

Abstract
An efficient synthetic route for ipomoeassin F and its tiglate-modified analogues was developed. The route features late-stage conformation-controlled highly regioselective esterification of the glucose diol in the disaccharide core. The results from the NCI-60 cell line screens of ipomoeassin F were reported for the first time. Moreover, two new C-3-cinnamoyl-Glcp analogues (2 and 3) were prepared. Their in-house cytotoxicity data convey an important message that both identity and positioning of the two α,β-unsaturated esters are crucial. They are not interchangeable.
Graphical Abstract
Many morning glory plants have been used as herbal medicines for centuries. Resin glycosides are considered active ingredients for the therapeutic effects of those plants.1 To date, more than 250 resin glycosides have been discovered. Among them, the ipomoeassin family isolated from the leaves of Ipomoea squamosa is unique.2 Whereas most resin glycosides exhibited only moderate (micromolar) inhibition activity against cancer cell growth, some ipomoeassins are exceptionally cytotoxic. The IC50 values of ipomoeassins D and F are as low as single digit nanomolar (Table 1).3,4 This suggests that the ipomoeassins could be promising leads for developing a new class of anticancer agents.
Table 1.
Structures and IC50 values of ipomoeassins A–F.
|
Ipomoeassin | Structure
|
IC50 (nM)
|
||||||||
| R1 | R2 | n | HeLa | L-929 | A2780 | U937 | HT-29 | MDAMB-435 | H522-T1 | ||
| A | H | Ac | 1 | 64 | 77.8 | 500 | 20.2 | 46.1 | 42.6 | 108.9 | |
| B | H | H | 1 | 2500 | 400 | 134 | 396 | 2700 | 1070 | ||
| C | OH | Ac | 1 | 1500 | > 1000 | 2900 | |||||
|
| |||||||||||
| D | OAc | Ac | 1 | 32 | 135 | 35 | 7.9 | 11.8 | 19.9 | 23.2 | |
|
| |||||||||||
| E | OAc | H | 1 | 4300 | > 1000 | 3300 | 163 | 393 | 1633 | 967 | |
|
| |||||||||||
| F | H | Ac | 3 | 16.4 | 7.4 | 36 | 2.6 | 4.2 | 9.4 | 12.9 | |
After careful comparison with other families of resin glycosides, we thought that coexistence of two α,β–unsaturated esters, i.e. tiglate and cinnamate (see the molecular structure in Table 1), is a very distinctive feature of the ipomoeassins. Although tiglate or cinnamate alone occasionally appears in some other resin glycosides,1,5–8 it is the ipomoeassins that have both of these two Michael acceptor systems in the same molecule. In our previous effort to identify the pharmacophore of ipomoeassin F, we unambiguously demonstrated the critical roles of the two Michael acceptor systems.9 Furthermore, one analogue (1, Figure 1) in which cinnamate is replaced with tiglate showed a dramatic activity loss. Because our biological data indicated that cinnamate seemed to be more important for cytotoxicity than tiglate,9 this raised a follow-up question regarding whether tiglate could be replaced with cinnamate. Would the two Michael systems have to be different, one aromatic and one aliphatic? To answer these questions, we designed two new analogues of ipomoeassin F (2 and 3, Figure 1).
Figure 1.

Structures of ipomoeassin F analogues 1–3.
To date, three total syntheses of ipomoeassin F have been reported; however, none of them would be productive for accessing analogues 2 and 3 or even more 3-OH-Glcp (glucopyranose) modified analogues of ipomoeassin F in the future. To elaborate further, in both strategies developed by Fürstner10 and our group11, the tiglate moiety was introduced in a very early stage, which would make medicinal chemistry studies of the C-3-Glcp position inefficient. In contrast, the route adopted by Postema and coworkers could be the most suitable because tiglate was introduced at the penultimate step.12 Unfortunately, the overall yield for this route is only ~0.4%. Therefore, a more efficient synthesis for ipomoeassin F analogues with 3-OH-Glcp modifications is highly desirable.
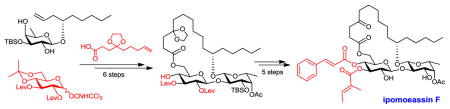
Because the high efficiency of ring closing metathesis (RCM) in constructing ring structures, we adopted this strategy in our studies.13–16 To postpone the introduction of an ester group to C-3-Glcp, we designed the diol precursors 4 and 5 (Scheme 1). Regioselective esterification of 3-OH-Glucp with tiglic or cinnamic acid followed by TBS deprotection would lead to the final products. We anticipated that 3-OH-Glucp would be favored over 2-OH-Glucp for esterification due to the low reactivity of the latter in β-glucopyranoside.17 This hypothesis was also supported by the fact that a ratio of 9:1 in favor of 3-OH-Glucp was observed for a similar but non-cyclic disaccharide substrate under Steglich conditions during the Fürstner total synthesis of ipomoeassins B and E.10 Compounds 4/5 could be obtained from a key diene intermediate 6 by a 5-step sequential transformation – RCM, hydrogenation, cinnamic/tiglic ester formation, removal of levulinoyl (Lev) groups, and hydrolysis of cyclic ketal. The Lev group was chosen to protect the 2-O-Glcp and 3-O-Glcp positions for desired β-glycosylation via neighbouring participation. Also, Lev could be removed selectively using hydrazine acetate. To avoid hydrazonization during the Lev removal, however, the C-4 ketone in aglycone would have to be protected as a ketal. The diene 6 could be prepared from the disaccharide diol 7 by chemoselective esterification of 6-OH-Glcp with the ketal protected 4-oxo-8-nonenoic acid 8. Disaccharide 7 could be assembled from glucosyl donor 9 and fucoside acceptor 10.
Scheme 1.
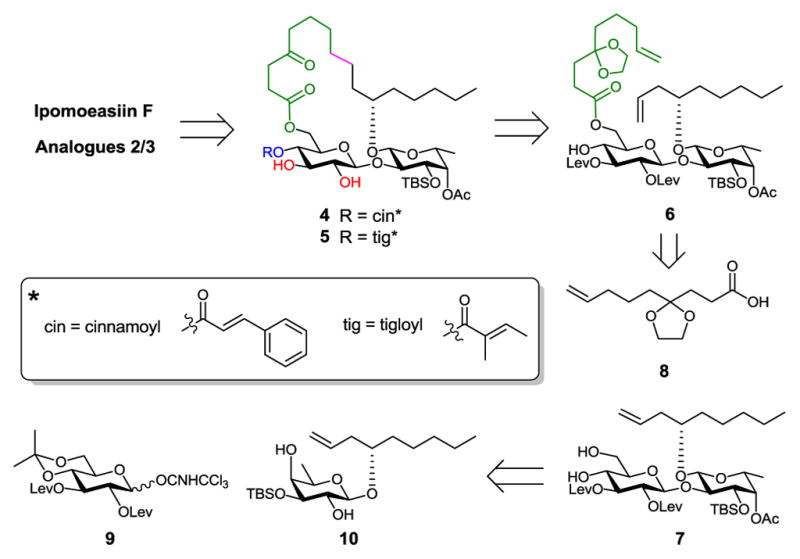
Retrosynthesis of Ipomoeassin F and Analogues 2 and 3.
In our previous studies, we developed a route to selectively introduce a tert-butyldimethylsilyl (TBS) group to 3-O-Fucp in triol 11 using DMF as solvent to form fucoside acceptor 10 in good yield (72%).11 In this paper, we optimized the reaction conditions by changing the solvent to CH2Cl2. Besides the convenience in workup, it was a pleasure to find that the yield also slightly increased (74%, Scheme 2). The glucosyl donor 9 was prepared from commercially available D-glucose penta-acetate in 6 steps with an overall yield of 35% according to a procedure we developed earlier11. With both glucosyl donor 9 and fucoside acceptor 10 in hand, we then built the β-(1→2)-linked disaccharide 12 efficiently (80% over two steps) through regioselective glycosylation followed by acetylation of the remaining 4-OH-Glcp. Removal of the isopropylidene group using camphor-10-sulfonic acid (CSA) gave the diol 7 smoothly.
Scheme 2.
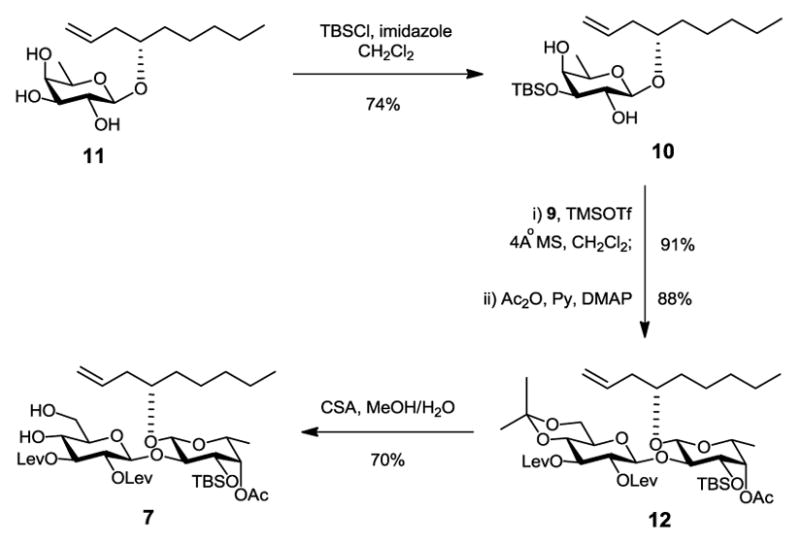
Synthesis of Disaccharide 7.
