

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a common clinicopathological condition, encompassing a range of conditions caused by lipid deposition within liver cells. To date, no approved drugs are available for the treatment of NAFLD, despite the fact that it represents a serious and growing clinical problem in the Western world. Identification of the molecular mechanisms leading to NAFLD-related fat accumulation, mitochondrial dysfunction and oxidative balance impairment facilitates the development of specific interventions aimed at preventing the progression of hepatic steatosis. In this review, we focus our attention on the role of dysfunctions in mitochondrial bioenergetics in the pathogenesis of fatty liver. Major data from the literature about the mitochondrial targeting of some antioxidant molecules as a potential treatment for hepatic steatosis are described and critically analysed. There is ample evidence of the positive effects of several classes of antioxidants, such as polyphenols (i.e., resveratrol, quercetin, coumestrol, anthocyanins, epigallocatechin gallate and curcumin), carotenoids (i.e., lycopene, astaxanthin and fucoxanthin) and glucosinolates (i.e., glucoraphanin, sulforaphane, sinigrin and allyl-isothiocyanate), on the reversion of fatty liver. Although the mechanism of action is not yet fully elucidated, in some cases an indirect interaction with mitochondrial metabolism is expected. We believe that such knowledge will eventually translate into the development of novel therapeutic approaches for fatty liver.
Keywords: Hepatic steatosis, Fatty liver, Lipogenesis, Mitochondria, Oxidative stress
Core tip: So far, there are no approved drugs for the treatment or the prevention of fatty liver and strategies mainly rely on dietary, physical activity and lifestyle modifications, as well as correction of hepatic steatosis-associated metabolic disturbances. This review mainly covers the biochemical mechanisms responsible for the dysfunctions in mitochondrial bioenergetics observed in fatty liver and analyses the most recent data evidencing the effects of new bioactive compounds on the preservation of mitochondrial function. New insight into biochemical details underlying fat accumulation in the liver could lead to more targeted and effective therapeutics for fatty liver.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is now considered the most prevalent form of chronic liver disease in the world, representing a serious and growing clinical problem in Western countries[1,2]. NAFLD is a clinicopathological condition, associated with a significant lipid deposition in hepatocytes and characterized by persistent defects in hepatic enzymes. Although NAFLD presents histological features of alcohol-induced liver injury, it affects patients who drink little to no alcohol. NAFLD is a histological disease which may progress from simple steatosis to nonalcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis and eventually hepatocellular carcinoma[3-6].
To date, there are no approved drugs to treat patients with fatty liver disease and clinical management strategies mainly rely on modifications of diet, physical activity and lifestyle, as well as correction of hyperglycemia, insulin resistance and hyperlipidemia, which are metabolic disturbances associated with NAFLD[7-9].
The understanding of the molecular mechanisms responsible for lipid accumulation, oxidative balance impairment and fibrosis in the liver could improve the therapeutic approach to lower the risk of fatty liver development. Antioxidant compounds, which modulate lipogenesis, lipid oxidation and peroxidation, and inflammation, represent a new attractive therapeutic approach for patients suffering from hepatic steatosis.
In this review, we focus our attention on the biochemical mechanisms by which selected antioxidant molecules exert their beneficial effect by targeting mitochondria function. Such knowledge may translate into novel treatment strategies for fatty liver.
BIOCHEMICAL EVENTS ASSOCIATED WITH HEPATIC STEATOSIS
Steatosis is the biochemical result of an imbalance between the rates of fatty acid input (uptake and synthesis with subsequent esterification to triglycerides, TGs) and output (oxidation and secretion)[10]. Therefore, TGs levels in hepatocytes derives from a complex interaction between hepatic fatty acid uptake, de novo synthesis, oxidation and export within very low density lipoprotein (VLDL)-TG[11].
Hepatic fatty acid uptake is the result of plasma free fatty acids (FFAs) released from hydrolysis of adipose tissue TGs, FFAs deriving from hydrolysis of lipoproteins, and dietary FFAs.
De novo fatty acid synthesis occurs via a complex series of reactions that take place in the mitochondrial matrix and in the cytosol of hepatocytes[12]. The rate of de novo lipogenesis is dependent on the transport activity of the mitochondrial citrate carrier (CIC)[13] and on enzymatic activities of acetyl-CoA carboxylase (ACC), fatty acid synthase, (FAS)[12], diacylglycerol acyltransferase (DGAT) 1 and 2, and stearoyl-CoA desaturase 1 (SCD1). Induction of lipogenic genes occurs in response to the combined actions of several nuclear transcription factors: sterol regulatory element binding proteins (SREBPs), carbohydrate responsive element binding protein (ChREBP), liver X receptor α (LXRα), farnesoid X receptor (FXR) and peroxisome proliferator-activated receptors (PPARs)[14].
