

Abstract
Background
The circadian clock regulates plant metabolic functions and is an important component in plant health and productivity. Rhizosphere bacteria play critical roles in plant growth, health, and development and are shaped primarily by soil communities. Using Illumina next-generation sequencing and high-resolution mass spectrometry, we characterized bacterial communities of wild-type (Col-0) Arabidopsis thaliana and an acyclic line (OX34) ectopically expressing the circadian clock-associated cca1 transcription factor, relative to a soil control, to determine how cycling dynamics affected the microbial community. Microbial communities associated with Brachypodium distachyon (BD21) were also evaluated.
Results
Significantly different bacterial community structures (P = 0.031) were observed in the rhizosphere of wild-type plants between light and dark cycle samples. Furthermore, 13% of the community showed cycling, with abundances of several families, including Burkholderiaceae, Rhodospirillaceae, Planctomycetaceae, and Gaiellaceae, exhibiting fluctuation in abundances relative to the light cycle. However, limited-to-no cycling was observed in the acyclic CCAox34 line or in soil controls. Significant cycling was also observed, to a lesser extent, in Brachypodium. Functional gene inference revealed that genes involved in carbohydrate metabolism were likely more abundant in near-dawn, dark samples. Additionally, the composition of organic matter in the rhizosphere showed a significant variation between dark and light cycles.
Conclusions
The results of this study suggest that the rhizosphere bacterial community is regulated, to some extent, by the circadian clock and is likely influenced by, and exerts influences, on plant metabolism and productivity. The timing of bacterial cycling in relation to that of Arabidopsis further suggests that diurnal dynamics influence plant-microbe carbon metabolism and exchange. Equally important, our results suggest that previous studies done without relevance to time of day may need to be reevaluated with regard to the impact of diurnal cycles on the rhizosphere microbial community.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0287-1) contains supplementary material, which is available to authorized users.
Keywords: Bacterial community structure, Diurnal rhythm, Rhizosphere, Arabidopsis
Background
The plant rhizosphere contains a complex microbial community that directly impacts plant growth, health, and development [1]. The rhizosphere bacterial community composition is predominantly determined by the soil community, and associated environmental factors (e.g., climate), but is also influenced to some extent by plant host genotype [2, 3]. Relative to bulk soil, a “rhizosphere effect” results in a plant-root-associated microbial community that is more numerous and shows greater metabolic activity that is shaped by exudation of carbon and other metabolites from the plant root [4]. Thus, mutations in genes related to plant nutrient metabolism may in turn influence the rhizosphere microbial community, with implications for plant growth and health [5]. Furthermore, manipulation of host-associated microbial communities is receiving increasing attention as a biological mechanism to improve plant growth and stress resistance [6].
Important to the determination of what constitutes a beneficial microbial community is the characterization of how diverse soil bacteria interact with host plant species [7]. The rhizosphere effect that exerts some control on the rhizosphere community composition is mediated by metabolic exchange between the roots and soil [4]. The partitioning of nutrients to the roots and their exchange with the soil environment is controlled by the response of plants to environmental signals, such as light and temperature [8]. These responses are modulated by a plant’s innate ability to estimate time within an approximately 24-h period and synchronize biological events via the circadian clock [8]. The importance of light in shaping the outcome of host-microbe interactions is becoming increasingly evident. Recent studies show that the intestinal microbiota of humans [9] and mice [10, 11] undergo diurnal oscillations under the control of host feeding time and diet, and silencing of a host’s molecular clock genes causes gut dysbiosis [12].
The circadian clock is an important regulator of numerous basic plant functions including central carbon metabolism [13], gene expression, stomatal function, and the timing component of photoperiodism, which regulates seasonal reproduction [14, 15]. The clock is also subject to extensive natural variation both within and between species, and this is reported to influence plant fitness and performance [16–20]. As a result, the circadian clock is considered a key regulator of plant physiology and adaptation to different geographic environments, enabling an organism to anticipate periodic environmental changes and adapt its physiological and developmental states accordingly [8, 21]. Indeed, the life cycles of pathogens are closely associated with diurnally regulated host plant metabolism, and the circadian clock has been suggested to contribute to enhanced plant fitness by balancing innate immune responses with cellular metabolism [22, 23].
In Arabidopsis, the circadian clock consists of a series of intertwined feedback loops, regulated both transcriptionally and post-transcriptionally, through post-translational modification and protein turnover [8]. This mechanism appears to be conserved across plant species [24]. The circadian clock has been found to influence a variety of metabolic functions in the plant including chlorophyll biosynthesis, transport photosystems, starch synthesis and degradation, and nitrogen and sulfur assimilation [25]. The concentrations of metabolites including nitrate, glutamate, glutamine, and sucrose have also been shown to alter clock timing [26, 27]. However, due to differences in methodology, these results are sometimes inconsistent across studies, highlighting a need to consider photoperiod duration and the time of sample collection when describing results [25].
While the circadian clock machinery and associated metabolic and physiological responses have been well characterized, especially in Arabidopsis [28], little is known about how the rhizosphere bacterial community responds to host circadian cycling. Here, we examine the rhizosphere community dynamics of Arabidopsis thaliana in natural soil using next-generation sequencing (NGS) of the 16S rRNA gene. Soil organic matter (SOM) composition in the rhizosphere was also characterized by high-resolution mass spectrometry, 21T Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS), to help elucidate microbial metabolism.
In this study, we assessed cycling dynamics: (1) among biological replicates of A. thaliana sampled during dark and light periods to determine whether microbial community composition changed due to a diel cycle associated with exposure to light; (2) over a 72-h period utilizing an acyclic Arabidopsis line in which the cca1 gene is ectopically over-expressed [29] to determine differences in rhizosphere communities between the wild-type and mutant genotypes; and (3) of rhizosphere communities of a second species, Brachypodium distachyon, to determine if this cycling phenomenon is conserved among physiologically diverse plant species. While current hypotheses suggest that microbial populations in the rhizosphere are relatively static [2, 3], our results suggest that the rhizosphere microbial community is surprisingly dynamic, responding to both biotic and abiotic factors, including the circadian clock.
Results
16S rRNA characterization of AM and PM rhizosphere communities
Sequencing of the V5–V6 hypervariable regions of the 16S rRNA gene from DNA obtained from dark (AM, 1 h before light exposure) and light (PM, 1 h before dark) wild-type (Col-0) rhizosphere and fallow soil samples produced a mean Good’s coverage of 94.2 ± 2.3% (mean ± standard deviation), following rarefaction to 20,000 reads per sample with OTU binning at 97% similarity. While differences in alpha diversity, measured as Shannon diversity and abundance-based coverage estimate (ACE) of richness, did not significantly differ between dark and light samples (9 h between time points) for a given host environment (Table 1), Shannon diversity was significantly greater in rhizosphere samples versus fallow soil (P = 0.001). Samples had similar taxonomic compositions and were predominantly comprised of members of the Proteobacteria, Actinobacteria, and Acidobacteria (Additional file 1: Figure S1). Soil communities harbored greater relative abundances of the Actinobacteria and Acidobacteria than were observed in the rhizosphere. Communities in both the rhizosphere and fallow soil consisted of a large number (265 to 274) of families present at relatively low abundances (Additional file 1: Figure S2). Rhizosphere communities contained approximately 10 more families at low abundance than those of fallow soil. Planctomycetaceae was the most abundant family in both environments at both time points (Additional file 1: Figure S2), and about a quarter of the community (23.3 ± 2.3% and 28.5 ± 4.7% of sequence reads in rhizosphere and soil samples, respectively) could not be classified to a family.
