

Abstract
The γ-proteobacterium Shewanella oneidensis strain MR-1 is a metabolically versatile organism that can reduce a wide range of organic compounds, metal ions, and radionuclides. Similar to most other sequenced organisms, ≈40% of the predicted ORFs in the S. oneidensis genome were annotated as uncharacterized “hypothetical” genes. We implemented an integrative approach by using experimental and computational analyses to provide more detailed insight into gene function. Global expression profiles were determined for cells after UV irradiation and under aerobic and suboxic growth conditions. Transcriptomic and proteomic analyses confidently identified 538 hypothetical genes as expressed in S. oneidensis cells both as mRNAs and proteins (33% of all predicted hypothetical proteins). Publicly available analysis tools and databases and the expression data were applied to improve the annotation of these genes. The annotation results were scored by using a seven-category schema that ranked both confidence and precision of the functional assignment. We were able to identify homologs for nearly all of these hypothetical proteins (97%), but could confidently assign exact biochemical functions for only 16 proteins (category 1; 3%). Altogether, computational and experimental evidence provided functional assignments or insights for 240 more genes (categories 2–5; 45%). These functional annotations advance our understanding of genes involved in vital cellular processes, including energy conversion, ion transport, secondary metabolism, and signal transduction. We propose that this integrative approach offers a valuable means to undertake the enormous challenge of characterizing the rapidly growing number of hypothetical proteins with each newly sequenced genome.
Keywords: computational biology, expression analysis, microarrays, proteomics, integrative microbiology
Shewanella oneidensis strain MR-1 is a facultatively anaerobic γ-proteobacterium that can use a broad range of electron acceptors for anaerobic respiration, including organic compounds, metal ions, and radionuclides (1). It is currently the subject of comprehensive study by the Shewanella Federation, a multiinstitutional consortium supported through the U.S. Department of Energy Genomics: GTL program (ref. 2; http://doegenomestolife.org). More than 2 years ago the S. oneidensis genome was sequenced and thoroughly annotated (3) with 4,931 predicted ORFs, 1,988 of which were considered uncharacterized “hypothetical” (40%). Since then, several publications have addressed issues regarding the S. oneidensis genome, including a correction of the total number of the predicted genes and analysis of genes designated as hypothetical (4–6). Our current estimate includes 4,467 predicted genes, 1,623 of which are annotated as hypothetical (36%).
Although this result represents an improvement, it also serves to point out one of the emerging challenges of modern biology: namely, the rapid accumulation of uncharacterized hypothetical genes (7–11). This assignment is given to genes that have not been experimentally characterized and whose functions cannot be deduced from simple sequence comparisons. Although analytical approaches are now available for comprehensive measurements of gene and protein expression, the lack of knowledge of the function of a large proportion of this genome limits our ability to take full advantage of capabilities for advancing biology to a more predictive science. Even the prediction that these genes encode proteins, that these proteins are intact (e.g., not truncated by errors in the genome sequence), and that they are expressed in living cells is uncertain. Nonetheless, every new sequencing project results in hundreds or even thousands of new hypothetical genes. For example, the recent sequencing of Sargasso Sea microbial communities resulted in a large number of uncharacterized genes (≈69,900) grouped into ≈15,600 families (12). Experimental characterizations of new proteins from one of the most extensively studied organisms, Escherichia coli strain K-12, are producing 20 to 30 new functional characterizations per year (13). At this rate, more than half of a century will be required to determine the biological functions of all S. oneidensis hypothetical genes. There is an obvious need for new approaches for rapid functional characterization of these hypothetical genes.
To address this problem, we have begun a four-phase program: first, to experimentally evaluate the expression of hypothetical genes under various conditions; second, to validate that these genes encode expressed proteins; third, to propose, by using various approaches, the most likely function of the expressed proteins; and fourth, to experimentally verify these functions. This study describes the first three phases of this program. The first and second phases herein involved a comprehensive experimental dataset that includes both microarray and proteomics expression data from multiple experiments. These analyses confidently identified 538 hypothetical genes as expressed in S. oneidensis cells both as mRNAs and as proteins (33% of 1,623). We then executed efforts to more fully understand the function of these hypothetical genes by combining sequence searches, statistical, computational, comparative, and structural genomics analyses and careful curation (phase three).
Materials and Methods
Gene Expression. S. oneidensis strain MR-1 (ATCC 700550) cultures were grown and sampled under aerobic and suboxic conditions and after UV irradiation. For the UV irradiation experiments, cells were aerobically grown to mid-log phase in Davis medium after exposure to different UV irradiation levels as described in ref. 14. Sample processing for the gene expression analysis followed standard protocols with minor modifications (14) and is described in Supporting Materials, which is published as supporting information on the PNAS web site. Bioflow model 110 fermentors (New Brunswick Scientific), either in fed-batch or in continuous-feed modes, were used for the aerobic and suboxic conditions, in which cells were grown in a modified defined medium M1 (15). Sample processing for the transcriptome analysis (16) as well as the experimental protocol are described in Supporting Materials. The same S. oneidensis arrays, consisting of ≈95% of the predicted genes (17), were used for all gene expression analyses. The expression values and corresponding standard errors were estimated by using the maximum likelihood analysis (18), and the resulting set included confidently expressed genes (11). Analysis of differential expression was not part of this study.
