

Abstract
Background
Atypical lipomatous tumor (ALT), well differentiated liposarcoma (WDLS) and dedifferentiated liposarcoma (DDLS) are cytogenetically characterized by near-diploid karyotypes with no or few other aberrations than supernumerary ring or giant marker chromosomes, although DDLS tend to have somewhat more complex rearrangements. In contrast, pleomorphic liposarcomas (PLS) have highly aberrant and heterogeneous karyotypes. The ring and giant marker chromosomes contain discontinuous amplicons, in particular including multiple copies of the target genes CDK4, HMGA2 and MDM2 from 12q, but often also sequences from other chromosomes.
Results
The present study presents a DDLS with an atypical hypertriploid karyotype without any ring or giant marker chromosomes. SNP array analyses revealed amplification of almost the entire 5p and discontinuous amplicons of 12q including the classical target genes, in particular CDK4. In addition, amplicons from 1q, 3q, 7p, 9p, 11q and 20q, covering from 2 to 14 Mb, were present. FISH analyses showed that sequences from 5p and 12q were scattered, separately or together, over more than 10 chromosomes of varying size. At RNA sequencing, significantly elevated expression, compared to myxoid liposarcomas, was seen for TRIO and AMACR in 5p and of CDK4, HMGA2 and MDM2 in 12q.
Conclusions
The observed pattern of scattered amplification does not show the characteristics of chromothripsis, but is novel and differs from the well known cytogenetic manifestations of amplification, i.e., double minutes, homogeneously staining regions and ring chromosomes. Possible explanations for this unusual distribution of amplified sequences might be the mechanism of alternative lengthening of telomeres that is frequently active in DDLS and events associated with telomere crisis.
Electronic supplementary material
The online version of this article (doi:10.1186/s13039-017-0325-5) contains supplementary material, which is available to authorized users.
Keywords: Liposarcoma, Chromosomes, Amplification, 5p, 12q, Gene expression
Background
Cytogenetic analyses of more than 3200 benign and malignant soft tissue tumors have revealed that different patterns of chromosomal aberrations exist among these lesions [1, 2]. Several tumor entities are characterized by specific, sometimes pathognomonic, structural rearrangements, mostly translocations, giving rise to oncogenic fusion genes, often with no or few other changes of chromosome number or morphology. Another set of tumors displays a moderate number of chromosomal imbalances, whereas still another set of tumors shows highly complex karyotypic rearrangements with extensive cytogenetic heterogeneity. Both losses and gains of sequences may be of pathogenetic importance. While losses affect one or both copies of one or more genes, gains can range from one to hundreds of extra gene copies. Moderate and high level gene amplification manifest cytogenetically as intrachromosomal homogeneously staining regions (hsr), extrachromosomal double minutes (dmin) or ring chromosomes (r); other mechanisms behind amplification are presumed to be rare and are not easily recognized by chromosome banding analysis. Among soft tissue tumors, ring chromosomes are much more abundant than dmin, and hsr is even more infrequent (Additional file 1). Ring chromosomes, allowing for gene amplification through breakage-fusion-bridge cycles [3], constitute the characteristic cytogenetic feature of some soft tissue tumors, including atypical lipomatous tumor/well differentiated liposarcoma (ALT/WDLS) and dedifferentiated liposarcoma (DDLS).
Modern array technologies have revealed that gene amplification is more common among neoplastic cells than detected by banding analyses. Such technologies, however, do not reveal the chromosomal organization of multiplied sequences, which might provide some clues about how they originated and their evolutionary potential. In the present study, amplification through scattering over many chromosomes is described in a case of dedifferentiated liposarcoma.
Methods
As part of a study of soft tissue sarcomas that at G-banding analysis showed aberrations including add(5)(p15), FISH analyses were performed in order to find out if the breakpoint in 5p was localized to a restricted position that could indicate the involvement of a particular gene. No consistent pattern was found, but one case showed a peculiar distribution of chromosome 5 sequences, which prompted further investigation.
The patient was a 67-year-old man with a deep-seated tumor in the left thigh. The largest diameter of the highly necrotic, infiltratively growing tumor was 24 cm. Two samples – an open biopsy and the resected specimen – were obtained with an interval of five weeks. The diagnosis was dedifferentiated liposarcoma with atypical fat cells, sclerosis, a spindle cell component, as well as a component of spindle cells with rhabdoid differentiation; no region compatible with a well-differentiated liposarcoma was seen. Postoperative radiation therapy was given. X-ray two years later revealed no apparent lung metastases. Five years after diagnosis, metastases to the lungs and soft tissues on the back appeared. The patient died soon after.
Chromosome preparations were made from short-term cultured cells obtained from disaggregated tumor tissue from both samples and stained for G-banding as previously described [4].
FISH analyses were performed using whole chromosome painting probes wcp5 (green) and wcp12 (blue) (Vysis, Downers Grove, IL). Site-specific probes were CTD-2074D8 (5p14.1–14.3), RP11-509B9 (5p15.1), RP11-35 K22 (5p15.32), CTD-3080P12 (5p15.33), hereafter referred to as D8, B9, K22 and P12, respectively, as well as RP11–1137 N1 (12q14.3–15) for detection of the MDM2 gene (BACPAC Resource Center; https://bacpacresources.org). The following fluorophores were used for labelling: red, Cy3 dUTP (VWR), green, Chromatide Alexa Fluor 488–5-dUTP (Thermo Fisher Scientific). Hybridizations were performed as described [5]. No material was available for further analyses.
