82 kDa and 66.9 kDa respectively.
Expression of recombinant proteins in Gram-positive bacteria
S. gordonii, B. subtilis, and Enterococcus faecalis were three Gram-positive hosts used to analyze the capability of PP to direct transcription of the heterologous genes expressing M6, M6/TTFC and M6/OVA recombinant proteins.
Expression of M6 in S. gordonii
pSMB139, bearing a transcriptional fusion of PP with emm6, was introduced by natural transformation in S. gordonii GP201, a strain with a single copy of Tn916 integrated into the chromosome. One of the transformants (GP1241), in which the integrative suicide vector drove the integration of the PP-emm6 fusion into Tn916, was isolated and analyzed for M6 protein expression. Envelope fractions (containing surface associated proteins) prepared from equal amounts of cells grown to mid-log, early and late stationary phase, were analyzed by Western-blotting with an anti-M6 monoclonal antibody. Multiple bands could be detected in fractions of cells grown to mid-log and early stationary phase, whereas no band was detected in the fraction of cultures grown to late stationary phase (Fig. 6A). The intensity of the signal was higher during the mid-log growth phase suggesting that either PP is more active during exponential growth or that the M6 protein is being degraded during stationary phase. The presence of multiple reactive bands of molecular masses close to the hypothetical size of M6 (predicted molecular weight, 49 kDa) is probably due to the fact that coiled-coil proteins like M6 run at aberrant sizes on denaturing gels [19].
Figure 6.
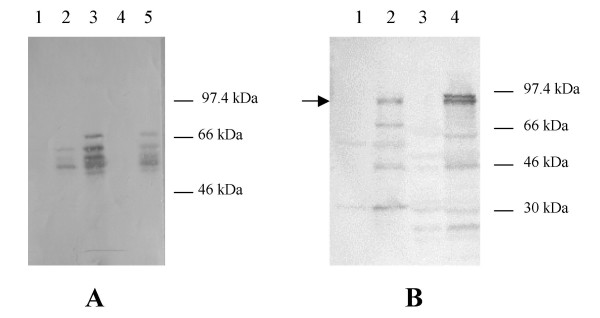
Western blot analysis of recombinant S. gordonii and B. subtilis strains expressing M6 protein and M6/TTFC fusion protein. (A) S. gordonii envelope fractions. Lane 1 through 3, GP1241 expressing M6 under the control of PP promoter. Lane 1, GP1241 harvested after overnight growth. Lane 2, GP1241 harvested after early stationary phase. Lane 3, GP1241 harvested after exponential phase. Lane 4, recipient strain GP201 (negative control). Lane 5, GP231 (positive control). Blot was developed with anti-M6 monoclonal antibody 10B6. (B) S. gordonii and B. subtilis envelope fractions. Lane 1, S. gordonii GP204 (negative control). Lane 2, S. gordonii GP1253 expressing M6/TTFC (positive control). Lane 3, B. subtilis recipient strain GP800.2 (negative control). Lane 4, B. subtilis GP848 expressing M6/TTFC under the control of PP promoter. Blot was developed with anti TTFC rabbit serum. Molecular weight markers are shown in the right side of panels.
Expression of M6/TTFC in B. subtilis
Competent cells of B. subtilis GP800.2, containing one copy of Tn916 integrated into the chromosome, were transformed with the insertion vector pSMB288, bearing the transcriptional fusion PP-emm6/ttfc. One transformant, GP848, was chosen for further studies. A culture of GP848 was grown to mid-exponential phase and analyzed by Western-blotting for the presence of recombinant M6/TTFC. As shown in Fig. 6B, two reactive bands could be detected in the envelope fraction. The lower band, indicated by an arrow, corresponds to the mature protein (predicted molecular weight, 82 kDa), while the upper band probably represents an unprocessed form (predicted molecular weight, 86.4 kDa).
