

Abstract
Alzheimer’s disease (AD) is the most common type of dementia afflicting the elderly. In addition to the presence of cortical senile plaques and neurofibrillary tangles, AD is characterized at autopsy by extensive degeneration of brainstem locus coeruleus (LC) neurons that provide noradrenergic innervation to cortical neuropil, together with relative stability of dopaminergic neuron number in substantia nigra (SN) and ventral tegmental area (VTA). The present study used design-based stereological methods to assess catecholaminergic neuronal loss in brains of double transgenic female mice that co-express two human mutations associated with familial AD, amyloid precursor protein (APPswe) and presenilin-1 (PS1ΔE9). Mice were analyzed at two age groups, 3–6 mos and 16–23 mos, when deposition of AD-type β-amyloid (Aβ) plaques occurs in cortical brain regions. Blocks of brain tissue containing the noradrenergic LC nucleus and two nuclei of dopaminergic neurons, the SN and VTA, were sectioned and sampled in a systematic-random manner and immunostained for tyrosine hydroxylase (TH), a specific marker for catecholaminergic neurons. Using the optical fractionator method we found a 24% reduction in the total number of TH-positive neurons in LC with no changes in SN-VTA of aged dtg APP/PS1 mice compared with non-transgenic controls. No significant differences were observed in numbers of TH-positive neurons in LC or SN-VTA in brains of young female dtg APP/PS1 mice compared to their age-matched controls. The findings of selective neurodegeneration of LC neurons in the brains of aged female dtg APP/PS1 mice mimic the neuropathology in the brains of AD patients at autopsy. These findings support the use of murine models of Aβ deposition to develop novel strategies for the therapeutic management of patients afflicted with AD.
Keywords: Substantia Nigra, Ventral Tegmental Area, Unbiased Stereology, APP/PS1 Mice, Tyrosine Hydroxylase
Alzheimer’s disease (AD) is a debilitating, age-related neurological disease characterized by progressive deterioration of cognitive function, including severe impairments in memory and learning; deposition of congophilic β-amyloid (Aβ) plaques and neurofibrillary tangles containing ubiquinated tau protein; and, neuronal loss (Alzheimer 1907; Aletrino et al., 1992; Mirra et al., 1993; Busch et al., 1997). The pathological markers for the diagnosis of AD, the deposition of amyloid plaques and tangles, appear first in hippocampus and surrounding temporal lobe of the brain, and in later stages spread to all the cortical areas (Hyman et al., 1984; Braak and Braak, 1991; West et al., 1994). AD appears to selectively affect neuronal systems associated with cognitive and sensory processes such as hippocampus and cortex (Vogels et al., 1990; Busch et al., 1997; Aletrino et al., 1997), while sparing motor systems and their underlying biological substrate. At autopsy the brains of AD patients have significant reductions in total numbers of locus coeruleus (LC) neurons, the major source of brain norepinephrine (NE) (Swanson and Hartman, 1976; Busch et al., 1997), compared to the relative stability of LC neurons in normal aging (Mouton, et al., 1994; Ohm et al., 1997). In contrast, the substantia nigra (SN) and ventral tegmental area (VTA), two mesencephalic nuclei that project to striatum and neocortex, respectively, are relatively spared in concert with stable motor function in AD.