Protein Expression. Several parallel proteomic approaches were implemented in this study (Supporting Materials) to analyze S. oneidensis protein expression: (i) liquid chromatography (LC) coupled to tandem MS (MS/MS) as described in refs. 11 and 19–24; (ii) LC coupled to Fourier transform ion cyclotron resonance MS (25, 26); (iii) LC coupled to quadruple-TOF MS (27); and (iv) 2D gel electrophoresis followed by LC-MS/MS (16, 23). Common approaches used in different proteome analyses included: LC-MS/MS implemented by using the LCQ platform (Thermo Electron, San Jose, CA), standard sample processing (Supporting Materials), standard top-down data-dependent ion selection (11, 19–24), use of the turbo-sequest (Thermo Electron) search engine, and use of the recently developed standard mixtures for proteome studies (24). These approaches, in turn, allowed implementation of the same stringent criteria (28) for all peptide and protein identifications included in this study (Supporting Materials).
Gene Selection and Annotation. Several criteria were used for gene selection, namely to identify expressed hypothetical genes to be further analyzed (11). Among all S. oneidensis genes that showed statistically significant expression levels in microarray experiments, we identified those whose products were confidently detected by at least two independent protein analysis approaches. We further focused on those proteins that were originally (3) annotated as hypothetical and were still listed as such in GenBank as of August 20, 2003 (Fig. 1). These proteins were compared against the Conserved Domain, Clusters of Orthologous Groups (COG), InterPro, Pfam, Protein Data Bank, and SwissProt databases by using their respective search tools (29–34), Homologous Bacterial Genes server (35), psi-blast (36), and rps-blast (37) as described in refs. 11 and 22 (see also Supporting Materials). Additionally, comparative genomic analyses were performed by using the seed (http://theseed.uchicago.edu/FIG) and phylogenetic footprinting (38) methods (Supporting Materials). The proteins were also searched against a collection of Structural Classification of Proteins-based superfamily profiles (39) as described in ref. 6. Manually validated functional annotations were made according to a seven-category schema (Table 1).
Fig. 1.

Venn diagrams of gene (mRNA) and protein expression profilings. Diagrams show all genes and proteins (Upper) and only hypothetical genes and proteins (Lower) expressed in S. oneidensis.
Table 1. Seven-category schema for annotating proteins.
| Category no. | Description | No. of genes (% of 538) |
|---|---|---|
| 1 | Exact biochemical function, based on high similarity to experimentally characterized closely related homolog | 16 (3) |
| 2 | Well defined biochemical function, unknown specificity | 16 (3) |
| 3 | General biochemical function, based on family/superfamily assignment and/or a conserved sequence motif | 66 (12) |
| 4 | General biological function derived from the domain organization, genome context (e.g., operons), experimental (e.g., protein-protein interactions), and/or structural genomics data (e.g., similarities to proteins with known 3D structures) | 86 (16) |
| 5 | Certain functional insights derived from the above data | 72 (13) |
| 6 | Widely conserved protein, expressed under certain growth condition(s); e.g., “Conserved expressed protein” | 190 (35) |
| 7 | Organism- or genus-specific protein, expressed under certain growth condition(s); e.g., “Expressed protein in Shewanella” | 94 (17) |
Results
Expression of S. oneidensis Hypothetical Genes. The choice of methods for statistical analysis of the global gene expression data critically depends on the study's goals (40). The focus of the analysis reported here was to demonstrate, on the whole-genome scale, evidence for the expression of predicted genes and their corresponding protein products. Expression data were combined from several different experiments, including steady-state aerobic and suboxic growth, as well as from aerobically grown cells after exposure to different UV wavelengths. Because the analysis concentrated on the expressed genes (without considering whether they were induced or repressed under a particular growth condition), conservative criteria (18) for establishing minimum levels of gene (mRNA) expression were implemented. Then, a three-step approach was chosen to produce highly conservative protein expression estimates. The thresholds implemented for XCorr and ΔCn scores from the LC-MS/MS peptide identifications have been shown to greatly reduce the probability of false positive predictions (24, 41) and to ensure high accuracy of these identifications (28). The third criterion, identification of a protein by two different proteomics approaches, ensured an additional reduction in the number of false protein identifications. The resulting set of 538 proteins was selected based on four independent criteria: expression of a given gene both as (i) its mRNA and (ii) its protein product, and its designation as hypothetical by both (iii) the original (3) and (iv) GenBank annotations (Fig. 1).
A surprising result of this study was the identification of orthologs in other genomes for nearly all uncharacterized hypothetical proteins expressed in S. oneidensis indicating that they can be now referred to as “conserved proteins.” Although a recent pilot study of 54 hypothetical proteins expressed in Haemophilus influenzae obtained similar results (11), in the study presented here there was a substantial (10-fold) increase in the number of expressed hypothetical proteins. In this study, we were able to find homologs to 520 proteins (97% of 538), which would facilitate further annotation and functional characterization of these proteins.
