

Abstract
During polyketide biosynthesis, acyltransferases (ATs) are the essential gatekeepers which provide the assembly lines with precursors and thus contribute greatly to structural diversity. Previously, we demonstrated that the discrete AT KirCII from the kirromycin antibiotic pathway accesses non-malonate extender units. Here, we exploit the promiscuity of KirCII to generate new kirromycins with allyl- and propargyl-side chains in vivo, the latter were utilized as educts for further modification by ‘click’ chemistry.
Keywords: antibiotic, kirromycin, polyketide synthase, trans-acyltransferase, engineering, click chemistry
Polyketides are important secondary metabolites widely used as antibiotics, antifungals, and drugs for other clinical applications1, 2. Erythromycin, mupirocin, rapamycin, FK506, lovastatin and epothilone B are examples of antimicrobial, immunosuppressant, hypocholesterolemic and anticancer drugs, respectively. All these compounds are biosynthesized by complex multifunctional enzymes with modular architecture, the polyketide synthases (PKSs).
In typical modular type I PKSs, acyltransferase domains (ATs) select and load malonyl-CoA derived precursors onto acyl carrier proteins (ACPs). The AT domains are either embedded in the PKS (cis-ATs) or encoded by distinct genes and function in trans as discrete enzymes (trans-ATs)3, 4. After the AT-dependent loading step, ketosynthases (KSs) catalyze the condensation of ACP-linked extender units. Optional domains, such as ketoreductases (KRs), dehydratases (DHs), enoylreductases (ERs) or methyltransferases (MTs) further process the polyketide chain. In most cases, the product is released from the assembly line by a thioesterase (TE) domain and modified by post-PKS tailoring enzymes.
Notably, the AT domain determines which extender unit is incorporated into the growing polyketide chain5-7. Consequently, the extender unit specificity of the AT cumulatively dictates large portions of the final polyketide structure, and could be leveraged to improve the pharmacological properties of polyketide-based chemical entities through alteration of the precursors and/or ATs8. These features make AT engineering attractive for rational modification of polyketide assembly lines. Directed engineering approaches such as AT domain swapping, AT site-directed mutagenesis9 and cross-complementation of an AT-inactivated cis-PKS have been described for various polyketides, including erythromycin, rapamycin, tylosin and geldanamycin10. However, most of the previous attempts to vary AT substrate specificity target cis-AT PKS pathways. Furthermore, all studies on extender unit variation involve non-alkyne precursors with the exception of 2-propargyl-erythromycin11 and propargyl-premonensin12, which were produced by feeding N-acetylcysteamine (SNAC) pre-activated substrates to a strain harboring the engineered AT6DEBS and to the wild-type producing strain, respectively.
For trans-AT type PKSs, only a few examples of assembly lines utilizing non-malonyl-CoA extender units have been reported. One of the best studied biosynthetic pathways, which involves a trans-AT type PKS and incorporates a branched unit into its product, is that of the antibiotic kirromycin (1) from Streptomyces collinus Tü 36513. The unusual and complex trans- and cis-PKS/NRPS assembly line recruits two discrete ATs, KirCI and KirCII (Figure 1). In a previous study we demonstrated that KirCI provides the biosynthetic machinery with malonate14. The second AT, KirCII, was shown to transfer ethyl malonate onto the kirromycin ACP5 (Kirr-ACP5) and thus introduces the C-28 ethyl branch into the backbone of the antibiotic5. More recently, in vitro studies revealed that this enzyme accepts other non-malonyl-CoA substrates such as allylmalonyl-CoA, propargylmalonyl-CoA and to a lesser extent azidoethyl- and phenylmalonyl-CoA15, 16.
Figure 1. Kirromycin assembly line and the production of new kirromycin derivatives.
