

Abstract
Prostate cancer is often slowly progressive, and it can be difficult to treat with conventional cytotoxic drugs. Nonsteroidal antiinflammatory drugs inhibit the development of prostate cancer, but the mechanism of chemoprevention is unknown. Here, we show that the R-enantiomer of the nonsteroidal antiinflammatory drug etodolac inhibited tumor development and metastasis in the transgenic mouse adenocarcinoma of the prostate (TRAMP) model, by selective induction of apoptosis in the tumor cells. This proapoptotic effect was associated with loss of the retinoid X receptor (RXRα) protein in the adenocarcinoma cells, but not in normal prostatic epithelium. R-etodolac specifically bound recombinant RXRα, inhibited RXRα transcriptional activity, and induced its degradation by a ubiquitin and proteasome-dependent pathway. The apoptotic effect of R-etodolac could be controlled by manipulating cellular RXRα levels. These results document that pharmacologic antagonism of RXRα transactivation is achievable and can have profound inhibitory effects in cancer development.
Keywords: cancer, prostate, R-etodolac, ubiquitin, chemoprevention
Several epidemiological studies have shown that the use of nonsteroidal antiinflammatory drugs (NSAIDs) is associated with a reduced incidence of clinically detectable prostate cancer (1–3). The side effects of cyclooxygenase (COX) inhibitors preclude the use of these agents in many elderly men (4). Thus, there is a major need to determine whether there are chemopreventative, and antimetastatic effects of NSAIDs that can be separated from COX inhibition. Controversy has permeated this field, because many of the COX-independent actions of NSAIDs are measurable only at concentrations that are not safely achievable in vivo, and because the members of this structurally diverse group of drugs are metabolized extensively and can exert different mechanisms of action (5, 6).
Various NSAIDs have been demonstrated to induce apoptosis in malignant cells (7–9). Etodolac is a commercially available NSAID containing a racemic mixture, in which the S-enantiomer has COX inhibitory activity, whereas the R-enantiomer does not (10). Unlike all other chiral NSAIDs, the two enantiomers of etodolac are not metabolically interconvertible. Moreover, the R-enantiomer is metabolized much more slowly than the S-enantiomer, and it accumulates to 10-fold higher concentrations than the S-enantiomer in plasma (11). In a recent study, sufficient plasma levels of R-etodolac were achieved after oral gavage in a xenograft prostate cancer model to diminish the growth of the transplanted tumor (12).
The in vivo effect of R-etodolac was associated with enhancement of peroxisome proliferator-activated receptor γ (PPARγ) transactivation (12). PPARγ, as well as other nuclear hormone receptors, forms heterodimers with the retinoid X receptor α (RXRα) (13, 14), which has been implicated in the pathogenesis of prostate cancer (15). The effect of RXRα may be due to its induction of apoptosis through its interaction with other proteins (16–19). An activating ligand of RXRα, 9-cis-retinoic acid (RA), is a lipophilic acid similar to R-etodolac. Thus, we hypothesized that the COX-independent effects of R-etodolac in malignant prostate cells might be attributed to its binding and modulation of RXRα activities.
Here, we show that R-etodolac induced apoptosis of prostate cancer cells, but not normal prostatic epithelial cells. The R-etodolac-induced apoptosis was associated with reduction of RXRα levels selectively in the tumor cells. Direct interaction of R-etodolac and RXRα was demonstrated by in vitro binding of radiolabeled R-etodolac with the purified recombinant ligand-binding domain (LBD) of RXRα and the ability of the drug to protect RXRα protein from trypsin digestion. In intact cells, R-etodolac antagonized RXRα transcriptional activity and induced its ubiquitination and degradation. Furthermore, suppression of RXRα expression reduced the apoptotic effect of R-etodolac. These results demonstrate that RXRα acts as a receptor that mediates the COX-independent anticancer effects of R-etodolac.
Materials and Methods
Drug Preparation. R-etodolac was prepared from pharmaceutical-grade tablets of racemic etodolac to a purity of >97% as described (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site) (20). The drug was tritiated by Sibtech (Newington, CT) and purified with HPLC. The resulting material had a specific activity of 20–25 Ci (1 Ci = 37 GBq)/mmol, and it was stored in acetonitrile at a concentration of 0.45 mCi/ml at -20°C. The RXR-selective retinoids SR11237, SR11345, and SR11246 were described in ref. 21 and provided by M. Dawson (The Burnham Institute). Staurosporine, MG-132 (Calbiochem) and 9-cis-RA (Sigma) were purchased commercially.
Cell Lines. LNCaP, PrEC, CV-1, ZR-75–1, and HEK 293T cells were maintained in standard media. The F9 murine embryonal carcinoma cell line with both alleles of RXRα disrupted was provided by P. Chambon (Institut de Genetique et de Biologie Moleculaire et Cellulaire, College de France, Illkirch, France) (22). See Supporting Materials and Methods for details.