SNP array analysis of the two samples was performed as described [6]. In brief, tumor DNA (250 ng) was extracted and analyzed using the Affymetrix CytoScan HD array (Affymetrix, Santa Clara, CA, USA). Genomic aberrations were identified by visual inspection using the Chromosome Analysis Suite version 1.2 (Affymetrix). The human reference sequence used for alignment was the GRCh37/hg19 assembly. Constitutional copy number variations were excluded through comparison with the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home). Further bioinformatic analysis regarding copy numbers and segmentation was performed using Rawcopy and the Tumor Aberration Prediction Suite (TAPS), as described [7, 8]. Since the chromosome number was at the triploid level, only copy numbers of at least 6 were considered true amplification. Mean and median copy numbers were calculated as well as the total length of amplified sequences.
RNA sequencing (RNA-Seq) was performed on the excised tumor biopsy, as described [9]. Identification of potential fusion transcripts was performed on fastq files using FusionCatcher [10]. The GRCh37/hg19 build was used as the human reference genome. Expression of some selected candidate target genes in 5p and known targets in 12q was compared with their expression in a set of myxoid liposarcomas.
Results
A hypertriploid, complex karyotype was found in both samples (Fig. 1). The only difference between the two samples was a slight variation in chromosome number, 70–74 and 73–76, respectively. Based on both samples the composite karyotype was interpreted as 70–76,XX,-Y, +1,del(1)(q12)×2,add(2)(p1?),+del(3)(q11),-4,-5,add(5)(p15),?add(5)(p11),-7,add(7)(p11)×2,-8,-10, −11,?add(11)(q22),?ins(12;?)(q13;?)×2,der(12)add(12)(p11)add(12)(q24),add(14)(p11),add(19) (q13)×2,?der(19)add(19)(p11)del(19)(q12),-20,+21,+22,inc[cp24].
Fig. 1.
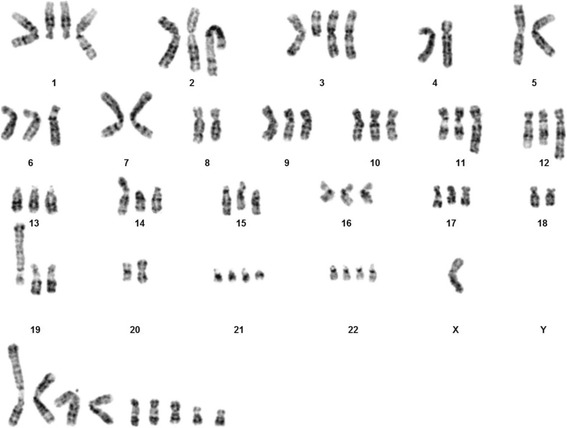
G-band karyogram showing fairly complex chromosomal aberrations
FISH analyses revealed that chromosome 5 sequences were spread to several chromosomes (Fig. 2). Using wcp5, no intact chromosome 5 was found, but wcp5-positive segments were detected in 14–19 chromosomes, at least 17 of which were clonal (Fig. 3a). Large segments were seen in four chromosomes (designated A-D in Fig. 3a) and three of these most likely contained the centromere of chromosome 5. Two identical chromosomes (L and M) could represent i(5)(p10). One chromosome (E) was identified, based on the DAPI staining pattern, as a derivative chromosome 9 with chromosome 5 material added to the truncated 9p. FISH using the more proximal probes D8 and B9 revealed 9–11 and 9–13 signals, respectively, per metaphase. The corresponding number of signals for K22 and P12 were 11–15 and 11–16 respectively. Similar signal patterns for all four probes of the two probe sets were seen in chromosomes E, H, L, M, and Q.
Fig. 2.
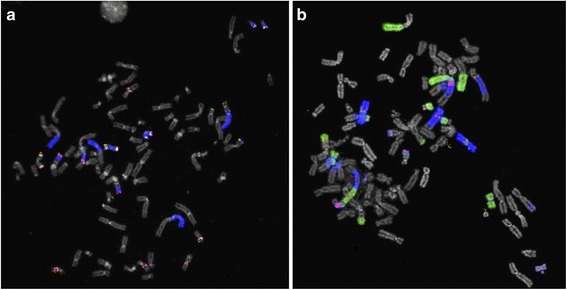
Metaphase FISH images showing multiple signals (a) for 5p site-specific probes (green and red) and wcp5 (blue), and (b) for MDM2 (red) as well as wcp5 (green) and wcp12 (blue)
Fig. 3.
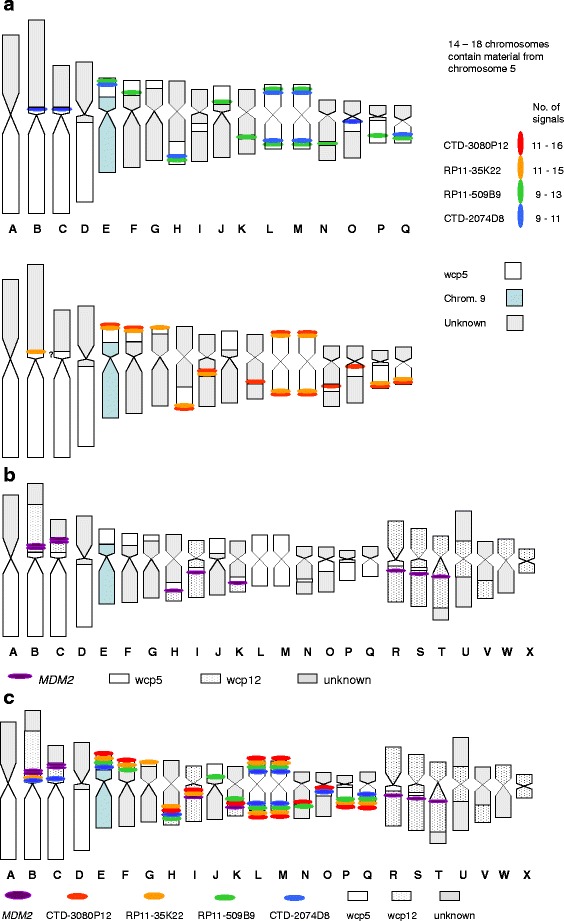
Schematic representations of the FISH results. a analysis using four site-specific 5p probes and wcp5; b analysis using a probe detecting MDM2 as well as wcp5 and wcp12; c summary of all FISH analyses. Letters A to X are used as identification of different aberrant chromosomes
Chromosome 12 sequences were identified in 12 chromosomes (Fig. 3b). In most metaphases there were 10 signals for MDM2 located in eight chromosomes. Sequences from both chromosomes 5 and 12 were present in seven chromosomes (B, C, H, I, K, R, and S); in R and S no signals for the site-specific 5p probes were detected. Twelve chromosomes were positive for wcp5 but not for wcp12 (A, D-G, J, and L-Q), whereas five chromosomes were wcp5 negative but wcp12 positive (T-X). A summary of several FISH analyses is shown in Fig. 3c.