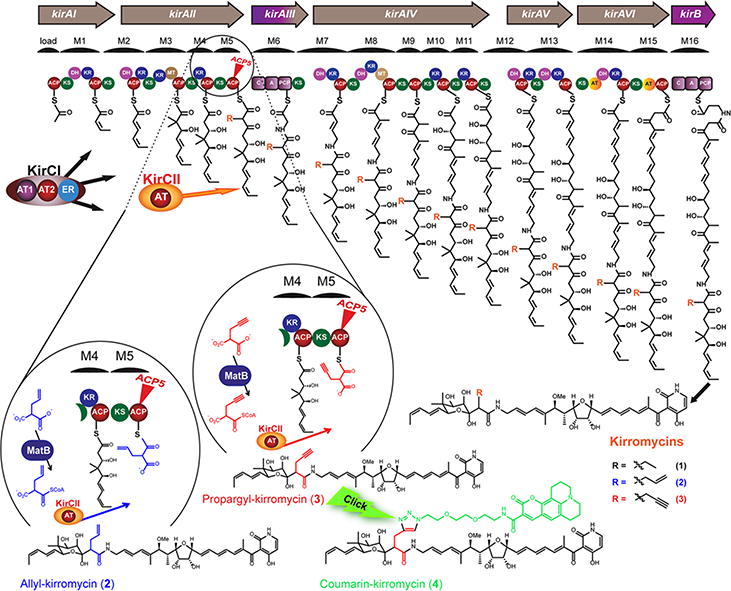
A: acyltransferase ACP: acyl carrier protein, KS: ketosynthase, DH: dehydrogenase, KR: ketoreductase, MT: methyltransferase, ER: enoylreductase, C: condensation domain, A: adenylation domain, PCP: peptidyl carrier protein, MatB: engineered malonyl-CoA synthetase (T207G/M306I), KirCII: discrete acyltransferase and R: functional group. Kirromycin (1), allyl-kirromycin (2), propargyl-kirromycin (3) and coumarin-kirromycin (4).
Alkyne-containing molecules, such as propargyl-derived compounds provide functional groups for further derivatization by “click chemistry”. The term “click chemistry” describes rapid, selective, high-yield reactions of chemical moieties under mild, aqueous conditions. Copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC) is the most commonly used click reaction, which fulfills those criteria. In recent years, different click chemistry reactions were developed, often resulting in new chemical structures and therefore, the approach become one of the most powerful tools in drug discovery, chemical biology, and proteomic applications17, 18.
To the best of our knowledge, in vivo production of allyl- and/or propargyl-modified polyketides, derived from trans-AT pathways has so far not been described. This and the fact that KirCII continues to be the only characterized discrete AT with such a promising spectrum of substrate selectivity, encouraged us to study the flexibility of KirCII in vivo and to exploit its features for polyketide engineering.
In our kirromycin bio-derivatization approach, the producer strain S. collinus Tü 365 was engineered to facilitate the utilization of non-natural polyketide precursors. The malonyl-CoA synthetase T207G/M306I MatB16, 19, which is able to activate non-natural extender units, was cloned into the vector pRM4.2 downstream of the strong ermE* promoter. The resulting plasmid pRM4.2-matB was transferred into S. collinus Tü 365 and an isolated S. collinus pRM4.2-matB clone was cultivated for feeding experiments. Separate cultures of the engineered strain were fed with allyl- and propargylmalonic acid and the production of new compounds was analyzed by HPLC-MS (Supplementary Figure 1). In both the allyl- and propargyl-derived extract, an ion consistent with kirromycin (1) (m/z = 795.5 [M-H]-) was present. Remarkably however, additional ions at m/z = 807.5 [M-H]- and m/z = 805.4 [M-H]- were detected in the allyl- and propargyl-derived samples, respectively (Supplementary Figure 1). These ions corresponded to the calculated masses of allyl-kirromycin (2) and propargyl-kirromycin (3) (Supplementary Table 1).
In order to characterize the new metabolites, fermentation and feeding experiments were carried out in five liter scale. As only allylmalonic acid is commercially available and propargylmalonic acid is not, the latter had to be chemically synthesized to set up serial experiments with both substrates. Crude extracts obtained from these feeding experiments were subjected to preparative HPLC. Notwithstanding the modest structural differences between kirromycin (1) and its derivatives and, as a consequence, the challenging chromatographic purification of kirromycin derivatives, the compounds could be separated and highly enriched. Thereby 3.8 mg of a sample containing 90% allyl-kirromycin (2) and 10% kirromycin (1) and 3.7 mg of a sample containing 22 % propargyl-kirromycin (3) and 78 % kirromycin (1) were obtained. Interestingly, these derivative:kirromycin ratios are in better agreement with the KirCII in vitro activity data compared to the MatB in vitro activity data. The engineered MatB enzyme showed similar in vitro CoA-activation rates toward ethylmalonic acid (91±3%), propargylmalonic acid (91±5%) and allylmalonic acid (89±2%),16 but the loading efficiency of KirCII differed significantly depending on the substrate. For example, both ethylmalonyl-CoA and allylmalonyl-CoA are loaded by KirCII onto the cognate Kir-ACP5 with equal efficiency (∼15% conversion), whereas propargylmalonyl-CoA is loaded at about half of this efficiency (8% conversion)16. Furthermore, the isolated yields of allyl- and propargyl-kirromycin indicate that in the in vivo system, the downstream kirromycin assembly line domains are more tolerant to the artificial substrate allylmalonyl-CoA than for propargyl-CoA.