Expression of M6/OVA in E. faecalis
Using a previously described genetic system [12], we constructed a derivative of the E. faecalis strain OG1SS [20] expressing the recombinant M6/OVA protein under PP control. pSMB156, bearing the PP-emm6/ova fusion, was first introduced in the Tn916 containing B. subtilis GP800.2 by natural transformation, to obtain a recombinant conjugative transposon containing the transcriptional fusion. The recombinant transposon was then transferred by conjugation into E. faecalis OG1SS. Transconjugants were detected at a frequency of 4 × 10-10 transconjugants/recipient. One of them (GP431) was analyzed for cell-surface expression of M6/OVA by flow-cytometric analysis using an anti-ovalbumin polyclonal antibody. The presence of recombinant M6/OVA on the surface of GP431 was clearly demonstrated by the increase of the fluorescence intensity in this strain, as compared to the parental control OG1SS (Fig. 7).
Figure 7.
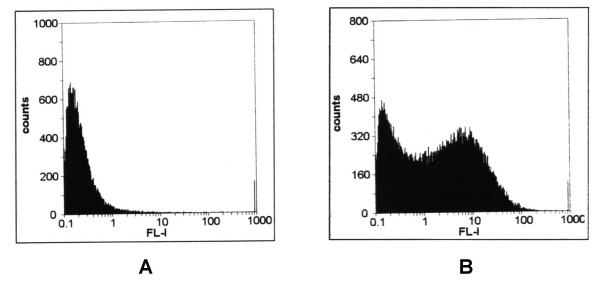
Flow-cytometric analysis of E. faecalis expressing M6/OVA. (A) OG1SS, recipient strain not expressing M6/OVA. (B) GP431, recombinant strain expressing M6/OVA on the surface. Bacterial cells were treated with anti-OVA rabbit serum and than with FITC-conjugated goat anti-rabbit IgG. x axis, arbitrary units (a.u.) of fluorescence intensity (log10); y axis, relative cell number.
Conclusions
We have isolated and characterized a promoter from the chromosome of S. gordonii, and demonstrated that it can be used to direct expression of heterologous genes in different Gram-positive bacteria when integrated in a single copy into the chromosome. This promoter, together with the genetic system based on suicide vectors able to integrate into conjugative transposons, represents a useful tool for the stable manipulation of a broad spectrum of Gram-positive bacteria.
Methods
Bacterial strains, plasmids and growth conditions
All strains and plasmids used in this work are listed in Table 1. E. coli strains DH5α and HB101 were cultured in Luria-Bertani (LB) broth. For maintenance of plasmids, ampicillin (100 μg/ml), chloramphenicol (20 μg/ml) or erythromycin (100 μg/ml) was added to the growth medium. Streptococcal strains were cultured in Brain Heart Infusion medium (BHI, Difco) or Tryptic Soy Broth (TSB, Difco) in the presence of chloramphenicol (5 μg/ml), erythromicin (100 μg/ml) or streptomycin (500 μg/ml) whenever required. Transformation of naturally competent cells of S. gordonii V288 and GP201, scoring and genetic analysis of transformants was carried out as already described [21,22]. B. subtilis strains were grown in LB broth with erythromicin (3 μg/ml) when it was required. Competent cells of B. subtilis GP800.2 were prepared and transformed according to described procedures [23]. Agar (1.5%) was added to LB, BHI or TSB to obtain solid media. All cultures were incubated at 37°C.
Table 1.