Murine Studies. Transgenic mouse adenocarcinoma of the prostate (TRAMP) and C57BL/6 mice were purchased from The Jackson Laboratory and bred at UCSD. All animal protocols received prior approval by the institutional review board. Plasma R-etodolac levels were measured on a group of seven 15-week-old C57BL/6 male mice who were fed R-etodolac (1.25 mg/kg) chow for 2 weeks by a bioanalytical LC/MS-based method developed by Maxxam Analytics (Mississauga, ON, Canada). Chiral HPLC (23) was used to confirm the lack of R- to S-etodolac in vivo interconversion.
We started 46 male TRAMP mice at 9–12 weeks of age on chow with R-etodolac 1.25 mg/kg or control food (prepared by Dyets, Bethlehem, PA) randomized by cage. At 30 weeks, or after appearance of a gross palpable tumor mass, the animals were sacrificed and necropsies were performed. The urogential system, the periaortic lymph nodes, and the major organs were removed and weighed. The prostatic tissues were dissected and separated into individual lobes and weighed. Tissues were fixed in 10% formalin embedded in paraffin, sectioned in step sections at 50-μm intervals, and stained with hematoxylin and eosin. The prostate sections were scored for carcinoma grade on a 1–6 scale (see Supporting Materials and Methods). The liver, lung, and lymph node sections were scored for the presence or absence of tumor. The weights of the different tissues, and the frequencies of metastases, in the drug treated and control animals were compared by the Mann–Whitney test or Fisher's exact test, with P < 0.05 considered significant.
Ligand Binding. The human RXRα LBD (223–462), prepared as a polyhistidine-tagged fusion protein in pET15b (Novagen) (1 μg), was incubated with radiolabeled ligand in the presence of different concentrations of unlabeled 9-cis-RA or R-etodolac at 4°C for 14 h. The RXRα LBD was captured by nickel-coated beads. Bound radiolabeled ligand was determined in a scintillation counter.
Immunohistochemistry and Apoptosis Assays. For 2 weeks, we fed 6- to 7-month-old TRAMP mice R-etodolac supplemented or control chow. The prostates were removed, and serial frozen sections were assayed for terminal deoxyribonucleotidyl transferase (TdT; TUNEL; Chemicon) or stained with anti-human RXRα (D20; Santa Cruz Biotechnology), followed by staining with DAPI (50 μg/ml; Sigma) containing DNase-free RNase A (100 μg/ml; Boehringer Mannheim) to visualize the nuclei and examined by fluorescence microscopy (18, 19, 24). Single-cell apoptosis was detected in vitro by removing adherent cells from the plate with 5 mM EDTA, incubating them with annexin-V–phycoerythrin (BD PharMingen) and analyzing by flow cytometry.
Transient-Transfection Assay. Expression vectors for RXRα, RA receptor β (RARβ), hemagglutinin (HA)-ubiquitin, and reporter gene βRARE-tk-chloramphenicol transferase (CAT) were prepared and transfected as described (25, 26). We mixed ≈300 ng of reporter plasmid, 50 ng of β-gal expression vector (pCH 110; Pharmacia), and vector expressing RXRα with carrier DNA (pBluescript) to give 1.0 μg of total DNA per well. CAT activity was normalized for transfection efficiency on the basis of cotransfected β-gal gene activity. Transfected cell lysates were separated by SDS/PAGE and immunoblotted (see Supporting Materials and Methods).
RXRα Small Interfering RNA (siRNA) Transfections. A SMARTpool of siRNAs specific for RXRα and GFP control siRNA were puchased from Dharmacon. A 10-μl aliquot of 20 μM siRNA per well was transfected into cells in six-well plates by using Lipofectamine Plus (Invitrogen) (17).
Results
Antiproliferative and Proapoptotic Effects of R-Etodolac. R-etodolac dose dependently inhibited the proliferation of LNCaP prostate cancer cells and PrEC normal human prostatic epithelial cells (see Fig. 7, which is published as supporting information on the PNAS web site). At 72 h, the ID50 values were ≈150 μM for LNCaP, compared with ≈400 μM for PrEC (Fig. 7a). Concentrations of R-etodolac of >500 μM induced apoptosis in primary prostate cancer explants (Fig. 7 b–e). In the latter instance, the malignant cells displayed shrunken and pyknotic nuclei, whereas the nuclei from the adjacent normal prostatic epithelium appeared morphologically normal.
Activity of R-Etodolac in the TRAMP Model. Preliminary dose-ranging pharmacokinetic data showed that plasma concentrations of 370 ± 30 μM could be achieved by supplementing a standard mouse chow diet with 1.25 mg/kg R-etodolac for 2 weeks. Chiral HPLC revealed no detectable conversion of R-etodolac to the S-stereoisomer. Therefore, in vivo experiments in the TRAMP mouse model were undertaken under these conditions. Male TRAMP mice develop histological intraepithelial neoplasia of the prostate by 8–12 weeks of age that progresses to adenocarcinoma with distant site metastases by 24–28 weeks of age (27, 28). Control chow or diets supplemented with R-etodolac were initiated at 9–12 weeks. By 30 weeks, nearly all of the prostates in the R-etodolac treated and untreated groups had macroscopic evidence of tumor (Table 1). However, both the average tumor mass (Fig. 1) and the frequencies of metastases (Table 1) were significantly lower in the R-etodolac-treated animals. Histological evaluation of the excised tissues confirmed the anti-metastatic effects of the drug and did not show evidence of drug toxicity (Fig. 2 a–l). Collectively, these data indicated that R-etodolac retarded the progression and metastasis of prostate cancer in the TRAMP system.