At SNP array analysis, amplified sequences were found on chromosome arms 1q, 3q, 5p, 7p, 9p, 11q, 12q, and 20q (Table 1, Additional file 2). Few differences were found between the two samples (Fig. 4). The chromosome 5 amplicons emanated from almost the entire short arm, with peak copy numbers from p15.33-p15.32, p15.31-p15.2, p14.3, and p14.1-p12. The major parts of 5q were estimated to 4 copies. In chromosome 12, discontinuous high level amplicons were found from q12 to q24.21. There were about 10 copies of sequences covering the HMGA2 and MDM2 genes, whereas CDK4 was estimated to 17 copies. In general, amplified sequences in 12q showed higher copy numbers than those in 5p. The size of increased copy numbers in 5p and 12q corresponded to 46.3 Mb and 17.2 Mb representing about 94% and 17% of the length of these chromosome arms, respectively. Gain/amplification in other chromosomes was found for sequences within 1q21.2-q22 and 1q24.1, 3q26.2, 7p15.2-p12.3, 9p21.3-p13.1, 11q13.2-q13.4 and 11q22.1, and 20q11.23-q13.33, representing about 7%, 2%, 23%, 32%, 5% and 37%, respectively, of the chromosome arms. Among these chromosomes, only chromosome 20 displayed more extensive high level amplification (12 copies), in particular confined to 20q13.2-q13.33.
Table 1.
Distribution and size of chromosome segments showing amplification
| Chromosome arm | Mean copy number | Median copy number | Extension (Mb) | Fraction of arm with amplification |
|---|---|---|---|---|
| 1q | 8.1 | 8 | 8.462 | 6.8% |
| 3q | 7.2 | 7 | 2.013 | 1.9% |
| 5p | 9.9 | 10 | 46.273 | 93.6% |
| 7p | 6.8 | 6 | 13.772 | 23.2% |
| 9p | 8.1 | 8 | 13.905 | 31.5% |
| 11q | 12.0 | 9 | 3.866 | 4.8% |
| 12q | 12.4 | 11 | 17.159 | 17.4% |
| 20q | 13.3 | 12.5 | 13.212 | 36.6% |
| Total | 118.662 |
Only copy numbers of at least 6 are included
Fig. 4.
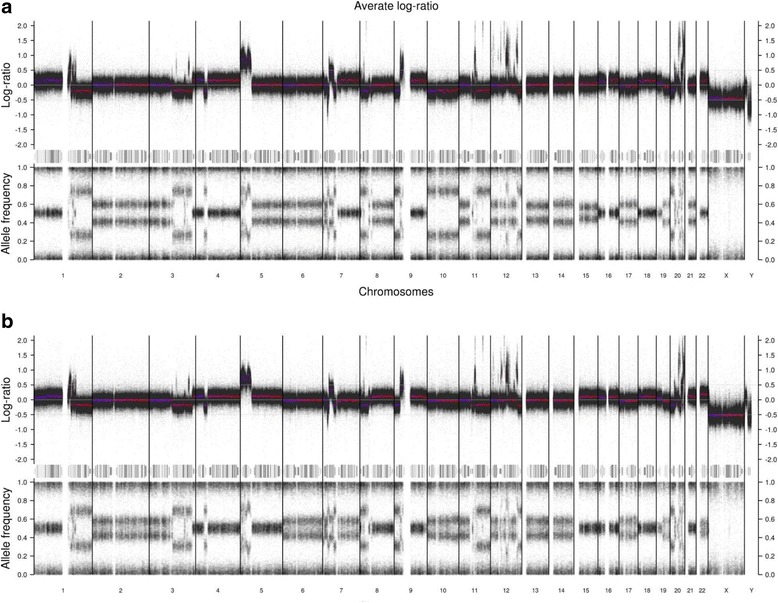
Log ratio and B-allele frequency from SNP array profiles of a the first and b the second sample of the DDLS. The log ratio was normalized to a near-triploid karyotype. Thus, log ratio 0.0 represents 3 copies and in the first sample 4 and 2 copies have log ratios 0.2 and −0.2, respectively. The corresponding shifts in allele frequencies (AF) could be exemplified by chromosome arms 1p (2 maternal +2 paternal copies; AF 0.5), most of 1q (2 + 0 copies; AF 0.75), and chromosome 2 (2 + 1 copies; AF 0.6) in the first sample. Both copy number and AF profiles are less distinct in the second sample, presumably due to larger fraction of stromal cells
None of the potential fusion transcripts that were detected at RNA-Seq was considered significant (Additional file 3). Of the selected target genes, AMACR and TRIO in 5p and CDK4, HMGA2 and MDM2 in 12q showed significantly (p < 0.05) elevated expression in relation to myxoid liposarcomas (Fig. 5).
Fig. 5.