Liver fatty acid oxidation occurs primarily within the mitochondria, and a little part in peroxisomes and microsomes. Fatty acids are transported into the mitochondrial matrix by a carnitine-dependent enzyme shuttle. Mitochondrial β-oxidation implies a series of dehydrogenation, hydration and cleavage reactions catalyzed by membrane-bound and soluble enzymes transcriptionally regulated by PPARα[15]. Each cycle of β-oxidation shortens the acyl-CoA molecule by two carbon units. The released acetyl-CoA then enters the Krebs cycle if the supply of oxaloacetate is sufficient; alternatively, it can be utilized for the production of ketone bodies. Reducing equivalents (FADH2 and NADH), that derived from the β-oxidation reactions then transfer their electrons to oxygen through the respiratory chain. Liver fatty acids may also be esterified to TGs and then stored as within hepatocytes or secreted into the blood as VLDL.
Metabolism of fatty acids plays a key role in the development of hepatic steatosis. Liver lipid accumulation is, therefore, the result of prolonged positive energy balance (de novo lipogenesis is the preferred mechanism to stock excess energy), adipose tissue dysfunction and insulin resistance, defects in fatty acid oxidation and mitochondrial metabolism, or imbalances in lipoprotein trafficking.
Hepatic TG accumulation (i.e., steatosis) is the “first hit” and the basis for hepatocyte damage that leads to a variety of “second hits”, such as cytokines, adipokines, bacterial endotoxins, mitochondrial dysfunction and/or endoplasmic reticulum stress. In fact, the traditional “two-hit” pathophysiological theory[16] is now being replaced by the “multiple parallel hits” hypothesis, which comprises lots of different parallel hits, represented by insulin resistance, oxidative stress, genetic and epigenetic mechanisms, cytokines and microbiota modifications, along with environmental elements[17].
DYSFUNCTIONAL MITOCHONDRIAL BIOENERGETICS IN THE FATTY LIVER
Several lines of evidence indicate that structural and functional alterations in mitochondria are crucial to the development of NAFLD[18-20]. The structural alterations include depletion of mitochondrial DNA (mtDNA), as well as morphological and ultrastructural changes, whereas the functional alterations include defects in mitochondrial β-oxidation and respiration[21]. The functional alterations lead to a decrease in ATP levels, leakage of deleterious reactive oxygen species (ROS) and excessive depots of fat[22]. A study describing the pathogenesis of NAFLD[23] proposed that the overflow of FFAs to hepatocytes leads to an increase in mitochondrial energetic metabolism, to support energy demand for TG storage and droplet growth. Later, it was hypothesized that accumulation of intermediate lipids could lead to mitochondrial stress and cell apoptosis.
Defects in the activities of key mitochondrial enzymes have been investigated using animal models[24,25], in which a diet rich in fat (high-fat diet, HF diet) has been shown to induce hyperglycemia, hyperinsulinemia and the onset of obesity and fatty liver. Carnitine palmitoyl-CoA transferase (CPT) activity, which is involved in fatty acid oxidation, was inhibited in animals fed the HF diet and showing NAFLD. The observed reduction in CPT activity was associated with the concomitant modulation of the activity of citrate synthase, a Krebs cycle enzyme that catalyses the reaction between acetyl-CoA and oxaloacetate to form citrate.
Reducing equivalents (NADH and FADH2) produced in the reactions of oxidation of fatty acids and Krebs cycle are used by the respiratory chain complexes to generate ATP. In fatty liver, mitochondrial respiration efficiency was decreased and this effect could be due to a possible uncoupling effect between ATP synthesis and transport through respiratory complexes. Expression of respiratory proteins are significantly increased to compensate the reduced activity of the respiratory complexes. Uncoupling between mitochondrial respiration and ATP synthesis could be the result of a different expression profile of the mitochondrial carriers responsible for proton conductance[25]. In accordance with this hypothesis, an increase in expression of the uncoupling protein (UCP) 2 isoform was found in rats with NAFLD[25,26]. In those animals, the expression of ATP/ATP carrier (AAC), another mitochondrial carrier which plays an important role in the mitochondrial permeability transition pore, was also found to be up-regulated[25]. Therefore, in uncoupled mitochondria, where proton gradient is dissipated by the entry of protons into the matrix via UCP2, an overexpression of AAC could compensate the reduced flux of ATP from mitochondria when membrane potential is reduced.