To confirm the product identity of allyl-kirromycin (2) and propargyl-kirromycin (3), the purified samples, as well as kirromycin (1) were analyzed by high resolution MS. Based on the kirromycin fragments20 previously detected and described by Edwards and coworkers, we proposed a fragmentation mechanism for kirromycin (1) and the derivatives, allyl-kirromycin (2) and propargyl-kirromycin (3) (Supplementary Table 1, Supplementary Figure 3 and Supplementary Figure 4a-c). The anticipated fragments A-C were detected for all three molecules, which confirms that the allyl and propargyl moieties were introduced in place of the original ethyl side chain at C-28 in kirromycin (1) (Supplementary Table 1, Supplementary Figure 4b-c). In addition, 1H, 13C, COSY, HSQC HMBC and NMR spectra were recorded for the allyl-kirromycin and for the propargyl-kirromycin sample (Supplementary Figure 5I-II and Supplementary Table 2). Complete NMR signal assignments of allyl-kirromycin (2) substantiated the presence of the allyl moiety. In the 1H spectrum of the enriched propargyl-kirromycin sample, a spin-system consisting of H-28 (δH 3.13, 1H, m), H-44 (δH 2.57, 2H, m) and H-46 (δH 2.30, 1H, t, J = 2.64 Hz) was detected in addition to “the collateral signals” from the residual traces of kirromycin (1). The corresponding HMBC correlations unambiguously confirm the identity of propargyl-kirromycin (2) (Figure 2).
Figure 2. New kirromycin derivatives.
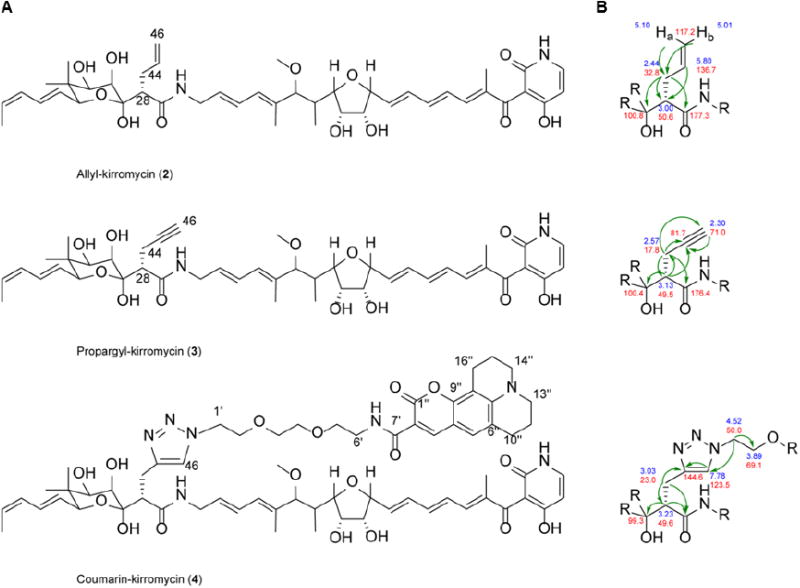
(A) Chemical structure of allyl-kirromycin (2), propargyl-kirromycin (3), coumarin-kirromycin (4). (B) Key HMBC correlations of the kirromycin derivatives.
The fact that the propargyl-kirromycin derivative (2) possesses an alkyne moiety attracted our particular attention, as alkynes are highly desired chemical structures for click reactions (i.e. CuAAC). Therefore, propargyl-kirromycin (2) was subjected to CuAAC reaction using the fluorescent dye coumarin-343-azide. The click-reaction product was purified by chromatographic methods. A total yield of 0.81 mg was obtained and analyzed by HPLC-MS in negative ion mode. The analytical data demonstrated the presence of a molecule ion with the sum formula C66H84N7O17, which is in accordance with the coumarin-modified compound. MS/MS fragmentation revealed signals corresponding to the fragments A-C of the 1,2,3-triazole compound (Supplementary Table 1 and Supplementary Figure 4d). Despite the small quantity of the click-reaction product, NMR analyses were carried out leading to a partial spectra recording and signal assignments (Supplementary Figure 5III and Supplementary Table 2). The triazole signal (δH 7.78, 1H, s) was observed and the respective HMBC correlations were detected (Figure 2 and Supplementary Figure 5III), confirming the identity of the click-reaction product coumarin-kirromycin (4). According to the HPLC-MS analysis this sample contained mainly coumarin-kirromycin (4) (96 %) and only traces of kirromycin (1) (4 %). Thus, taking advantage of the newly introduced propargyl-anchor, we could not only synthesize a new click product, but we also exploited its structural features as a strategy to separate coumarin-kirromycin (2) from kirromycin (1). The results support the application of the KirCII system for synthesizing biomolecules that are provided with functional groups, which can be used in highly selective click chemistry (Figure 1). This allows the production of further chemically modified valuable compounds as well as their straightforward purification. Moreover, coupling of azidofluorescent dyes can be used for fluorescent labeling of terminal alkynes in bioactive molecules, which is highly sought-after for studying drug-target interactions and rational drug design.