Strains and plasmids used in this work
| Strain | Relevant genotypea | Source/reference |
| S. gordonii | ||
| V288 | Recipient in transformation | [35] |
| GP201 | ΩTn5253, SmR, CmR, TcR | [13] |
| GP204 | str-204, SmR | [13] |
| GP214 | pKT random in V288, CmR | This work |
| GP215 | pKT random in V288, CmR | This work |
| GP216 | pKT random in V288, CmR | This work |
| GP231 | emm6, ErR, SmR | [21] |
| GP1241 | ΩTn5253 (ΔtetM::pSMB139) ErR, SmR, CmR, TcS | This work |
| GP1253 | emm6.1::TTFC | [28] |
| E. faecalis | ||
| OG1SS | Recipient in conjugation, SmR, SpR | [20] |
| GP431 | ΩTn916, ErR, TcS (Δ tetM::pSMB156) | This work |
| B. subtilis | ||
| GP800.2 | ΩTn916, TcR | [12] |
| GP847 | ΩTn916, ErR, TcS (Δ tetM::pSMB156) | This work |
| GP848 | ΩTn916, ErR, TcS (Δ tetM::pSMB288) | This work |
| Plasmid | Description | Source/reference |
| pKT | ApR, CmR | [25] |
| pBluescript | ApR | Stratagene |
| pVMB5 | pBluescript::PP-cat, ApR, CmR | This work |
| pVMB20 | pBluescript::emm6.1::ermC, ApR, ErR | [21] |
| pVA891 | CmR, ErR | [27] |
| pSMB47 | pVA891 derivative containing DNA sequences from Tn5253, CmR, ErR | [12] |
| pSMB89 | pVA891::emm6, CmR, ErR | This work |
| pSMB128 | pVA891::PP-emm6, CmR, ErR | This work |
| pSMB139 | pSMB47::PP-emm6, CmR, ErR | This work |
| pSMB148 | pSMB47::PP-emm6/55, CmR, ErR | This work |
| pSMB156 | pSMB47::PP-emm6/55::ova, CmR, ErR | This work |
aSm, streptomycin; Cm, chloramphenicol; Tc, tetracyclin; Er, erythromycin; spectinomycin; Ap, ampicillin.
DNA manipulation
Total DNA preparation of S. gordonii was performed as previously described [21]. Plasmid DNA was prepared using the Qiagen Plasmid Kit (Qiagen) according to the manufacturer's instructions. All recombinant techniques were performed following standard procedures [24], using E. coli HB101 or DH5α as a host. DNA restriction enzymes were obtained from Roche and used according to the manufacturer's instructions.
Promoter selection by chromosomal integration
A 1.6 kb HindIII-BamHI fragment of plasmid pKT [25], containing a cat gene, was ligated with random chromosomal DNA fragments of S. gordonii V288 previously cut with both HindIII and BamHI. The initiation codon of cat was 34 bp downstream of the HindIII site, therefore HindIII cleavage would leave cat promoterless and preceded by an intact ribosome binding site. The ligation mixture was introduced in S. gordonii V288 and transformants were selected for CmR.
MIC determination
The MIC of chloramphenicol for S. gordonii was determined following standard procedures [26].
Construction of recombinant vectors
A 1648 bp fragment containing the emm6 gene (encoding M6, a fibrillar surface protein of S. pyogenes) was amplified by PCR from plasmid pVMB20 [22] using the oligonucleotides 5'-ATGGATCCATCATATGGCTAAAAATAACACGAAT-3' (upstream primer, containing a BamHI site and introducing a NdeI site at the ATG translation initiation codon) and 5'-GCATGTCGACCATAATCATTAAATGTATCTCAT-3' (downstream primers containing a SalI site). This 1648 bp PCR fragment was digested with BamHI and SalI and cloned in pVA891 [27] previously digested with BamHIand SalI, resulting in plasmid pSMB89. A 451 bp region containing the streptococcal promoter PP was amplified from pVMB5 using the following primers: 5'-CGAGGATCCTTTAATCGATACTCATG-3' (upstream primer, containing a BamHI and a ClaI site) and 5'-CCGCATATGGTTCTCCTTTTTATTTGT-3' (downstream primers containing a NdeI