Table 1. Incidence of primary tumors and metastases.
| Measurement | Control | Treated |
|---|---|---|
| Primary tumor incidence* | 24/24 (100%) | 16/17 (94%) |
| Metastasis incidence† | 14/24 (58%) | 5/17 (29%) |
| Animals with gross masses‡ | 6/24 (25%) | 2/17 (12%) |
Percentages of mice are given in parentheses.
No. of mice with histologic evidence of carcinoma (grade ≥4)
No. of mice found to have histologic evidence of metastasis to lymph node, lung, or liver tissues. P < 0.05 by Fischer's exact test
No. of mice found to have a gross urogenital mass at postmortem laparotomy
Fig. 1.

Inhibition of prostate cancer progression in the TRAMP model. Male TRAMP mice were fed control chow or chow with R-etodolac (1.25 mg/kg). (a)At 30 weeks of age, or after development of a gross palpable mass, the mice were sacrificed, and the urogenital systems were removed and weighed. (b–f) The prostate lobes (b) were separated from the other organs and weighed separately; anterior (c), ventral (d), lateral (e), and posterior (f). The prostates were not dissectible in mice that had gross tumor masses (two in the treatment group and six in the control group). The weights of the control urogenital tracts, lateral, and dorsal prostates were significantly higher than those of the treated group, as determined by the Mann–Whitney test.
Fig. 2.

Histological evaluation of R-etodolac-treated TRAMP prostate tissues. Examples of ventral (a), dorsal (c), and lateral (e) lobes of the prostate in an untreated 30-week-old TRAMP mouse are shown. For comparison, histology sections of the ventral (b), dorsal (d), and lateral (f) prostate from an R-etodolac treated TRAMP mouse at 30 weeks of age are shown. Metastases in the lymph-nodes (g), lungs (i), and liver (k) of an untreated TRAMP mouse at 30 weeks of age are shown also. Examples of lymph nodes (h), lung (j), and liver (l)in R-etodolac treated TRAMP mice at 30 weeks of age are shown also. (m) Induction of tumor cell apoptosis by R-etodolac. For 2 weeks, we fed 6- to 7-month-old TRAMP mice with R-etodolac-supplemented chow or control chow, and they were then sacrificed. The prostate lobes were removed and frozen in OCT, sectioned, and subjected to TUNEL staining (red) and DAPI (to visualize nuclei) (×200). Extensive apoptosis (TUNEL-positive) was detected in the prostates of R-etodolac-fed TRAMP mice compared with control chow-fed mice.
R-Etodolac Selectively Induces Apoptosis in Cancerous Prostates. To determine whether R-etodolac treatment resulted in apoptosis in vivo, 6- to 7-month-old male TRAMP and nontransgenic littermates were fed with R-etodolac or control chow for 2 weeks and sacrificed, and the prostates were examined for apoptosis. Extensive apoptosis in the prostates of drug-treated TRAMP mice was seen on TUNEL staining of frozen sections, whereas no apoptosis was seen in mice fed with control chow (Fig. 2m). No apoptosis was detectable in prostates of nontransgenic mice fed with R-etodolac.
RXRα Is Required for the Apoptotic Effect of R-Etodolac. A previous study demonstrated that PPARγ was associated with the apoptosis induced by R-etodolac (12). PPARγ heterodimerizes with RXRα, and thus, we examined its possible role. Transfection of RXRα- specific siRNA, but not control siRNA, almost completely inhibited RXRα expression in LNCaP cells (Fig. 3a). The apoptotic effect of R-etodolac in RXRα siRNA-transfected cells was substantially diminished (Fig. 3b). CV-1 cells lack detectable levels of RXRα (25, 26), and they were relatively resistant to R-etodolac-induced apoptosis. However, transfection of CV-1 cells with RXRα, but not a control vector, reversed this drug-resistant phenotype (Fig. 3c). Similarly RXRα-/- F9 embryonal cells did not undergo apoptosis with drug treatment compared with the extensive apoptosis in wild-type F9 cells (Fig. 3d). Thus, RXRα is required for the apoptotic effect of R-etodolac.
Fig. 3.