Table 1.
Coverage and alpha diversity (mean ± standard deviation) among AM and PM rhizosphere samples based on 16S rRNA gene sequencing
| Host environment | Time | Number | Coverage (%) | S obs a | Shannonb | ACE |
|---|---|---|---|---|---|---|
| Wild-type | AM (dark) | 10 | 94.5 ± 3.3 | 3120 ± 887 | 7.18 ± 0.22A | 4771 ± 2620 |
| PM (light) | 9 | 93.4 ± 1.9 | 3543 ± 587 | 7.29 ± 0.14A | 4995 ± 1150 | |
| Fallow soil | AM (dark) | 10 | 95.2 ± 1.9 | 2823 ± 536 | 6.89 ± 0.30B | 4072 ± 1558 |
| PM (light) | 10 | 93.5 ± 1.4 | 3290 ± 344 | 7.07 ± 0.11A,B | 5272 ± 1335 | |
| P value | 0.001 | 0.486 | ||||
Values sharing the same capital letter subscripts did not differ significantly by Tukey's post-hoc test (P < 0.05)
a S obs: number of OTUs observed
bIndices sharing the same superscript did not differ significantly by Tukey’s post hoc test (P > 0.05)
Principal coordinate analysis of Bray-Curtis distances revealed significant separation (AMOVA, F s = 1.28, P = 0.003) of dark (AM) communities from those characterized from light (PM) samples in the Arabidopsis rhizosphere (Additional file 1: Figure S3). Thus, while alpha diversity (species richness and evenness within a single sample) did not differ, community composition changed significantly between samples collected during dark and light conditions in the rhizosphere. In contrast, separation was not significant in fallow soil (F s = 1.48, P = 0.076). Similarly, phylogenetic differences in community composition (phylobetadiversity, described further in the “Methods” section), assessed using unweighted UniFrac distances, showed significant differences (P = 0.031) between the dark and light communities in the rhizosphere, but not in soil alone (P = 0.217), suggesting changes in the soil were evolutionary driven.
Community compositions (beta diversity) between dark and light samples were significantly different in both environments (P = 0.018 and 0.031, for rhizosphere and soil communities, respectively), as evaluated by analysis of similarity (ANOSIM), but as stated above, phylobetadiversity did not differ in fallow soil. Among rhizosphere samples, <10% of the community showed variation in the relative abundances of operational taxonomic units (OTUs) between sampling times, with a significantly lower abundance of Acidobacteria Gp 6, and greater relative abundances of Burkholderiales and Myxococcales, among other orders (Fig. 1) between dark and light time points. The soil community showed greater variability between light and dark time points (13.3–17.5% of the community), with the most dynamic shifts observed within orders of the Acidobacteria (Fig. 1). Taken together, these results suggest that while beta diversity changed in both the rhizosphere and fallow soil between dark and light samples, only rhizosphere communities, and not those in soil, showed a significant change in phylobetadiversity, despite a greater percentage of community variation in soil.
Fig. 1.
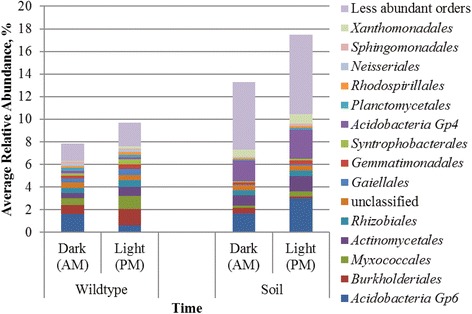
Order-level classification and relative abundances of OTUs that differed significantly between time points. More specific taxonomic classifications were not performed due to a large number of unclassified sequences found at family level. Significance was evaluated by using the Kruskal-Wallis test (P < 0.05). Analyses were performed separately for rhizosphere and fallow soil communities
SOM characterization of AM and PM rhizosphere organic matter
The SOM composition was significantly different in the water fraction of rhizosphere samples between the dark (AM) and light (PM) samples (Fig. 2). Approximately two-fold formula-assigned compounds were more abundant among light samples compared to dark samples. Ordination of chemical compounds by principal components analysis (PCA, Additional file 1: Figure S4, Panel A and Figure S5, Panel A) revealed that dark and the light samples were mainly separated along PC1, whereas the different biological and technical replicates within each cluster were separated along PC2.
Fig. 2.
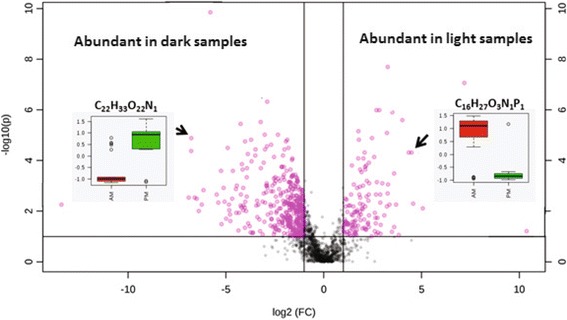
Relative abundances of different organic compounds identified by 21T FT-ICR-MS. The volcano plot was obtained by plotting the log2 fold change on the x-axis and –log10(P) on the y-axis. Compounds that changed twofold or more with a P value <0.05 are indicated in pink
The organic compounds responsible for such separation were extracted from the loading plots and plotted on a van Krevelen diagram (Additional file 1: Figure S4, Panel B and Figure S5, Panel B). The majority of the compounds present in the light samples had high oxygen/carbon (O/C) ratios that fell in the aromatic-like, lignin-like, and tannin-like region of the van Krevelen diagram and represent potential alkaloids, ketones, terpenoids, flavonoids, organic acids, and condensed tannins. Additionally, the majority of these compounds included heteroatoms such as S, N, and P. On the contrary, compounds with low O/C and high hydrogen/carbon (H/C) ratios dominated the dark samples and mainly fit in the lipid-like and unsaturated H/C region of the van Krevelen diagrams. The majority of these compounds contained only C, H, and O. The increase in the abundance of lipid-like compounds in the dark samples compared to the light samples was significant and primarily drove the separation between the dark and light in methanol extracts (Additional file 1: Figure S5).
The average mass-to-charge ratio (m/z) range of the compounds identified in AM samples was lower than that in the PM samples, where the PM samples appeared to have significantly higher m/z values. Similarly, organic compounds in the PM samples had significantly higher nominal oxidation state of carbon (NOSC) values. Expressing the average oxidation state of all carbons in one formula, NOSC provides information on the biogeochemical reactivity of a compound. An increase in the NOSC values during the day is therefore consistent with increased oxidation and the production of soluble compounds that are available for microbial metabolism.