site). After digestion with BamHI and NdeI, this PCR product was inserted between the BamHI and NdeI site of pSMB89 to obtain a transcriptional fusion of PP with emm6. The resulting plasmid was named pSMB128. This plasmid was first cut with BamHI, treated with Klenow enzyme to generate blunt ends, and finally cut with SalI to obtain a 2.0 kbfragment containing PP-emm6 fusion. This fragment was gel-purified and ligated to the suicide integrative plasmid pSMB47 [12] previously cut with HindIII, treated with Klenow enzyme to generate blunt ends, and finally cut with SalI. The resulting plasmid was named pSMB139 (Fig. 4). To introduce a multiple cloning site in the emm6 gene contained in pSMB139, the 900 bp AvrII-HindIII fragment internal to emm6 was replaced with the 390 bp AvrII-HindIII fragment of emm6 from pSMB55 [10] (Fig. 4). The resulting plasmid was named pSMB148. A 1016 bp DNA region encoding 339 aa of the chicken ovalbumin (OVA) (Gene Bank accession number: V00383) was amplified with the following primers: 5'-CTAGATCTGACAGCA CCAGGACAC-3' (upstream primer containing a BglII site) and 5'-TAAAGCTTTAGGGG AAACACATCTG-3' (downstream primer containing a HindIII site). After digestion with BglII and HindIII, this segment was introduced in pSMB148 previously digested with BglII and HindIII. The resulting plasmid, named pSMB156, contained a translational fusion of M6 with OVA. To create a translational fusion of M6 with the tetanus toxin fragment C (TTFC) a 1374 bp BglII-HindIII fragment encoding 458 aa of TTFC was isolated from pSMB158 [28] and cloned between the BglII and HindIII sites of pSMB148. The resulting plasmid was named pSMB288.
Western-blot analysis
Preparation of S. gordonii and B. subtilis cell envelope fractions (representing the protoplast surface containing the cell membrane together with cell wall fragments associated to the protoplasts) was performed as already described [29,30]. The monoclonal antibody 10B6 [31] diluted 1:1000 was used to detect the presence of M6 protein. M6/TTFC fusion protein was visualized with an anti-TTFC rabbit serum (Calbiochem-Novabiochem Corporation) diluted 1:1000.
Flow-cytometric analysis
Flow-cytometric analysis of E. faecalis was performed as already described [28,32] using an anti-OVA rabbit serum diluted 1:300 [29].
DNA sequence determination
The promoter containing fragment cloned in pVMB5 was sequenced by dideoxy chain termination method [33] as already described [34]. Denatured plasmid DNA was used as template.
RNA isolation
Total RNA was isolated from a 50 ml cell culture of S. gordonii and E. coli grown to late exponential phase (OD590 ≌ 0.5). Cells were harvested by centrifugation at 6000 × g at 4°C and lysed according to the following procedures. E. coli cells were first resuspended in hot (100°C) lysis buffer (50 mM Tris/HCl pH8, 1 mM EDTA, 1% SDS) and lysed by boiling the suspension for 5 minutes. S. gordonii cells were resuspended in lysozyme buffer (25 mM Tris/HCl (pH8), 10 mM EDTA, 50 mM glucose) and subjected to three cycles of freezing in liquid nitrogen and thawing at 52°C. After incubating with 0.2 mg/ml of lysozyme for 30 min at 37°C, one volume of hot (100°C) lysis buffer (100 mM Tris/HCl (pH8), 2 mM EDTA, 2% SDS) was added, and complete lysis was obtained by boiling cells for 5 minutes. After boiling, all lysates were cooled on ice for 5 min and total RNA was purified using the SV Total RNA Isolation System (Promega).