Alteration in mitochondrial function (Figure 1) leads to increased ROS production. The accumulation of lipid peroxidation and inactivation of key enzymatic activities leads to ROS-induced oxidative damage to cardiolipin. This phospholipid, which is almost exclusively present in the inner mitochondrial membrane, plays an important role in mitochondrial bioenergetics, as well as in the apoptosis[27,28].
Figure 1.
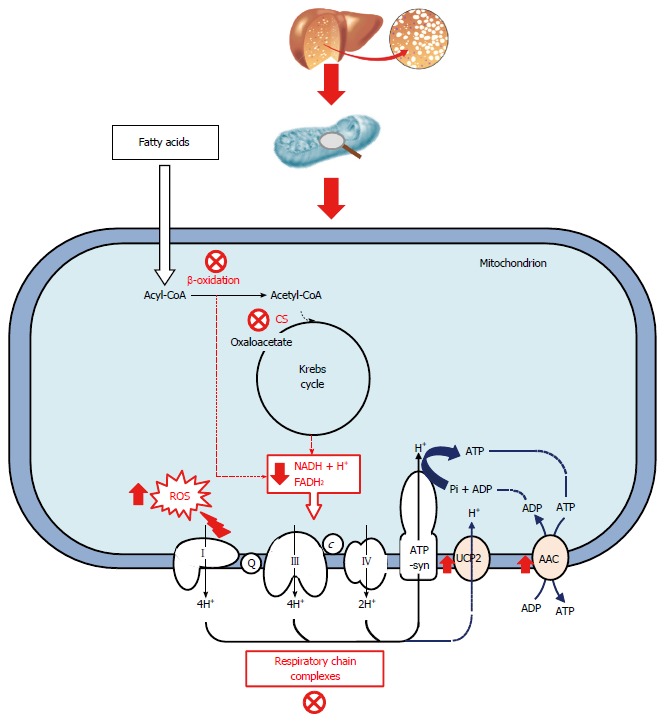
Dysfunctional mitochondrial bioenergetics in the fatty liver. In fatty liver, inhibition of β-oxidation and the reduction in citrate synthase (CS) activity implies a reduction in the flux of reducing equivalents to mitochondria. Mitochondrial respiration efficiency was decreased and this effect could be due to a possible uncoupling effect between ATP synthesis and transport through respiratory complexes. Alteration in mitochondrial function leads to increased ROS production. c: Cytochrome c; Q: Ubiquinone.
As cytosolic fatty acids accumulate as a consequence of an impairment of the oxidative capacity of the mitochondria, alternative pathways are activated in the peroxisomes (β-oxidation) and microsomes (ω-oxidation) and additional ROS are produced[29]. In fact, in the initial step of peroxisomal β-oxidation, acyl-CoA oxidase donates electrons directly to molecular oxygen, producing hydrogen peroxide; ROS are also formed during the microsomal ω-oxidation of fatty acids via flavoprotein-mediated donation of electrons to molecular oxygen. In addition, during fatty acids ω-oxidation, the corresponding dicarboxylic acids of the metabolized fatty acids can uncouple oxidative phosphorylation. Therefore, extramitochondrial fatty acid oxidation leads to a further increase in oxidative stress, contributing to mitochondrial dysfunction.
CONTRIBUTION OF OXIDATIVE STRESS TO STEATOSIS PATHOGENESIS
Oxidative stress is one of the key mediators of hepatic damage and is a major contributor to the progression from simple steatosis to steatohepatitis[30]. The oxidative stress condition encompasses the various deleterious processes that result from an imbalance between the excessive formation of pro-oxidants (i.e., ROS and/or reactive nitrogen species, RNS) and the counteracting antioxidant mechanisms. In mitochondria, increased ROS production can cause mtDNA depletion, attack biomolecules (i.e., proteins, carbohydrates and lipids), and damage the mitochondrial membrane. It is worth noting that mitochondria have a substantial concentration of phospholipids containing polyunsaturated fatty acids (PUFAs). The PUFAs are more prone to oxidative damage, because of the double bonds in their chemical structure, which lead to lipid peroxidation. In turn, peroxidation of these mitochondrial membrane components, especially docosahexaenoic acid which is necessary for a functional assembly of the mitochondrial complexes[31], could lead to further impairment of the activity of the respiratory chain, with consequent decreased ATP synthesis and overproduction of ROS. PUFA peroxidation seems to be able to enhance post-endoplasmic reticulum presecretory proteolysis of ApoB, thereby reducing VLDL secretion[32]; this may further contribute to TG accumulation in the liver.