The ability to generate and isolate the allyl-kirromycin (2) and coumarin-kirromycin (4) raised the question of whether the modifications at C-28 affect the antibiotic activity compared to kirromycin (1). The mode of action of kirromycin (1) is well understood. The antibiotic binds to the elongation factor Tu (EF-Tu)21, 22 and thereby inhibits the protein biosynthesis in some species of gram positive and gram negative bacteria (e.g. Neisseria gonorrhoeae, Haemophilus influenzae, Streptococci and some Enterococci strains). In order to examine and compare the efficiency of the new derivatives, a GFP-based in vitro translation assay23, 24 was used (Supplementary Figure 7). Increasing concentrations of the compounds were added to the assay reactions and changes in eGFP synthesis were monitored by measuring the eGFP fluorescence (Figure 3, Supplementary Tables 3a-b). In this system, allyl-kirromycin (IC50 = 1.0 μM) inhibited the eGFP protein synthesis comparably to the native compound kirromycin (1) (IC50 = 0.9 μM) (Figure 3). The derivative coumarin-kirromycin (4) also inhibited the system (IC50= 7.3 μM), albeit to a lesser extent. The data showed that the replacement of the ethyl moiety at C-28 of kirromycin (1) with an allyl moiety has no negative effect on the interactions with its target. According to the crystal structure of the complex of Thermus thermophiles EF-Tu with aurodox, an N-methyl derivative of kirromycin (1), the antibiotic mainly interacts through the pyridine, the tetrahydrofuran rings, and the aliphatic tail on the goldinoic acid with the EF-Tu residues25, 26. These structural moieties do not include the branch at C-28 of kirromycin (1). Thus, our results are in agreement with the binding site studies obtained from the crystal structure of the EF-Tu-aurodox complex25, 26 and confirm that the allyl-moiety is not directly involved in the interaction of the antibiotic with the target. In case of coumarin-kirromycin (4), where the ethyl group is conjugated to the relatively large coumarin moiety, the molecule is still able to inhibit protein biosynthesis, but with an approximately seven-fold lower inhibitory activity, indicating that the bulky coumarin residue affects the drug-target interaction. Nevertheless, our results demonstrate that kirromycin (1) is robust to variations at the C-28 atom and highlights that this position is suitable for further modifications.
Figure 3. In vitro activity assay.
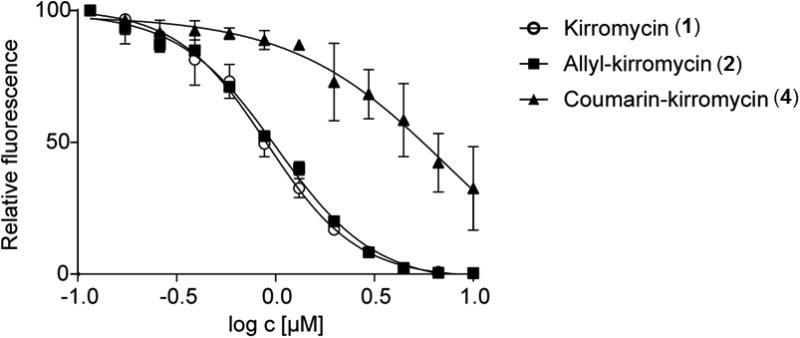
Inhibition of eGFP translation using kirromycin and kirromycin derivatives.
The modification to propargyl-kirromycin is of particular interest as it enables the “fusion” of diverse moieties to the kirromycin-backbone. The attachment of chemical groups, which would alter the physicochemical properties of the molecule, could be used to generate derivatives with optimized import into the cell and thereby extend the spectrum of antimicrobial activity for this family of antibiotic compounds. Furthermore, labelling of the structure with fluorescent agents would allow the monitoring of the antibiotic penetrating the cell and provides tools for studying protein biosynthesis (e.g. interactions with the elongation factor EF-Tu, which can be easily visualized by fluorescence detection methods).
This work is the first example that leverages a tailored malonyl-CoA synthetase and a trans-AT for rational and regioselective engineering of a complex polyketide antibiotic in order to introduce artificial alkene and alkyne sidechains in vivo. Our results demonstrate that after the extender unit substitution, which is accomplished relatively early in the assembly process, the downstream NRPS and PKS modules accept the un-natural sidechains and continue the product biosynthesis. These findings uncover the flexibility of a mixed PKS/NRPS assembly line towards the un-natural building blocks and encourage the further exploration of trans-AT-based polyketide engineering. The MatB-KirCII/ACP5 system may have the potential to become a valuable tool, enabling the regiospecific incorporation of chemical handles into polyketides, which can serve as anchors for “click” chemistry. The combination of this “bio-derivatization” approach and “click” chemistry methods enables the generation of novel molecular probes and analogues of compounds, which would be otherwise difficult to access by using exclusively conventional organic chemistry.