Primer-extention analysis
Primer extention analysis was performed with a synthetic oligonucleotide 5'-GTTCTTTACGATGCC-3' (position 47 to 61 relative to the initiation of the cat gene). Two pmol of the oligonucleotide, labeled with [γ-32P] ATP (3000 Ci/mmol, Amersham) using T4 polynucleotide kinase (New England Biolabs), were precipitated with 10 μg of RNA and the pellet was resuspended in 8 μl of Moloney Murine Leukemia Virus (M-MuLV) Reverse Transcriptase buffer (50 mM Tris/HCl (pH8.3), 8 mM MgCl2, 10 mM DTT). The mixture was heated at 65°C for 3 min, cooled rapidly at -80°C for 1 min and then transfered on ice until it was completely thawed. 1 μl of a 3.75 mM deoxynucleoside triphosphate solution and 10 U of M-MuLV Reverse Transcriptase (New England Biolabs) were added to the RNA-primer hybrid. The reaction mixture was incubated at 48°C for 30 min and terminated with 10 ml of stop solution (95% formamide, 20 mM EDTA pH8.0, 0.05% bromophenol blue, and 0.05% xylene cyanol FF). The reverse transcriptase reactions were analyzed by electrophoresis on a 6% polyacrylamide-7 M urea gel with sequencing reaction obtained with the same primer used as size standards.
Conjugation
Conjugation experiments were performed on solid media as previously described [12].
Authors' contributions
RP, characterization of promoter, engineering of S. gordonii, writing of manuscript.
TM, engineering of B. subtilis and E. faecalis
MRO, participation in experimental work, data evaluation
RM, participation in experimental work, data evaluation, writing of manuscript
GP, design and coordination of the study, data evaluation, direct supervision of experimental work, writing of manuscript
All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The work was supported in part by the European Commission grant QLK2-CT2000-00543 to GP, a grant from MIUR (COFIN 2002) to GP and from MIUR (FIRB RBAU01X9TB) to MRO.
The authors wish to thank Claudio Gualerzi for help and advice with the primer extention analysis.
Contributor Information
Roberta Provvedi, Email: roberta.provvedi@unipd.it.
Tiziana Maggi, Email: tiziana_maggi@chiron.com.
Marco R Oggioni, Email: oggioni@unisi.it.
Riccardo Manganelli, Email: riccardo.manganelli@unipd.it.
Gianni Pozzi, Email: pozzi@unisi.it.
References
- Samuelson P, Gunneriusson E, Nygren PA, Stahl S. Display of proteins on bacteria. J Biotechnol. 2002;96:129–154. doi: 10.1016/S0168-1656(02)00043-3. [DOI] [PubMed] [Google Scholar]
- Seegers JF. Lactobacilli as live vaccine delivery vectors: progress and prospects. Trends Biotechnol. 2002;20:508–515. doi: 10.1016/S0167-7799(02)02075-9. [DOI] [PubMed] [Google Scholar]
- Medaglini D, Rush CM, Sestini P, Pozzi G. Commensal bacteria as vectors for mucosal vaccines against sexually transmitted diseases: vaginal colonization with recombinant streptococci induces local and systemic antibodies in mice. Vaccine. 1997;15:1330–1337. doi: 10.1016/S0264-410X(97)00026-1. [DOI] [PubMed] [Google Scholar]
- Oggioni MR, Ciabattini A, Cuppone AM, Pozzi G. Bacillus spores for vaccine delivery. Vaccine. 2003;21 Suppl 2:S96–101. doi: 10.1016/S0264-410X(03)00207-X. [DOI] [PubMed] [Google Scholar]
- Magliani W, Conti S, Frazzi R, Pozzi G, Oggioni M, Polonelli L. Engineered commensal bacteria as delivery systems of anti-infective mucosal protectants. Biotechnol Genet Eng Rev. 2002;19:139–156. doi: 10.1080/02648725.2002.10648027. [DOI] [PubMed] [Google Scholar]