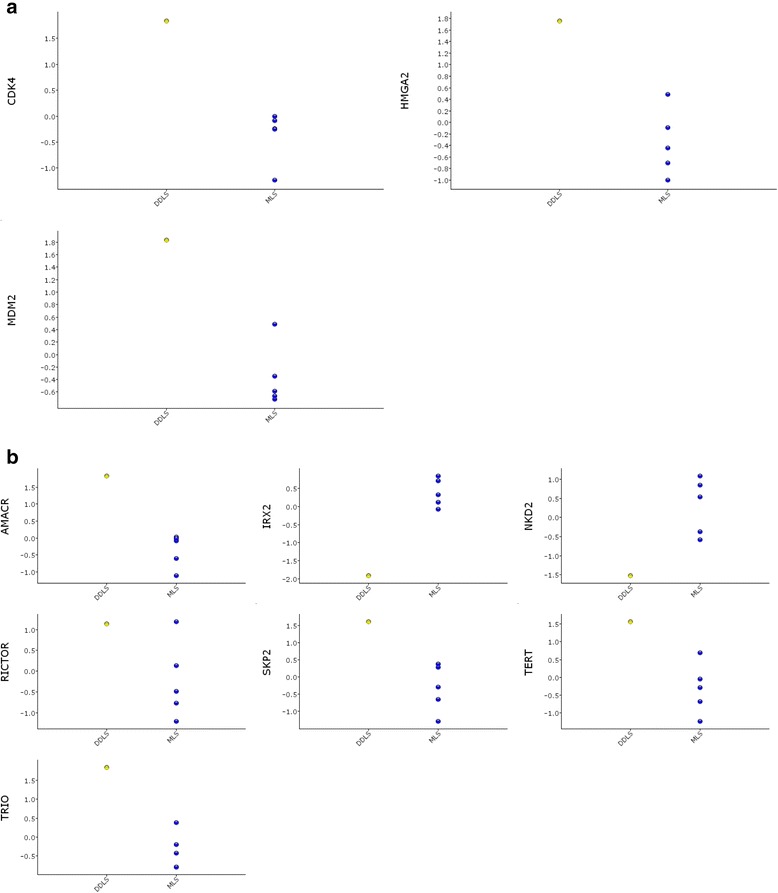
Scatter plots of the expression of some selected genes in a 12q and b 5p in the present case of DDLS compared with 5 samples of myxoid liposarcoma (MLS)
Discussion
ALT/WDLS share many cytogenetic characteristics with DDLS - supernumerary ring chromosomes and/or giant marker chromosomes constitute the hallmark of the reported karyotypes from ALT/WDLS (n = 174) and DDLS (n = 27). On average, DDLS tend to have somewhat more complex karyotypes than ALT/WDLS, whereas the 15 published karyotypes of pleomorphic liposarcomas (PLS) are distinctly more complex and ring chromosomes are much less frequent [2] (Additional file 1). Similarly, near-diploid stemline chromosome numbers predominate in ALT/WDLS and DDLS, but are rare in PLS. The ring and giant marker chromosomes in ALT/WDLS and DDLS always contain sequences from 12q, typically with several separate amplicons that invariably include MDM2 (and often also CDK4 and HMGA2), and frequently also segments from one or more other chromosomes [11–17]. Available data show that sequences from almost all chromosome arms may be co-amplified with 12q; the only exceptions - Yp, Yq and the p-arms of the acrocentric chromosomes - could be due to the methods of detection. The most commonly involved chromosome arms are 1q (46%), 6q (22%), 7p, 8q, 9q (13%), 1p, 4p, 14q (12%), 5p, 12p, 20q (11%) and 16q (10%). The non-random co-amplification of certain regions suggests that they harbour genes of potential pathogenetic significance, or that they contain sequences prone to recombine with 12q amplicons.
DDLS usually also show more copy number changes than ALT/WDLS [16]. A comparison between the well-differentiated and dedifferentiated components of the same tumor revealed more aberrations among the latter, but no particular sequence(s) could account for the dedifferentiation process [12, 18]. Even more extensive genomic reorganization is found among PLS. The dominating (>15% of cases) amplifications include sequences from 1q and 12q in WDLS, from 1p, 1q, 6q, 8q and 12q in DDLS, and from 5p and 20q in PLS (Table 2). A conspicuous difference is the paucity of 12q amplification in PLS. A clear trend of increasing frequencies in WDLS to DDLS to PLS is seen for amplifications in 5p and 20q – 3%, 13%, 23% and 0%, 6%, 23%, respectively. Such a trend is not seen for amplifications in any other chromosome arm. Possibly, these differences indicate that some gene(s) in 5p and 20q are of importance for tumor aggressiveness.
Table 2.