Direct ketal formation by treating 4-oxo-8-nonenoic acid 1318 with ethylene glycol under acidic conditions was not successful (Scheme 3). Therefore, we employed a three-step process, including esterification of 13 with ethanol, the ketal formation of 14 with ethylene glycol, and the final step of base hydrolysis of 15. The overall yield for making ketal protected aglycone 8 was 54%.
Scheme 3.
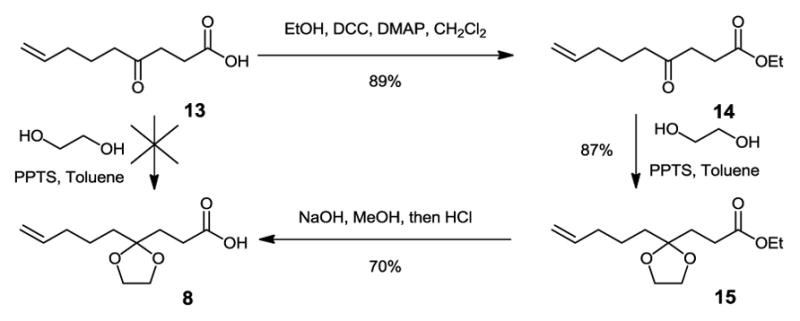
Synthesis of Ketal Protected Aglycone 8.
Subsequently, chemoselective esterification of the 6-OH-Glcp in diol 7 with the carboxylic acid 8 in the presence of DCC and DMAP gave diene 6 as the RCM precursor (Scheme 4). Treatment of the diene 6 with Hoveyda-Grubbs catalyst (II)19,20 (10 mol%) gave macrolides as a mixture of E/Z isomers, which were not separated but directly subjected to hydrogenation (Pd/C, EtOH) to give the ring structure 16. The cinnamate moiety was then introduced using Steglich esterification to furnish disaccharide 17. Removal of the Lev groups with hydrazine acetate followed by cleavage of ketal in the presence of CSA gave the key intermediate 4. To prevent conversion of the ketone in the aglycone into a methyl ketal in the presence of MeOH under acidic conditions, water was added to the reaction mixture. Diol 5 was prepared in a very similar manner from 16. A TBS-deprotected byproduct 5′ was obtained besides diol 5 after prolonged exposition of 20 to CSA in MeOH-H2O.
Scheme 4.
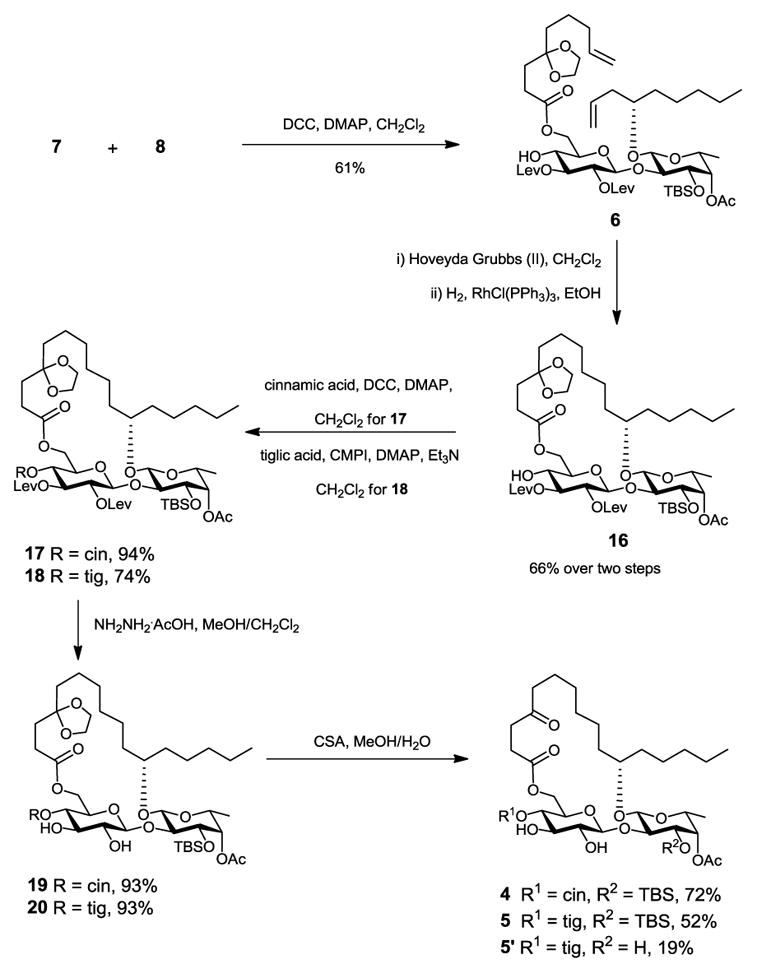
Syntheses of diols 4 and 5.
Regioselective introduction of the tigloyl group or the cinnamoyl group to the 3-O-Glcp position in diol 4 or 5 is a crucial step for the success of our synthesis. To our delight, Steglich or Mukaiyama esterification of 4 or 5 with tiglic or cinnamic acid yielded the desired 3-O-tigloyl product 21 or the 3-O-cinnamoyl products 22 and 23 in high yield (Scheme 5). Mukaiyama esterification was favored because of its relatively simpler workup and purification process. We speculated that in the stable conformation of 4 or 5, the bulky TBS group at 3-O-Fucp might be close to 2-OH-Glcp, thereby shielding it from reacting with an incoming carboxylic acid. The regioselectivity was confirmed by 1H, 13C, COSY and HMBC NMR spectra (see the supporting information). The typical COSY and HMBC correlations in 21 are illustrated in Figure 2. The final removal of the TBS group was achieved by treating 21 with tetra-n-butylammonium fluoride (TBAF) and AcOH to give the natural product ipomoeassin F in 89% yield. Its 1H and 13C NMR spectra were identical to the published literature data.4,10–12 Similarly, analogues 2 and 3 were obtained without any difficulty (Scheme 5).
Scheme 5.
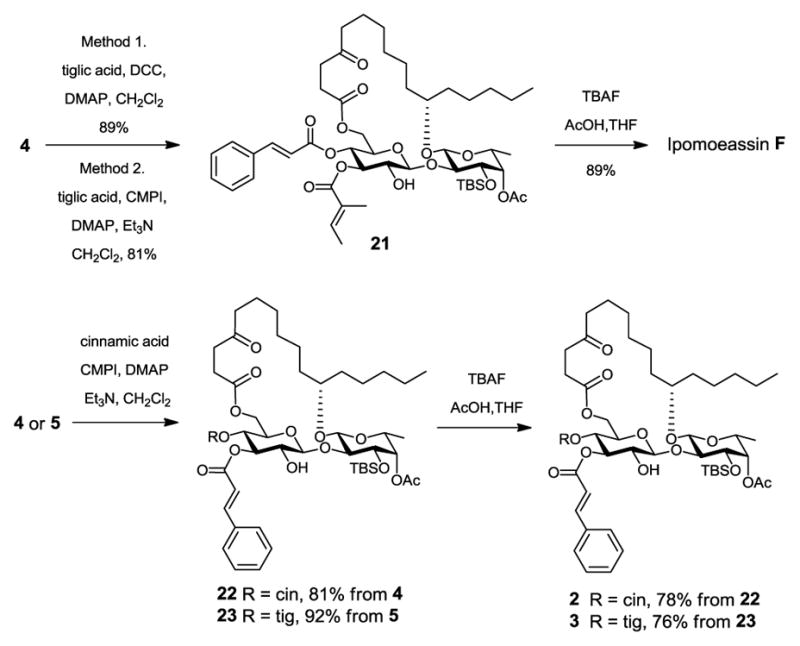
Completion of Total Syntheses.
Figure 2.