Moreover, aldehydes formed through PUFA peroxidation impair cellular homeostasis[33], because these molecules affect nucleotide and protein synthesis, reduce hepatic glutathione content and increase production of the proinflammatory cytokine TNF-α. These effects lead to hepatocyte death and necrosis, inflammation, and liver fibrosis.
NUTRITIONAL MANAGEMENT OF HEPATIC STEATOSIS
Elucidation of the molecular mechanisms leading to fat accumulation, mitochondrial dysfunction and oxidative balance impairment facilitates the development of specific interventions aimed at preventing the progression of hepatic steatosis. Food bioactive compounds, which modulate the activation of genes and the function of proteins involved in steatosis pathogenesis, represent a new attractive therapeutic approach for this condition[34].
General recommendations include reducing intakes of total fat, saturated and trans fatty acids and fructose[35,36]. Conversely, increasing intakes of PUFAs, especially of the long-chain n-3 fatty acids, and monounsaturated fatty acids is advised[12,37,38].
Many dietary natural compounds isolated from fruits, vegetables and edible plants may be suggested as promising agents for treatment of fatty liver, since they are able to target mitochondria and to restore their function. Other molecules have not yet been tested for their potential effects on liver mitochondria, but they show potential for improving or restoring defective mitochondrial function in cells from different tissues. Since mitochondrial function is compromised in hepatic steatosis, in the following paragraphs we summarize the most recent data evidencing the mitochondrial targeting capabilities of new bioactive compounds.
POLYPHENOLS
Polyphenols are secondary metabolites of plants that have been studied primarily for their potential roles to combat oxidative stress and inflammatory processes. They contain at least one aromatic ring in their structure that is/are linked to different chemical groups, including phenolic, hydroxyl or carbon groups. Polyphenols can be broadly divided into flavonoids and nonflavonoids, based on their chemical structure. The flavonoids include flavonols (i.e., quercetin), flavones (i.e., luteolin), flavanols (i.e., catechins), flavanones (i.e., hesperetin, hesperidin naringenin and naringin), isoflavones (i.e., genistein and daidzein), anthocyanidins (i.e., cyanidin, malvidin, pelargonidin and delphinidin) and proanthocyanidins (i.e., condensed tannins), while the nonflavonoids include stilbenes, phenolic acids and hydroxycinnamates[39].
Flavonoids are the most abundant natural antioxidants present in the human diet, and their antioxidant activity is due to the presence of hydroxyl groups in the phenolic structure of flavonoids[40]. These compounds exert their numerous beneficial and antioxidative effects through several mechanisms; indeed, they are able to target different pathways that are possibly involved in the pathogenesis of liver diseases. Flavonoids are able to control de novo lipogenesis, inhibiting lipogenic proteins (i.e., ACC, SREBP-1, FAS and LXRα) and increasing lipolytic proteins (i.e., AMPK, PPARα and CPT-1). They are also effective scavengers of ROS and RNS (i.e., superoxide, hydrogen peroxide, hydroxyl radicals and peroxynitrite) that are elevated in pathological states and metabolic disorders such as NAFLD[41].
As mitochondria are an important source of ROS, redox-active compounds can be targeted to these organelles to counteract ROS production and its associated oxidative damage. However, as antioxidants, polyphenolic compounds may act directly by scavenging reactive species; alternatively may act indirectly by controlling the redox environment. In particular, findings from studies in several cellular models have indicated the abilities of polyphenols to modulate mitochondrial biogenesis and to control the mitochondrial membrane potential and oxidative phosphorylation[42].
Resveratrol
Resveratrol (trans-3,4’,5-trihydroxystilbene) is a stilbene naturally found in various food stuffs, such as grapes, berries, red wine and nuts. This molecule has been shown to control energetic metabolism in obese mice, improving the glucose homeostasis, increasing the fatty acid oxidation and inducing the expression of genes associated with the regulation of mitochondrial biogenesis[43,44]. The effects of high resveratrol doses (10-100 μmol/L) on mitochondrial metabolism have been evaluated in different models where it has been shown that resveratrol is able to increase the number of mitochondria in the tissues studied, and that this occurs via a sirtuin-dependent mechanism.