Amplification of sequences from chromosome arms in WDLS, DDLS and PLS, based on literature data
| Chromosome arm | Ampl. WDLS (%) n = 79 | Ampl. DDLS (%) n = 32 | Ampl. PLS (%) n = 22 | Chromo-some arm | Ampl. WDLS (%) n = 79 | Ampl. DDLS (%) n = 32 | Ampl. PLS (%) n = 22 |
|---|---|---|---|---|---|---|---|
| 1p | 0 | 19 | 9 | 11q | 0 | 6 | 0 |
| 1q | 30 | 28 | 9 | 12p | 3 | 6 | 0 |
| 2p | 3 | 0 | 0 | 12q | 76 | 88 | 0 |
| 2q | 0 | 9 | 0 | 13q | 0 | 9 | 5 |
| 3p | 0 | 3 | 0 | 14q | 4 | 13 | 5 |
| 3q | 3 | 3 | 5 | 15q | 0 | 9 | 0 |
| 4p | 5 | 9 | 0 | 16p | 0 | 0 | 0 |
| 4q | 3 | 3 | 0 | 16q | 0 | 3 | 0 |
| 5p | 3 | 13 | 23 | 17p | 0 | 0 | 5 |
| 5q | 0 | 0 | 0 | 17q | 0 | 0 | 0 |
| 6p | 0 | 6 | 0 | 18p | 1 | 0 | 0 |
| 6q | 5 | 31 | 14 | 18q | 3 | 3 | 0 |
| 7p | 3 | 9 | 5 | 19p | 1 | 3 | 0 |
| 7q | 1 | 3 | 0 | 19q | 4 | 9 | 0 |
| 8p | 4 | 6 | 0 | 20p | 0 | 3 | 5 |
| 8q | 4 | 16 | 0 | 20q | 0 | 6 | 23 |
| 9p | 0 | 3 | 5 | 21q | 4 | 0 | 5 |
| 9q | 3 | 9 | 0 | 22q | 0 | 0 | 5 |
| 10p | 0 | 9 | 0 | Xp | 0 | 3 | 0 |
| 10q | 0 | 0 | 0 | Xq | 0 | 0 | 0 |
| 11p | 1 | 9 | 0 | Yp, Yq | 0 | 0 | 0 |
Amplification of 5p segments is not confined to adipocytic tumors, but has been reported in other soft tissue sarcomas, as well as in epithelial neoplasms. Among sarcomas, it is preferentially seen in tumors typically characterized by complex chromosomal aberrations, such as myxofibrosarcomas, undifferentiated pleomorphic sarcomas, leiomyosarcomas and angiosarcomas, some of which are difficult to distinguish from PLS [19–28]. Also some other non-mesenchymal tumors, such as urinary bladder cancer, non-small cell lung cancer, cervical cancer and multiple myeloma, show 5p amplification with amplicons to some extent overlapping those found in sarcomas (e.g., [29–32]). These data further support the suggestion that amplification of genes in 5p may be associated with aggressive tumor growth. Information on concomitant amplification of 5p and 12q sequences is only available in some of the tumor types listed above, but data indicate that it is not common among PLS, leiomyosarcoma, or myxofibrosarcoma. Findings from array analyses support the paucity of extra copies of both 5p and 12q in PLS; it is rare in WDLS, but found in about one-fourth of DDLS (Table 3).
Table 3.
Fraction of borderline and malignant adipose tissue tumors with copy number changes in 5p and 12q13–21
| Chromosome segmenta | WDLS | DDLS | PLS | |
|---|---|---|---|---|
| 5p | 12q13–21 | |||
| 0 | 0 | 0.01 | 0 | 0.14 |
| 0 | G | 0.21 | 0.09 | 0.09 |
| 0 | A | 0.73 | 0.63 | 0 |
| G | 0 | 0 | 0.03 | 0.36 |
| A | 0 | 0 | 0 | 0.23 |
| G | G | 0.01 | 0 | 0.18 |
| G | A | 0.01 | 0.13 | 0 |
| A | A | 0.03 | 0.13 | 0 |
| Summary of the figures above, making no distinction between gain and amplification | ||||
| 0 | G/A | 0.94 | 0.72 | 0.09 |
| G/A | 0 | 0 | 0.03 | 0.59 |
| G/A | G/A | 0.05 | 0.26 | 0.18 |
These calculations are based on available literature data
a0 = no copy number change; G = gain; A = amplification; G/A = gain or amplification
Apart from amplified 12q sequences, regularly confined to ring and giant marker chromosomes, the chromosomal distribution of other amplified sequences is less well documented. Co-amplified chromosomal material, in particular from 1q, has, however, been shown to be intermingled with 12q sequences in rings and giant markers [13, 33–35]. The present case, showing both 5p and 12q amplicons, fits well with a minor subset of DDLS, but is atypical in the sense that it displays a near-triploid chromosome complement without any ring or giant marker chromosomes. Moreover, the complex pattern of amplification of 5p and 12q sequences, together in the same chromosomes and separately in different chromosomes, is unusual. Admittedly, there is no definition of what should be regarded as a giant marker, but those described in the literature are typically at least twice as large as chromosome 1. The size of the largest aberrant chromosome (B) containing 5p and 12q sequences in the present case was 1.5 times the length of chromosome 1, as estimated from G-banding (Fig. 3c). The vast majority of the 24 chromosomes with wcp5 and/or wcp12 signals were much smaller than chromosome 1. Only rare cases of ALT/WDLS with amplification in medium-sized linear chromosomes have been reported [36, 37]. No similar pattern of amplified sequences scattered over so many chromosomes has been reported before.
Genes located in 5p that have been suggested as possible amplicon targets in sarcomas include NKD2, TERT, IRX2, TRIO, AMACR, SKP2 and RICTOR (e.g., [24, 38–40]). In the present case, these genes were amplified at similar levels (about 10 copies), but with a slightly lower level for IRX2 and a slightly higher level for TRIO. On average, the amplification levels of sequences covering almost the entire 5p were lower than the levels seen in 12q and 20q, In particular, one of the well documented targets in 12q, CDK4, was highly amplified (about 17 copies). Also, in 5p there were very few and short intervening sequences showing a copy number corresponding to the ploidy level, in contrast to 12q and 20q where such intervening sequences were more abundant and mostly much larger, resulting in a more discontinuous amplicon pattern. This could indicate that the gene gains in 5p are less important, or merely passengers, or that 5p contains no or few genes that, if amplified, would counteract cell survival or proliferative advantages conferred by the amplified target genes. Negatively acting genes, from the tumor cell’s perspective, would be selected against. Also neutral passenger genes would gradually be lost since they represent a replication cost affecting the tumor cells’ fitness. However, it is not necessarily so that higher copy numbers are a sign of pathogenetic impact. First, the copy number is not always directly correlated with expression at the protein level [41]. Second, the tuning of protein co-expression is delicate. Amplified genes can affect the activity of many non-amplified genes and too many copies of some genes could be counterproductive for the cancer cell fitness.