Methods
General methods and strain cultivation conditions
E. coli strains, the wild type and mutants of S. collinus Tü 365 were cultivated in liquid or on solid media as described previously5. Detailed information on strains, media composition, antibiotic concentrations, plasmids, oligonucleotides and PCR conditions can be obtained from Supporting Information.
Chemical synthesis of 2-(Prop-2-yn-1-yl)malonic acid
Dimethyl propargylmalonate (0.94 ml, 6.2 mmol) and NaOH (2.4 g, 60 mmol) were added to a round bottom flask containing 10 ml H2O and the mixture was stirred for 6 hours at 65°C. The solution was cooled on ice and 10 ml ice cold 12.1 N HCl was added. The reaction was extracted 6x with diethyl ether. The organic extractions were pooled, dried using MgSO4 and filtered. The solvent was removed under vacuum to give 2-(prop-2-yn-1-yl)malonic acid as a white solid (0.298 g, 34%). 1H-NMR (D2O, 300 MHz): δH 3.59 (1H, t, J = 7.2 Hz), 2.6 (3H, dd, J = 7.2, 2.7 Hz), 2.26 (1H, t, J = 2.7 Hz).
Cloning of the malonyl-CoA synthetase expression construct
To activate the substrates allylmalonic acid and propargylmalonic acid, the engineered malonyl-CoA synthetase (WP_011650729.1)16, 19 from Rhizobium leguminosarum (MatB T207G/M306I) was transferred into S. collinus Tü 365. Therefore, the engineered matB gene from the plasmid pET28a-matB, was cloned into the integrative vector pRM4.2 via the restriction sites NdeI and HindIII. The resulting construct pRM4.2-matB was introduced into S. collinus Tü 365 by intergeneric conjugation using E. coli ET12567 pUB307. Exconjugants were confirmed by control PCR with primers matB-test-up and matB-test-low (Supporting Information). The obtained mutant S. collinus pRM4.2-matB was applied to feeding experiments with allylmalonic acid (Fluka/Sigma-Aldrich) and the synthesized propargylmalonic acid.
Feeding of allylmalonic acid or propargylmalonic acid to S. collinus Tü 365 and S. collinus pRM4.2-matB
Pre-cultures of S. collinus Tü 365 WT and S. collinus pRM4.2-matB were cultivated for three days in TSB medium under antibiotic selection, if required (for antibiotic concentration, see Supporting Information). A main culture of 90 ml SGG medium was inoculated with 10% of the pre-culture (for media, see Supporting Information) and supplemented with 50 mg of allylmalonic acid and propargylmalonic acid (t0 = 0 h, final concentration 0.5 mg ml-1). After five days of incubation, each 100 ml culture was extracted 1:1 with ethyl acetate (1 h, RT). The samples were centrifuged (10 min, RT, 4270 g, Sorvall RC6+ Centrifuge, Thermo Scientific) and the solvent fraction was concentrated to dryness under vacuum. Extracts were re-dissolved in 1 ml methanol and analyzed by HPLC/ESI-MS (Supporting Information).
Scale-up of feeding and extraction of kirromycin derivatives
Four and a half liters of SGG medium, complemented with apramycin (50 μg ml-1) were inoculated with 10% pre-culture of S. collinus pRM4.2-matB and fed with allymalonic acid or propargylmalonic acid (final concentration, 0.5 mg ml-1). The cultures were incubated in 1 L shaking flasks for five days at 29°C. After incubation each allymalonic- or propargylmalonic-fed culture was extracted 1:1 with ethyl acetate at RT for 1 h, using an extractor. Phase separation was achieved by incubation of the extract/ethyl acetate mixture over night at RT. The ethyl acetate phase was collected, evaporated and the residue dissolved in ethyl acetate and precipitated by addition of petroleum ether (40-60°C) and a subsequent centrifugation. The precipitant was dried in a desiccator. The samples were redissolved in methanol and analyzed by HPLC/ESI-MS (Supporting Information).
Purification of allyl-kirromycin and propargyl-kirromycin
The allyl-kirromycin and propargyl-kirromycin sample were dissolved in DMSO:MeOH (5:2). Multiple chromatography runs were conducted, each with a solution of 750 μl, containing approximately 50 mg of the respective extract. The samples were separated on a Kromasil C18, 7 μm 250 × 20 mm HPLC column (Dr. Maisch, Ammerbuch, Germany) using MeCN:H2O (43:57) as mobile phase and a flow of 16 ml min-1. The purity of the obtained yields was determined by NMR analysis. In case of allyl-kirromycin, the product of 3.8 mg contained 10% residual kirromycin and for propargyl-kirromycin, the pooled fractions resulted in a mixture of 78 % kirromycin and 22 % propargyl-kirromycin. The propargyl-kirromycin sample was directly utilized in CuAAC reaction without further purification.