Seven-Category Schema for Annotating Proteins. To provide the best possible functional annotation for this high number of uncharacterized genes, it was imperative to be comprehensive and include all available information. As in other genome-scale analyses, there was a necessity to assess the resulting annotations with respect to both their confidence and precision. The seven-category general schema introduced here (Table 1) attempts to address these issues, classifying functional assignments based on the degree of sequence similarity to the experimentally characterized homologs and the availability of supporting data. The first category includes genes that have high levels of sequence identity to well characterized proteins from other species and can be confidently predicted to have the same function (Table 2). The second category (Table 3) includes proteins with lower, but significant, levels of sequence similarity to experimentally characterized homologs, so that their biochemical function can be well defined, although the substrate (or ligand) specificity remains unclear. The third category includes proteins that share only low-level sequence similarity (typically limited to the common active site motifs) to experimentally studied homologs and can be given only a general biochemical functional assignment (Table 5, which is published as supporting information on the PNAS web site), common for all of the proteins of a given (super)family. The fourth category contains genes whose products cannot be assigned a biochemical function but participate in a known biological process, such as cell division, membrane transport (Table 6, which is published as supporting information on the PNAS web site). The fifth category consists of uncharacterized proteins for which only certain functional insights can be obtained (Table 7, which is published as supporting information on the PNAS web site). The sixth category consists of widely conserved expressed proteins (Table 8, which is published as supporting information on the PNAS web site). Finally, the seventh category includes all of the remaining organism- or genus-specific (herein Shewanella-specific) expressed proteins (Table 9, which is published as supporting information on the PNAS web site).
Table 2. Annotations of proteins with exact biochemical function: Category 1.
| SO ID no. | Upgraded annotation | COG no. | Pfam no. |
|---|---|---|---|
| SO0332 | Homoserine kinase, type II | 2334 | 06111 |
| SO0342 | PrpF protein required for repair/synthesis of Fe-S center of AcnD | 2828 | 04303 |
| SO0506 | 3-octaprenyl-4-hydroxybenzoate decarboxylase UbiD | 0043 | 01977 |
| SO0887 | Peptidylarginine deiminase | 2957 | 04371 |
| SO1523 | NAD kinase | 0061 | 01513 |
| SO1597 | ω-3 polyunsaturated fatty acid synthase PfaD subunit | 2070 | 03060 |
| SO1789 | UDP-2,3-diacylglucosamine hydrolase | 2908 | 00149 |
| SO1963 | Homogenetisate 1,2-dioxygenase | 3508 | 04209 |
| SO2593 | NAD-specific glutamate dehydrogenase | 2902 | 05088 |
| SO2614 | Aminodeoxychorismate lyase | 1559 | 02618 |
| SO2627 | ATP-dependent Clp protease adaptor protein ClpS | 2127 | 02617 |
| SO3340 | Mechanosensitive ion channel protein MscS | 0668 | 00924 |
| SO3436 | tRNA pseudouridine synthase TruD | 0585 | 01142 |
| SO4413 | Kynureninase | 0520 | 00266 |
| SO4680 | CDP-glycerol:poly(glycerophosphate) glycerophosphotransferase | 1887 | 04464 |
| SO4719 | Periplasmic tungstate-binding protein TupA, component of an ABC-type transporter | 2998 | 01547 |
Table 3. Annotations of proteins with well defined biochemical function: Category 2.
| SO ID no. | Upgraded annotation | COG no. | Pfam no. |
|---|---|---|---|
| SO0265 | Cytochrome c-type biogenesis factor CycH | 4235 | 00515 |
| SO0337 | Endoribonuclease L-PSP | 0251 | 01042 |
| SO0363 | Nucleoside-diphosphate-sugar pyrophosphorylase | 1208 | - |
| SO0455 | TRAP-type dicarboxylate transporter, permease component with fused DctQM subunit | 4666 | 06808 |
| SO0471 | Flavin-dependent dioxygenase | 2070 | 03060 |
| SO0783 | Superfamily I DNA and RNA helicase | 3972 | - |
| SO1007 | Na+/H+ antiporter NhaC | 1757 | 03553 |
| SO1267 | Glutamine synthetase-associated glutamine amidotransferase | 2071 | 00117 |
| SO1742 | 3-oxoacyl-acyl-carrier-protein | 0332 | 00195 |
| SO1981 | Nicotinic acid phosphoribosyltransferase | 1488 | 04095 |
| SO3051 | Mo-dependent oxidoreductase maturation factor | 1975 | - |
| SO3542 | Phosphoketolase | 3957 | 03894 |
| SO3667 | Heme iron utilization protein HugZ | 0748 | 01243 |
| SO3668 | Heme iron utilization protein HugX | 3721 | 06228 |
| SO4227 | S-adenosylmethionine-dependent methyltransferase MraW, involved in cell division | 0275 | 01795 |
| SO4690 | Undecaprenyl phosphate-sugar: lipid A glycosyltransferase | 1807 | 02366 |
Exact Biochemical Functions. The original genome sequence analysis of S. oneidensis (3) resulted in the annotation of all 538 genes of interest as hypothetical, but one would expect that, with time, additional evidence for the function of some of these genes would become available. Indeed, searches of public databases (29–32, 34) revealed some proteins that can now be functionally annotated based on the experimental characterization of close homologs in other organisms. Unfortunately, this set represents only 16 of these 538 proteins that have been assigned exact biochemical functions [Tables 1 (category 1) and 2]. In a >2-year period of experimental efforts by the entire community, the exact biochemical characterization was determined for only 3% of the hypothetical proteins of interest.