The introduction of transgenic strategies for the in-vivo expression of genetic mutations associated with familial AD, including the amyloid precursor protein (APP) and presenilin-1 (PS1), have provided important tools for understanding neural reactions to the deposition of mutant Aβ proteins in the mouse brain and developing novel approaches for the therapeutic management of AD in humans (Games et al., 1995; Hsiao et al., 1995; Malherbe et al., 1996; Hardy, 1997; Johnson-Wood et al., 1997; Strurchler-Pierrat et al., 1997; Morgan et al., 2000; Wang et al. 2003). In line with the view of gender differences reported in AD (Molsa et al., 1982; Jorm et al., 1987; Hagnell et al., 1992; Letenneur et al., 1994; Brayne et al., 1995; Fratiglioni et al., 1997, 2000), female dtg APP/PS1 mice appear to accumulate Aβ at an earlier age and to deposit more amyloid plaques in the hippocampus than the age-matched males (Wang et al. 2003; Callahan et al., 2001). One of these mouse lines co-express the so-called Swedish APP mutation (APPswe) and the E9 presenilin-1 (PS1ΔE9) mutations (Borchelt et al., 1997). By 3–4 months of age these mice express high levels of mutant APP, PS1 and Aβ, and by 5 months of age deposit substantial numbers of Aβ–containing amyloid plaques which, like other lines of single and double transgenic mice, closely resemble the histological appearance of those found in AD (Frautschy et al. 1998; Holcomb et al., 1998; McGowan et al., 1999; Gordon et al., 2000; Selko, 2001). To help characterize the neuropathological similarity between AD and dtg APP/PS1 mice, we used unbiased stereological approaches to quantify total neuron numbers in the noradrenergic LC and dopaminergic SN-VTA in two groups of young and aged dtg APP/PS1 female mice and age-matched wild-type littermate controls.
Materials and Methods
Mice
Brains from 27 female mice were raised at the Laboratory of Experimental Gerontology at the Gerontology Research Center (GRC, NIA/NIH) in Baltimore, MD. The numbers per group, ages, and genotypes of the mice were as follows: 3–6 mos old dtg APP/PS1 [(APPswe, PS1dE9)85Dbo/J; PrP promoter] (n=6, young dtg APP/PS1); and 16–23 months old (n=7, aged dtg APP/PS1); and, age-matched, non-transgenic littermate controls (n=5 young, n=9 aged).
Mice were group housed (2–5) in plastic cages with corncob bedding, with ad lib. access to food (NIH formula 07) and filtered water in a vivarium maintained on a 12:12 h light:dark cycle, and a temperature of 22 +/− 2°C. The colony was certified as specific pathogen free for the following pathogens: mouse pneumonia virus, sendai virus, hepatitis virus, reovirus, lymphocytic choriomeningitis, Theiler Martin encephalomyelitis virus, ectromelia (poxvirus), minute virus of mouse and mocplasma pulmonis. The GRC is accredited and animal care and treatment followed the guidelines of the American Association for the Accreditation of Laboratory Animal Care.
Tissue Preparation
Mice were sacrificed by perfusion with 4% paraformaldehyde, and brains were removed and stored at −80°C until sectioning. Each brain was serially sectioned in the coronal plane on a cryostat at an instrument setting of 50μm. For stereological studies the LC and SN-VTA were sampled in a systemic-random manner, as detailed elsewhere (Mouton 2002).
Immunohistochemistry
For visualization of TH-positive neurons, systematic-random sections through the LC, SN and VTA, a total 8–10 sections per region per brain, were collected in 12-well plates and washed in 0.1 M PBS, incubated in 1% hydrogen peroxide for 30 minutes at room temperature (RT), washed again in PBS 0.1 M, and placed in 0.3% Triton X-100 for ten minutes RT. Sections were washed in PBS 0.1 M then transferred into 5% normal goat serum in 0.1 M PBS for 30 minutes at RT to block non-specific binding. Sections were incubated overnight in rabbit anti-tyrosine hydroxylase antibody (polyclonal, Chemicon International, Temecula, CA, USA) diluted to 1:3000 with 2% normal goat serum and 0.3% Triton X-100 in PBS at 4°C. After incubation, sections were washed in PBS 0.1 M and incubated in biotinylated secondary anti-rabbit antibody (Vector Laboratories, Burlingame, CA, USA) with normal goat serum in PBS 0.1 M for 90 minutes at RT. Sections were washed in PBS 0.1 M and re-incubated for another 90 minutes in ABC solution from the Vectorstain Kit (Vector Laboratories, Burlingame, CA, USA) at RT. Sections were rinsed in PBS 0.1M and colorized using DAB (10 mg DAB, 40 ml PBS 0.1M) for six to ten minutes. Additional sections from each group were immunostained for qualitative assessment of TH-immunopositive fibers in hippocampus, and cortex. All TH-immunostained sections were lightly counterstained in a 0.1% solution of cresyl violet, rinsed, dehydrated through an ascending graded series of alcohol, and cleared in xylene and coverslipped with DPX. To visualize amyloid deposits, an adjacent set of sections through each brain was counterstained with Congo red, as reported previously (Lee et al., 2005).