The origin of the observed scattered pattern of amplified 5p and 12q sequences is obscure. Most probably, there was an early rearrangement involving substantial parts of chromosomes 5 and 12 resulting in a mitotically unstable, possibly dicentric, structure. Through further structural reorganization, MDM2/CDK4 and 5p genes may have been positioned close to each other and then spread to other chromosomes, sometimes together and sometimes separately. The many chromosomes involved indicate a stage of karyotypic instability, which may have been transient. Although the observed karyotype may represent a sideline in the tumor cell population that was preferentially dividing in vitro, or biased sampling, it did not, despite its complexity, show signs of extensive heterogeneity. A possible initial event could be chromothripsis, a phenomenon that in itself does not result in copy number changes, but can be a starting point [42, 43]. However, some of the aberrant chromosomes, in particular B but also C, containing large segments of both chromosomes 5 and 12 are hardly compatible with amplification following chromothripsis. An alternative scenario might be related to the mechanism of telomere length maintenance active in the tumor. Instead of activation of the telomerase-associated mechanism many sarcomas use alternative lengthening of telomere mechanisms. This is rare in WDLS and myxoid liposarcomas, but fairly common in DDLS and the dominating mechanism in PLS [44]. Part of the alternative lengthening of telomeres confers a destabilization of the genome through nuclear receptor binding to telomeres resulting in multiple inserted interstitial telomere sequences that are fragile and thus recombination prone [45]. This mechanism alone or, more likely, combined with an early mitotically unstable structure including 5p and 12q sequences as alluded to above could lead to amplification and a spreading of these sequences to a variety of chromosomes. Indeed, other mechanisms may be responsible for the observed pattern of scattered amplification. Recent studies have demonstrated that telomere crisis and telomere healing can have dramatic and multiple effects on the genome [46, 47]. These include polyploidization as well as chromosome instability that may lead to kataegis or chromothripsis-like aberrations.
Whether the present tumor represents an exceptional case remains unknown since few similar studies of liposarcomas without ring or giant marker chromosomes have been reported.
Conclusions
The finding of genomic amplification through distribution of 5p and 12q sequences, together and separately, to many chromosomes in a DDLS lesion represents a novel cytogenetic pattern of copy number gains. This contrasts with amplification through formation of ring or giant marker chromosomes commonly seen in WDLS and DDLS. The amplicons of 12q were discontinuous, whereas those of 5p comprised almost the entire arm. Apart from CDK4, HMGA2 and MDM2 in 12q, candidate target genes in 5p contributing to pathogenesis include TRIO and AMACR that showed elevated expression.
Additional files
Fraction of lesions with cytogenetically detectable structures associated with gene amplification among soft tissue tumors. (DOC 104 kb)
SNP array results from the two tumor samples. (ZIP 720 kb)
Putative fusion transcripts detected at mRNA sequencing. (XLSX 26 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by the Swedish Cancer Society.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
NM and FM designed the study and wrote the manuscript. LM and NM performed karyotyping and FISH analyses. FVvS provided patient data. JN, FM, BV, AI and NM performed the SNP array analyses. FM and EA performed the RNA-seq analyses. All authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Written informed consent was obtained from the patient for the publication of their details.
Ethics approval and consent to participate
This research was approved by the Local ethics committee at the Lund University Hospital. The patient provided informed consent.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13039-017-0325-5) contains supplementary material, which is available to authorized users.
Contributor Information
Nils Mandahl, Email: nils.mandahl@med.lu.se.
Linda Magnusson, Email: linda.magnusson@med.lu.se.
Jenny Nilsson, Email: jenny.nilsson@med.lu.se.
Björn Viklund, Email: bjorn.viklund@medsci.uu.se.
Elsa Arbajian, Email: elsa.arbajian@med.lu.se.
Fredrik Vult von Steyern, Email: fredrik.vult_von_steyern@med.lu.se.
Anders Isaksson, Email: anders.isaksson@medsci.uu.se.
Fredrik Mertens, Email: fredrik.mertens@med.lu.se.
References
- 1.Mandahl N, Mertens F. Soft tissue tumors. In: Heim S, Mitelman F, editors. Cancer cytogenetics: chromosomal and molecular genetic aberrations of tumor cells, 4th ed. Oxford: Wiley Blackwell; 2015. p. 583-614.
- 2.Mitelman F, Johansson B, Mertens F (Eds.). Mitelman database of chromosome aberrations and Gene fusions in cancer. 2017. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 3.Gisselsson D, Pettersson L, Höglund M, Heidenblad M, Gorunova L, Wiegant J, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A. 2000;97:5357–5362. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandahl N, Heim S, Arheden K, Rydholm A, Willén H, Mitelman F. Three major cytogenetic subgroups can be identified among chromosomally abnormal solitary lipomas. Hum Genet. 1988;79:203–208. doi: 10.1007/BF00366238. [DOI] [PubMed] [Google Scholar]
- 5.Jin Y, Möller E, Nord KH, Mandahl N, Vult von Steyern F, Domanski HA, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer. 2012;51:510–520. doi: 10.1002/gcc.21939. [DOI] [PubMed] [Google Scholar]
- 6.Walther C, Mayrhofer M, Nilsson J, Hofvander J, Jonson T, Mandahl N, et al. Genetic heterogeneity in rhabdomyosarcoma revealed by SNP array analysis. Genes Chromosomes Cancer. 2016;55:3–15. doi: 10.1002/gcc.22285. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen M, Sundström M, Göransson Kultima H, Botling J, Micke P, Birgisson H, et al. Allelespecific copy number analysis of tumor samples with aneuploidy and tumor heterogeneity. Genome Biol. 2011;12:R108. doi: 10.1186/gb-2011-12-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayrhofer M, Viklund B, Isaksson A, Rawcopy: improved copy number analysis with Affymetrix arrays http://www.nature.com/articles/srep36158 [DOI] [PMC free article] [PubMed]
- 9.Hofvander J, Tayebwa J, Nilsson J, Magnusson L, Brosjö O, Larsson O, et al. RNA sequencing of sarcomas with simple karyotypes: identification and enrichment of fusion transcripts. Lab Investig. 2015;95:603–609. doi: 10.1038/labinvest.2015.50. [DOI] [PubMed] [Google Scholar]
- 10.Nicorici D, Satalan M, Edgren H, Kangaspeska S, Murumagi A, Kallioniemi O, Virtanen S, Kilkku O. Fusion catcher - a tool for finding somatic fusion genes in paired-end RNA-sequencing data. 2014. http://dx.doi.org/10.1101/011650.