RXRα is required for R-etodolac-induced apoptosis. (a) Inhibition of RXRα expression by RXRα siRNA. LNCaP cells were untransfected or transfected with a pool of RXRα siRNAs or control GFP siRNA for 48 h. Cell lysates were analyzed by immunoblotting with antibodies against RXRα or α-tubulin. (b) RXRα siRNA suppresses the apoptotic effect of R-etodolac. LNCaP cells were transfected with RXRα or control GFP siRNA for 48 h and then treated with 500 μM or 1 mM R-etodolac for 24 h in medium containing 0.5% FBS. Apoptotic cells were quantified by annexin-V binding and flow cytometry. Similar results were obtained in two separate experiments. The percentage of apoptosis represents the percentage of annexin-V-positive drug-treated cells minus the percentage of annexin-V-positive untreated transfected cells. (c) Expression of RXRα confers apoptotic sensitivity of R-etodolac in CV-1 cells. CV-1 cells were transfected with GFP or GFP-RXRα in six-well plates. After overnight transfection, cells were treated with either 0.1% DMSO (vehicle) or 1 mM R-etodolac for 28 h, harvested, and fixed, and nuclei were stained by DAPI. Nuclear morphology of the GFP-positive cells was visualized by fluorescence microscopy and cells showing nuclear condensation and fragmentation were scored as apoptotic cells. Shown are averages ± means from two independent evaluations of at least 200 transfected cells. (d) RXRα-/- F9 cells are resistant to the apoptotic effect of R-etodolac. F9 cells or F9 cells without RXR (F9 cellsΔRXR) were treated with DMSO or R-etodolac as indicated. Apoptosis was determined after 44 h of treatment. Bars represent mean + SD from three independent experiments.
R-Etodolac Binds to RXRα. To investigate whether R-etodolac could bind RXRα, a ligand competition assay with [3H]9-cis-RA was used. Both unlabeled 9-cis-RA and R-etodolac displaced [3H]9-cis-RA bound to RXRα LBD (Fig. 4a), with an IC50 value of ≈200 μM for R-etodolac. Furthermore, [3H]R-etodolac directly bound the RXRα LBD, and this binding was competitively inhibited by both unlabeled R-etodolac and 9-cis-RA (Fig. 4 b and c).
Fig. 4.

R-etodolac binds to RXRα. (a) R-etodolac competes with 9-cis-RA for binding to RXRα. Human RXRα LBD was incubated with 1 nM [3H]9-cis-RA in the presence of different concentrations of unlabeled 9-cis-RA (•) or R-etodolac (□) at 4°C for 14 h. [3H]9-cis-RA bound to RXRα was measured after capturing the RXRα LBD by nickel-coated beads. The data represent the average of total bound cpm ± SEM. One of five experiments is shown. (b) R-etodolac binds directly to RXRα. The indicated concentrations of [3H]R-etodolac were incubated with or without polyhistidine-tagged-RXRα LBD. RXRα-bound [3H]R-etodolac was separated with nickel-coated beads and measured. One of three independent experiments is shown. (c) 9-cis-RA competes with R-etodolac for binding of RXRα. Purified RXRα LBD (1 μg) was incubated with 1 mM [3H]R-etodolac in the presence of unlabeled 9-cis-RA (10-6 M) or R-etodolac (1 mM) as indicated. RXRα-bound [3H]R-etodolac was separated and assayed as described above.
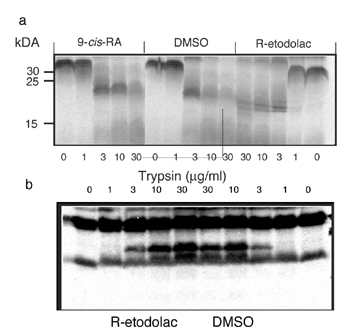
R-Etodolac Binding Induces Conformational Change in RXRα. Binding of ligands to their receptors often induces changes in susceptibility to proteolysis (13, 14). Digestion of the RXRα LBD with a low concentration of trypsin (3 μg/ml) yielded a proteolytic fragment of ≈20 kDa, whreeas higher concentrations of trypsin (10 or 30 μg/ml) completely digested the LBD (see Fig. 8, which is published as supporting information on the PNAS web site). Preincubation of the RXRα LBD with 9-cis-RA did not alter its sensitivity to trypsin digestion, consistent with previous studies (29). However, incubation of the RXRα LBD with R-etodolac before trypsin digestion (3 μg/ml) resulted in a different digestion pattern, with two new proteolytic fragments of ≈18 kDa, in contrast to the lack of protection detected with PPARγ (Fig. 8).
R-Etodolac Modulates RXRα Transcriptional Activity. To test whether R-etodolac binding modulated RXRα transcriptional activity, a reporter gene containing RXRα homodimer-responsive elements, (TREpal)2-tk-CAT (26), was transfected with a RXRα expression vector into CV-1 cells. Treatment of cells with 9-cis-RA strongly induced reporter gene activity, whereas treatment with R-etodolac did not (Fig. 5a). However, when transfected cells were treated with 9-cis-RA, the addition of R-etodolac dose dependently reduced the transcriptional activity of RXRα.
Fig. 5.