Stereology
Total numbers of TH-positive in two reference spaces (LC and SN-VTA) were quantified using the optical fractionator method (West et al., 1991) as previously detailed (Long et al., 1998; Mouton et al., 2002; Lei et al., 2003; for review of relevant stereological techniques, see Mouton 2002). For both reference spaces, the sampling fractions were as follows: section sampling fraction for LC (ssf=1) and for SN/VTA (ssf=0.333), the number of sections sampled divided by the total number of sections; the area sampling fraction for LC (asf=0.2844) and for SN/VTA (asf=0.4444), the area of the sampling frame divided by the area of the x-y sampling step; and, the thickness sampling fraction for LC (tsf=0.5882) and for SN/VTA (tsf=0.625), the height of the disector divided by the section thickness. The reference space on each section was outlined under low power magnification (4X NA=0.10), and TH-positive cells were visualized under oil immersion magnification (60X, NA=1.4). The counting item was the presence of TH immunoreactivity in a cell with a neuronal phenotype. To avoid artifacts (e.g., lost caps) at the sectioning surface, a guard volume of 2–3 um was used. Sampling was continued to 100 to 150 neurons per reference space until a mean coefficient of error (CE) for each group was between 5 and 10% (Gundersen et al. 1999). The average section thickness for both regions was 16 ± 0.27 (SEM) μm.
Statistical Analysis
Differences between groups were assessed using the Student’s two-tailed t-test, with statistical significance at the p<0.05 level.
Results
To expand our initial qualitative examination of TH-positive neurons in dtg APP/PS1 and control mice (Figure 1), total numbers of TH immunopositive neurons in LC, SN-VTA of female dtg APP/PS1 mice and non-tg littermate controls were quantified by design-based stereology. In the group of older dtg APP/PS1 mice and age-matched, non-transgenic littermate controls there were significantly fewer TH-positive neurons (24%) in the LC (p < 0.01; Figure 2). In a follow-up study in young mice there was no genotype effect on the total number of TH-positive neurons in the LC (Figure 3). For both young and old mice there were no differences in the total numbers of TH-positive neurons in SN-VTA in dtg APP/PS1 compared to age-matched controls (Figure 4). Qualitative assessment of sections through the hippocampus, and cortex indicated a robust loss of TH-positive fiber staining in these regions (data not shown).
Figure 1.
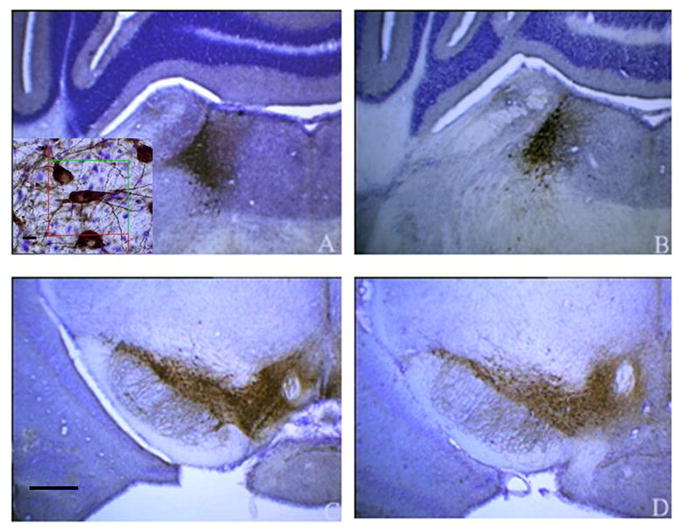
Low magnification (4X) photomicrographs of TH-positive neurons in (A) LC from non-tg littermate controls aged 3–6 mos; (B) LC from dtg APP/PS1 mice aged 16–23 mos; (C) SN-VTA from non-tg littermate controls aged 3–6 mos; (D) SN-VTA from dtg APP/PS1 mice aged 16–23 mos. Insert: High magnification (60 x) with an optical disector of TH positive cells. Scale bar= 50μm
Figure 2.