Diurnal cycling of bacterial communities in the Arabidopsis rhizosphere
To further investigate potential cycling dynamics, bacterial communities were characterized every 6 h over a 72-h period from the rhizosphere of wild-type (Col-0) Arabidopsis, the rhizosphere of an acyclic line (OX34), and fallow soil. Samples were collected during dark conditions (1 am), 2 h before light (7 am), during light exposure (1 pm), and 1 h after dark (7 pm). Alpha diversity among samples from the wild-type rhizosphere, by both Shannon and ACE indices, was significantly greater (P = 0.018 and 0.024) during dark periods (1 am and 7 am samples), compared to when samples were exposed to light or shortly thereafter (1 pm and 7 pm samples) (Table 2). This apparent discrepancy from the earlier AM/PM experiment may reflect greater statistical power in this experiment due to a greater number of samples collected (9–10 compared to 17 in dark and light groups) or slight differences in the times when samples were collected relative to the photoperiod. Differences in alpha diversity between light and dark exposures were not significant among communities in the OX34 rhizosphere (P ≥ 0.813). In soil alone, differences in the Shannon index were also not significant (P = 0.091). In contrast, ACE richness was significantly greater among dark samples (P < 0.0001), which may reflect greater variation in soil over a longer time periods compared to single-time point AM/PM samples. Differences in ACE index at individual time points were also observed for wild-type rhizosphere and fallow soil (Table 2).
Table 2.
Coverage and alpha diversity (mean ± standard deviation) among microbiota from the rhizosphere of wild-type and OX34 mutant Arabidopsis plants and fallow soil samples
| Host environment | Time | N a | Coverage (%) | S obs | Shannonb | ACE |
|---|---|---|---|---|---|---|
| Wild-type | 1 am | 6 | 96.4 ± 1.5 | 2656 ± 429 | 6.72 ± 0.15A | 3420 ± 957A,B |
| 7 am | 11 | 95.0 ± 1.0 | 3035 ± 230 | 6.83 ± 0.08A | 4455 ± 1195A | |
| 1 pm | 8 | 97.5 ± 1.4 | 2278 ± 553 | 6.67 ± 0.21A | 2714 ± 916B | |
| 7 pm | 9 | 97.2 ± 2.0 | 2372 ± 579 | 6.62 ± 0.22A | 3185 ± 1990A,B | |
| OX34 mutant | 1 am | 8 | 93.7 ± 1.1 | 3317 ± 308 | 6.87 ± 0.14A | 5915 ± 1489A |
| 7 am | 11 | 93.9 ± 1.2 | 3340 ± 327 | 6.89 ± 0.14A | 5695 ± 1573A | |
| 1 pm | 9 | 93.7 ± 1.1 | 3357 ± 287 | 6.87 ± 0.11A | 6020 ± 1348A | |
| 7 pm | 9 | 94.2 ± 1.8 | 3246 ± 388 | 6.87 ± 0.09A | 5562 ± 2489A | |
| Fallow soil | 1 am | 7 | 94.3 ± 1.8 | 3081 ± 483 | 6.82 ± 0.14A | 5699 ± 2183A,B |
| 7 am | 5 | 92.6 ± 0.9 | 3533 ± 218 | 6.93 ± 0.12A | 7673 ± 1391A | |
| 1 pm | 6 | 96.3 ± 2.1 | 2540 ± 730 | 6.72 ± 0.20A | 3449 ± 1354B | |
| 7 pm | 6 | 96.2 ± 1.3 | 2747 ± 328 | 6.82 ± 0.14A | 3571 ± 769B |
a N refers to the total numbers of samples (replicates) collected at the time point over a 3-day period. Three samplings each were performed in triplicate at 1 am, 1 pm, and 7 pm, and four samplings were performed at 7 am. Samples that could not be rarefied to 22,380 sequences were removed from the dataset
bTimes sharing the same superscript did not differ significantly by post hoc test for a given host environment (P > 0.05)
Similar to the AM/PM experiment, bacterial communities among rhizosphere and soil samples were primarily comprised of members of the phyla Proteobacteria, Bacteroidetes, Acidobacteria, and Actinobacteria (Fig. 3 and Additional file 1: S6). Differences in beta diversity among host environments were significant by ANOSIM (P < 0.001), with most of the variation attributable to OTU-level shifts in relative abundance within the same predominant families in all environments (Additional file 1: Figure S7).
Fig. 3.

Distribution of phyla and Proteobacteria classes in rhizosphere samples from wild-type Arabidopsis plants. Percent relative abundance refers to cumulative abundance and 42 less abundant taxa that are not shown
Increasing the duration of sampling revealed that bacterial communities collected from the wild-type rhizosphere (Fig. 3) appeared to show cycling in the relative abundances of phyla that corresponded to the 9-h photoperiod. Cycling patterns were generally characterized by increases in the Proteobacteria, except for the Betaproteobacteria, the Actinobacteria, and the Firmicutes, and decreases in the relative abundance of Acidobacteria and Bacteroidetes during light exposure. In contrast, less apparent diel variation was observed in the rhizosphere microbial communities obtained from the acyclic OX34 mutant (Additional file 1: Figure S6, Panel A).
Cycling dynamics among the rhizosphere genotypes and fallow soil were further statistically interrogated using the Jonckheere-Terpstra-Kendall (JTK) algorithm [30], with OTUs assigned to families. By this analysis, a significantly greater proportion of the community (P < 0.0001) exhibited cycling dynamics in the wild-type rhizosphere (13.2 ± 1.1%), compared to that obtained from the OX34 mutant rhizosphere (3.6 ± 0.7%) or fallow soil (1.3 ± 0.7%). Burkholderiaceae, Rhodospirillaceae, Planctomycetaceae, and Gaiellaceae were among the most abundant cycling families identified. Similarly, the Kruskal-Wallis test indicated that OTUs that varied between light and dark periods, without determination of cycling dynamics, accounted for up to 9.5%, on average, of the community in the wild-type rhizosphere (Fig. 4), and these OTUs were classified to several families found to exhibit cycling dynamics by the JTK algorithm. The microbial community in the OX34 rhizosphere and fallow soil showed greater amounts of variability independent of cycling by Kruskal-Wallis test, with ~25% of the community varying in the mutant rhizosphere and 30–35% varying in soil alone. Thus, the key feature between the wild type, OX34, and soil was that, although some variability was seen in each sample type, the wild type showed a greater statistically significant cycling effect, encompassing a greater proportion of the community.
Fig. 4.
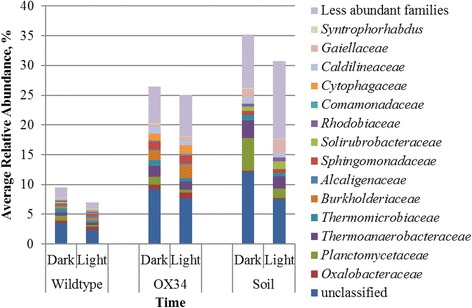
Family-level classification of OTUs that differed significantly among light and dark periods. Significance was evaluated by Kruskal-Wallis test (P < 0.05). Analyses were performed separately for each genotype in the Arabidopsis rhizosphere and fallow soil
As described in detail in Additional file 1, a similar experiment was performed in which significant (P < 0.05) diurnal cycling of the rhizosphere bacterial community associated with B. distachyon was also observed. In contrast to Arabidopsis, there was no significant difference in alpha diversity between light and dark periods (P = 0.658). However, 3.5% of the community showed significant (P < 0.05) cycling dynamics using the JTK algorithm, the most abundant of which was Gaiellaceae. In contrast, only 0.2% of the community, on average, in fallow soil showed significant cycling dynamics. Similar to Arabidopsis, a similar percentage of the community (<12%) also varied between light and dark periods by Kruskal-Wallis test.