R-etodolac inhibits 9-cis-RA-induced transcriptional activity of RXRα. (a) Inhibition of RXRα homodimer activity by R-etodolac. CV-1 cells were cotransfected with or without an expression vector for RXRα (25 ng), a CAT reporter vector containing RXR homodimer responsive elements [(TREpal)2-tk-CAT; 300 ng] and a β-gal expression vector (50 ng). After subsequent treatment with 9-cis-RA (10-8 M) and the indicated concentrations of R-etodolac for 24 h CAT activities were determined and normalized relative to the β-gal activity. (b) R-etodolac modulation of RXRα/RAR heterodimer activity. ZR-75-1 cells were transfected with βRARE-tk-CAT reporter plasmid (300 ng) and a β-gal expression vector (50 ng), and they were then treated with all-trans-RA (10-7 M) and the indicated concentrations of R-etodolac. CAT activities were determined as described above. (c) Inhibition of trans-RA-induced RARβ protein expression by R-etodolac. ZR-75-1 cells were treated with or without trans-RA (10-6 M), R-etodolac (1 mM), or their combination for 24 h. RARβ protein expression was determined by Western blot analysis. (d) Inhibitory effect of R-etodolac on RXRα/PPARγ heterodimer activity. ZR-75-1 cells were transfected with βRARE-tk-CAT reporter plasmid (300 ng) and a β-gal expression vector (50 ng), treated with RXR ligand SR11237 (10-6 M) and PPARγ ligand ciglitazone (10-5 M) as well as R-etodolac and then CAT activities were determined. (e) R-etodolac inhibits RARβ protein expression induced by the combination of RXR and PPARγ ligands. ZR-75-1 cells were treated with or without SR11237 (10-6 M), ciglitazone (10 μM), R-etodolac (1 mM) in the indicated combinations. Cell lysates were immunoblotted and probed for relative levels of RARβ and β-actin.
We investigated whether R-etodolac inhibited transactivation of endogenous RXRα. An RA-response element (βRARE) in the RARβ promoter, which binds various RXR-containing heterodimers including RXR/RAR and RXR/PPARγ (26, 30, 31), was transfected into ZR-75–1 breast cancer cells. Reporter activity was induced by all-trans-RA, presumably because of binding of endogenous RXR/RAR heterodimer. Cotreatment with R-etodolac suppressed all-trans-RA-induced reporter activity in a R-etodolac concentration-dependent manner, suggesting that R-etodolac inhibited transcriptional activity of endogenous RXR/RAR heterodimers (Fig. 5b). The transcriptional effect of R-etodolac on RXRα was confirmed by analyzing its effect on the expression of RARβ. Treatment of ZR-75-1 cells with all-trans-RA strongly induced the endogenous expression of RARβ (Fig. 5c), which was completely inhibited by R-etodolac, consistent with the inhibitory effect of R-etodolac on the βRARE reporter gene (Fig. 5b). To determine the effect of R-etodolac on RXR/PPARγ heterodimer activity, ZR-75-1 cells were stimulated with the PPARγ ligand ciglitazone and the RXRα ligand SR11237 in the presence or absence of R-etodolac (Fig. 5 d and e) (30). The induction of endogenous RARβ expression by the two drugs in combination was abolished by R-etodolac cotreatment (Fig. 5e).
Loss of RXRα Expression After R-Etodolac Treatment. Subcellular localization of RXRα plays a role in the regulation of apoptosis (17). To analyze whether R-etodolac treatment altered RXRα subcellular localization, in vivo immunostaining of RXRα was performed on the prostate tissues from male TRAMP and nontransgenic littermates fed with R-etodolac or control chow. RXRα was predominantly localized in the nucleus in the prostates of the nontransgenic mice fed with or without R-etodolac chow (Fig. 6a). TRAMP mice fed with control chow displayed similar RXRα nuclear staining. However, RXRα staining was greatly reduced in prostates of TRAMP mice fed with R-etodolac.
Fig. 6.

R-etodolac induces RXRα degradation. (a) Diminished RXRα staining in prostate of R-etodolac-fed TRAMP mice. For 2 weeks, we fed 6- to 7-month-old TRAMP mice R-etodolac-supplemented chow or control chow. The prostate tissues were immunostained for human RXRα (red), and the nuclei were stained with DAPI (×200). (b) R-etodolac induces RXRα degradation. LNCaP cells were untreated, treated with 1 mM R-etodolac for 18 h, treated with R-etodolac with pretreatment for 1 h with 20 μM MG132, or treated with MG132 alone. Cell lysates were immunoblotted with antibodies to human RXRα and α-tubulin. (c) R-etodolac induces ubiquitination of RXRα. Expression vectors for myc-tagged RXRα or RARβ, and ubiquitin (HA-tagged) were transfected into HEK 293T cells. After 24 h, the cells were treated with R-etodolac (1 mM), lysed after 24 h, and immunoprecipitated with an anti-myc antibody. The precipitated proteins were immunoblotted with an anti-HA antibody to detect ubiquitinated protein. (d) R-etodolac induces ubiquitination of endogenous RXRα. HEK 293T cells were transfected with a vector expressing HA-ubiquitin and then treated with R-etodolac as described above and 1 μM synthetic retinoids as indicated. The lysates were immunoprecipitated with an anti-HA antibody and immunoblotted with an antibody to detect RXRα. (e) Synthetic RXRα ligands inhibit R-etodolac-induced ubiquitination of RXRα. Expression vectors for RXRα (myc-tagged) and ubiquitin (HA-tagged) were transfected into HEK 293T cells. Cells were treated with 1 μM synthetic retinoids, SR11237, SR11345, or SR11246, as indicated. Cell lysates were immunoprecipitated with an anti-myc antibody and the precipitated proteins were immunoblotted with an anti-HA antibody to detect ubiquitinated protein.