Homology-Based Searches and Biochemical Functions. Homologs were detected by exhaustive psi-blast and rps-blast searches (36, 37) for a significant fraction (97%) of the expressed conserved proteins. Even when these homologs had experimentally characterized functions, the relatively low degree of sequence identity made such functional annotations somewhat uncertain. Nevertheless, in many cases formerly hypothetical proteins could be confidently assigned to a known protein family by using the domain-specific profile search tools provided by the Conserved Domain, COG, or Pfam databases. This approach resulted in a dramatic improvement in the search sensitivity and allowed well defined, or at least general, biochemical functional assignments [Tables 1 (categories 2 and 3) 3, and 5, respectively]. For example, even with the presence of a clearly defined biochemical activity for protein SO0471 (Flavin-dependent dioxygenase), its exact substrate specificity remained uncertain, resulting in a category 2 assignment for this gene (Table 3).
In one notable example, S. oneidensis gene SO1267, predicted to be a glutamine amidotransferase, was found in the same operon with a gene encoding glutamine synthetase (SO1268). Clearly, the combined activities of these two enzymes provides for the conversion of ammonia into an amide group of some unknown substrate at the expense of one ATP molecule, whereas glutamate is recycled:
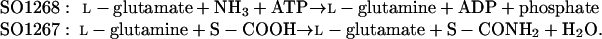 |
[1] |
Unfortunately, the exact nature of the substrate S remains unknown, resulting in assignment of SO1267 (Glutamine synthetase-associated glutamine amidotransferase) to category 2 (Table 3).
Those proteins that could not be assigned to known protein families could sometimes be functionally annotated based on conserved protein sequence motifs or by using other data from the literature. Thus, for the pair of proteins SO0110 and SO0578, both annotated as “metalloprotease,” our revised annotations distinguished the two. SO0110 was reannotated as “metalloprotease, family M48” (Table 5, category 3), which corresponds to a known protein family (COG0501). In contrast, SO0578 was reannotated as “Zn-dependent metalloprotease domain-containing protein,” based on the presence of a signal peptide and a conserved “HExxH” Zn2+-binding motif but only a limited similarity to COG1913 (Table 6, category 4).
General Biological Activity Prediction and Certain Functional Insights. Combining all sources of information in public databases and experimental data can be used to predict general biological function [Tables 1 (category 4) and 5] or at least to get certain functional insights [Tables 1 (category 5) and 7] for many expressed hypothetical proteins. To this end, two genome context data analysis methods were implemented. The first approach focused on operons and chromosomal clusters of genes conserved between diverse species (seed). The second method focused on predicting regulatory binding sites (38) and is summarized in Table 10, which is published as supporting information on the PNAS web site. Information regarding protein–protein interactions was also used but with limited success, as was the case in the recent pilot study (11).
In contrast, structural genomics data made an important complementary contribution toward annotating hypothetical genes. As far as we are aware, S. oneidensis has not been a subject of a focused structural genomics project, so very few solved structures have been deposited in the Protein Data Bank (33). Nonetheless, information on structural domains from the Structural Classification of Proteins-based superfamily database (39) searches by using darwin were valuable for the functional assignments for ≈200 genes (6). In many cases, this structure-based information provided additional supporting evidence for annotations obtained through other means, typically from similarities to proteins with known structure (Table 6, category 4). One such example is SO1195, earlier annotated as a “putative RNA-binding protein (30).” We were able to identify a closely related homolog with known 3D structure and further predict its regulatory binding site (Table 10). As a result, SO1195 has been reannotated as “RNA-binding protein with a KH domain” (Table 6, category 4). To the extent possible at this point, these structure-based results seem to accurately reflect the somewhat-limited contribution of structural genomics to the understanding of protein function on the genome scale (10, 11, 42).
Finally, combining all available data provided only certain functional insights for numerous proteins of interest. For example, no clear functional annotation was obtained for SO0066 and SO0308, so they have been annotated as “conserved extracellular α-helical protein” and “DUF1212 family domain-containing membrane protein with eight transmembrane segments,” respectively [Tables 1 (category 5) and 7]. Altogether, 256 proteins with significantly or modestly improved annotations were identified (categories 1–5; 48%) and are listed in Tables 2, 3, and 5–7.
Proteins Expressed Under Certain Growth Condition(s). Despite comprehensive analysis and careful curation performed by several groups of researchers, we were unable to significantly improve annotations for 52% of the expressed hypothetical proteins. These proteins are listed in Tables 8 and 9 (categories 6 and 7). The key characteristic of the sixth and seventh categories is the distinction between proteins that are conserved in different phylogenetic lineages or at least different genera [Tables 1 (category 6) and 8], and organism- or genus-specific (herein Shewanella-specific) proteins [Tables 1 (category 7) and 9]. For some of these proteins, SO0046 for example, numerous homologs have been identified in diverse organisms, resulting in annotation of this gene as “conserved expressed protein” (Table 8, category 6). For some other proteins, homolog(s) were found only in Shewanella species. For example, the only homolog of SOA0042 was found in Shewanella SAR-1, whose genome sequence was obtained by assembly of Sargasso Sea microbial community DNA sequences (12). Hence, SOA0042 has been annotated as “plasmid-encoded expressed protein in Shewanella” (Table 8, category 7).