High Resolution Mass Spectrometry (HRMS) analysis of kirromycins
The HR-MS analysis of allyl- and propargyl-kirromycin was carried out on MaXis 4G instrument (Bruker Daltonics) coupled to a Ultimate 3000 HPLC (Thermo Fisher Scientific). An HPLC-method was applied as follows: 0.1% formic acid in water as solvent A and 0.06% formic acid in methanol as solvent B, with gradients from 10% to 100% B in 20 min and 100% B for 5 min, using a flow rate of 0.3 ml min-1. The separation was carried out on a Nucleoshell 2.7 μm 150 × 2mm column (Macherey-Nagel).
The ESI source was operated at a nebulizer pressure of 2.0 bar and dry gas was set to 8.0 L min-1 at 200°C. MS/MS spectra were recorded in Auto MS/MS mode resulting in a collision energy stepping from 32.1 to 52.0 eV. Sodium formate was used as internal calibrant and Hexakis(2,2-difluoroethoxy)phosphazene (Apollo Scientific Ltd, Stockport, UK) as lock mass.
Nuclear Magnetic Resonance (NMR) spectroscopy of kirromycins
The 1H, 13C, 1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC NMR spectra were recorded on Bruker Avance III HD spectrometers operating at a proton frequency of 600 MHz equipped with a Prodigy BBO CryoProbe or 700 MHz equipped with a TCI CryoProbe, respectively (Bruker Biospin). CD3OD as solvent and an internal standard were used. All spectra were recorded at room temperature. Chemical shifts were expressed in parts per million (ppm, δ) relative to TMS.
CuAAC reaction of propargyl-kirromycin and coumarin-343-azide and product purification
The propargyl-kirromycin sample (3.7mg) was dissolved in 0.9 ml MeOH and transferred to a Schlenk flask containing 3.75 ml t-BuOH and 3.75 ml H2O. The solution was mixed with 90 μL of 50 mM coumarin-343-azid (Lumiprobe GmbH, Hannover, Germany) in DMSO, 1 ml of 5 mM aqueous ascorbic acid and 1 ml of 2 M aqueous triethylammmonium acetate. The reaction was flushed with argon for 5 min and subsequently treated with 0.5 ml of 10 mM Cu(II)-TBTA, resolved in DMSO:H2O (55:45). The solution was stirred in a sealed flask, at room temperature for 16h. The reaction mixture was extracted with ethyl acetate and the solvent evaporated using a rotary evaporator. The remaining crude extract was dissolved in MeOH and applied to size exclusion chromatography using Sephadex LH-20 (75 × 2.6 cm, Methanol, 0.5 ml min-1). Coumarin-kirromycin-containing fractions were pooled and subjected to preparative HPLC using 43 % MeCN for 25 min, followed by a gradient to 100% in 5 minutes. To remove the residual tris-(benzyltriazolylmethyl)amine (TBTA) and kirromycin from the sample, the chromatographic separation was repeated using the same column and the following conditions: 12 ml min-1, gradient from 40(A):60(B) to 100(A):0(B) with MeOH(A):H2O(B) within 30 minutes. The final product was analyzed by LC-MS and a yield of 0.81 mg coumarin-kirromycin containing 4 % kirromycin was determined.
In vitro activity testing of kirromycin and kirromycin derivatives
The in vitro translation assay was developed based on a combination of protocols of Kim et al.23, 24 and Calhoun and Swartz27. Briefly, an E. coli S12 cell extract was prepared and mixed with nucleotides and amino acid as substrates for transcription and translation, fructose-1,6-bisphosphate for energy supply and the reporter plasmid pET28-egfp in wells of a 96 well microtiter plate to a final volume of 100 μL (Supporting Information). An assay reaction contained: 57 mM Hepes-KOH (pH 8.2), 90 mM potassium glutamate, 80 mM ammonium acetate, 12 mM magnesium acetate, 1.67 % (w/v) PEG(8000), 5 mM DTT, 1.2 mM AMP, 0.85 mM each of CMP, GMP and UMP, 0.5 mM IPTG, 34 μg ml-1 L-5-formyl-THF, 2 mM each of all 20 proteinogenic amino acids, 33 mM fructose-1,6-bisphosphate, 25% (v/v) E. coli S12 extract and approximately 5 μg ml-1 of the reporter plasmid pET28-egfp (Supporting Information, all chemicals were obtained from Sigma, all given values represent the final concentrations). The plasmid pET28-egfp was generated by cloning of the eGFP gene from pRM4.328 with NdeI and HindIII into pET28a (Novagen) to take advantage of the strong inducible T7 promoter. The S-12 extract was prepared from E. coli BL21 strain (DE3) (Invitrogen) as described by Kim et al.29, except that 1 L of 2xYTPG medium30 was used for cultivation.