Cycling of inferred functional genes
Changes in potential metabolic functions as a result of taxonomic cycling were assessed using PICRUSt [31] to infer abundances of functional genes. Twelve inferred tier 3 KEGG orthology (KO) categories differed between time points among dark (AM) and light (PM) wild-type Arabidopsis rhizosphere samples, predominantly within the broader category of metabolism (Fig. 5). Generally, only one or two taxa were found to contribute at high abundances to these functional categories, particularly members of the families Nitrospiraceae and Comamonadaceae (Additional file 1: Table S1). In contrast, no functional categories differed between dark and light time points in fallow soil.
Fig. 5.
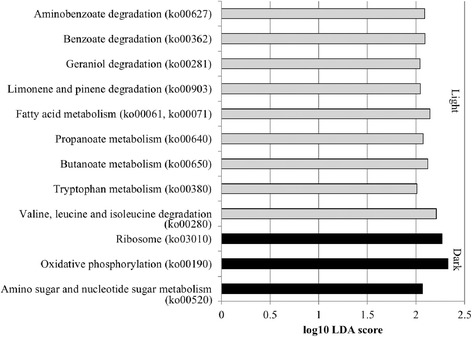
Tier 3 KO inferred functional annotations that differed by LEfSe analysis. Inferred functions differed between bacterial communities from wild-type Arabidopsis rhizosphere samples harvested in AM and PM time points
The JTK algorithm showed that 23 tier 3 KO functional categories had significant cycling among wild-type Arabidopsis rhizosphere bacterial communities throughout the 72-h experiment (inferred abundances supplied in Additional file 1: Table S2). Functional predictions were assigned within the broader category of metabolism (Additional file 1: Figure S8), with genes encoding functions associated with carbohydrate metabolism showing the greatest abundances 1 h before exposure to light (7 am). Due to the high diversity of taxa associated with carbohydrate metabolism among Col-O samples, taxonomic affiliations of OTUs associated with these fluctuations could not be definitively determined. In contrast, the lowest abundances were observed in the middle of the dark period (1 am). Cycling of “ether lipid metabolism” (ko00565), “glycosphingolipid biosynthesis - lacto and neolacto series” (ko00564), and “neuroactive ligand-receptor interaction” (ko04080) were also identified using the JTK algorithm in the OX34 rhizosphere, albeit at low abundance. No significant cycling of functional genes in fallow soil could be identified using the JTK algorithm.
Discussion
Results of this study showed that rhizosphere microbiota composition and predicted function varied according to a diel cycle and differed between the acyclic OX34 line and wild-type plants. Approximately 10% of the bacterial community in the wild-type Arabidopsis rhizosphere, determined by the JTK, exhibited diel cycling that was not seen in the acyclic Arabidopsis line. This strongly suggests that there is a relationship between changes in plant metabolism and bacterial community composition. Similar to previous studies [2, 3], the same predominant phyla were observed in rhizosphere and soil samples, and the relatively high percentage of taxonomically unclassified OTUs is also similar to previous findings [2]. Notably, many families previously found to be associated with root communities, for which the plant would likely select [2] (e.g., Flavobacteriaceae, Rhizobiaceae, Comamonadaceae, and Oxalobacteraceae), were differentially abundant between light and dark periods or showed significant cycling dynamics. This suggests that these potentially selected taxa are responding to host circadian-induced changes or may be contributing to them.
Species showing cyclical variation in abundances may also serve as pioneer microorganisms colonizing newly emerging root tissue. Root growth and elongation is controlled by the circadian clock in Arabidopsis, with faster growth reported under light and during longer light conditions [32]. Similarly, previously reported root-associated taxa, including members of the Actinomycetales, Burkholderiales, and Flavobacteriales [2, 33], among others, were observed to increase in abundance during light periods. Furthermore, variation in beta diversity was reduced during light conditions (demonstrated by closer clustering, shown in Additional file 1: Figure S3), which may indicate a selective pressure exerted by the root community to promote a community composition that facilitates plant growth. While this suggestion cannot be definitively concluded from the data presented here, follow-up studies using fluorescent in situ hybridization may help elucidate the dynamics of the root microbiota as they relate to regions of greater root growth (root tips).
Plant carbon partitioning may directly influence microbial associations, and this partitioning also shows diel fluctuations and is regulated by the circadian clock [34]. This likely defines a “metabolic dawn,” where sugar from photosynthesis may feed back to the clock to set its rhythm on a daily basis [35]. Soluble sugars [36], as well as genes associated with starch synthesis, chloroplast biosynthesis, and photosystems [25, 34], peak at or approximately 4 h after dawn, presumably to allow maximal production of sugars during the light period, a fraction of which are released to soil [37]. These results are consistent with SOM composition derived from high-resolution mass spectra that indicated an increase in labile compounds such as sugars during the light period. In contrast, genes regulating transport, breakdown, and storage of sugars, glycolysis, and the pentose phosphate pathway reach their maximum toward dusk to maximize the mobilization of starch and maintain carbon homeostasis [34]. Interestingly, among the rhizosphere bacterial community, genes involved in glycolysis and the pentose phosphate pathway were similarly inferred to show cycling dynamics (Additional file 1: Figure S8) in opposite phase to that previously observed in Arabidopsis [25, 34]. Thus, one attractive hypothesis is that the plant and rhizosphere communities are communicating to serve complementary functions, mediated in large part by circadian cycling.
A very small percentage of the rhizosphere bacterial community showed cycling dynamics in fallow soil samples. Both photosynthetic and non-photosynthetic bacteria have previously been shown to exhibit their own circadian rhythms, entrained by environmental conditions [38–40]. Thus, this fluctuation may represent independent cycling of these bacteria or changes in water potential due to plant transpiration.
The magnitude of community variation determined by Kruskal-Wallis test, unrelated to cycling dynamics, varied considerably between wild-type and mutant rhizosphere samples and fallow soil. Mutant rhizosphere and soil communities showed greater variation, independent of cycling, than wild-type rhizosphere communities, yet a smaller proportion of the community showed significant cycling dynamics by the JTK algorithm, suggesting that variation in mutant rhizosphere and soil samples is likely stochastic, rather than cyclical. Furthermore, while the composition of dark and light communities in fallow soil was significantly different, phylogenetic structure of the community was not significantly altered, although it was in wild-type rhizosphere samples. This result suggests that shifts in the wild-type rhizosphere are more likely to be directed rather than as a result of stochastic variation, resulting in a phylogenetic restructuring of the community. Lower variation between light and dark sampling points in both wild-type and mutant rhizosphere samples may also likely reflect moderation of the community by rhizosphere effects [4]. Fewer cycling taxa were observed in the rhizosphere community of Brachypodium, relative to Arabidopsis, although a significantly greater percentage of the rhizosphere community showed cycling dynamics relative to fallow soil. Differences between Arabidopsis and Brachypodium cycling communities are not surprising since species-specific microbial assemblages are determined based on the nature of root exudates and plant-microbe signaling molecules [2, 4]. Thus, difference in assemblage may, in large part, help explain differences in cycling dynamics observed between species.