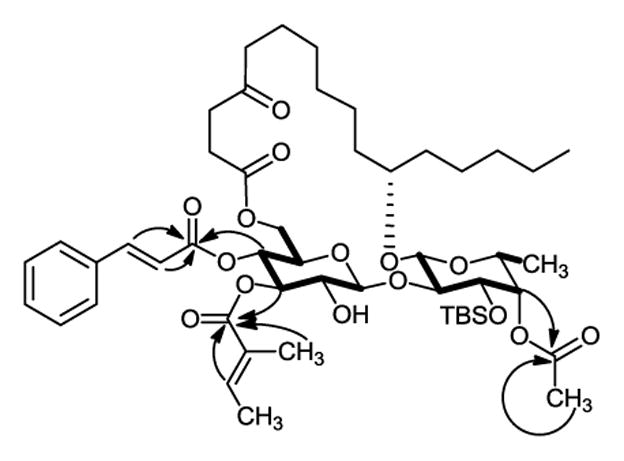
Key COSY (bold) and HMBC (arrows) correlations in 21.
With more ipomoeassin F in hand, we submitted it to NCI for the 60 cell line screen. The results (see the supporting information) confirmed its potent and selective cytotoxicity with the average GI50 of ~30 nM. In general, breast, renal and melanoma cell lines are very sensitive to ipomoeassin F (GI50 < 10 nM), whereas some ovarian and leukemia cell lines are quite resistant to the natural product (GI50 > 1,000 nM). Subsequently, we conducted the COMPARE analyses against three databases (standard agents, marketed drugs, and diversity set). The highest correlation indices are 0.462, 0.446, and 0.435, respectively. Because all the index numbers are smaller than 0.6, it implies that ipomoeassin F has a functional mechanism different from all the existing agents in the three NCI databases. This also supports the same conclusion derived from the previous NCI screen of ipomoeassin A, the naturally most abundant member of the family.2
Next, using the fluorescent alarmarBlue or colorimetric MTT assay, we performed in-house cytotoxicity evaluation of analogues 1, 2 and 3 side-by-side against two human breast cancer cell lines (MDA-MB-231 and MCF7) with ipomoeassin F as the positive control. The results presented in Table 2 illustrate the concentrations required for 50% cell death (IC50 values) when compared to the vehicle-treated negative control. Compared to ipomoeassin F, all the three analogues are significantly less potent. While 1 and 3 lost bioactivity almost completely, the IC50 values of 2 decreased by 150 and 161 fold against MDA-MB-231and MCF7 cells, respectively. Our results suggest that the combination of an aromatic α,β-unsaturated ester at C-4-Glcp and an aliphatic α,β-unsaturated ester at C-3-Glcp is optimal for high potency.
Table 2.
Cytotoxicity (IC50, μM) of ipomoeassin F and analogues 1–3 and 5′.a
| MDA-MB-231 | MCF7 | |
|---|---|---|
| Ipomoeassin F | 0.014 | 0.070 |
| 1 | 17.0 | > 25 |
| 2 | 2.1 | 11.3 |
| 3 | > 25 | > 25 |
| 5′ | > 10 | –b |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
“–” means “not tested”.
In conclusion, a more efficient route for the synthesis of ipomoeassin F was developed with an overall yield of 4.0% over 17 steps of the longest linear sequence from commercially available D-glucose penta-acetate. It is particularly suitable for efficient preparation of ipomoeassin F analogues with modifications at 3-OH-Glcp. Compared to the previous synthesis developed by Postema and his team members12, our synthesis increased the overall yield by at least 10-fold. To exemplify, by following this route, two new analogues 2 and 3 in which tiglate is replaced with cinnamate at the C-3-Glcp position were synthesized and their biological activities were evaluated. For the first time, it was revealed that tiglate and cinnamate work together at the right location to make a synergistic contribution to the cytotoxicity of the natural product. In the future, we will apply this newly-developed route to assemble a library of analogues with different acyl groups at 3-O-Glcp. Systematic structure–activity relationship (SAR) studies with those analogues will help extend ipomoeassin research to new/advanced areas of drug discovery and chemical biology.
EXPERIMETNAL SECTION
General Methods
Reactions were carried out in oven-dried glassware. All reagents were purchased from commercial sources and were used without further purification unless noted. Unless stated otherwise, all reactions were carried out under a nitrogen atmosphere and monitored by thin layer chromatography (TLC) using Silica Gel GF254 plates (Agela) with detection by charring with 5% (v/v) H2SO4 in EtOH or by visualizing in UV light (254 nm). Column chromatography was performed on silica gel (230–450 mesh, Sorbent). The ratio between silica gel and crude product ranged from 100 to 50:1 (w/w). NMR data were collected on a Bruker 300 or 400 MHz NMR spectrometer and a Bruker 300 or 400 MHz system. 1H NMR spectra were obtained in deuterochloroform (CDCl3) with chloroform (CHCl3, δ = 7.27 for 1H) as an internal reference. 13C NMR spectra were proton decoupled and were in CDCl3 with CHCl3 (δ = 77.0 for 13C) as an internal reference. Chemical shifts are reported in ppm (δ). Data are presented in the form: chemical shift (multiplicity, coupling constants, and integration). 1H NMR data are reported as though they were first order. The errors between the coupling constants for two coupled protons were less than 0.5 Hz, and the average number was reported. Proton assignments, when made, were done so with the aid of COSY NMR spectra. For some compounds, HSQC and HMBC NMR were also applied to assign the proton signals. Optical rotations were measured on an Autopol III Automatic Polarimeter at 25 ± 1 °C for solutions in a 1.0 dm cell. High resolution mass spectra (HRMS) were performed with an ion cyclotron resonance analyzer by electrospray ionization.
Compound 1011
To a cold (0 °C) solution of triol 11 (1.51 g, 5.24 mmol) and 1H-Imidazole (1.07 g, 15.7 mmol) in CH2Cl2 (30 mL) was added t-butyldimethylsilyl chloride (1.42 g, 9.43 mmol). The reaction was stirred at 0 °C for 30 min and then was allowed to warm to ambient temperature and stirred for a further 2 h. At this point, TLC (silica, 1:9 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was washed with water (30 mL). The aqueous layer was extracted with CH2Cl2 (30 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:30 → 1:20) to afford compound 10 (1.56 g, 74%) as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 5.90 – 5.75 (m, 1H, CH2=CH-CH2-), 5.14 – 4.95 (m, 2H, CH2=CH-CH2-), 4.20 (d, J = 7.6 Hz, 1H, H-1-Fucp), 3.73 – 3.43 (m, 5H, -CH2-CH-CH2-, H-2-Fucp, H-3-Fucp, H-4-Fucp, H-5-Fucp), 2.62 (br, 1H, OH), 2.35 – 2.21 (m, 2H), 2.17 (d, J = 2.0 Hz, 1H, OH), 1.67 – 1.45 (m, 2H), 1.40 – 1.18 (m, 9H), 0.94 – 0.80 (m, 12H), 0.15 (s, 3H, CH3-Si), 0.13 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 135.1, 117.4, 102.2, 78.9, 74.9, 72.1 (×2), 70.0, 38.5, 34.7, 31.8, 25.7 (×3), 24.7, 22.5, 18.1, 16.4, 14.1, −4.4, −5.0. The 1H, 13C NMR data were in accordance with the literature.11
Compound 7
β-(1→2)-Linked disaccharide 12 was obtained by coupling glucosyl donor 99 and fucoside acceptor 10 followed by acetylation of the remaining 4-OH-Glcp according to our previously developed procedure.9 CSA (261 mg, 1.12 mmol) was added in one portion to a solution of 12 (4.74 g, 5.62 mmol) in MeOH (50 mL) at room temperature. The reaction mixture was stirred for 3 h at which point TLC (silica, 1:1 EtOAc–hexanes) showed the starting material was gone. Water (0.5 mL) was added to the reaction mixture and stirred for another 20 min. The reaction was quenched with Et3N (0.3 mL, 2.2 mmol) and concentrated. The residue was purified by column chromatography (silica, EtOAc: hexanes, 1:2 to 1:1) gave diol 7 (3.18 g, 70%) as a colorless syrup. [α]D25 −13.6° (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.01 – 5.86 (m, 1H, CH2=CH-CH2-), 5.12 – 4.95 (m, 5H, CH2=CH-CH2-, H-1-Glup, H-3-Glup, H-4-Fucp), 4.89 (dd, J = 9.6, 7.6 Hz, 1H, H-2-Glup), 4.26 (d, J = 7.6 Hz, 1H, H-1-Fucp), 3.96 – 3.83 (m, 2H, H-6-Glup, H-2-Fucp), 3.82 – 3.68 (m, 3H, H-4-Glup, H-6-Glup, H-3-Fucp), 3.64 – 3.55 (m, 2H, H-5-Fucp, -CH2-CH-CH2-), 3.52 (d, J = 3.6 Hz, 1H, OH), 3.44 – 3.35 (m, 1H, H-5-Glup), 2.88 – 2.48 (m, 9H), 2.34 – 2.24 (m, 2H), 2.17 (s, 3H, CH3C(O)CH2CH2C=O), 2.15 (s, 3H, CH3C(O)CH2CH2C=O), 2.13 (s, 3H, CH3-C=O), 1.60 – 1.42 (m, 2H), 1.37 – 1.20 (m, 6H), 1.11 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.89 – 0.81 (m, 12H), 0.12 (s, 3H, CH3-Si), 0.09 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 208.2, 206.1, 173.0, 171.4, 170.8, 135.1, 116.7, 101.3, 98.7, 80.6, 76.2, 74.9, 74.8, 73.5, 73.3, 72.1, 69.6, 68.8, 61.9, 38.3 (×2), 37.6, 34.3, 31.7, 29.7 (×2), 28.0, 27.9, 25.7 (×3), 24.7, 22.5, 20.9, 17.7, 16.5, 14.0, −4.4, −4.6. HRMS (ESI) m/z calcd for C39H66NaO15Si [M+Na]+ 825.4063, found: 825.4060.