S. oneidensis Physiological Capabilities. To further illustrate improvements in our understanding of how the S. oneidensis proteins relate to its physiological capabilities and metabolism, we analyzed new functional assignments of former hypothetical genes. Former hypothetical proteins reannotated in this study were classified according to the COG functional categories by comparing each protein to the COG-based profiles with rps-blast (37) with default parameters. Then, these functional category assignments were manually validated by independent experts and summarized in Table 11, which is published as supporting information on the PNAS web site. We compared these assignments against the COG functional category assignments made for all originally annotated proteins. The distributions of the functional categories among former hypothetical proteins and previously characterized proteins were found to be significantly different (P = 0.0005; Table 11). For example, fewer DNA replication, recombination, and repair proteins (COG functional category L) were found among the former hypothetical genes. This result was expected because these proteins are among the most conserved ones and are therefore easier to characterize by standard similarity approaches. In contrast, proteins involved in secondary metabolism (COG category Q) and posttranslational modification (COG category O) as well as outer membrane proteins (COG category M) were more common among the former hypothetical genes (Table 11).
It is worth noting that S. oneidensis gene distribution among general functional categories is consistent with that observed for other bacteria with relatively large genomes (43, 44). Indeed, S. oneidensis appears to be enriched in genes that code for transcription and its regulation (COG functional category K), signal transduction (COG category T), secondary metabolism (COG category Q), cell motility (COG category N), and energy production and conversion (COG category C) systems, consistent with the data of a recent study (44). For example, several proteins involved in energy production and conversion were previously identified and annotated, including Na+/H+ antiporters and ubiquinone oxidoreductases. Our analyses revealed additional proteins in this class (SO1007, “Na+/H+ antiporter NhaC,” and SO2512, “Na+-translocating ubiquinone oxidoreductase, subunit RnfG”; see Table 4). In addition to the sole flavodoxin (SO2330) found in the original annotation (3), this study added another one (SO1622, “Flavodoxin; chromosomally linked to PTS system glucose transporter PtsG”). We also confirmed the expression of two formerly hypothetical proteins that were subsequently reannotated as FeS center-containing oxidoreductases (SO0311 and SO1520; Table 4). For SO1520, an upstream regulatory binding site was also detected (Table 10).
Table 4. Annotations of representative energy-related ion transport and membrane proteins.
| SO ID no. | Annotation category | COG no. | COG category | Upgraded annotation |
|---|---|---|---|---|
| SO0311 | 3 | 1032 | C | FeS center-containing oxidoreductase |
| SO0363 | 2 | 1210 | M | Nucleoside-diphosphate-sugar pyrophosphorylase |
| SO1007 | 3 | 1757 | C | Na+/H+ antiporter NhaC |
| SO1309 | 3 | 1629 | P | TonB-dependent outer membrane receptor |
| SO1520 | 3 | 0247 | C | FeS center-containing oxidoreductase |
| SO1622 | 3 | 0716 | C | Flavodoxin; chromosomally linked to PTS system glucose transporter PtsG |
| SO2512 | 3 | 4659 | C | Na+-translocating ubiquinone oxidoreductase, subunit RnfG |
| SO2523 | 3 | 1629 | P | TonB-dependent outer membrane receptor; chromosomally linked to phytase |
| SO3340 | 1 | 0668 | M | Mechanosensitive ion channel protein MscS |
| SO3514 | 3 | 1629 | P | TonB-dependent outer membrane receptor |
| SO3667 | 2 | 0748 | P | Heme iron utilization protein HugZ |
| SO3668 | 2 | 3721 | P | Heme iron utilization protein HugX |
| SO4227 | 2 | 0275 | M | S-adenosylmethionine-dependent methyltransferase MraW, involved in cell division |
| SO4680 | 1 | 1887 | M | CDP-glycerol:poly(glycerophosphate) glycerophosphotransferase |
| SO4690 | 2 | 1807 | M | Undecaprenyl phosphate-sugar: lipid A glycosyltransferase |
COG categories: C, energy production and conversion; M, cell envelope biogenesis, outer membrane; and P, inorganic ion transport and metabolism.
The unique respiratory versatility of S. oneidensis may be traced not only to its exceptionally high number of c-type cytochromes (currently estimated to be 42; see ref. 4) but also to the existence of several copies of numerous specialized genes, as noted above. Additionally, we were able to find new genes that code for ion transport proteins (COG category P). For example, this study detected two copies of heme iron utilization proteins (HugZ, SO3667; and HugX, SO3668; Table 4), neither of which was previously recognized. We also were able to improve the annotation of several outer membrane proteins (COG category M), previously described as hypothetical genes. Among others, they include three TonB-dependent outer membrane receptors (SO1309, SO2523, and SO3514; Table 4).