- Giomarelli B, Provvedi R, Meacci F, Maggi T, Medaglini D, Pozzi G, Mori T, McMahon JB, Gardella R, Boyd MR. The microbicide cyanovirin-N expressed on the surface of commensal bacterium Streptococcus gordonii captures HIV-1. Aids. 2002;16:1351–1356. doi: 10.1097/00002030-200207050-00006. [DOI] [PubMed] [Google Scholar]
- Wells JM, Wilson PW, Le Page RW. Improved cloning vectors and transformation procedure for Lactococcus lactis. J Appl Bacteriol. 1993;74:629–636. doi: 10.1111/j.1365-2672.1993.tb05195.x. [DOI] [PubMed] [Google Scholar]
- Samuelson P, Hansson M, Ahlborg N, Andreoni C, Gotz F, Bachi T, Nguyen TN, Binz H, Uhlen M, Stahl S. Cell surface display of recombinant proteins on Staphylococcus carnosus. J Bacteriol. 1995;177:1470–1476. doi: 10.1128/jb.177.6.1470-1476.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouwels PH, Leer RJ, Boersma WJ. The potential of Lactobacillus as a carrier for oral immunization: development and preliminary characterization of vector systems for targeted delivery of antigens. J Biotechnol. 1996;44:183–192. doi: 10.1016/0168-1656(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Oggioni MR, Pozzi G. A host-vector system for heterologous gene expression in Streptococcus gordonii. Gene. 1996;169:85–90. doi: 10.1016/0378-1119(95)00775-X. [DOI] [PubMed] [Google Scholar]
- Isticato R, Cangiano G, Tran HT, Ciabattini A, Medaglini D, Oggioni MR, De Felice M, Pozzi G, Ricca E. Surface display of recombinant proteins on Bacillus subtilis spores. J Bacteriol. 2001;183:6294–6301. doi: 10.1128/JB.183.21.6294-6301.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Provvedi R, Berneri C, Oggioni MR, Pozzi G. Insertion vectors for construction of recombinant conjugative transposons in Bacillus subtilis and Enterococcus faecalis. FEMS Microbiol Lett. 1998;168:259–268. doi: 10.1016/S0378-1097(98)00449-2. [DOI] [PubMed] [Google Scholar]
- Pozzi G, Musmanno RA, Renzoni EA, Oggioni MR, Cusi MG. Host-vector system for integration of recombinant DNA into chromosomes of transformable and nontransformable streptococci. J Bacteriol. 1988;170:1969–1972. doi: 10.1128/jb.170.4.1969-1972.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan SE, Zitzow LA, Su YA, Clewell DB. Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid. 1994;32:350–354. doi: 10.1006/plas.1994.1077. [DOI] [PubMed] [Google Scholar]
- Stassi DL, Dunn JJ, Lacks SA. Nucleotide sequence of DNA controlling expression of genes for maltosaccharide utilization in Streptococcus pneumoniae. Gene. 1982;20:359–366. doi: 10.1016/0378-1119(82)90204-9. [DOI] [PubMed] [Google Scholar]
- Debabov DV, Kiriukhin MY, Neuhaus FC. Biosynthesis of lipoteichoic acid in Lactobacillus rhamnosus: role of DltD in D-alanylation. J Bacteriol. 2000;182:2855–2864. doi: 10.1128/JB.182.10.2855-2864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi RH, Wang LF. Multiple procaryotic ribonucleic acid polymerase sigma factors. Microbiol Rev. 1986;50:227–243. doi: 10.1128/mr.50.3.227-243.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivula T, Sibakov M, Palva I. Isolation and characterization of Lactococcus lactis subsp. lactis promoters. Appl Environ Microbiol. 1991;57:333–340. doi: 10.1128/aem.57.2.333-340.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingshead SK, Fischetti VA, Scott JR. Complete nucleotide sequence of type 6 M protein of the group A Streptococcus. Repetitive structure and membrane anchor. J Biol Chem. 1986;261:1677–1686. [PubMed] [Google Scholar]
- Dunny GM, Craig RA, Carron RL, Clewell DB. Plasmid transfer in Streptococcus faecalis: production of multiple sex pheromones by recipients. Plasmid. 1979;2:454–465. doi: 10.1016/0147-619X(79)90029-5. [DOI] [PubMed] [Google Scholar]