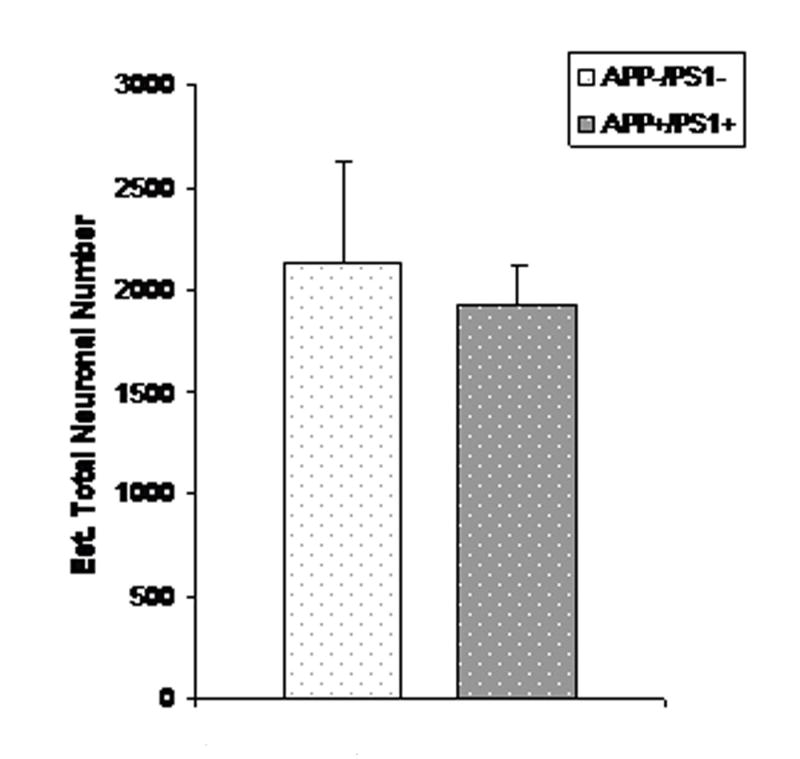
Mean (SEM) total number of TH-positive neurons in LC of 16–23 month old female APP/PS1 mice (n=7) and age-matched non-tg littermate controls. (n=9); p<0.01.
Figure 3.
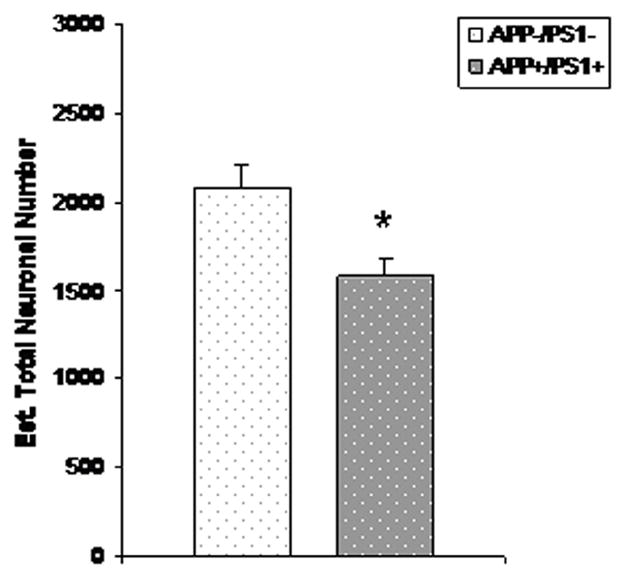
Mean (SEM) total number of TH-positive neurons in LC of 3–6 month old female APP/PS1 mice (n=7) and age-matched non-tg littermate controls (n=9).