Discussion
Approximately 50–70% of the proteins encoded in any given genome are homologous to genes already annotated in current databases, but each newly sequenced genome adds hundreds to thousands of uncharacterized genes (7–12). This study focused on S. oneidensis strain MR-1 that has recently begun serving as a model microorganism for determining the genetic basis of the metabolic respiratory versatility in metal-reducing Shewanella (3, 4, 14, 16, 17, 23). Transcriptome and proteome analyses resulted in the identification of 538 hypothetical genes as expressed in S. oneidensis cells (Fig. 1). Special emphasis was placed on robust, reproducible, and statistically validated expression results rather than optimizing coverage. It is becoming an imperative for any high-throughput, whole-genome, integrative study to implement proper experimental designs for different types of analyses and develop standards and statistical models, tailored specifically toward different platforms, approaches, and data types (40). These methods, in turn, will help quantify the certainty of identifications enabling researchers to differentiate between clear-cut cases (conservative estimates), less-certain findings or indeterminate data, and clearly noisy data, allowing them to more easily extract biologically relevant information (11, 22, 28, 40).
A seven-category schema for classifying the annotation level of proteins was developed and applied (Table 1) by using experimental data from transcriptome and proteome analyses and a variety of publicly available analysis tools to achieve an improved annotation of the targeted genes. We were also able to identify homologs for nearly all (520, 97% of 538) expressed proteins. The existence of such a large and uncharacterized group of genes that are conserved among a variety of organisms is disconcerting in that it highlights major gaps in our understanding of basic biology (2, 7–13, 22). Using a tiered-based annotation approach, this study significantly or modestly improved annotations for 256 proteins (categories 1–5; 48% of 538). An important advantage of this tiered approach is its ability to prioritize the functional interrogation and validation of these proteins. Formerly hypothetical proteins assigned to categories 2–4 (168, 31% of 538) will have high priority for inclusion in screens for characterization and validation of specific function(s). Only a small number of proteins, i.e., those in category 1 with confident assignments of exact biochemical function (16, 3% of 538), will not require additional analyses.
The new functional annotations for these proteins will help in our understanding of S. oneidensis biology and enhance our ability to deduce the complex networks and pathways responsible for this organism's metabolic versatility through global expression analyses. The newly annotated proteins are predicted to be involved in fundamental metabolic processes, including energy conversion and ion transport, as well as in biosynthesis of specific cell components such as membranes (Table 4). The extensive respiratory versatility of S. oneidensis is likely due, at least in part, to its exceptionally high number of c-type cytochromes (4). Additionally, the S. oneidensis genome contains one of the highest numbers of sensor proteins among all sequenced prokaryotes (3, 45). The ensemble of these sensor proteins includes, among others, 45 histidine kinases, 26 methyl-carrier chemotaxis proteins, and 52 diguanylate cyclase domain proteins (45). The number of two-component response regulators in the S. oneidensis genome is also among the highest observed among sequenced prokaryotes. The information obtained in this study is ultimately expected to lead to a deeper understanding of how S. oneidensis is able to sense and use such a wide range of electron acceptors for anaerobic respiration, including solid phase metal oxides such as Fe2O3, Mn2O3, and MnO2 (1).
Several limitations of the approach used in this study are worth noting. When both high-throughput gene and protein expression data were combined by using stringent criteria, the resulting overlapping dataset (538 proteins) was highly correlated (91%) with the protein expression dataset alone (592 proteins). Careful examination of 53 “dropped” genes showed that (i) some of these genes were not included on the arrays (e.g., SO3039), and (ii) half of these genes were expressed as mRNAs (when less stringent criteria were used), resulting in the overlapping dataset of 566 (96% of 592). Clearly, the protein expression dataset was found to be the most critical in this study. Nongel-based (MS) proteomics methods are highly preferred with regard to protein identifications, as supported by both recent estimates of protein identifications obtained for several microorganisms by applying different proteomics platforms (e.g., 22). The data obtained herein lead to the same observation: 95% of protein identifications were obtained by nongel-based methods. Altogether, with the above limitations applied, when analyzing genes encoding proteins, MS-based proteomics approaches were found to be most effective in this study.
The increasing amount of data obtained from genome sequencing projects and growing databases of sequence profiles enhance the sensitivity of programs such as psi-blast and rps-blast. These factors, along with a variety of comparative genomic approaches, allow researchers to significantly improve genome annotation. This study demonstrated how these approaches and expression data were integrated to gain additional insight into the functions of >250 previously uncharacterized S. oneidensis proteins.
As the list of the hypothetical genes continues to grow, the challenge they pose to our understanding of how genomes actually function also increases (9–11). Integrative studies that combine expression analysis, computational biology, comparative genomics, and careful curation can help to achieve a more comprehensive and accurate annotation of sequenced genomes. These factors, in turn, will create reasonable and verifiable hypotheses for further experimental work toward better understanding and prediction of cellular function and physiology.