Compound 14
DCC (1.75 g, 8.47 mmol) was added in one portion to a 0 °C CH2Cl2 (20 mL) solution of 4-oxo-8-nonenoic acid18 (13, 1.20 g, 7.06 mmol), EtOH (2.1 mL, 35.3 mmol) and 4-dimethylaminopyridine (86 mg, 0.071 mmol). The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, 1: 2 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was diluted with ether (20 mL) and hexanes (10 mL), stirred for 20 minutes then filtered through a pad of Celite using ether (10 mL) as the eluent and the filtrate concentrated in vacuo. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:8) gave 14 (1.25 g, 89%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.83 – 5.70 (m, 1H, CH2=CH-CH2-), 5.08 – 4.94 (m, 2H, CH2=CH-CH2-), 4.13 (dd, J = 14.4, 7.2 Hz, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.58 (t, J = 6.4 Hz, 2H), 2.47 (t, J = 7.4 Hz, 2H), 2.10 – 2.02 (m, 2H), 1.75 – 1.65 (m, 2H), 1.26 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 208.9, 172.8, 137.9, 115.2, 60.6, 41.9, 37.1, 33.0, 28.0, 22.7, 14.1. HRMS (ESI) m/z calcd for C11H18NaO3 [M+Na]+ 221.1148, found: 221.1151.
Compound 15
Ethyl 4-oxo-8-nonenoic ester (14, 914 mg, 4.61 mmol), ethylene glycol (2.32 mL, 41.5 mmol) and PPTS (174 mg, 0.69 mmol) were dissolved in toluene (20 mL). The reaction mixture was heated to reflux under Dean-Stark conditions overnight. The mixture was cooled to RT and extracted with sat. aq. NaHCO3 (2 × 50 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:5 → 1:4) gave compound 15 (966 mg, 87%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.85 – 5.70 (m, 1H, CH2=CH-CH2-), 5.04 – 4.90 (m, 2H, CH2=CH-CH2-), 4.11 (dd, J = 14.0, 7.2 Hz, 2H), 3.91 (s, 4H), 2.37 – 2.31 (m, 2H), 2.07 – 2.00 (m, 2H), 2.00 – 1.94 (m, 2H), 1.62 – 1.55 (m, 2H), 1.50 – 1.40 (m, 2H), 1.24 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.5, 138.4, 114.7, 110.7, 65.0 (×2), 60.2, 36.7, 33.7, 32.0, 28.9, 23.0, 14.2. HRMS (ESI) m/z calcd for C13H22NaO4 [M+Na]+ 265.1410, found: 265.1414.
Compound 8
To a solution of ethyl ester 15 (738 mg, 3.05 mmol) in MeOH (10 mL) was added NaOH (2 M solution in H2O, 6.1 mL, 12.2 mmol) at 0 °C. The mixture was allowed to slowly warm to RT and was stirred overnight. TLC analysis (silica, 1:1 EtOAc–hexanes) showed it was complete. The mixture was concentrated under reduced pressure and the residue was dissolved in sat. aq. NaHCO3/H2O (3:1 v/v, 20 mL) and extracted twice with EtOAc. Next, the aqueous layer was acidified with 10% w/v aq. HCl until pH 4 and extracted again twice with EtOAc. The latter organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave compound 8 (456 mg, 70%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 11.59 (br, 1H, COOH), 5.84 – 5.72 (m, 1H, CH2=CH-CH2-), 5.05 – 4.91 (m, 2H, CH2=CH-CH2-), 3.95 (s, 4H), 2.41 (t, J = 7.6 Hz, 2H), 2.10 – 1.96 (m, 4H), 1.64 – 1.55 (m, 2H), 1.52 – 1.40 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 179.8, 138.4, 114.8, 110.7, 65.1 (×2), 36.8, 33.7, 31.8, 28.6, 23.0. HRMS (ESI) m/z calcd for C11H18NaO4 [M+Na]+ 237.1097, found: 237.1101.
Diene 6
DCC (980 mg, 4.75 mmol) was added in one portion to a 0 °C CH2Cl2 (150 mL) solution of 7 (3.18 g, 3.96 mmol), acid 8 (1.02 g, 4.75 mmol) and 4-dimethylaminopyridine (48 mg, 0.40 mmol). The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, 1:1 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was concentrated to a volume of around 20 mL, then the residue was diluted with ether (20 mL) and hexanes (5 mL), stirred for 20 minutes then filtered through a pad of Celite using ether (10 mL) as the eluent and the filtrate concentrated in vacuo. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:3 → 1:2) gave diene 6 (2.42 g, 61%) as a colorless syrup. [α]D25 −18.0° (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.10 – 5.95 (m, 1H, CH2=CH-CH2-), 5.85 – 5.71 (m, 1H, CH2=CH-CH2-), 5.12 – 4.84 (m, 8H, 2 × CH2=CH-CH2-, H-1-Glup, H-2-Glup, H-3-Glup, H-4-Fucp), 4.46 (dd, J = 12.0, 3.6 Hz, 1H, H-6-Glup), 4.32 – 4.23 (m, 2H, H-6-Glup, H-1-Fucp), 4.00 – 3.85 (m, 5H, H-2-Fucp, -OCH2CH2O-), 3.80 (dd, J = 9.6, 3.6 Hz, 1H, H-3-Fucp), 3.71 – 3.55 (m, 3H, H-4-Glup, H-5-Fucp, -CH2-CH-CH2-), 3.52 – 3.42 (m, 2H, H-5-Glup, OH), 2.86 – 2.50 (m, 8H), 2.45 – 2.36 (m, 2H), 2.33 – 2.27 (m, 2H), 2.17 (2s, 6H, 2 × CH3C(O)CH2CH2C=O), 2.13 (s, 3H, CH3-C=O), 2.09 – 1.97 (m, 4H), 1.62 – 1.41 (m, 6H), 1.38 – 1.20 (m, 6H), 1.12 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.91 – 0.83 (m, 12H), 0.13 (s, 3H, CH3-Si), 0.10 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 207.4, 206.1, 174.1, 172.9, 171.3, 170.7, 138.4, 135.6, 116.4, 114.8, 110.8, 101.5, 98.9, 80.7, 75.4, 75.0, 73.9, 73.6, 73.5, 72.4, 68.8, 68.7, 65.1, 65.0, 62.9, 38.5, 38.1, 37.7, 36.9, 34.2, 33.7, 32.1, 31.8, 29.8 (×2), 28.7, 28.0, 27.8, 25.8 (×3), 24.6, 23.0, 22.6, 20.9, 17.7, 16.5, 14.0, −4.4, −4.5. HRMS (ESI) m/z calcd for C50H82NaO18Si [M+Na]+ 1021.5163, found: 1021.5148.
RCM Product
To a solution of diene 6 (2.41 g, 2.41 mmol) in CH2Cl2 (800 mL) was added Hoveyda-Grubbs catalyst 2nd generation (151 mg, 0.24 mmol) in one portion at room temperature. The reaction mixture was refluxed for 4 h. At this point, TLC (silica, 1:1 EtOAc–hexanes) showed the reaction was complete. The reaction was cooled to ambient temperature and then concentrated. Flash chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave RCM products (1.91 g, 82%) as a colorless syrup which was not fully characterized. The obtained isomers were subjected to hydrogenation in the next step and the product was fully characterized.