It is also important to note that functional genes reported here were inferred from taxonomic data [31]. Nevertheless, the taxa associated with diurnal cycling in this study are among those previously found to be selected for in Arabidopsis root communities [2, 3, 33] and the functional genes inferred corresponded reasonably well to previously reported gene expression data for Arabidopsis [34]. This further supports the contention that there is a true rhizosphere cycling dynamic that is important to the health and growth of the plant.
It is likely that there is two-way communication between the plant and its microbiota and that the rhizosphere microbial community influences circadian rhythm, gene expression, and metabolic functioning of the host plant. Microbial regulation of host genes has been observed in rhizobial-legume interactions and in the Euprymna scolopes squid, in which Vibrio fischeri bioluminescence regulates expression of a host cryptochrome [41]. Similarly, the gut microbiota regulates rhythmic signaling pathways in intestinal epithelial cells that coordinate glucocorticoid production in the intestine [42], and the bacterial community is responsible for normal clock functioning in the liver and hypothalamus in mice [9]. Several groups have begun investigating how rhizosphere and endophytic communities might be used to improve plant biomass and stress resistance [6], and future studies in Arabidopsis are necessary to understand how the microbiota influences host plant circadian functioning and productivity.
Conclusions
The extent of change of the rhizosphere microbiota over a 24-h period equals or exceeds those previously attributed to factors such as plant genotype [43–45]. While current hypotheses suggest that microbial populations in the rhizosphere are relatively static and primarily related to soil communities [2, 3, 33], our work suggests otherwise. The rhizosphere microbial community indeed appears to be very dynamic in time, responding to both biotic and abiotic factors. This additional complexity may shape a better understanding of plant-microbe interactions and how rhizosphere dynamics may affect plant productivity in a changing environment. Perhaps, more importantly, our results highlight that temporal factors including light exposure, in addition to other sources of experimental error, should be considered in the interpretation of data from future studies, since in several previous studies, the time of sampling is either not noted or not well controlled. Expanding upon the scope of the present work, which was conducted using an annual dicot, to include perennial plants and most particularly bioenergy grasses, will allow us to consider new possibilities of plant-microbial interactions.
Methods
Rhizosphere sample harvesting
Rhizosphere soil samples were obtained as previously described [46] after plants had grown in soil for 5 weeks. Three experiments were conducted: (1) a biological replicate experiment in which A. thaliana or fallow soil (n = 10 for each) was collected 1 h prior to light exposure (AM) and 1 h prior to turning off the lights (PM), with a 9-h photoperiod (9 am–6 pm); (2) a 72-h cycling experiment in which wild-type Arabidopsis, the acc1-ox34 (OX34) acyclic mutant strain of Arabidopsis [47], or fallow soil was grown with a 9-h light period (9 am–6 pm) and harvested in triplicate every 6 h (7 am, 1 pm, 7 pm, and 1 am) for 72 h; and (3) a 72-h B. distachyon BD21 experiment that employed the same sampling regime as experiment 2, except using Brachypodium and fallow soil with a 12-h light period (8 am–8 pm). A 9-h photoperiod was used for Arabidopsis experiments to avoid flowering prior to sample collection, while a 12-h photoperiod was used for the Brachypodium experiment to allow optimal growth.
For the AM/PM experiment, 10 replicates were harvested 1 h prior to light exposure (AM) and 1 h prior to turning off the lights (PM). Each replicate contained rhizosphere soil from 25 plants (fallow soil replicates were collected from 10 individual pots containing only soil located in the same growth chamber with plants). For the 72-h cycling experiments of Arabidopsis or Brachypodium, three replicates were collected for each time point and each replicate was collected from ≥30 plants. Loose soil was manually removed from the roots by shaking with sterile gloves. Roots were placed in a clean and sterile 50-ml tube containing 22.5 ml of ice-cold extraction buffer (0.1% sodium pyrophosphate, pH 7.0, and 0.1% Tween 20) and 2.5 ml of RNA stop solution (5% water-saturated phenol, pH 4.3, in 95% ethanol). Tubes were vortexed at maximum speed for 20 s, which released most of the rhizosphere soil from the roots and turned the water turbid. The turbid solution was transferred onto a petri dish, and sterile forceps were used to remove broken plant parts followed by filtering the mixture through a Miracloth strainer into a new 50-ml tube. The turbid filtrate was centrifuged for 15 min at 3200×g to form a pellet containing fine sediment and microorganisms. Most of the supernatant was removed and the loose pellets were resuspended and transferred to 2-ml microfuge tubes. The tubes were centrifuged at 10,000×g for 5 min to form tight pellets, from which all supernatant was removed. The final rhizosphere pellets were flash-frozen in liquid nitrogen and stored at −80 °C until used.
SOM extraction and characterization
Two different solvents with different polarities in the order of (1) water-H2O and (2) methanol-CH3OH were used to sequentially extract organic matter (OM) from the rhizosphere soil, as described previously [48]. Briefly, samples were prepared by adding 0.6 ml of solvent to 50 mg soil and shaking for 2 h at room temperature followed by mixing at 800 rpm on an Eppendorf Thermomixer (Eppendorf, Hauppauge, NY, USA) in 2 ml capped glass vials. Samples were removed from the shaker and centrifuged for 16 min at 3500 rpm, and the supernatant was removed. The soil residue was dried with nitrogen gas and the second solvent was added and processed in the same way. The extracts were then injected directly into the 21T Agilent FTICR mass spectrometer (magnet from Agilent Technologies, Oxford, England; spectrometer built at the Environmental Molecular Sciences Laboratory (EMSL)) using a Hamilton 250 μl glass syringe (Hamilton Company, Reno, Nevada, USA) at a flow rate of 0.5 μl min−1. The H2O extracts were diluted in MeOH at a ratio of 1:2 to improve electrospray ionization (ESI) efficiency.
Mass spectrometry was performed by the EMSL. Samples were introduced directly to a 21T FTICR mass spectrometer (Thermo Scientific, San Jose, CA, USA) outfitted with a custom ESI interface. Electrospray emitters were custom made using 360 μm outer diameter × 50 μm inner diameter, chemically etched, fused silica, as described previously [49]. The ion transfer tube temperature and spray voltage were 300 °C and 3.0 kV, respectively. Mass spectra (AGC 3 × 106) were collected from 240 to 1200 m/z with a mass measurement accuracy of less than 500 ppb. Four hundred individual scans were averaged for each sample and internally calibrated using organic matter homologous series separated by 14 Da (–CH2 groups). Chemical formulas were assigned using in-house software based on the compound identification algorithm (CIA), described by Kujawinski and Behn [50] and modified by Minor et al. [51]. Chemical formulas were assigned based on the following criteria: S/N >2, and mass measurement error <0.5 ppm, taking into consideration the presence of C, H, O, N, S, and P and excluding other elements.