- Pozzi G, Oggioni MR, Manganelli R, Fischetti VA. Expression of M6 protein gene of Streptococcus pyogenes in Streptococcus gordonii after chromosomal integration and transcriptional fusion. Res Microbiol. 1992;143:449–457. doi: 10.1016/0923-2508(92)90090-B. [DOI] [PubMed] [Google Scholar]
- Pozzi G, Contorni M, Oggioni MR, Manganelli R, Tommasino M, Cavalieri F, Fischetti VA. Delivery and expression of a heterologous antigen on the surface of streptococci. Infect Immun. 1992;60:1902–1907. doi: 10.1128/iai.60.5.1902-1907.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D, Davidoff-Abelson R. Fate of transforming DNA following uptake by competent Bacillus subtilis. I. Formation and properties of the donor-recipient complex. J Mol Biol. 1971;56:209–221. doi: 10.1016/0022-2836(71)90460-8. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, John Wiley and Sons; 1995. [Google Scholar]
- Shiau A, Smith JM. Improved cat gene cassette for promoter analysis and genetic constructions. Gene. 1988;67:295–299. doi: 10.1016/0378-1119(88)90406-4. [DOI] [PubMed] [Google Scholar]
- Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 1989;33:1588–1591. doi: 10.1128/aac.33.9.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrina FL, Tobian JA, Jones KR, Evans RP, Clewell DB. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene. 1982;19:345–353. doi: 10.1016/0378-1119(82)90025-7. [DOI] [PubMed] [Google Scholar]
- Medaglini D, Ciabattini A, Spinosa MR, Maggi T, Marcotte H, Oggioni MR, Pozzi G. Immunization with recombinant Streptococcus gordonii expressing tetanus toxin fragment C confers protection from lethal challenge in mice. Vaccine. 2001;19:1931–1939. doi: 10.1016/S0264-410X(00)00434-5. [DOI] [PubMed] [Google Scholar]
- Pozzi G, Oggioni MR, Manganelli R, Medaglini D, Fischetti VA, Fenoglio D, Valle MT, Kunkl A, Manca F. Human T-helper cell recognition of an immunodominant epitope of HIV-1 gp120 expressed on the surface of Streptococcus gordonii. Vaccine. 1994;12:1071–1077. doi: 10.1016/0264-410X(94)90175-9. [DOI] [PubMed] [Google Scholar]
- Oggioni MR, Medaglini D, Romano L, Peruzzi F, Maggi T, Lozzi L, Bracci L, Zazzi M, Manca F, Valensin PE, Pozzi G. Antigenicity and immunogenicity of the V3 domain of HIV type 1 glycoprotein 120 expressed on the surface of Streptococcus gordonii. AIDS Res Hum Retroviruses. 1999;15:451–459. doi: 10.1089/088922299311204. [DOI] [PubMed] [Google Scholar]
- Jones KF, Khan SA, Erickson BW, Hollingshead SK, Scott JR, Fischetti VA. Immunochemical localization and amino acid sequences of crossreactive epitopes within the group A streptococcal M6 protein. J Exp Med. 1986;164:1226–1238. doi: 10.1084/jem.164.4.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci S, Medaglini D, Rush CM, Marcello A, Peppoloni S, Manganelli R, Palu G, Pozzi G. Immunogenicity of the B monomer of Escherichia coli heat-labile toxin expressed on the surface of Streptococcus gordonii. Infect Immun. 2000;68:760–766. doi: 10.1128/IAI.68.2.760-766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provvedi R, Manganelli R, Pozzi G. Characterization of conjugative transposon Tn5251 of Streptococcus pneumoniae. FEMS Microbiol Lett. 1996;135:231–236. doi: 10.1016/0378-1097(95)00455-6. [DOI] [PubMed] [Google Scholar]
- Macrina FL, Wood PH, Jones KR. Genetic transformation of Streptococcus sanguis (Challis) with cryptic plasmids from Streptococcus ferus. Infect Immun. 1980;28:692–699. doi: 10.1128/iai.28.3.692-699.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]