Hydrogenation Product 16
To a solution of the obtained RCM products (1.91 g, 1.97 mmol) in EtOH (40 mL) was added 10% Pd/C (100 mg) in one portion at room temperature. The reaction was then stirred under an atmosphere of hydrogen overnight at the same temperature. At this point, TLC (silica, 1:1 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was filtered through a pad of Celite using EtOAc (10 mL) as the eluent and the resulting filtrate concentrated. Flash chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave 16 (1.55 g, 81 %) as a white foam. [α]D25 −26.7° (c 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 5.11 (d, J = 7.8 Hz, 1H, H-1-Glup), 5.05 (t, J = 9.6 Hz, 1H, H-3-Glup), 5.01 – 4.96 (m, 1H, H-4-Fucp), 4.91 (dd, J = 12.6, 2.4 Hz, 1H, H-6-Glup), 4.84 (dd, J = 9.6, 8.1 Hz, 1H, H-2-Glup), 4.20 (d, J = 7.8Hz, 1H, H-1-Fucp), 4.13 – 3.83 (m, 8H, H-4-Glup, H-6-Glup, H-2-Fucp, H-3-Fucp, -OCH2CH2O-), 3.68 – 3.31 (m, 4H, H-5-Glup, H-5-Fucp, OH, -CH2-CH-CH2-), 2.84 – 2.71 (m, 4H), 2.68 – 2.35 (m, 6H), 2.32 – 2.20 (m, 1H), 2.16 (s, 6H, CH3CO), 2.10 (s, 3H, CH3CO), 2.09 – 1.87 (m, 1H), 1.71 – 1.20 (m, 20H), 1.11 (d, J = 6.3 Hz, 3H, H-6-Fucp), 0.95 – 0.81 (m, 12H), 0.17 (s, 3H, CH3-Si), 0.13 (s, 3H, CH3-Si). 13C NMR (75 MHz, CDCl3) δ 206.9, 206.3, 175.5, 172.3, 171.3, 170.8, 111.0, 102.0, 98.9, 82.4, 75.0, 74.8, 74.6, 73.6, 73.5, 72.1, 68.7, 67.3, 64.6, 64.5, 62.3, 37.9, 37.7, 35.9, 34.5, 33.9, 31.9, 31.5, 29.8, 29.7, 29.0, 28.5, 27.9, 25.8 (×3), 25.0, 23.3, 22.6, 20.9, 17.6, 16.6, 14.0, −4.1, −4.6. HRMS (ESI) m/z calcd for C48H80NaO18Si [M+Na]+ 995.5006, found: 995.5002.
Compound 17
DCC (364 mg, 1.76 mmol) was added in one portion to a 0 °C CH2Cl2 (15 mL) solution of 16 (1.43 g, 1.47 mmol), cinnamic acid (261 mg, 1.76 mmol) and 4-dimethylaminopyridine (18.0 mg, 0.15 mmol). The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was diluted with ether (15 mL) and hexanes (3 mL), stirred for 20 minutes then filtered through a pad of Celite using ether (10 mL) as the eluent and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:3 → 1:2) gave 17 (1.53 g, 94%) as a colorless syrup. [α]D25 −16.9° (c 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 15.9 Hz, 1H, Ph-CH=C-), 7.56 – 7.47 (m, 2H, 2 × ArH), 7.43 – 7.35 (m, 3H, 3 × ArH), 6.35 (d, J = 15.9 Hz, 1H, Ph-CH=CH-), 5.32 – 5.18 (m, 3H, H-3-Glup, H-4-Glup, H-1-Glup), 5.04 – 4.92 (m, 2H, H-4-Fucp, H-2-Glup), 4.35 (dd, J = 12.3, 3.0 Hz, 1H, H-6-Glup), 4.27 (d, J = 7.5 Hz, 1H, H-1-Fucp), 4.12 (dd, J = 12.0, 2.4 Hz, 1H, H-6-Glup), 4.05 – 3.83 (m, 6H, H-2-Fucp, H-3-Fucp, -OCH2CH2O-), 3.76 – 3.53 (m, 3H, H-5-Glup, H-5-Fucp, -CH2-CH-CH2-), 2.83 – 2.75 (m, 2H), 2.70 – 2.31 (m, 8H), 2.17 (s, 3H, CH3COCH2CH2COO), 2.11 (s, 3H, CH3-COO), 2.08 (s, 3H, CH3COCH2CH2COO), 2.05 – 1.85 (m, 2H), 1.73 – 1.20 (m, 20H), 1.13 (d, J = 6.3 Hz, 3H, H-6-Fucp), 0.95 – 0.83 (m, 12H), 0.19 (s, 3H, CH3-Si), 0.14 (s, 3H, CH3-Si). 13C NMR (75 MHz, CDCl3) δ 206.2, 206.1, 173.0, 171.9, 171.2, 170.8, 165.0, 146.1, 134.1, 130.5, 128.8 (×2), 128.3 (×2), 116.7, 111.3, 103.5, 101.4, 98.9, 81.9, 74.5, 74.1, 73.6, 73.0, 72.2, 71.9, 68.3, 68.5, 64.5, 61.7, 37.8, 37.7, 34.6, 34.3, 33.7, 32.0, 31.2, 30.0, 29.8, 29.6, 29.0, 28.3, 27.9, 27.8, 25.9 (×3), 24.6, 24.0, 22.9, 22.6, 20.9, 17.8, 16.6, 14.1, −4.3, −4.5. HRMS (ESI) m/z calcd for C57H86NaO19Si [M+Na]+ 1125.5425, found: 1125.5414.
Compound 19
Hydrazine acetate (1.00 g, 10.9 mmol) was added to a solution of compound 17 (1.50 g, 1.36 mmol) in 2:1 CH2Cl2/MeOH (30 mL) at room temperature. The reaction mixture was stirred for 2 h, at which point TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. Then it was quenched with aqueous NaHCO3 (30 mL) and extracted with CH2Cl2 (40 mL × 2). The combined organic extracts were dried over Na2SO4. Evaporation and purification by column chromatography (silica, EtOAc–hexanes, 1:3 → 1:2) to afford compound 19 (1.15 g, 93%) as a colorless syrup. [α]D25 0.8° (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.56 – 7.50 (m, 2H, 2 × ArH), 7.42 – 7.37 (m, 3H, 3 × ArH), 6.46 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.15 – 5.06 (m, 2H, H-4-Glup, H-4-Fucp), 4.65 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.41 (d, J = 7.6 Hz, 1H, H-1-Fucp), 4.26 (dd, J = 12.0, 4.0 Hz, 1H, H-6-Glup), 4.18 (dd, J = 12.0, 4.0 Hz, 1H, H-6-Glup), 3.98 – 3.60 (m, 11H, H-3-Glup, H-5-Glup, H-2-Fucp, H-3-Fucp, H-5-Fucp, OH, -OCH2CH2O-, -CH2-CH-CH2-), 3.50 – 3.41 (m, 1H, H-2-Glup), 2.64 (d, J = 2.4 Hz, 1H, OH), 2.40 – 2.25 (m, 2H), 2.13 (s, 3H, CH3-C=O), 2.05 – 1.86 (m, 2H), 1.61 – 1.48 (m, 6H), 1.41 – 1.20 (m, 14H), 1.15 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.95 – 0.86 (m, 12H), 0.22 (s, 3H, CH3-Si), 0.18 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 173.1, 170.7, 166.2, 146.2, 134.1, 130.5, 128.9 (×2), 128.2 (×2), 117.1, 111.2, 103.5, 101.1, 79.8, 78.5, 76.0, 73.6, 73.0, 72.7, 72.5, 71.3, 68.7, 64.6, 64.5, 63.5, 35.1, 33.9, 33.3, 32.0, 31.3, 29.7, 28.9, 28.4, 25.9 (×3), 24.3, 24.0, 22.6 (×2), 20.8, 17.9, 16.5, 14.1, −4.3, −4.6. HRMS (ESI) m/z calcd for C47H74NaO15Si [M+Na]+ 929.4689, found: 929.4682.
Compound 4
CSA (58.9 mg, 0.254 mmol) was added in one portion to a solution of 19 (1.15 g, 1.27 mmol) in MeOH (30 mL) and H2O (0.3 mL) at room temperature. The reaction mixture was stirred for 12 h at which point TLC (silica, 1:1 EtOAc–hexanes) showed it was complete. The reaction was quenched with Et3N (70 μL) and concentrated. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1: 3 → 1:2) gave compound 4 (0.79g, 72%) as a colorless syrup. [α]D25 −13.4° (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.58 – 7.51 (m, 2H, 2 × ArH), 7.43 – 7.37 (m, 3H, 3 × ArH), 6.51 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.16 – 5.08 (m, 2H, H-4-Glup, H-4-Fucp), 4.59 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.40 (d, J = 7.6 Hz, 1H, H-1-Fucp), 4.34 (dd, J = 12.0, 2.4 Hz, 1H, H-6-Glup), 4.19 (dd, J = 12.0, 5.2 Hz, 1H, H-6-Glup), 4.02 (d, J = 1.6 Hz, 1H, OH), 3.96 – 3.89 (m, 1H, H-2-Fucp), 3.85 (dd, J = 9.6, 3.2 Hz, 1H, H-3-Fucp), 3.83 – 3.75 (m, 1H, H-3-Glup), 3.73 – 3.60 (m, 3H, H-5-Glup, H-5-Fucp, -CH2-CH-CH2-), 3.48 – 3.40 (m, 1H, H-2-Glup), 2.75 – 2.40 (m, 6H), 2.14 (s, 3H, CH3-C=O), 1.70 – 1.61 (m, 2H), 1.59 – 1.45 (m, 4H), 1.39 – 1.20 (m, 12H), 1.15 (d, J = 6.0 Hz, 3H, H-6-Fucp), 0.95 – 0.84 (m, 12H), 0.22 (s, 3H, CH3-Si), 0.19 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 210.3, 171.6, 170.7, 166.2, 146.1, 134.2, 130.6, 129.0 (×2), 128.2 (×2), 117.2, 104.0, 101.2, 80.3, 79.0, 76.1, 73.5, 73.1, 72.8, 72.2, 71.6, 68.8, 63.3, 42.0, 37.3, 34.1, 33.3, 32.0, 29.5, 29.1, 28.5, 25.9 (×3), 25.1, 24.4, 23.6, 22.6, 20.9, 17.9, 16.5, 14.1, −4.3, −4.6. HRMS (ESI) m/z calcd for C45H70NaO14Si [M+Na]+ 885.4427, found: 885.4422.