To interpret the large data set, the chemical character of thousands of data points for each sample spectrum were evaluated on van Krevelen diagrams generated from the complex mass spectra obtained by ESI FTICR MS [52]. Van Krevelen diagrams provide a means to visualize and compare the average properties of OM and enable identification of the major biochemical classes (i.e., lipids, proteins, lignin, carbohydrates, and condensed aromatics) of compounds present in samples. Compounds were assigned on the van Krevelen diagram on the basis of their molar H/C ratios (y-axis) and molar O/C ratios (x-axis). The stoichiometry of each assigned formula was used to calculate nominal oxidation state of carbon (NOSC) for each compound [53]. NOSC of individual compounds present in each sample was averaged to give the NOSC of soluble organic matter in that sample. In addition, MetaboAnalyst 3.0 [54] was used to generate volcano and principal component analysis (PCA) plots. We only considered compounds with assigned molecular formulae because we wanted to extract information with respect to SOM chemical characteristics. Additionally, only peaks that were present in more than 50% of the replicates were included in such analysis, and the ICR-MS data were both normalized (by median) and scaled (pareto scaling) to emphasize the importance of the compounds with smaller relative intensities using MetaboAnalyst.
DNA extraction and sequencing
DNA was extracted from 200 to 300 mg rhizosphere or soil samples using the PowerSoil® DNA Isolation Kit (MoBio Laboratories, Inc, Carlsbad, CA, USA) according to the manufacturer’s instructions. The V5 + V6 regions of the 16S rRNA gene were amplified using the barcoded BSF784/1064R primer set [55, 56] with a negative (sterile water) control by the University of Minnesota Genomics Center (UMGC, Minneapolis, MN, USA). Amplicons were gel purified, pooled in equal amounts, and paired-end sequenced at a read length of 300 nt on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA) by UMGC.
Bioinformatics
All sequence processing was done using mothur ver. 1.34.0 [57]. Sequences were trimmed to the first 150 nt to remove low-quality regions at the ends of reads, and reads were paired-end joined using fastq-join software [58]. Joined reads were trimmed to maintain an average quality score of at least 35 over a sliding 50 nt window, and sequences with more than two mismatches from primer sequences, >8 nt homopolymers, and those with ambiguous bases were removed. High-quality reads were aligned against the SILVA database ver. 119 [59] and subjected to a 2% pre-cluster step to remove probable sequence errors [60]. Chimeric sequences were identified and removed using UCHIME software [61]. Samples were rarefied to 20,000, 22,380, and 33,685 sequence reads, with respect to experiments 1, 2, and 3, respectively, by random subsample for further comparison and analysis [62].
Operational taxonomic units were assigned at 97% identity using the furthest-neighbor algorithm and taxonomic assignments were performed against ver. 14 of the Ribosomal Database Project [63]. For functional predictions, taxonomic classification was performed against the GreenGenes database ver. 13.5 [64]. Functional inferences within the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology were made using PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) software [31], with normalization to 16S rRNA gene copy number.
Statistical analyses
Statistical analyses of diversity, community composition (beta diversity), phylogenetic structures (phylobetadiversity), and ordination were performed using mothur. Alpha diversity was calculated using the Shannon and abundance-based coverage (ACE) indices. Differences in beta diversity were evaluated using analysis of similarity (ANOSIM) [65], phylobetadiversity was analyzed using unweighted Unifrac distances [66], and cluster analysis was performed using analysis of molecular variance (AMOVA) [67] using Bray-Curtis dissimilarity matrices [68]. In this study, we used the terminology of Graham and Fine [69] to define beta diversity as a change in the species composition between samples while phylobetadiversity referred to a change in phylogenetic relatedness of communities.
Ordination of samples was performed via principal coordinate analysis. To determine which members of the community exhibited significant cycling in relative abundance during light and dark cycles, community composition was evaluated at the family level using the JTK algorithm [30] in R ver. 3.2.2 [70]. Zeit geber time 0 was defined as the first sample collected at 7 am. Differences in functional inferences were evaluated using LEfSe [71], which employs consecutive Kruskal-Wallis and Wilcoxon rank-abundance tests and then utilizes linear discriminant analysis (LDA) to estimate effect sizes of features. Differences in OTUs were evaluated by Kruskal-Wallis test [72]. Other statistical analyses were performed using XLSTAT ver. 2015.01.0 (Addinsoft, Belmont, MA). All statistics were performed at α = 0.05.
Acknowledgements
Special thanks to Dr. Rob McClung (Dartmouth Univ.) for providing seeds of the CCA1ox line. Sequence processing and analysis was performed, in part, using the resources of the Minnesota Supercomputing Institute. Mass spectrometry was performed using the Environmental and Molecular Sciences Laboratory (EMSL), a US Department of Energy (DOE) Office of Science User Facility sponsored by Biological and Environmental Research (BER) and located at Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for DOE.
Funding
The work conducted by the DOE Joint Genome Institute and EMSL is supported by the Office of Science of the DOE under contract no. DE-AC02-05CH11231 (JGI) and DE-AC05-76RL01830 (EMSL). Work was also funded, in part, by a grant from the DOE Office of Science, Biological and Environmental Research (Grant No. DESC0013978 to GS).
Availability of data and materials
Sequence data are deposited in the Sequence Read Archive at the National Center for Biotechnology Information under BioProject accession number SRP072358.
Authors’ contributions
CS performed processed and analyzed sequence data and drafted the manuscript. APF helped develop the concept and contributed to the design of the study, helped with sample preparation for MS analyses, data acquisition, and analysis, and contributed to writing and editing the manuscript. MMT, RKC, and JBS performed the mass spectrometry experiments including data acquisition and data analysis. YC conducted plant experiments and harvested rhizosphere and soil samples. PW performed DNA extractions on samples and assisted with sequence analysis. CKA and RF helped develop the concept and contributed to the design of the study. HB prepared samples for mass spectrometry. ADN and MM performed data analysis. FdA, TT, TP, and BA assisted with sample collection. SGT, LP-T, GS, and MJS conceived the study, participated in its design, and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ACE
Abundance-based coverage estimate
- ANOSIM
Analysis of similarity
- FTICR-MS
Fourier transform ion cyclotron resonance mass spectrometry
- JTK
Jonckheere-Terpstra-Kendall algorithm
- KO
Kyoto Encyclopedia of Genes and Genomes orthology
- NGS
Next-generation sequencing
- OTU
Operational taxonomic unit
- PCA
Principal component analysis
- SOM
Soil organic matter
Additional file
Taxonomic classification of OTUs predominantly associated with tier 3 KO inferred functional annotations that differed between bacterial communities from wild-type Arabidopsis rhizosphere samples harvested in dark (AM) and light (PM) time points (Fig. 5). Functions for which contributing taxa could not be determined are not shown. Table S2. Functional annotations of inferred genes that show significant cycling in abundance in wild-type Arabidopsis rhizosphere communities. Functional categories are arranged by KEGG orthology tiers, with each tier increasing in specificity. Table S3. Alpha diversity indices for Brachypodium distachyon BD21 experiment samples. Figure S1 Distribution of abundant phyla among AM and PM samples in the rhizosphere of wild-type Arabidopsis and fallow soil. Figure S2. Distribution of abundant families among AM and PM samples in the rhizosphere of wild-type Arabidopsis and fallow soil. Figure S3. Principal coordinate analysis of Bray-Curtis dissimilarities among AM and PM bacterial communities characterized by 16S rRNA sequencing in A) the rhizosphere of Arabidopsis (r 2 = 0.689) and B) fallow soil (r 2 = 0.876). Points represent individual samples. Figure S4. PCA plot of the relative abundance of the different organic compounds identified in each sample by water extraction (A) and van Krevelen diagram of elemental H/C (hydrogen-to-carbon) vs O/C (oxygen-to-carbon) ratios of the organic compounds extracted from the loading plot of the PCA plot (B). Figure S5. 3D PCA plot of the relative abundance of the different compounds extracted by MeOH (A) and van Krevelen diagram of elemental H/C (hydrogen-to-carbon) vs O/C (oxygen-to-carbon) ratios of the organic compounds extracted from the loading plot of the PCA plot (B)). Figure S6. Distributions of phyla in (A) Arabidopsis OX34 mutant rhizosphere and (B) fallow soil samples, omitting less abundant phyla. Percent abundances are cumulative. Sequence data from later time points for soil could not be obtained, and less abundant taxa are not shown. Figure S7. Family-level classification of OTUs that differed significantly among Arabidopsis rhizosphere genotypes and soil groups by Kruskal-Wallis test (P < 0.05). Figure S8. Inferred carbohydrate metabolism genes that showed significant cycling (P < 0.05) in abundance among wild-type Arabidopsis rhizosphere bacterial communities by the JTK algorithm. Figure S9. Distribution of phyla among samples from (A) the Brachypodium rhizosphere and (B) fallow soil. Forty-one less abundant phyla are not shown. Figure S10. Family-level classification of OTUs that differed significantly among light and dark periods by Kruskal-Wallis test (P < 0.05) in the Brachypodium rhizosphere. (DOCX 980 kb).