To test the susceptibility of the in vitro translation system, a microtiter plate was preloaded with different concentrations of kirromycin, allyl-kirromycin and coumarin-kirromycin prior to the addition of the assay master mix. All measurements have been performed in triplicate using the same preparation of the S12 extract. The assay was incubated for approximately 4 hours at 37°C. Thereafter, the fluorescence deriving from eGFP was measured at 485/520 nm excitation/emission (POLARstar Galaxy, BMG) (Supporting Information). The data evaluation and IC50 calculation was performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com, with non-linear fit regression.
Supplementary Material
Acknowledgments
This work was supported in part by the German Center for Infection Research (Deutsches Zentrum für Infektionsforschung, DZIF, TTU09.802 and TTU09.704 to T.W. and W.W.), the Novo Nordisk Foundation to SYL and TW (NNF15OC0016226 to TW), and the National Institute of Health (GM104258-01 to G.J.W).
Footnotes
The Supporting Information is available free of charge on ACS Publications website.
Author Contributions: E.M.M-K., F.Z. contributed equally to this work.
S.G., G.J.W., W.W. and T.W. designed research. E.M.M-K., F.Z., T.S., T.H., and A.K. performed research and analyzed data. J.M. and I.K. contributed reagent. E.M.M-K., T.S., S.Y.L, G.J.W. and T.W. wrote the paper.
Notes: The authors declare no competing financial interests.
References
- 1.Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT, Wencewicz TA. Prospects for new antibiotics: a molecule-centered perspective. J Antibiot (Tokyo) 2014;67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 3.Musiol EM, Weber T. Discrete acyltransferases involved in polyketide biosynthesis. MedChemComm. 2012;3:871–886. [Google Scholar]
- 4.Helfrich EJ, Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2016;33:231–316. doi: 10.1039/c5np00125k. [DOI] [PubMed] [Google Scholar]
- 5.Musiol EM, Härtner T, Kulik A, Moldenhauer J, Piel J, Wohlleben W, Weber T. Supramolecular templating in kirromycin biosynthesis: the acyltransferase KirCII loads ethylmalonyl-CoA extender onto a specific ACP of the trans-AT PKS. Chem Biol. 2011;18:438–444. doi: 10.1016/j.chembiol.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Zhao C, Coughlin JM, Ju J, Zhu D, Wendt-Pienkowski E, Zhou X, Wang Z, Shen B, Deng Z. Oxazolomycin biosynthesis in Streptomyces albus JA3453 featuring an “acyltransferase-less” type I polyketide synthase that incorporates two distinct extender units. J Biol Chem. 2010;285:20097–20108. doi: 10.1074/jbc.M109.090092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irschik H, Kopp M, Weissman KJ, Buntin K, Piel J, Muller R. Analysis of the sorangicin gene cluster reinforces the utility of a combined phylogenetic/retrobiosynthetic analysis for deciphering natural product assembly by trans-AT PKS. Chembiochem. 2010;11:1840–1849. doi: 10.1002/cbic.201000313. [DOI] [PubMed] [Google Scholar]
- 8.Mo S, Kim DH, Lee JH, Park JW, Basnet DB, Ban YH, Yoo YJ, Chen SW, Park SR, Choi EA, Kim E, Jin YY, Lee SK, Park JY, Liu Y, Lee MO, Lee KS, Kim SJ, Kim D, Park BC, Lee SG, Kwon HJ, Suh JW, Moore BS, Lim SK, Yoon YJ. Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J Am Chem Soc. 2011;133:976–985. doi: 10.1021/ja108399b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koryakina I, Kasey C, McArthur JB, Lowell AN, Chemler JA, Li S, Hansen DA, Sherman DH, Williams GJ. Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem Biol. 2017;12:114–123. doi: 10.1021/acschembio.6b00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunn BJ, Khosla C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J R Soc Interface. 2013;10:20130297. doi: 10.1098/rsif.2013.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- 12.Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, Sanchez-Garcia E. Predicted incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives. ChemBioChem. 2014;15:1991–1997. doi: 10.1002/cbic.201402206. [DOI] [PubMed] [Google Scholar]