Figure 4.
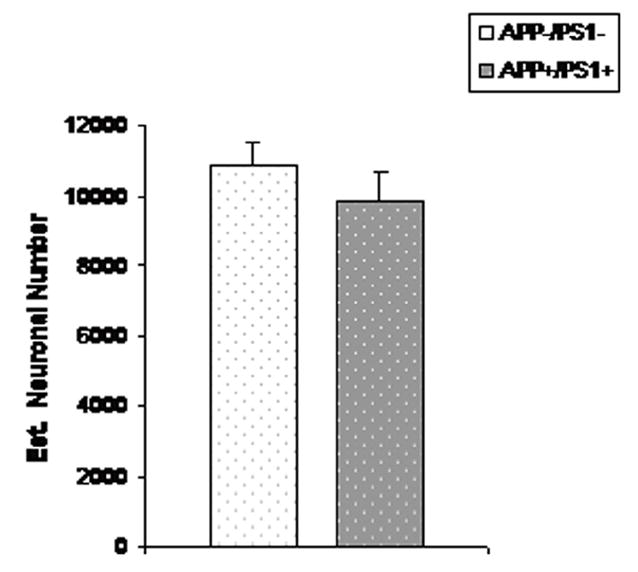
Mean (SEM) total number of TH-positive neurons in SN-VTA of 16–23 month old female APP/PS1 mice (n=7) and age-matched non-tg littermate controls. (n=9).
Discussion
The underlying neurobiological basis for the severe, progressive cognitive decline and underlying neuropathological changes in AD remains unknown. To date, studies to characterize the neuropathological effects in transgenic mouse models of AD have focused on neurodegeneration in hippocampus (Calhoun et al., 1998; Casa et al., 2004; Schmitz et al., 2004; Dickson 2004), the brain region thought to contain the critical memory circuits that underlie short-term memory in the mammalian brain. The LC provides tonic noradrenergic innervation to hippocampus and other cortical brain regions, and is thought to play an important role in the regulation of normal cognitive abilities such as attention, learning and memory (German et al. 2005, Aston-Jones et al., 1999). During normal aging these cognitive functions remain generally stable (Kawas et al., 2000) in concert with stability in the total number of TH-positive neurons in LC (Mouton et al., 1994; Ohm et al. 1997). In contrast, cognitive abilities diminish in AD in association with degeneration of transmitter-specific subcortical nuclei, including noradrenergic (Busch et al., 1997; Swaab et al., 1997), cholinergic (Vogels et al., 1990) and serotinergic neurons (Aletrino et al., 1992). Importantly, the catecholaminergic neuron loss in AD is selective for TH-positive neurons in LC that synthesize and release noradrenaline, while dopaminergic SN and VTA neurons are generally spared in concert with the extrapyramidal motor abilities mediated by these nuclei (Lyness et al., 2003).
The initial purpose of this study was to test whether expression of human APP and PS1 mutations was associated with loss of TH positive neurons in LC. To test this idea, we applied design-based stereology to a group of dtg APP/PS1 female mice aged 16–23 months and their age- and gender-matched, non-tg littermate controls. The results of these studies showed a significant 24% reduction of TH-positive neurons in the LC of dtg APP/PS1 mice compared to their controls, which confirmed our hypothesis. Careful immunostaining in the cortical projection regions from the LC revealed a loss of TH fibers in the hippocampus and cortex. To test whether this genotype effect was selective for the LC, we used the same stereology approaches to quantify the total number of TH-positive neurons in the SN-VTA region. This study revealed no differences in SN-VTA, supporting the view that the observed loss of TH-positive neurons in the aged female dtg APP/PS1 mice was limited to the LC.