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0287-1) contains supplementary material, which is available to authorized users.
Contributor Information
Ljiljana Paša-Tolić, Phone: (509)-371-6003, Email: Ljiljana.PasaTolic@pnnl.gov.
Michael J. Sadowsky, Phone: (612)-624-2706, Email: sadowsky@umn.edu
References
- 1.Hardoim PR, van Overbeek LS, van Elsas JD. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 2008;16:463–71. doi: 10.1016/j.tim.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Bulgarelli D, Rott M, Schlaeppi K, Van Themaat EV, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–5. [DOI] [PubMed]
- 3.Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nat. 2012;488:86–90. [DOI] [PMC free article] [PubMed]
- 4.Berg G, Smalla K, Ahrenholtz I, Harms K, De VJ, Wackernagel W, et al. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68:1–13. doi: 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- 5.Haney CH, Samuel BS, Bush J, Ausubel FM, Hospital MG. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat Plants. 2015;1:1–22. doi: 10.1038/nplants.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mei C, Flinn BS. The use of beneficial microbial endophytes for plant biomass and stress tolerance improvement. Recent Pat Biotechnol. 2010;4:81. doi: 10.2174/187220810790069523. [DOI] [PubMed] [Google Scholar]
- 7.Bisseling T, Dangl JL, Schulze-Lefert P. Next-generation communication. Science. 2009;324:691. doi: 10.1126/science.1174404. [DOI] [PubMed] [Google Scholar]
- 8.Harmer SL. The circadian system in higher plants. Annu Rev Plant Biol. 2009;60:357–77. doi: 10.1146/annurev.arplant.043008.092054. [DOI] [PubMed] [Google Scholar]
- 9.Leone V, Gibbons SM, Martinez K, Hutchison AL, Huang EY, Cham CM, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17:681–9. doi: 10.1016/j.chom.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X, Bushman FD, FitzGerald GA. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc Natl Acad Sci. 2015;112:10479–84. doi: 10.1073/pnas.1501305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarrinpar A, Chaix A, Yooseph S, Panda S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014;20:1006–17. doi: 10.1016/j.cmet.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell. 2014;159:514–29. doi: 10.1016/j.cell.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 13.Kölling K, Thalmann M, Müller A, Jenny C, Zeeman SC. Carbon partitioning in Arabidopsis thaliana is a dynamic process controlled by the plants metabolic status and its circadian clock. Plant Cell Environ. 2015;38:1965–79. doi: 10.1111/pce.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michael TP, Salomé PA, Yu HJ, Spencer TR, Sharp EL, McPeek MA, et al. Enhanced fitness conferred by naturally occurring variation in the circadian clock. Science. 2003;302:1049–53. doi: 10.1126/science.1082971. [DOI] [PubMed] [Google Scholar]
- 15.Yanovsky MJ, Kay SA. Signaling networks in the plant circadian system. Curr Opin Plant Biol. 2001;4:429–35. doi: 10.1016/S1369-5266(00)00196-5. [DOI] [PubMed] [Google Scholar]
- 16.Yerushalmi S, Yakir E, Green RM. Circadian clocks and adaptation in Arabidopsis. Mol Ecol. 2011;20:1155–65. doi: 10.1111/j.1365-294X.2010.04962.x. [DOI] [PubMed] [Google Scholar]
- 17.Sulpice R, Flis A, Ivakov AA, Apelt F, Krohn N, Encke B, et al. Arabidopsis coordinates the diurnal regulation of carbon allocation and growth across a wide range of photoperiods. Mol Plant. 2014;7:137–55. doi: 10.1093/mp/sst127. [DOI] [PubMed] [Google Scholar]
- 18.Kinmonth-Schultz HA, Golembeski GS, Imaizumi T. Circadian clock-regulated physiological outputs: dynamic responses in nature. Semin Cell Dev Biol. 2013;24:407–13. doi: 10.1016/j.semcdb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anwer MU, Davis SJ. An overview of natural variation studies in the Arabidopsis thaliana circadian clock. Semin Cell Dev Biol. 2013;24:422–9. doi: 10.1016/j.semcdb.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Dodd AN, Salathia N, Hall A, Kévei E, Tóth R, Nagy F, et al. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science. 2005;309:630–3. doi: 10.1126/science.1115581. [DOI] [PubMed] [Google Scholar]
- 21.Graf A, Schlereth A, Stitt M, Smith AM. Circadian control of carbohydrate availability for growth in Arabidopsis plants at night. Proc Natl Acad Sci. 2010;107:9458–63. doi: 10.1073/pnas.0914299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo PJ, Mas P. STRESSing the role of the plant circadian clock. Trends Plant Sci. 2015;20:230–7. doi: 10.1016/j.tplants.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Roden LC, Ingle RA. Lights, rhythms, infection: the role of light and the circadian clock in determining the outcome of plant–pathogen interactions. Plant Cell. 2009;21:2546–52. doi: 10.1105/tpc.109.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song YH, Ito S, Imaizumi T. Similarities in the circadian clock and photoperiodism in plants. Curr Opin Plant Biol. 2010;13:594–603. doi: 10.1016/j.pbi.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farré EM, Weise SE. The interactions between the circadian clock and primary metabolism. Curr Opin Plant Biol. 2012;15:293–300. doi: 10.1016/j.pbi.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Gutierrez RA, Stokes TL, Thum K, Xu X, Obertello M, Katari MS, et al. Systems approach identifies an organic nitrogen-responsive gene network that is regulated by the master clock control gene CCA1. Proc Natl Acad Sci. 2008;105:4939–44. doi: 10.1073/pnas.0800211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight H, Thomson AJW, McWatters HG. SENSITIVE TO FREEZING6 integrates cellular and environmental inputs to the plant circadian clock. Plant Physiol. 2008;148:293–303. doi: 10.1104/pp.108.123901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atamian HS, Harmer SL. Circadian regulation of hormone signaling and plant physiology. Plant Mol Biol. 2016;91:1–12. doi: 10.1007/s11103-016-0477-4. [DOI] [PubMed] [Google Scholar]
- 29.Daniel X, Sugano S, Tobin EM. CK2 phosphorylation of CCA1 is necessary for its circadian oscillator function in Arabidopsis. Proc Natl Acad Sci. 2004;101:3292–7. [DOI] [PMC free article] [PubMed]
- 30.Hughes ME, Hogenesch JB, Kornacker K. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms. 2010;25:372–80. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yazdanbakhsh N, Sulpice R, Graf A, Stitt M, Fisahn J. Circadian control of root elongation and C partitioning in Arabidopsis thaliana. Plant Cell Environ. 2011;34:877–94. doi: 10.1111/j.1365-3040.2011.02286.x. [DOI] [PubMed] [Google Scholar]