Compound 20
2-Chloro-1-methylpyridinium iodide (CMPI, 120.9 mg, 0.473 mmol), N,N-dimethylaminopyridine (DMAP, 14.5 mg, 0.118 mmol) and Et3N (330 μL, 2.37 mmol) were added to a solution of 16 (180.0 mg, 0.237 mmol) and tiglic acid (35.5 mg, 0.355 mmol) in dry DCM (5 mL) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred for 36 h. At this point, TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. Evaporation of the solvent followed by purification of the residue by column chromatography (silica, EtOAc–hexanes, 1:3 → 1:2) to give 18 (144 mg, 74%) as a colorless syrup. Hydrazine acetate (96.3 mg, 1.05 mmol) was added to a solution of compound 18 (138 mg, 0.131 mmol) in 2:1 CH2Cl2/MeOH (5 mL) at room temperature. The reaction mixture was stirred for 1 h, at which point TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. Then it was quenched with aqueous NaHCO3 (20 mL) and extracted with CH2Cl2 (20 mL × 2). The combined organic extracts were dried over Na2SO4. Evaporation and purification by column chromatography (silica, EtOAc–hexanes, 1:4 → 1:2) to afford compound 20 (99.0 mg, 88%) as a colorless syrup. [α]D25 4.5° (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.00 – 6.81 (m, 1H, Me-CH-C(Me)-C=O), 5.10 – 5.07 (m, 1H, H-4-Fucp), 5.02 (t, J = 9.6 Hz, 1H, H-4-Glup), 4.63 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.40 (d, J = 7.6 Hz, 1H, H-1-Fucp), 4.21 (dd, J = 12.0, 4.4 Hz, 1H, H-6-Glup), 4.15 (dd, J = 12.0, 4.4 Hz, 1H, H-6-Glup), 3.97 – 3.81 (m, 7H, H-2-Fucp, H-3-Fucp, -OCH2CH2O-, OH), 3.79 – 3.59 (m, 4H, H-3-Glup, H-5-Glup, H-5-Fucp, -CH2-CH-CH2-), 3.49 – 3.37 (m, 1H, H-2-Glup), 2.68 (d, J = 2.4 Hz, 1H, OH), 2.45 – 2.24 (m, 2H), 2.13 (s, 3H, CH3-C=O), 2.06 – 1.89 (m, 2H), 1.87 – 1.76 (m, 6H, CH3-CH-C(CH3)-C=O), 1.63 – 1.44 (m, 6H), 1.41 – 1.20 (m, 14H), 1.15 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.94 – 0.80 (m, 12H), 0.21 (s, 3H), 0.17 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 173.2, 170.7, 167.3, 138.7, 128.0, 111.2, 103.5, 101.0, 79.6, 78.4, 76.0, 73.7, 73.1, 72.8, 72.7, 71.2, 68.8, 64.6, 64.5, 63.5, 35.1, 34.0, 33.2, 32.0, 31.3, 29.6, 28.9, 28.4, 25.9 (×3), 24.4, 24.0, 22.6, 22.6, 20.8, 17.9, 16.5, 14.5, 14.1, 12.1, −4.3, −4.6. HRMS (ESI) m/z calcd for C43H74NaO15Si [M+Na]+ 881.4689, found: 881.4680.
Compound 5
CSA (5.0 mg, 0.021 mmol) was added in one portion to a solution of 20 (92.0 mg, 0.107 mmol) in MeOH (10 mL) and H2O (0.1 mL) at room temperature. The reaction mixture was stirred for 16 h. The reaction was quenched with Et3N (10 μL) and concentrated. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1: 5 → 1:3) gave compound 5 (45.4 mg, 52%) as a colorless syrup and a TBS removed byproduct 5′ (14.3 mg, 19%).
Compound 5
[α]D25 −5.9° (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.96 – 6.90 (m, 1H, Me-CH-C(Me)-C=O), 5.10 – 5.07 (m, 1H, H-4-Fucp), 5.02 (t, J = 9.6 Hz, 1H, H-4-Glup), 4.56 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.38 (d, J = 7.2 Hz, 1H, H-1-Fucp), 4.30 (dd, J = 11.6, 2.8 Hz, 1H, H-6-Glup), 4.13 (dd, J = 12.0, 4.8 Hz, 1H, H-6-Glup), 3.97 – 3.81 (m, 3H, H-2-Fucp, H-3-Fucp, OH), 3.76 – 3.58 (m, 4H, H-3-Glup, H-5-Glup, H-5-Fucp, -CH2-CH-CH2-), 3.46 – 3.37 (m, 1H, H-2-Glup), 2.78 – 2.34 (m, 7H), 2.13 (s, 3H, CH3-C=O), 1.90 – 1.77 (m, 6H), 1.67 – 1.42 (m, 6H), 1.38 – 1.20 (m, 12H), 1.14 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.95 – 0.79 (m, 12H), 0.21 (s, 3H), 0.18 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 210.2, 171.6, 170.7, 167.2, 138.6, 128.1, 104.1, 101.1, 79.9, 79.0, 76.1, 73. 7, 73.1, 72.8, 72.4, 71.2, 68.8, 63.1, 42.0, 37.4, 34.2, 33.2, 31.9, 29.4, 29.1, 28.4, 25.9 (×3), 25.1, 24.4, 23.6, 22.6, 20.8, 17.9, 16.5, 14. 5, 14.1, 12.1, −4.3, −4.7. HRMS (ESI) m/z calcd for C41H70NaO14Si [M+Na]+ 837.4427, found: 837.4423.
Compound 5′
[α]D25 −11.0° (c 0.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.95 – 6.85 (m, 1H, Me-CH-C(Me)-C=O), 5.16 – 5.11 (m, 1H, H-4-Fucp), 4.94 (t, J = 9.6 Hz, 1H, H-4-Glup), 4.49 (d, J = 8.0 Hz, 1H, H-1-Fucp), 4.45 – 4.35 (m, 2H, H-1-Glup, H-6-Glup), 4.14 – 4.06 (m, 1H, H-6-Glup), 3.90 (dd, J = 9.6, 3.6 Hz, 1H, H-3-Fucp), 3.74 – 3.48 (m, 8H, H-2-Glup, H-3-Glup, H-5-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-, 2×OH), 2.87 – 2.34 (m, 6H), 2.17 (s, 3H, CH3-C=O), 1.88 – 1.74 (m, 6H), 1.71 – 1.20 (m, 18H), 1.17 (t, J = 6.4 Hz, 3H, H-6-Fucp), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 210.1, 171.7, 171.7, 167. 8, 139.1, 127. 9, 104.7, 100.1, 82.0, 79.7, 75.1(×2), 72.9, 72.6, 72.4, 70.2, 68.8, 62.1, 41.8, 37.6, 34.4, 33.0, 31.9, 29.0, 29.0, 28.2, 24.6, 24.3, 23.5, 22.6, 21.0, 16.3, 14.5, 14.1, 12.1. HRMS (ESI) m/z calcd for C35H56NaO14 [M+Na]+ 723.3562, found: 723.3561.