Supplementary Material
Acknowledgments
We thank Marvin Frazier, Eugene Koonin, David Lipman, Andrei Osterman, Ross Overbeek, Richard Roberts, and many other people for helpful discussions; David Anderson, William Cannon, Tim Cherny, James Cole, Loren Hauser, Kim Hixson, Ron Moore, Heather Mottaz, and Andrei Osterman for technical assistance; and the reviewers for numerous suggestions, which significantly improved the manuscript. This work was carried out by the Shewanella Federation, supported by the Genomics: GTL program of the Department of Energy's Offices of Biological and Environmental Research and Advanced Scientific Computing Research (Grants DE-FG02-03ER63527, DE-FG02-02ER63427, DE-FG02-01ER63202, and DE-FG02-02ER63342). Argonne, Oak Ridge, and Pacific Northwest National Laboratories are managed for the U.S. Department of Energy by the University of Chicago (Contract W-31-109-ENG-38), the University of Tennessee-Battelle LLC (Contract DE-AC05-00OR22725), and the Battelle Memorial Institute (Contract DE-ACO6-76RLO1830), respectively.
Author contributions: E.K., M.Y.G., M.F.R., F.R.C., R.D.S., C.S.G., J.R.Y., K.H.N., J.K.F., and J.M.T. designed research; E.K., A.F.P., M.Y.G., M.F.R., R.H., K.S.M., N.K., G.A.A., X.Q., K.J.A., G.B., A.S.B., P.E., D.A.E., Y.A.G., T.H., J.A.K., K.T.K., M.L.L., M.S.L., L.-A.M., M.M., L.P.-T., G.P., S.P., M.H.S., S.T., B.A.Z., W.Z., F.W.L., C.E.L., M.R., F.R.C., J.R.Y., R.D.S., C.S.G., K.H.N., J.K.F., and J.M.T. performed research; J.Z. contributed new reagents/analytic tools; E.K., A.F.P., M.Y.G., M.F.R., R.H., K.S.M., N.K., K.H.N., and J.M.T. analyzed data; and E.K., A.F.P., M.Y.G., M.F.R., R.H., K.S.M., X.Q., A.S.B., F.R.C., R.D.S., C.S.G., K.H.N., J.K.F., and J.M.T. wrote the paper.
Abbreviations: COG, Clusters of Orthologous Groups; LC, liquid chromatography.
References
- 1.Myers, C. R. & Nealson, K. H. (1988) Science 240, 1319-1321. [DOI] [PubMed] [Google Scholar]
- 2.Frazier, M. E., Johnson, G. M., Thomassen, D. G., Oliver, C. E. & Patrinos, A. (2003) Science 300, 290-293. [DOI] [PubMed] [Google Scholar]
- 3.Heidelberg, J. F., Paulsen, I. T., Nelson, K. E., Gaidos, E. J., Nelson, W. C., Read, T. D., Eisen, J. A., Seshadri, R., Ward, N., Methe, B., et al. (2002) Nat. Biotechnol. 20, 1118-1123. [DOI] [PubMed] [Google Scholar]
- 4.Meyer, T. E., Tsapin, A. I., Vandenberghe, I., de Smet, L., Frishman, D., Nealson, K. H., Cusanovich, M. A. & van Beeumen, J. J. (2004) OMICS 8, 57-77. [DOI] [PubMed] [Google Scholar]
- 5.Romine, M. F., Elias, D. A., Monroe, M. E., Auberry, K., Fang, R., Fredrickson, J. K., Anderson, G. A., Smith, R. D. & Lipton, M. S. (2004) OMICS 8, 239-254. [DOI] [PubMed] [Google Scholar]
- 6.Serres, M. H. & Riley, M. (2005) OMICS 8, 306-321. [DOI] [PubMed] [Google Scholar]
- 7.Bork, P. (2000) Genome Res. 10, 398-400. [DOI] [PubMed] [Google Scholar]
- 8.Koonin, E. V. & Galperin, M. Y. (2002) Sequence-Evolution-Function. Computational Approaches in Comparative Genomics (Kluwer, Boston). [PubMed]
- 9.Roberts, R. J. (2004) PLoS Biol. 2, E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galperin, M. Y. & Koonin, E. V. (2004) Nucleic Acids Res. 32, 5452-5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolker, E., Makarova, K. S., Shabalina, S., Picone, A. F., Purvine, S., Holzman, T., Cherny, T., Armbruster, D., Munson, R. S., Jr., Kolesov, G., et al. (2004) Nucleic Acids Res. 32, 2353-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venter, J. C., Reminton, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., et al. (2004) Science 304, 66-74. [DOI] [PubMed] [Google Scholar]
- 13.Thomas, G. H. (1999) Bioinformatics 15, 860-861. [DOI] [PubMed] [Google Scholar]
- 14.Qiu, X., Sundin, G. W., Chai, B. & Tiedje, J. M. (2004) Appl. Environ. Microbiol. 70, 6435-6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kostka, J. & Nealson, K. H. (1998) in Techniques in Microbial Ecology, eds. R. S. Burlage, Atlas, R., Stahl, D., Geesey, G. & Sayler, G. (Oxford Univ. Press, New York), pp. 58-78.