- 33.Schlaeppi K, Dombrowski N, Oter RG, Van Themaat EV, Schulze-Lefert P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc Natl Acad Sci U S A. 2014;111:585–92. [DOI] [PMC free article] [PubMed]
- 34.Harmer SL, Hogenesch JB, Straume M, Chang HS, Han B, Zhu T, et al. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science. 2000;290:2110–3. [DOI] [PubMed]
- 35.Haydon MJ, Mielczarek O, Robertson FC, Hubbard KE, Webb AAR. Photosynthetic entrainment of the Arabidopsis thaliana circadian clock. Nature. 2013;502:689–92. [DOI] [PMC free article] [PubMed]
- 36.Lu Y, Gehan JP, Sharkey TD. Daylength and circadian effects on starch degradation and maltose metabolism. Plant Physiol. 2005;138:2280–91. doi: 10.1104/pp.105.061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol. 2006;57:233–66. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- 38.Johnson CH. Circadian rhythms: as time glows by in bacteria. Nature. 2004;430:23–4. doi: 10.1038/430023a. [DOI] [PubMed] [Google Scholar]
- 39.Min H, Guo H, Xiong J. Rhythmic gene expression in a purple photosynthetic bacterium, Rhodobacter sphaeroides. FEBS Lett. 2005;579:808–12. [DOI] [PubMed]
- 40.Soriano MI, Roibás B, García AB, Espinosa-Urgel M. Evidence of circadian rhythms in non-photosynthetic bacteria? J Circadian Rhythms. 2010;8:8. doi: 10.1186/1740-3391-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heath-Heckman EAC, Peyer SM, Whistler CA, Apicella MA, Goldman WE, McFall-Ngai MJ. Bacterial bioluminescence regulates expression of a host cryptochrome gene in the squid-vibrio symbiosis. MBio. 2013;4:e00167–13. doi: 10.1128/mBio.00167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013;153:812–27. doi: 10.1016/j.cell.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 43.Weinert N, Piceno Y, Ding G-C, Meincke R, Heuer H, Berg G, et al. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: many common and few cultivar-dependent taxa. FEMS Microbiol Ecol. 2011;75:497–506. doi: 10.1111/j.1574-6941.2010.01025.x. [DOI] [PubMed] [Google Scholar]
- 44.Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–4. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 45.Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, et al. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015;17:392–403. doi: 10.1016/j.chom.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schortemeyer M, Hartwig UA, Hendrey GR, Sadowsky MJ. Microbial community changes in the rhizospheres of white clover and perennial ryegrass exposed to free air carbon dioxide enrichment (FACE) Soil Biol Biochem. 1996;28:1717–24. doi: 10.1016/S0038-0717(96)00243-X. [DOI] [Google Scholar]
- 47.Wang Z-Y, Tobin EM. Constitutive expression of the circadian clock associated 1 (cca1) gene disrupts circadian rhythms and suppresses its own expression. Cell. 1998;93:1207–17. doi: 10.1016/S0092-8674(00)81464-6. [DOI] [PubMed] [Google Scholar]
- 48.Tfaily MM, Chu RK, Tolić N, Roscioli KM, Anderton CR, Paša-Tolić L, et al. Advanced solvent based methods for molecular characterization of soil organic matter by high-resolution mass spectrometry. Anal Chem. 2015;87:5206–15. doi: 10.1021/acs.analchem.5b00116. [DOI] [PubMed] [Google Scholar]
- 49.Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, et al. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem. 2006;78:7796–801. doi: 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kujawinski EB, Behn MD. Automated analysis of electrospray ionization fourier transform ion cyclotron resonance mass spectra of natural organic matter. Anal Chem. 2006;78:4363–73. doi: 10.1021/ac0600306. [DOI] [PubMed] [Google Scholar]
- 51.Minor EC, Steinbring CJ, Longnecker K, Kujawinski EB. Characterization of dissolved organic matter in Lake Superior and its watershed using ultrahigh resolution mass spectrometry. Org Geochem. 2012;43:1–11. doi: 10.1016/j.orggeochem.2011.11.007. [DOI] [Google Scholar]
- 52.Tfaily MM, Hamdan R, Corbett JE, Chanton JP, Glaser PH, Cooper WT. Investigating dissolved organic matter decomposition in northern peatlands using complimentary analytical techniques. Geochim Cosmochim Acta. 2013;112:116–29. doi: 10.1016/j.gca.2013.03.002. [DOI] [Google Scholar]
- 53.LaRowe DE, Van Cappellen P. Degradation of natural organic matter: a thermodynamic analysis. Geochim Cosmochim Acta. 2011;75:2030–42. doi: 10.1016/j.gca.2011.01.020. [DOI] [Google Scholar]
- 54.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–7. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Claesson MJ, Wang QO, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38:gkq873. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Staley C, Gould TJ, Wang P, Phillips J, Cotner JB, Sadowsky MJ. Evaluation of water sampling methodologies for amplicon-based characterization of bacterial community structure. J Microbiol Methods. 2015;114:43–50. doi: 10.1016/j.mimet.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 57.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aronesty E. Comparison of sequencing utility programs. Open Bioinforma J. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 59.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–98. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gihring TM, Green SJ, Schadt CW. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol. 2012;14:285–90. doi: 10.1111/j.1462-2920.2011.02550.x. [DOI] [PubMed] [Google Scholar]
- 63.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–5. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clarke KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–43. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 66.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27:325–49. doi: 10.2307/1942268. [DOI] [Google Scholar]
- 69.Graham CH, Fine PVA. Phylogenetic beta diversity: linking ecological and evolutionary processes across space in time. Ecol Lett. 2008;11:1265–77. doi: 10.1111/j.1461-0248.2008.01256.x. [DOI] [PubMed] [Google Scholar]
- 70.R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2012.
- 71.Segata N, Huttenhower C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acar EF, Sun L. A generalized Kruskal-Wallis test incorporating group uncertainty with application to genetic association studies. Biometrics. 2013;69:427–35. doi: 10.1111/biom.12006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequence data are deposited in the Sequence Read Archive at the National Center for Biotechnology Information under BioProject accession number SRP072358.