Compound 21
Method 1
DCC (6.8 mg, 0.033 mmol) was added in one portion to a 0 °C CH2Cl2 (2 mL) solution of 4 (26 mg, 0.030 mmol), tiglic acid (3.3 mg, 0.033 mmol) and 4-dimethylaminopyridine (0.4 mg, 0.003 mmol). The reaction was allowed to warm to ambient temperature and stirred for 24 h. At this point, TLC (silica, 1:3 EtOAc–hexanes) showed the reaction was complete. The reaction mixture was diluted with ether (2 mL) and hexanes (1 mL), stirred for 20 minutes then filtered through a pad of Celite using ether (5 mL) as the eluent and the filtrate concentrated in vacuo. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:6 → 1:3) gave 21 (25.4 mg, 89%) as a colorless syrup. [α]D25 −38.7° (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.55 – 7.48 (m, 2H, 2 × ArH), 7.43 – 7.36 (m, 3H, 3 × ArH), 6.87 – 6.78 (m, 1H, Me-CH-C(Me)-C=O), 6.38 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.33 (t, J = 9.6 Hz, 1H, H-3-Glup), 5.23 (t, J = 9.6 Hz, 1H, H-4-Glup), 5.10 – 5.08 (m, 1H, H-4-Fucp), 4.70 (d, 1H, J = 8.0 Hz, H-1-Glup), 4.39 (d, 1H, J = 7.6 Hz, H-1-Fucp), 4.30 – 4.18 (m, 2H, H-6-Glup), 3.94 (dd, J = 7.6, 10 Hz, 1H, H-2-Fucp), 3.89 – 3.80 (m, 2H, OH, H-3-Fucp), 3.78 – 3.72 (m, 1H, H-5-Glup), 3.71 – 3.55 (m, 3H, H-2-Glup, H-5-Fucp, -CH2-CH-CH2-), 2.70 – 2.42 (m, 6H), 2.11 (s, 3H, CH3-C=O), 1.79 – 1.63 (m, 8H), 1.60 – 1.43 (m, 4H), 1.40 – 1.20 (m, 12H), 1.14 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.94 – 0.83 (m, 12H), 0.22 (s, 3H, CH3-Si), 0.18 (s, 3H, CH3-Si). 13C NMR (100 MHz, CDCl3) δ 210.1, 171.4, 170.8, 167.5, 165.6, 146.0, 138.1, 134.0, 130.6, 128.9 (×2), 128.2 (×2), 127.9, 116.8, 104.3, 101.2, 80.2, 79.0, 74.5, 72.9, 72.8, 72.0, 69.9, 68.8, 63.3, 42.0, 37.4, 34.1, 33.2, 32.0, 29.6, 29.1, 28.6, 25.8 (×3), 25.1, 24.4, 23.7, 22.6, 20.9, 17.9, 16.5, 14.4, 14.1, 12.0, −4.4, −4.6. HRMS (ESI) m/z calcd for C50H76NaO15Si [M+Na]+ 967.4846, found: 967.4834.
Method 2
2-Chloro-1-methylpyridinium iodide (CMPI, 7.1 mg, 0.028 mmol), N,N-dimethylaminopyridine (DMAP, 0.8 mg, 0.007 mmol) and Et3N (19 μL, 0.14 mmol) were added to a solution of 4 (12.0 mg, 0.014 mmol) and tiglic acid (2.8 mg, 0.028 mmol) in dry DCM (2 mL) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred for 4 h. At this point, TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. Evaporation of the solvent followed by purification of the residue by column chromatography (silica, EtOAc–hexanes, 1:5 → 1:3) to give 21 (10.6 mg, 81%) as a colorless syrup. The 1H and 13C NMR data (SI, pages S60–S61) of 21 obtained via method 2 were identical to those of 21 obtained via method 1 (SI, pages S53–S54).
Ipomoeassin F
To a solution of 21 (11.0 mg, 0.0116 mmol) in THF (2 mL) was added AcOH (67 μL, 1.16 mmol) and TBAF (1M solution in THF, 0.58 mL, 0.58 mmol) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, EtOAc-hexanes, 1:1) showed the reaction was complete. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave synthetic Ipomoeassin F (8.6 mg, 89%) as a colorless film. [α]D25 −51.2° (c 0.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.55 – 7.45 (m, 2H, 2 × ArH), 7.45 – 7.35 (m, 3H, 3 × ArH), 6.93 – 6.87 (m, 1H, Me-CH-C(Me)-C=O), 6.35 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.32 (t, J = 9.6 Hz, 1H, H-4-Glup), 5.18 – 5.10 (m, 2H, H-3-Glup, H-4-Fucp), 4.62 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.56 (d, J = 1.6 Hz, 1H, OH), 4.47 (dd, J = 12.4, 3.6 Hz, 1H, H-6-Glup), 4.41 (d, J = 8.0 Hz, 1H, H-1-Fucp), 4.15 (dd, J = 12.4, 2.4 Hz, 1H, H-6-Glup), 3.96 – 3.88 (m, 2H, OH, H-3-Fucp), 3.78 – 3.73 (m, 1H, H-5-Glup), 3.73 – 3.60 (m, 4H, H-2-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-), 2.85 – 2.38 (m, 6H), 2.18 (s, 3H, CH3-C=O), 1.80 – 1.72 (m, 6H, CH3-CH-C(CH3)-C=O), 1.72 – 1.62 (m, 2H), 1.58 – 1.46 (m, 4H), 1.41 – 1.22 (m, 12H), 1.19 (t, J = 6.4 Hz, 3H, H-6-Fucp), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 210.0, 171.8, 171.7, 168.9, 165.4, 146.2, 139.9, 134.0, 130.6, 128.9 (×2), 128.3 (×2), 127.5, 116.7, 105.7, 100.2, 82.9, 79.8, 75.9, 74.0, 72.7, 72.6, 72.5, 68.8, 67.4, 61.8, 41.8, 37.6, 34.4, 33.1, 31.9, 29.1, 29.0, 28.3, 24.7, 24.5, 23.5, 22.6, 20.9, 16.3, 14.6, 14.1, 12.0. The 1H, 13C NMR data were in accordance with the literature.11
Analogue 2
2-Chloro-1-methylpyridinium iodide (CMPI, 13.0 mg, 0.051 mmol), N,N-dimethylaminopyridine (DMAP, 9.3 mg, 0.076 mmol) and Et3N (36 μL, 0.25 mmol) were added to a solution of 4 (22.0 mg, 0.025 mmol) and cinnamic acid (7.6 mg, 0.051 mmol) in dry CH2Cl2 (2 mL) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred for 1 h. At this point, TLC (silica, 1:2 EtOAc–hexanes) showed the reaction was complete. Evaporation of the solvent followed by purification of the residue by column chromatography (silica, EtOAc–hexanes, 1:5 → 1:3) to give 22 (20.4 mg, 81%) as a colorless syrup. Then to a solution of 22 (11.6 mg, 0.0117 mmol) in THF (2 mL) was added AcOH (67 μL, 1.17 mmol) and TBAF (1M solution in THF, 0.58 mL, 0.58 mmol) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, EtOAc-hexanes, 1:1) showed the reaction was complete. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave compound analogue 2 (8.0 mg, 78%) as a white powder. [α]D25 −127° (c 0.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.63 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.53 – 7.44 (m, 4H, 4 × ArH), 7.39 – 7.31 (m, 6H, 6 × ArH), 6.41 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 6.36 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.33 (t, J = 9.4 Hz, 1H, H-4-Glup), 5.28 (t, J = 9.4 Hz, 1H, H-3-Glup), 5.14 (d, J = 3.2 Hz, 1H, H-4-Fucp), 4.65 (d, J = 8.0 Hz, 1H, H-1-Glup), 4.55 – 4.40 (m, 3H, H-6-Glup, H-1-Fucp, OH), 4.18 (dd, J = 12.4, 2.0 Hz, 1H, H-6-Glup), 3.95 (dd, J = 9.6, 3.6 Hz, 1H, H-3-Fucp), 3.83 – 3.62 (m, 6H, H-2-Glup, H-5-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-, OH), 2.85 – 2.40 (m, 6H), 2.19 (s, 3H, CH3-C=O), 1.72 – 1.22 (m, 18H), 1.20 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 210.0, 172.0, 171.7, 167.3, 165.5, 146.6, 146.3, 134.1, 134.0, 130.6, 130.6, 128.8 (×2), 128.8 (×2), 128.3 (×2), 128.3 (×2), 116.8, 116.6, 105.5, 100.1, 82.4, 79.7, 75.5, 73.7, 72.8, 72.7, 72.5, 68.8, 67.8, 61.9, 41.9, 37.6, 34.3, 33.0, 31.9, 29.1, 29.0, 28.3, 24.6, 24.5, 23.4, 22.7, 20.9, 16.3, 14.1. HRMS (ESI) m/z calcd for C48H62NaO15 [M+Na]+ 901.3981, found: 901.3978.