In a follow-up study we addressed the question of whether the loss of LC neurons in the dtg APP/PS1 mice represents a degenerative vs. developmental disorder, i.e., an effect that is present in older but not younger mice, rather than an effect present at birth. To test this idea we used design-based stereology to quantify numbers of TH-positive neurons in a group of young dtg APP/PS1 mice and non-tg controls. The lack of a genotype effect in these young mice indicates that the observed differences reflect a degenerative, rather than development effect.
The finding of selective catecholaminergic neurodegeneration bears an apparent similarity to autopsy findings in AD patients compared to age-matched controls (Manaye et al., 1995; Busch et al., 1997; Swaab et al., 1997). It would be premature, however, to speculate that this apparent similarity arises from the same mechanism in AD patients and dtg APP/PS1 mice. Among the possible mechanisms are that the observed LC degeneration stems from retrograde degeneration of cortical projections arising in the LC. In this scenario an initial degeneration in the terminal projections in cortical tissue undergoing reactive inflammation to Aβ peptides could lead to the eventual degeneration of TH-positive neurons in the LC. Alternatively, loss of noradrenergic innervation from the pontine brainstem, secondary to primary loss of TH neurons in LC, could be an early event in the pathogenic cascade that includes neuroglia proliferation and activation and the formation of the characteristic amyloid plaques in AD. Further studies in the dtg APP/PS1 mice are needed to carefully characterize the temporal pattern of LC degeneration in association with the appearance of Aβ deposits. In one recent study, Heneka et al. (2006) used chronic treatment with N-(2-chloroethyl)-N-ethyl-bromo-benzylamine (DSP-4) to mice that express a single APP mutation to induce LC degeneration. Interestingly, though these mice normally show relatively light deposition of Aβ peptides in cortical tissue, after chronic DSP-4 treatment there was substantially greater deposition of Aβ plaques, robust neurogliosis in cortical regions, and reduced spatial memory (Heneka et al., 2006). Thus, loss of noradrenergic innervation to cortical tissue appeared to stimulate deposition of insoluble Aβ peptides, possibly in association with activation of adrenergic receptors on neuroglial cells.
Previous studies have reported AD-type pathology in single (APP) and double (APP/PS1) transgenic mouse models of AD (Guela et al., 1998, Guenette and Tanzi, 1999, Callahan et al., 2001, Reilly et al., 2003), including deficits in cognitive performance (Richard et al., 2003, Huang et al., 2003, Palop et al., 2003), increased neuroglial activation (Bradt et al., 1998, Schwab et al. 1997, Meda et al., 1995) and degeneration of neurons in hippocampal formation but not in LC (Casas et al., 2004; Schmitz et al., 2004; for review, see German and Eisch 2004). Here we present evidence that aged female dtg APP/PS1 mice show an AD-type loss of noradrenergic neurons in LC. Among the possible explanations for this difference is variability in the individual APP and PS1 mutations; differences in the resultant deposition of Aβ; and differences in the ages of mice examined across these studies. Our future research will continue to examine possible mechanisms that underlie this degeneration in APP/PS1 mice. Understanding these mechanisms could help to better appreciate the biological basis of age-related neurogeneration, and possibly lead to novel strategies for the therapeutic management of AD in the elderly population.