- 13.Weber T, Laiple KJ, Pross EK, Textor A, Grond S, Welzel K, Pelzer S, Vente A, Wohlleben W. Molecular analysis of the kirromycin biosynthetic gene cluster revealed beta-alanine as precursor of the pyridone moiety. Chem Biol. 2008;15:175–188. doi: 10.1016/j.chembiol.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Musiol EM, Greule A, Hartner T, Kulik A, Wohlleben W, Weber T. The AT (2) domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem. 2013;14:1343–1352. doi: 10.1002/cbic.201300211. [DOI] [PubMed] [Google Scholar]
- 15.Ye Z, Musiol EM, Weber T, Williams GJ. Reprogramming acyl carrier protein interactions of an acyl-CoA promiscuous trans-acyltransferase. Chem Biol. 2014;21:636–646. doi: 10.1016/j.chembiol.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koryakina I, McArthur J, Randall S, Draelos MM, Musiol EM, Muddiman DC, Weber T, Williams GJ. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem Biol. 2013;8:200–208. doi: 10.1021/cb3003489. [DOI] [PubMed] [Google Scholar]
- 17.Haritos VS. Biosynthesis: A terminal triple bond toolbox. Nat Chem Biol. 2015;11:98–99. doi: 10.1038/nchembio.1731. [DOI] [PubMed] [Google Scholar]
- 18.Thirumurugan P, Matosiuk D, Jozwiak K. Click chemistry for drug development and diverse chemical-biology applications. Chem Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 19.Koryakina I, Williams GJ. Mutant Malonyl-CoA Synthetases with Altered Specificity for Polyketide Synthase Extender Unit Generation. Chembiochem. 2011;12:2289–2293. doi: 10.1002/cbic.201100383. [DOI] [PubMed] [Google Scholar]
- 20.Edwards DMF, Selva E, Stella S, Zerilli LF, Gallo GG. Mass spectrometric techniques for structure and novelty determination of kirromycin-like antibiotics. Biol Mass Spectrom. 1992:51–59. [Google Scholar]
- 21.Wolf H, Chinali G, Parmeggiani A. Kirromycin, an inhibitor of protein biosynthesis that acts on elongation factor Tu. Proc Natl Acad Sci USA. 1974;71:4910–4914. doi: 10.1073/pnas.71.12.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf H, Chinali G, Parmeggiani A. Mechanism of the inhibition of protein synthesis by kirromycin. Role of elongation factor Tu and ribosomes. Eur J Biochem. 1977;75:67–75. doi: 10.1111/j.1432-1033.1977.tb11504.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim HC, Kim TW, Park CG, Oh IS, Park K, Kim DM. Continuous cell-free protein synthesis using glycolytic intermediates as energy sources. J Microbiol Biotechnol. 2008;18:885–888. [PubMed] [Google Scholar]
- 24.Kim TW, Keum JW, Oh IS, Choi CY, Kim HC, Kim DM. An economical and highly productive cell-free protein synthesis system utilizing fructose-1,6-bisphosphate as an energy source. J Biotechnol. 2007;130:389–393. doi: 10.1016/j.jbiotec.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Vogeley L, Palm GJ, Mesters JR, Hilgenfeld R. Conformational change of elongation factor Tu (EF-Tu) induced by antibiotic binding. Crystal structure of the complex between EF-Tu GDP and aurodox. J Biol Chem. 2001;276:17149–17155. doi: 10.1074/jbc.M100017200. [DOI] [PubMed] [Google Scholar]
- 26.Olsthoorn-Tieleman LN, Palstra RJ, van Wezel GP, Bibb MJ, Pleij CW. Elongation factor Tu3 (EF-Tu3) from the kirromycin producer Streptomyces ramocissimus is resistant to three classes of EF-Tu-specific inhibitors. J Bacteriol. 2007;189:3581–3590. doi: 10.1128/JB.01810-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calhoun KA, Swartz JR. An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnol Prog. 2005;21:1146–1153. doi: 10.1021/bp050052y. [DOI] [PubMed] [Google Scholar]
- 28.Menges R, Muth G, Wohlleben W, Stegmann E. The ABC transporter Tba of Amycolatopsis balhimycina is required for efficient export of the glycopeptide antibiotic balhimycin. Appl Microbiol Biotechnol. 2007;77:125–134. doi: 10.1007/s00253-007-1139-x. [DOI] [PubMed] [Google Scholar]
- 29.Kim TW, Keum JW, Oh IS, Choi CY, Park CG, Kim DM. Simple procedures for the construction of a robust and cost-effective cell-free protein synthesis system. J Biotechnol. 2006;126:554–561. doi: 10.1016/j.jbiotec.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Kim RG, Choi CY. Expression-independent consumption of substrates in cell-free expression system from Escherichia coli. J Biotechnol. 2001;84:27–32. doi: 10.1016/s0168-1656(00)00326-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.