- 16.Beliaev, A. S., Thompson, D. K., Khare, T., Lim, H., Brandt, C. C., Li, G., Murray, A. E., Heidelberg, J. F., Giometti, C. S., Yates, J., III, et al. (2002) OMICS 6, 39-60, and erratum (2003) 7, 138-139. [DOI] [PubMed] [Google Scholar]
- 17.Gao, H., Wang, Y., Liu, X., Yan, T., Wu, L., Alm, E., Arkin, A., Thompson, D. K. & Zhou, J.-Z. (2004) J. Bacteriol. 186, 7796-7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ideker, T. E., Thorsson, V., Siegel, A. & Hood, L. (2000) J. Comp. Biol. 7, 805-817. [DOI] [PubMed] [Google Scholar]
- 19.Washburn, M. P., Wolters, D. & Yates, J. R., III (2001) Nat. Biotechnol. 19, 242-247. [DOI] [PubMed] [Google Scholar]
- 20.Keller, A., Purvine, S., Nesvizhskii, A. I., Stolyar, S., Goodlett, D. R. & Kolker, E. (2002) OMICS 6, 207-212. [DOI] [PubMed] [Google Scholar]
- 21.Lipton, M. S., Pasa-Tolic, L., Anderson, G. A., Anderson, D. J., Auberry, D. L., Battista, J. R., Daly, M. J., Fredrickson, J., Hixson, K. K., Kostandarithes, H., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 11049-11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolker, E., Purvine, S., Galperin, M. Y., Stolyar, S., Goodlett, D. R., Nesvizhskii, A. I., Keller, A., Xie, T., Eng, J. K., Yi, E., et al. (2003) J. Bacteriol. 185, 4593-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giometti, C. S., Khare, T., Tollaksen, S. L., Tsapin, A., Zhu, W., Yates, J. R., III, & Nealson, K. H. (2003) Proteomics 3, 777-785. [DOI] [PubMed] [Google Scholar]
- 24.Purvine, S., Picone, A. F. & Kolker, E. (2004) OMICS 8, 79-92. [DOI] [PubMed] [Google Scholar]
- 25.Masselon, C., Anderson, G. A., Harkewicz, R., Bruce, J. E., Pasa-Tolic, L. & Smith, R. D. (2000) Anal. Chem. 72, 1918-1924. [DOI] [PubMed] [Google Scholar]
- 26.Belov, M. E., Anderson, G. A., Wingerd, M. A., Udseth, H. R., Tang, K., Prior, D. C., Swanson, K. R., Buschbach, M. A., Strittmatter, E. F., Moore, R. J., et al. (2004) J. Am. Soc. Mass. Spectrom. 15, 212-232. [DOI] [PubMed] [Google Scholar]
- 27.Strittmatter, E. F., Ferguson, P. L., Tang, K. & Smith, R. D. (2003) J. Am. Soc. Mass Spectrom. 14, 980-991. [DOI] [PubMed] [Google Scholar]
- 28.Higdon, R., Kolker, N., Picone, A. F., van Belle, G. & Kolker, E. (2005) OMICS 8, 356-369. [DOI] [PubMed] [Google Scholar]
- 29.Marchler-Bauer, A., Anderson, J. B., DeWeese-Scott, C., Fedorova, N. D., Geer, L. Y., He, S., Hurwitz, D. I., Jackson, J. D., Jacobs, A. R., Lanczycki, C. J., et al. (2003) Nucleic Acids Res. 31, 383-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278, 631-637. [DOI] [PubMed] [Google Scholar]
- 31.Mulder, N. J., Apweiler, R., Attwood, T. K., Bairoch, A., Barrell, D., Bateman, A., Binns, D., Biswas, M., Bradley, P., Bork, P., et al. (2003) Nucleic Acids Res. 31, 315-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002) Nucleic Acids Res. 30, 276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourne. P. E., Westbrook. J. & Berman, H. M. (2004) Brief. Bioinformatics 5, 23-30. [DOI] [PubMed] [Google Scholar]
- 34.Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., Martin, M. J., Michoud, K., O'Donovan, C., Phan, I., et al. (2003) Nucleic Acids Res. 31, 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larimer, F. (2000) Brief. Bioinformatics 1, 415-416. [DOI] [PubMed] [Google Scholar]
- 36.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zheng, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchler-Bauer, A. & Bryant, S. H. (2004) Nucleic Acids Res. 32, 327-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCue, L. A., Thompson, W., Carmack, C. S. & Lawrence, C. E. (2002) Genome Res. 12, 1523-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gough, J., Karplus, K., Hughey, R. & Chothia, C. (2001) J. Mol. Biol. 313, 903-919. [DOI] [PubMed] [Google Scholar]
- 40.Holzman, T. & Kolker, E. (2004) Curr. Opin. Biotechnol. 15, 52-57. [DOI] [PubMed] [Google Scholar]
- 41.Peng, J., Elias, J. E., Thoreen, C. C., Licklider, L. J. & Gygi, S. P. (2003) J. Proteome Res. 2, 43-50. [DOI] [PubMed] [Google Scholar]
- 42.Frishman, D. (2003) OMICS 7, 211-224. [DOI] [PubMed] [Google Scholar]
- 43.van Nimwegen, E. (2003) Trends Genet. 19, 479-484. [DOI] [PubMed] [Google Scholar]
- 44.Konstantinidis, K. T. & Tiedje, J. M. (2004) Proc. Natl. Acad. Sci. USA 101, 3160-3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galperin, M. Y. (2004) Environ. Microbiol. 6, 552-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.