Analogue 3
2-Chloro-1-methylpyridinium iodide (CMPI, 11.0 mg, 0.043 mmol), N,N-dimethylaminopyridine (DMAP, 1.3 mg, 0.011 mmol) and Et3N (30.0 μL, 0.22 mmol) were added to a solution of 5 (17.5 mg, 0.021 mmol) and cinnamic acid (6.4 mg, 0.043 mmol) in dry DCM (4 mL) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, 1:3 EtOAc–hexanes) showed the reaction was complete. Evaporation of the solvent followed by purification of the residue by column chromatography (silica, EtOAc–hexanes, 1:5 → 1:3) to give 23 (18.7 mg, 92%) as a colorless syrup. To a solution of 23 (13.6 mg, 0.0144 mmol) in THF (2 mL) was added AcOH (82 μL, 1.44 mmol) and TBAF (1M solution in THF, 0.72 mL, 0.72 mmol) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, EtOAc-hexanes, 1:1) showed the reaction was complete. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes, 1:2 → 1:1) gave analogue 3 (9.1 mg, 76%) as a white film. [α]D25 −55.8° (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.56 – 7.46 (m, 2H, 2 × ArH), 7.44 – 7.34 (m, 3H, 3 × ArH), 6.85 – 6.73 (m, 1H, Me-CH-C(Me)-C=O), 6.40 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.31 – 5.18 (m, 2H, H-3-Glup, H-4-Glup), 5.15 – 5.09 (m, 1H, H-4-Fucp), 4.63 (d, J = 7.6 Hz, 1H, H-1-Glup), 4.48 – 4.37 (m, 3H, H-6-Glup, H-4-Fucp, OH), 4.13 (dd, J = 12.0, 2.0 Hz, 1H, H-6-Glup), 3.98 – 3.90 (m, 1H, H-3-Fucp), 3.81 – 3.58 (m, 6H, H-2-Glup, H-5-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-, OH), 2.88 – 2.38 (m, 6H), 2.19 (s, 3H, CH3-C=O), 1.78 – 1.44 (m, 12H), 1.42 – 1.21 (m, 12H), 1.19 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 210.0, 171.9, 171.7, 167.3, 166.6, 146.5, 138.9, 134.1, 130.6,128.9 (×2), 128.3 (×2), 127.7, 116.9, 105.4, 100.1, 82.3, 79.6, 75.6, 73.6, 72.8, 72.8, 72.5, 68.8, 67.6, 61.8, 41.9, 37.7, 34.4, 33.0, 31.9, 29.1, 29.0, 28.3, 24.6, 24.5, 23.4, 22.6, 20.9, 16.3, 14.5, 14.1, 12.1. HRMS (ESI) m/z calcd for C44H62NaO15 [M+Na]+ 853.3981, found: 853.3980.
Biology
Cell culture
Two breast cancer cell lines MCF7 and MDA-MB-231 were maintained in a DMEM/HIGH culture medium supplemented with 10% bovine calf serum (BCS) and 2 mM L-glutamine, so-called complete medium. Cell cultures were grown in monolayers in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. The culture medium was changed every 2–3 days. Cell cultures were passaged once or twice a week using trypsin-EDTA (0.25%) to detach the cells from their culture flasks/dishes.
MTT Cytotoxicity Assay
Counting of viable cells was performed before each experiment. Experiments were done in triplicate. First, 100 μL of cell suspension at the density of 25,000 cells/mL was seeded in a 96-well plate (2,500 cells/well), which was incubated at 37 °C in 5% CO2 for 24 hours. The compounds were dissolved in DMSO (dimethyl sulfoxide) to make drug stocks (10 mM). The stock solutions were diluted with the complete DMEM/HIGH medium to make a series of gradient fresh working solutions right before each test. Subsequently, the cells were treated with 100 μL of the freshly made gradient working solution in the total volume of 200 μL/well for 72 hours. After that, the media were discarded and 200 μL of the fresh complete medium containing 10% of MTT stock solution (5 mg/mL) was added to each well. The plate was then incubated at 37 °C in 5% CO2 atmosphere for another 3 hours. Next, 180 μL of the medium was discarded from each well. The formed formazan crystals were dissolved with 180 μL of DMSO. Absorbance of formazan was detected by a microplate reader (BioTek Synergy H1) at 570 nm with 650 nm as the reference wavelength. The percentage of viability compared to the negative control (DMSO-treated cells) was determined.
Activities of the synthesized compounds against breast cancer cell line MCF7 were tested by the MTT cytotoxicity assay. GraphPad Prism 6 software was used to make a plot of % viability versus sample concentration and to calculate the concentration at which a compound exhibited 50% cytotoxicity (IC50).
AlamarBlue Cytotoxicity Assay
Counting of viable cells was performed before each experiment. Experiments were done in triplicate. First, 100 μL of cell suspension at the density of 25,000 cells/mL was seeded in a 96-well plate (2,500 cells/well), which was incubated at 37 °C in 5% CO2 for 24 hours. The compounds were dissolved in DMSO (dimethyl sulfoxide) to make drug stocks (10 mM). The stock solutions were diluted with the complete DMEM/HIGH medium to make a series of gradient fresh working solutions right before each test. Subsequently, the cells were treated with 100 μL of the freshly made gradient working solution in the total volume of 200 μL/well for 72 hours. After that, the media were discarded and 200 μL of the fresh complete medium containing 10% of AlamarBlue (resazurin) stock solution (3 mg/27.15mL) was added to each well. The plate was then incubated at 37 °C in 5% CO2 atmosphere for another 3 hours. Next, 180 μL of the medium was discarded from each well. The formed formazan crystals were dissolved with 180 μL of DMSO. Absorbance of formazan was detected by a microplate reader (BioTek Synergy H1) at excitation 580 nm with emission 620 nm as the reference wavelength. The percentage of viability compared to the negative control (DMSO-treated cells) was determined.
Activities of synthesized compounds against breast cancer cell line MDA-MB-231 were tested by the alamarBlue cytotoxicity assay. GraphPad Prism 6 software was used to make a plot of % viability versus sample concentration and to calculate the concentration at which a compound exhibited 50% cytotoxicity (IC50).
Supplementary Material
Acknowledgments
This work was primarily supported by the startup funds from the University of Arkansas and also in part by Grant Number P30GM103450 and R15GM116032 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and by seed money from the Arkansas Biosciences Institute (ABI). We wish to thank Dr. Jianjun Zhang at CAU (China Agricultural University) for providing per-acetylated D-fucopyranose (NKT R&D Program of China, 2015BAK45B01).
Footnotes
Notes: The authors report no conflicts of interest.
The results of the NCI-60 cell line screen of ipomoeassin F, the IC50 curves of 2 and 3 against two breast cancer cell lines, and the 1H, 13C NMR and HRMS spectra of all synthesized new compounds (COSY, HSQC and HMBC NMR for representative compounds). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pereda-Miranda R, Rosas-Ramirez D, Castaneda-Gomez J. Prog Chem Org Nat Prod. 2010;92:77. doi: 10.1007/978-3-211-99661-4_2. [DOI] [PubMed] [Google Scholar]
- 2.Kingston DGI. J Org Chem. 2008;73:3975. doi: 10.1021/jo800239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao S, Guza RC, Wisse JH, Miller JS, Evans R, Kingston DGI. J Nat Prod. 2005;68:487. doi: 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]
- 4.Cao S, Norris A, Wisse JH, Miller JS, Evans R, Kingston DGI. Nat Prod Res. 2007;21:872. doi: 10.1080/14786410600929576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono M, Takigawa A, Kanemaru Y, Kawakami G, Kabata K, Okawa M, Kinjo J, Yokomizo K, Yoshimitsu H, Nohara T. Chem Pharm Bull. 2014;62:97. doi: 10.1248/cpb.c13-00610. [DOI] [PubMed] [Google Scholar]
- 6.Takigawa A, Muto H, Kabata K, Okawa M, Kinjo J, Yoshimitsu H, Nohara T, Ono M. J Nat Prod. 2011;74:2414. doi: 10.1021/np2006378. [DOI] [PubMed] [Google Scholar]
- 7.Noda N, Kogetsu H, Kawasaki T, Miyahara K. Phytochemistry. 1992;31:2761. doi: 10.1016/0031-9422(92)83626-a. [DOI] [PubMed] [Google Scholar]
- 8.Escalante-Sanchez E, Pereda-Miranda R. J Nat Prod. 2007;70:1029. doi: 10.1021/np070093z. [DOI] [PubMed] [Google Scholar]
- 9.Zong G, Aljewari H, Hu Z, Shi WQ. Org Lett. 2016;18:1674. doi: 10.1021/acs.orglett.6b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagano T, Pospisil J, Chollet G, Schulthoff S, Hickmann V, Moulin E, Herrmann J, Mueller R, Füerstner A. Chem - Eur J. 2009;15:9697. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]
- 11.Zong G, Barber E, Aljewari H, Zhou J, Hu Z, Du Y, Shi WQ. J Org Chem. 2015;80:9279. doi: 10.1021/acs.joc.5b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Postema MHD, TenDyke K, Cutter J, Kuznetsov G, Xu Q. Org Lett. 2009;11:1417. doi: 10.1021/ol900086b. [DOI] [PubMed] [Google Scholar]
- 13.Füerstner A, Mueller T. J Org Chem. 1998;63:424. doi: 10.1021/jo971918k. [DOI] [PubMed] [Google Scholar]
- 14.Füerstner A, Mueller T. J Am Chem Soc. 1999;121:7814. [Google Scholar]
- 15.Füerstner A, Jeanjean F, Razon P. Angew Chem, Int Ed. 2002;41:2097. [PubMed] [Google Scholar]
- 16.Füerstner A. Eur J Org Chem. 2004:943. [Google Scholar]
- 17.Haines AH. Adv Carbohydr Chem Biochem. 1976;33:11. [Google Scholar]
- 18.Killen JC, Leonard J, Aggarwal VK. Synlett. 2010:579. [Google Scholar]
- 19.Gessler S, Randl S, Blechert S. Tetrahedron Lett. 2000;41:9973. [Google Scholar]
- 20.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.