- 11.Szymanska J, Tarkkanen M, Wiklund T, Virolainen M, Blomqvist C, Asko-Seljavara S, et al. Gains and losses of DNA sequences in liposarcomas evaluated by comparative genomic hybridization. Genes Chromosomes Cancer. 1996;15:89–94. doi: 10.1002/(SICI)1098-2264(199602)15:2<89::AID-GCC2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Chibon F, Mariani O, Derre J, Malinge S, Coindre J-M, Guillou L, et al. A subgroup of malignant fibrous histiocytomas is associated with genetic changes similar to those of well-differentiated liposarcomas. Cancer Genet Cytogenet. 2002;139:24–29. doi: 10.1016/S0165-4608(02)00614-3. [DOI] [PubMed] [Google Scholar]
- 13.Micci F, Teixeira MR, Bjerkehagen B, Heim S. Characterization of supernumerary rings and giant marker chromosomes in well-differentiated lipomatous tumors by a combination of G-banding, CGH, M-FISH, and chromosome- and locus specific FISH. Cytogenet Genome Res. 2002;97:13–19. doi: 10.1159/000064038. [DOI] [PubMed] [Google Scholar]
- 14.Rieker RJ, Joos S, Bartsch C, Willeke F, Schwarzbach M, Otano-Joos M, et al. Distinct chromosomal imbalances in pleomorphic and in high-grade dedifferentiated liposarcomas. Int J Cancer. 2002;99:68–73. doi: 10.1002/ijc.10287. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt H, Bartel F, Kappler M, Würl P, Lange H, Bache M, et al. Gains of 13q are correlated with a poor prognosis in liposarcoma. Modern Pathol. 2005;18:638–644. doi: 10.1038/modpathol.3800326. [DOI] [PubMed] [Google Scholar]
- 16.Tap WD, Eilber FC, Ginther C, Dry SM, Reese N, Barzan-Smith K, et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2011;50:95–112. doi: 10.1002/gcc.20835. [DOI] [PubMed] [Google Scholar]
- 17.Garsed DW, Marshall OJ, Corbin VDA, Hsu A, Di Stefano L, Schröder J, et al. The architecture and evolution of cancer neochromosomes. Cancer Cell. 2014;26:653–667. doi: 10.1016/j.ccell.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Horvai AE, DeVries S, Roy R, O’Donnell RJ, Waldman F. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Modern Pathol 2009;22:1477-1488. [DOI] [PubMed]
- 19.Wang R, Titley JC, Lu Y-J, Summersgill BM, Bridge JA, Fisher C, et al. Loss of 13q14-q21 and gain of 5p14-pter in the progression of leiomyosarcoma. Mod Pathol. 2003;16:778–785. doi: 10.1097/01.MP.0000083648.45923.2B. [DOI] [PubMed] [Google Scholar]
- 20.Idbaih A, Coindre J-M, Derre J, Mariani O, Terrier P, Ranchere D, et al. Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances. Lab Investig. 2005;85:176–181. doi: 10.1038/labinvest.3700202. [DOI] [PubMed] [Google Scholar]
- 21.Adamowicz M, Radlwimmer B, Rieker RJ, Mertens D, Schwarzbach M, Schraml P, et al. Frequent amplifications and abundant expression of TRIO, NKD2, and IRX2 in soft tissue sarcomas. Genes Chromosomes Cancer. 2006;45:829–838. doi: 10.1002/gcc.20343. [DOI] [PubMed] [Google Scholar]
- 22.Larramendy ML, Gentile M, Soloneski S, Knuutila S, Böhling T. Does comparative genomic hybridization reveal distinct differences in DNA copy number sequence patterns between leiomyosarcoma and malignant fibrous histiocytoma? Cancer Genet Cytogenet. 2008;187:1–11. doi: 10.1016/j.cancergencyto.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Ohguri T, Hisaoka M, Kawauchi S, Sasaki K, Aoki T, Kanemitsu S, et al. Cytogenetic analysis of myxoid liposarcoma and myxofibrosarcoma by array-based comparative genomic hybridization. J Clin Pathol. 2009;59:978–983. doi: 10.1136/jcp.2005.034942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barretina J. And 45other authors. Subtype-specific genomic alterations define new targets for soft tissue sarcoma therapy. Nat Genet. 2010;42:715–721. doi: 10.1038/ng.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guillou L, Aurias A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2010;456:201–217. doi: 10.1007/s00428-009-0853-4. [DOI] [PubMed] [Google Scholar]
- 26.Gibault L, Perot G, Chibon F, Bonnin S, Lagarde P, Terrier P, et al. New insights in sarcoma oncogenesis: a comprehensive analysis of a large series of 160 soft tissue sarcomas with complex genomics. J Pathol. 2011;223:64–71. doi: 10.1002/path.2787. [DOI] [PubMed] [Google Scholar]
- 27.Li C-F, Wang J-M, Kang H-Y, Huang C-K, Wang J-W, Fang F-M, et al. Characterization of amplification-driven SKP2 overexpression in myxofibrosarcoma: potential implications in tumor progression and therapeutics. Clin Cancer Res. 2012;18:1598–1610. doi: 10.1158/1078-0432.CCR-11-3077. [DOI] [PubMed] [Google Scholar]
- 28.Nord KH, Macchia G, Tayebwa J, Nilsson J, Vult von Steyern F, Brosjö O, et al. Integrative genome and transcriptome analyses reveal two distinct types of ring chromosome in soft tissue sarcomas. Hum Mol Genet. 2014;23:878–888. doi: 10.1093/hmg/ddt479. [DOI] [PubMed] [Google Scholar]