Acknowledgments
The authors wish to acknowledge support from the Public Health Service (NIH extramural grants SNRP2U54NS039407-07, MH076541-02A1), the NIA Intramural Program, and the American Health Assistance Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alzheimer A. Ueber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie. 1907;64:146–148. [Google Scholar]
- Aletrino MA, Vogels OJ, Van Domburg PH, Ten Donkelaar HJ. Cell loss in the nucleus raphes dorsalis in Alzheimer’s disease. Neurobiol Aging. 1992;13(4):461–8. doi: 10.1016/0197-4580(92)90073-7. [DOI] [PubMed] [Google Scholar]
- Astony-Jones G, Rajkowski J, Cohen J. Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry. 1999;46:1309–1320. doi: 10.1016/s0006-3223(99)00140-7. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Busch C, Bohl J, Ohm TG. Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging. 1997;18:401–406. doi: 10.1016/s0197-4580(97)00035-3. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J Exp Med. 1998 Aug 3;188(3):431–8. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayne C, Gill C, Paykel ES, Huppert F, O’Connor DW. Cognitive decline in an elderly population--a two wave study of change. Psychol Med. 1995;25:673–683. doi: 10.1017/s0033291700034930. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der KN, Vingtdeux V, van de SE, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW. Building a more perfect beast: APP transgenic mice with neuronal loss. Am J Pathol. 2004;164:1143–1146. doi: 10.1016/S0002-9440(10)63202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratiglioni L, Viitanen M, von Strauss E, Tontodonati V, Herlitz A, Winblad B. Very old women at highest risk of dementia and Alzheimer’s disease: incidence data from the Kungsholmen Project, Stockholm. Neurology. 1997;48:132–138. doi: 10.1212/wnl.48.1.132. [DOI] [PubMed] [Google Scholar]
- Fratiglioni L, Launer LJ, Andersen K, Breteler MM, Copeland JR, Dartigues JF, Lobo A, Martinez-Lage J, Soininen H, Hofman A. Incidence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S10–S15. [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irizzary M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPswe transgenic mice. AM J Pathol. 1998;152:307–17. [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373(6514):523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- German DC, Eisch AJ. Mouse models of Alzheimer’s disease: insight into treatment. Rev Neurosci. 2004;15:353–369. doi: 10.1515/revneuro.2004.15.5.353. [DOI] [PubMed] [Google Scholar]
- German DC, Nelson O, Liang F, Liang CL, Games D. The PDAPP mouse model of Alzheimer’s disease: locus coeruleus neuronal shrinkage. J Comp Neurol. 2005;492:469–76. doi: 10.1002/cne.20744. [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O’Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol Feb. 2002;173:183–95. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Guenette SY, Tanzi RE. Progress toward valid transgenic mouse models for Alzheimer’s disease. Neurobiol Aging. 1999;20:201–211. doi: 10.1016/s0197-4580(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology--reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Hagnell O, Ojesjo L, Rorsman B. Incidence of dementia in the Lundby Study. Neuroepidemiology. 1992;11(Suppl 1):61–66. doi: 10.1159/000110981. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus coeruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb I, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzaen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada C, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Huang XG, Yee BK, Nag S, Chan ST, Tang F. Behavioral and neurochemical characterization of transgenic mice carrying the human presenilin-1 gene with or without the leucine-to-proline mutation at codon 235. Exp Neurol. 2003;183:673–681. doi: 10.1016/s0014-4886(03)00242-5. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and AB42 deposition in a transgenic mouse model of Alzheimers disease. Proc Natl Acad Sci. 1997;94:1550–5. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorm AF, Korten AE, Henderson AF. The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand. 1987;76:465–79. doi: 10.1111/j.1600-0447.1987.tb02906.x. [DOI] [PubMed] [Google Scholar]
- Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: the Baltimore Longitudinal Study of Aging. Neurology. 2000;54:2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