RXRα protein levels in LNCaP cells were also markedly reduced after treatment with R-etodolac (Fig. 6b). R-etodolac-induced degradation of RXRα levels was completely prevented by the proteosome inhibitor MG132 (Fig. 6b). Proteins are often ubiquitinated before degradation by proteasomes (32). Thus, we determined whether R-etodolac induced ubiquitination of RXRα. Myc-tagged RXRα was cotransfected into HEK 293T cells with or without an expression vector for HA-tagged ubiquitin, followed by treatment with R-etodolac in the presence of MG132. Immunoprecipitation with anti-myc antibody, followed by immunoblotting with an anti-HA antibody, revealed that RXRα was extensively ubiquitinated after R-etodolac treatment (Fig. 6c) but not after treatment with the synthetic RXRα ligands SR11345 and SR11246 (Fig. 6d). Instead, these ligands abrogated R-etodolac-induced RXRα ubiquitination (Fig. 6e), probably because of their competition for binding to RXRα. Collectively, these results demonstrate that R-etodolac binds RXRα and induces its degradation in a proteasome-dependent manner.
Discussion
The standard therapy for progressive prostate cancer is androgen ablation. However, many patients become unresponsive and develop metastatic disease (33). Thus, there is a compelling need for the development of unconventional agents that can delay the progression of prostate cancer. In this article, we report that chronic oral administration of the COX-inactive R-stereoisomer of the common NSAID etodolac inhibited tumor expansion and metastasis in the TRAMP model. By analogy, R-etodolac could be a prospective agent for the treatment of human prostate cancer.
In the TRAMP model, treatment with the COX-2 selective agent celecoxib or the R-enantiomer of the NSAID flurbiprofen resulted in a significantly lower primary-tumor incidence and a reduced incidence of metastases (34, 35). However, both of these drugs may have exerted their effect by active COX inhibition because 15% of the R-flurbiprofen was converted to the active COX inhibitor S-flurbiprofen by 2–4 h after administration. In contrast, the stereoisomers of the conformationally rigid etodolac molecule, unlike all other approved racemic NSAIDs, cannot undergo chiral transformation under physiologic conditions. Indeed, S-etodolac was undetectable in the plasmas of the mice given diets supplemented with the R-stereoisomer. Hence, the cytostatic and antimetastatic effects of R-etodolac in the TRAMP model must be attributed to the drug or to a metabolite.
The results presented here reveal an unexpected function of RXRα as a mediator of the apoptotic effect of R-etodolac. A recent study (12) demonstrated that inhibition of prostate tumor growth by R-etodolac was associated with initial enhancement of PPARγ transcriptional activity, followed by degradation of the receptor (12). However, ligand competition and direct binding assays using PPARγ recombinant protein failed to demonstrate any direct binding of R-etodolac to PPARγ (data not shown). Because PPARγ activity depends on heterodimerization with RXRα, it is possible that modulation of PPARγ and degradation by R-etodolac is mediated by its binding to RXRα. Antagonists of RXR homodimers are known to function as agonists of RXR/PPARγ heterodimers (36, 37).
Exactly how RXRα mediates the apoptotic effects of R-etodolac remains unknown. It is unlikely that R-etodolac exerts its anticancer effect through its inhibition of RXRα transactivation (Fig. 5) because many RXRα agonists potently inhibit the growth of prostate cancer cells. However, the binding of RXRα by R-etodolac could affect the function and stability of several nuclear receptors that dimerize with RXRα, including PPARγ and Nur77.
Our results demonstrate that R-etodolac induced apoptosis of prostate cancer, but not normal epithelium (Fig. 2 and 7). The contrasting effects might be attributable to differences in RXRα posttranslational processing in cancer and normal cells (38). In this regard, it was reported recently that RXRα was phosphorylated by MAP kinase in surgically resected hepatocellular carcinoma samples but not in the corresponding noncancerous surrounding tissues (39).
A recent population-based study of NSAID use and prostate cancer revealed that the relative odds of prostate cancer among the drug users was 0.2 (95% confidence interval 0.1–0.5) in men during the eighth decade of life but only 0.9 in men during the sixth decade, compared with similarly aged men who did not use NSAIDs (3). The stronger effect among older men raised the possibility that NSAIDs may prevent the progression of prostate cancer from latent to clinical disease, rather than reduce the frequency of primary lesions. The experimental results with R-etodolac in the TRAMP model of prostate cancer display many parallels with the human population data. Thus, it is possible that R-etodolac could represent a potential approach toward preventing the progression of hormone refractory prostate cancer, especially in the very elderly patients who are not candidates for cytotoxic therapy.