- 29.Zheng M, Simon R, Mirlacher M, Maurer R, Gasser T, Forster T, et al. TRIO amplification and abundant mRNA expression is associated with invasive growth and rapid tumor cell proliferation in urinary bladder cancer. Am J Pathol. 2004;165:63–69. doi: 10.1016/S0002-9440(10)63275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet Cytogenet. 2008;182:1–11. doi: 10.1016/j.cancergencyto.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008;7:58. doi: 10.1186/1476-4598-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tapper W, Chiecchio L, Dagrada GP, Konn ZJ, Stocley DM, Szubert AJ, et al. 2011. Heterogeneity in the prognostic significance of 12p deletion and chromosome 5 amplification in multiple myeloma. J Clin Oncol. 2011;29:e37–e39. doi: 10.1200/JCO.2010.31.0516. [DOI] [PubMed] [Google Scholar]
- 33.Pedeutour F, Forus A, Coindre J-M, Berner J-M, Nicolo G, Michiels J-F, et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer. 1999;24:30–41. doi: 10.1002/(SICI)1098-2264(199901)24:1<30::AID-GCC5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 34.Meza-Zepeda LA, Berner J-M, Henriksen J, South AP, Pedeutour F, Dahlberg AB, et al. Ectopic sequences from truncated HMGIC in liposarcomas are derived from various amplified chromosomal regions. Genes Chromosomes Cancer. 2001;31:264–273. doi: 10.1002/gcc.1143. [DOI] [PubMed] [Google Scholar]
- 35.Snyder EL, Sandstrom DJ, Law K, Fiore C, Sicinska E, Brito J, et al. C-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol. 2009;218:292–300. doi: 10.1002/path.2564. [DOI] [PubMed] [Google Scholar]
- 36.Forus A, Bjerkehagen B, Sirvent N, Meza-Zepeda LA, Coindre J-M, Berner J-M, et al. A well-differentiated liposarcoma with a new type of chromosome 12-derived markers. Cancer Genet Cytogenet. 2001;131:13–18. doi: 10.1016/S0165-4608(01)00516-7. [DOI] [PubMed] [Google Scholar]
- 37.Nilsson M, Domanski H, Mertens F, Mandahl N. Atypical lipomatous tumor with rare structural rearrangements involving chromosomes 8 and 12. Oncol Rep. 2005;13:649–652. [PubMed] [Google Scholar]
- 38.Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 39.Gibault L, Ferreira C, Perot G, Audebourg A, Chibon F, Bonnin S, et al. From PTEN loss of expression to RICTOR role in smooth muscle differentiation: complex involvement of the mTOR pathway in leiomyosarcomas and pleomorphic sarcomas. Modern Pathol. 2012;25:197–211. doi: 10.1038/modpathol.2011.163. [DOI] [PubMed] [Google Scholar]
- 40.Okada T, Lee AY, Qin L-X, Agaram N, Mimae T, Shen Y, et al. Integrin-α10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov. 2016;6:1148–1165. doi: 10.1158/2159-8290.CD-15-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dürrbaum M, Storchova Z. Effects of aneuploidy on gene expression: implications for cancer. FEBS J. 2016;283:791–802. doi: 10.1111/febs.13591. [DOI] [PubMed] [Google Scholar]
- 42.Kloosterman WP, Koster J, Molenaar JJ. Prevalence and clinical implications of chromotripsis in cancer genomes. Current Opinion. 2014;26:64–72. doi: 10.1097/CCO.0000000000000038. [DOI] [PubMed] [Google Scholar]
- 43.Rode A, Maass KK, Willmund KV, Lichter P, Emst A. Chromothripsis in cancer cells: an update. Int J Cancer. 2015;138:2322–2333. doi: 10.1002/ijc.29888. [DOI] [PubMed] [Google Scholar]
- 44.Lee J-C, Jeng Y-M, Liau J-Y, Tsai J-H, Hsu H-H, Yang C-Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Modern Pathol. 2015;28:1064–1073. doi: 10.1038/modpathol.2015.67. [DOI] [PubMed] [Google Scholar]
- 45.Marzec P, Armenise C, Perot G, Roumelioti F-M, Basyuk E, Gagos S, et al. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell. 2015;160:913–927. doi: 10.1016/j.cell.2015.01.044. [DOI] [PubMed] [Google Scholar]
- 46.Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. 2017;18:175–186. doi: 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hannes F, Van Houdt J, Quarrell OW, Poot M, Hochstenbach R, Fryns J-P, et al. Telomere healing following DNA polymerase arrest-induced breakages is likely the main mechanism generating chromosome 4p terminal deletions. Hum Mut. 2010;31:1343–1351. doi: 10.1002/humu.21368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fraction of lesions with cytogenetically detectable structures associated with gene amplification among soft tissue tumors. (DOC 104 kb)
SNP array results from the two tumor samples. (ZIP 720 kb)
Putative fusion transcripts detected at mRNA sequencing. (XLSX 26 kb)
Data Availability Statement
The datasets analysed during the current study are available from the corresponding author on reasonable request.