- Lee GD, Aruna JH, Barret PM, Lei D-L, Ingram DK, Mouton PR. Stereological analysis of microvascular parameters in a double transgenic model of Alzheimer’s disease. Brain Research Bulletin. 2005;65:317–322. doi: 10.1016/j.brainresbull.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Lei DL, Long JM, Hengemihle J, O’Neill J, Manaye KF, Ingram DK, Mouton PR. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience. 2003;121:659–666. doi: 10.1016/s0306-4522(03)00245-8. [DOI] [PubMed] [Google Scholar]
- Letenneur L, Commenges D, Dartigues JF, Barberger-Gateau P. Incidence of dementia and Alzheimer’s disease in elderly community residents of south-western France. Int J Epidemiol. 1994;23:1256–1261. doi: 10.1093/ije/23.6.1256. [DOI] [PubMed] [Google Scholar]
- Long JM, Kalehua AN, Muth NJ, Hengemihle JM, Jucker M, Calhoun ME, Ingram DK, Mouton PR. Stereological estimation of total microglia number in mouse hippocampus. J Neurosci Methods. 1998;84:101–108. doi: 10.1016/s0165-0270(98)00100-9. [DOI] [PubMed] [Google Scholar]
- Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. NeurobiolAging. 2003;24:1–23. doi: 10.1016/s0197-4580(02)00057-x. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Richards JG, Martin JR, Bluethmann H, Maggio J, Huber G. Lack of beta-amyloidosis in transgenic mice expressing low levels of familial Alzheimer’s disease missense mutations. Neurobiol Aging. 1996;17:205–214. doi: 10.1016/0197-4580(95)02070-5. [DOI] [PubMed] [Google Scholar]
- Manaye KF, McIntire DD, Mann DM, German DC. Locus coeruleus cell loss in the aging human brain: a non-random process. J Comp Neurol. 1995;358:79–87. doi: 10.1002/cne.903580105. [DOI] [PubMed] [Google Scholar]
- McGowan E, Sanders S, Iwatsubo T, Takeuchi A, Saido T, Zehr C, Yu X, Uljon S, Wang R, Mann D, Dickson D, Duff K. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis. 1999;6:231–244. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Hart MH, Terry RD. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists. Arch Pathol Lab Med. 1993;117:132–144. [PubMed] [Google Scholar]
- Molsa PK, Marttila RJ, Rinne UK. Epidemiology of dementia in a Finnish population. Acta Neurol Scand. 1982;65:541–552. doi: 10.1111/j.1600-0404.1982.tb03109.x. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Mouton PR. Principles And Practices Of Unbiased Stereology: An Introduction For Bioscientists. The Johns Hopkins University Press; Baltimore, MD: 2002. [Google Scholar]
- Mouton PR, Long JM, Lei DL, Howard V, Jucker M, Calhoun ME, Ingram DK. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Res. 2002;956:30–35. doi: 10.1016/s0006-8993(02)03475-3. [DOI] [PubMed] [Google Scholar]
- Mouton PR, Pakkenberg B, Gundersen HJ, Price DL. Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J Chem Neuroanat. 1994;7:185–90. doi: 10.1016/0891-0618(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Busch C, Bohl J. Unbiased estimation of neuronal numbers in the human nucleus coeruleus during aging. Neurobiol Aging. 1997;18:393–399. doi: 10.1016/s0197-4580(97)00034-1. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci. 2003;100:9572–7. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JF, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M, Biacabe AG, Streichenberger N, Ironside JW, Mohr M, Kopp N, Perret-Liaudet A. Immunohistochemical localization of 14.3.3 zeta protein in amyloid plaques in human spongiform encephalopathies. Acta Neuropathol (Berl) 2003;105:296–302. doi: 10.1007/s00401-002-0642-5. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2004;164:1495–1502. doi: 10.1016/S0002-9440(10)63235-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc Natl Acad Sci. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab DF, Salehi A. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1997;56:216. [PubMed] [Google Scholar]
- Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. 1975;163:467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- Vogels OJ, Broere CA, ter Laak HJ, ten Donkelaar HJ, Nieuwenhuys R, Schulte BP. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puolivali J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloid-beta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318–327. doi: 10.1016/j.nbd.2003.08.009. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Ross CA, Hedreen JC, Steele C, Cardillo JE, Whitehouse PJ, Folstein MF, Price L. The neuropathology of aminergic nuclei in Alzheimer’s disease. Ann Neurol. 1988;24:233–242. doi: 10.1002/ana.410240210. [DOI] [PubMed] [Google Scholar]