Supplementary Material
Acknowledgments
We thank M. Rosenbach, B. Crain, S. Wu, K. Pekny, P. Charos, J. Wei, R. Tawatao, Y. Zhao, J. Stebbins, F. Lin, N. Bruey-Sedano, A. A. Bhattacharya, J. Kim, and J. Town for technical assistance. We also thank N. Noon for secretarial support. This work was supported by grants from CaPCURE (Association for the Cure of Cancer of the Prostate), the National Institutes of Health, the U.S. Army Medical Research and Materiel Command, the University of California Biotechnology Strategic Targets for Alliances in Research Project (BioSTAR), the Susan B. Komen Foundation, and the California Tobacco-Related Disease Research Program. Some of the histologic sections were performed by the Cancer Center Histology Core Facility, which is supported in part by a National Institutes of Health grant.
Author contributions: M.C., X.-k.Z., and D.A.C. designed research; S.J. and H.B.C. contributed new reagents/analytic tools; S.K., M.C., S.J., M.B., D.L., W.L., and L.M.L. performed research; S.K., M.C., M.B., D.L., W.L., H.B.C., L.M.L., X.-k.Z., and D.A.C. analyzed data; and S.K., M.C., X.-k.Z., and D.A.C. wrote the paper.
Abbreviations: HA, hemagglutinin; NSAID, nonsteroidal antiinflammatory drug; COX, cyclooxygenase; RXR, retinoid X receptor; TRAMP, transgenic mouse adenocarcinoma of the prostate; LBD, ligand-binding domain; PPARγ, peroxisome proliferator-activated receptor γ; RA, retinoic acid; CAT, chloramphenicol transferase; siRNA, small interfering RNA; RAR, RA receptor; RARE, RA-response element.
References
- 1.Norrish, A. E., Jackson, R. T. & McRae, C. U. (1998) Int. J. Cancer 77, 511-515. [DOI] [PubMed] [Google Scholar]
- 2.Nelson, J. E. & Harris, R. E. (2000) Oncol. Rep. 7, 169-170. [DOI] [PubMed] [Google Scholar]
- 3.Roberts, R. O., Jacobson, D. J., Girman, C. J., Rhodes, T., Lieber, M. M. & Jacobsen, S. J. (2002) Mayo Clin. Proc. 77, 219-225. [DOI] [PubMed] [Google Scholar]
- 4.Allison, M. C., Howatson, A. G., Torrance, C. J., Lee, F. D. & Russell, R. I. (1992) N. Engl. J. Med. 327, 749-754. [DOI] [PubMed] [Google Scholar]
- 5.Grosch, S., Tegeder, I., Niederberger, E., Brautigam, L. & Geisslinger, G. (2001) FASEB. J. 15, 2742-2744. [DOI] [PubMed] [Google Scholar]
- 6.Raz, A. (2002) Biochem. Pharmacol. 63, 343-347. [DOI] [PubMed] [Google Scholar]
- 7.Sheng, H., Shao, J., Morrow, J. D., Beauchamp, R. D. & DuBois, R. N. (1998) Cancer Res. 58, 362-366. [PubMed] [Google Scholar]
- 8.Shiff, S. J., Koutsos, M. I., Qiao, L. & Rigas, B. (1996) Exp. Cell Res. 222, 179-188. [DOI] [PubMed] [Google Scholar]
- 9.Sawaoka, H., Kawano, S., Tsuji, S., Tsujii, M., Gunawan, E. S., Takei, Y., Nagano, K. & Hori, M. (1998) Am. J. Physiol. 274, G1061-G1067. [DOI] [PubMed] [Google Scholar]
- 10.Demerson, C. A., Humber, L. G., Abraham, N. A., Schilling, G., Martel, R. R. & Pace-Asciak, C. (1983) J. Med. Chem. 26, 1778-1780. [DOI] [PubMed] [Google Scholar]
- 11.Brocks, D. R. & Jamali, F. (1994) Clin. Pharmacokinet. 26, 259-274. [DOI] [PubMed] [Google Scholar]
- 12.Hedvat, M., Jain, A., Carson, D. A., Leoni, L. M., Huang, G., Holden, S., Lu, D., Corr, M., Fox, W. & Agus, D. B. (2004) Cancer Cell 5, 565-574. [DOI] [PubMed] [Google Scholar]
- 13.Kastner, P., Mark, M. & Chambon, P. (1995) Cell 83, 859-869. [DOI] [PubMed] [Google Scholar]
- 14.Mangelsdorf, D. J. & Evans, R. M. (1995) Cell 83, 841-850. [DOI] [PubMed] [Google Scholar]
- 15.Huang, J., Powell, W. C., Khodavirdi, A. C., Wu, J., Makita, T., Cardiff, R. D., Cohen, M. B., Sucov, H. M. & Roy-Burman, P. (2002) Cancer Res. 62, 4812-4819. [PubMed] [Google Scholar]
- 16.Liu, B., Lee, H. Y., Weinzimer, S. A., Powell, D. R., Clifford, J. L., Kurie, J. M. & Cohen, P. (2000) J. Biol. Chem. 275, 33607-33613. [DOI] [PubMed] [Google Scholar]
- 17.Cao, X., Liu, W., Lin, F., Li, H., Kolluri, S. K., Lin, B., Han, Y.-H., Dawson, M. I. & Zhang, X.-k. (2004) Mol. Cell. Biol. 24, 9705-9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, H., Kolluri, S. K., Gu, J., Dawson, M. I., Cao, X., Hobbs, P. D., Lin, B., Chen, G., Lu, J., Lin, F., et al. (2000) Science 289, 1159-1164. [DOI] [PubMed] [Google Scholar]
- 19.Lin, B., Kolluri, S. K., Lin, F., Liu, W., Han, Y. H., Cao, X., Dawson, M. I., Reed, J. C. & Zhang, X. K. (2004) Cell 116, 527-540. [DOI] [PubMed] [Google Scholar]
- 20.Lu, D., Zhao, Y., Tawatao, R., Cottam, H. B., Sen, M., Leoni, L. M., Kipps, T. J., Corr, M. & Carson, D. A. (2004) Proc. Natl. Acad. Sci. USA 101, 3118-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dawson, M. I. & Zhang, X. K. (2002) Curr. Med. Chem. 9, 623-637. [DOI] [PubMed] [Google Scholar]
- 22.Clifford, J., Chiba, H., Sobieszczuk, D., Metzger, D. & Chambon, P. (1996) EMBO J. 15, 4142-4155. [PMC free article] [PubMed] [Google Scholar]
- 23.Jamali, F., Mehvar, R., Lemko, C. & Eradiri, O. (1988) J. Pharm. Sci. 77, 963-966. [DOI] [PubMed] [Google Scholar]
- 24.Kolluri, S. K., Bruey-Sedano, N., Cao, X., Lin, B., Lin, F., Han, Y. H., Dawson, M. I. & Zhang, X. K. (2003) Mol. Cell. Biol. 23, 8651-8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang, X. K., Lehmann, J., Hoffmann, B., Dawson, M. I., Cameron, J., Graupner, G., Hermann, T., Tran, P. & Pfahl, M. (1992) Nature 358, 587-591. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, X. K., Hoffmann, B., Tran, P. B., Graupner, G. & Pfahl, M. (1992) Nature 355, 441-446. [DOI] [PubMed] [Google Scholar]
- 27.Greenberg, N. M., DeMayo, F., Finegold, M. J., Medina, D., Tilley, W. D., Aspinall, J. O., Cunha, G. R., Donjacour, A. A., Matusik, R. J. & Rosen, J. M. (1995) Proc. Natl. Acad. Sci. USA 92, 3439-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gingrich, J. R., Barrios, R. J., Morton, R. A., Boyce, B. F., DeMayo, F. J., Finegold, M. J., Angelopoulou, R., Rosen, J. M. & Greenberg, N. M. (1996) Cancer Res. 56, 4096-4102. [PubMed] [Google Scholar]
- 29.Leid, M. (1994) J. Biol. Chem. 269, 14175-14181. [PubMed] [Google Scholar]
- 30.James, S. Y., Lin, F., Kolluri, S. K., Dawson, M. I. & Zhang, X. K. (2003) Cancer Res. 63, 3531-3538. [PubMed] [Google Scholar]
- 31.Wu, Q., Li, Y., Liu, R., Agadir, A., Lee, M. O., Liu, Y. & Zhang, X. (1997) EMBO J. 16, 1656-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nawaz, Z. & O'Malley, B. W. (2004) Mol. Endocrinol. 18, 493-499. [DOI] [PubMed] [Google Scholar]
- 33.Culine, S. & Droz, J. P. (2000) Ann. Oncol. 11, 1523-1530. [DOI] [PubMed] [Google Scholar]
- 34.Gupta, S., Adhami, V. M., Subbarayan, M., MacLennan, G. T., Lewin, J. S., Hafeli, U. O., Fu, P. & Mukhtar, H. (2004) Cancer Res. 64, 3334-3343. [DOI] [PubMed] [Google Scholar]
- 35.Wechter, W. J., Leipold, D. D., Murray, E. D., Jr., Quiggle, D., McCracken, J. D., Barrios, R. S. & Greenberg, N. M. (2000) Cancer Res. 60, 2203-2208. [PubMed] [Google Scholar]
- 36.Lala, D. S., Mukherjee, R., Schulman, I. G., Koch, S. S., Dardashti, L. J., Nadzan, A. M., Croston, G. E., Evans, R. M. & Heyman, R. A. (1996) Nature 383, 450-453. [DOI] [PubMed] [Google Scholar]
- 37.Cesario, R. M., Klausing, K., Razzaghi, H., Crombie, D., Rungta, D., Heyman, R. A. & Lala, D. S. (2001) Mol. Endocrinol. 15, 1360-1369. [DOI] [PubMed] [Google Scholar]
- 38.Matsushima-Nishiwaki, R., Shidoji, Y., Nishiwaki, S., Yamada, T., Moriwaki, H. & Muto, Y. (1996) Mol. Cell. Endocrinol. 121, 179-190. [DOI] [PubMed] [Google Scholar]
- 39.Matsushima-Nishiwaki, R., Okuno, M., Adachi, S., Sano, T., Akita, K., Moriwaki, H., Friedman, S. L. & Kojima, S. (2001) Cancer Res. 61, 7675-7682. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}