The sirtuins are a family of proteins that act as NAD-dependent deacetylases; three sirtuins are exclusively located in the mitochondrial compartment: SIRT3, -4 and -5[45]. However, the main target of resveratrol appears to be SIRT1. Although not found in the mitochondria, SIRT1 plays an important role in the regulation of metabolism, since it is able to stimulate both mitochondrial biogenesis and energetic metabolic fluxes via allosteric post-translational modifications of key enzymes[43]. A possible link between activation of SIRT1 and mitochondria could be the modulation of NAD+ concentration[46,47], because a significant portion of the cellular NAD+ pool is concentrated in the mitochondria[48]. In accordance with that hypothesis, a recent study[49] demonstrated that resveratrol can induce a mitochondrial complex I-dependent increase in NADH oxidation resulting in sirtuin activation in hepatocytes; moreover, this finding was obtained in different experimental models, including in vitro studies of isolated enzymes and HepG2 cells treated with resveratrol, as well as in vivo study of an aging model of mice fed with resveratrol. In particular, in the HepG2 cells (resveratrol administration at 1-5 μmol/L) and in the liver mitochondria from resveratrol-fed animals (administration of 50 mg/kg per day), complex I activation increased the mitochondrial NAD+/NADH ratio and, in turn, the higher NAD+ concentration initiated a SIRT3-dependent stimulation of the mitochondrial substrate supply pathways (i.e., the Krebs cycle and β-oxidation of fatty acids)[49]. We can therefore hypothesize - as already has been suggested for the aging process[50] - that mitochondrial complex I activity may be involved in fatty liver through at least two mechanisms: a ROS-dependent mechanism, which is related to the control of complex I activity, and a ROS-independent mechanism via the regulation of the ratio NAD+/NADH.
Quercetin
Quercetin is a type of flavonoid antioxidant that is found in plant foods, including leafy greens, tomatoes, berries and broccoli. Recent studies have shown that the beneficial effects of quercetin include the activation of mitochondrial biogenesis[42,51] through PGC-1α, which is a transcriptional coactivator of genes associated with oxidative phosphorylation and mtDNA replication. In HepG2 cells, the observed increase in mitochondrial biogenesis is associated with cytochrome oxidase subunit IV (COXIV) overexpression[52]. Interestingly, an increase in cytochrome c concentration is typically associated with similar increases in other mitochondrial proteins involved in the respiratory chain, Krebs cycle and fatty acid oxidation pathway. The result is an overall increase in mitochondrial capacity[52,53].
Results obtained in animal models have suggested that 50 mg/kg and 100 mg/kg doses of quercetin significantly affect the mitochondrial biogenesis; moreover, mice treated with up to 3000 mg/kg quercetin did not show any toxic effects[53]. However, the effect of oral administration of quercetin on liver mitochondria has not been tested. This is an intriguing aspect, because there are several reports indicating that quercetin exhibits both antioxidant and pro-oxidant activity, in a concentration dependent manner[54,55]. Therefore, further studies are needed to determine the therapeutic efficacy of oral quercetin administration.
Coumestrol
Coumestrol is the most commonly studied coumestan, and is a natural organic compound present in some plants, including alfalfa, legumes, Brussels sprouts, spinach, clover and soybeans. Recent data[56] have suggested that coumestrol induces an increase in mitochondria number and function, by activating SIRT1. By promoting mitochondrial content, coumestrol appears to be able to increase cellular ATP levels and glucose uptake, thereby improving energy metabolism at the same time. Moreover, coumestrol stimulation has been shown to augment the protein expression of respiratory chain components, such as NADH dehydrogenase 1α subcomplex 9 (NDUFA9), succinate dehydrogenase complex subunit A (SDHA), ubiquinol cytochrome c reductase core protein 2 (UQCRC2), cytochrome oxidase subunit 1 (COX1) and ATP synthase mitochondria F1 complex α subunit 1 (ATP5a), and of the transcriptional regulators that are responsible for mitochondrial biogenesis[56].
Coumestrol has also been shown to decrease the mRNA expression of lipogenic genes in adipocytes and hepatocytes and to increase fatty acid oxidation in myocytes. It has been proposed that SIRT1 and AMPK might reciprocally activate each other. AMPK enhances SIRT1 activity by increasing cellular NAD+ levels[57], whereas SIRT1 mediates the deacetylation of liver kinase B1[58] (an upstream regulator of AMPK), thereby assembling a positive feedback loop that serves to enhance energy catabolism. It might, therefore, be possible for coumestrol to activate AMPK, itself a regulator of SIRT1; further studies are needed to determine which one - SIRT1 or AMPK - is the upstream regulator of the other[56].
Anthocyanins
Anthocyanidins are water-soluble pigments of coloured berries, fruits and vegetables. They can protect hepatocytes against injury caused by high glucose-induced oxidative damage, by improving antioxidant status and inhibiting the mitochondria pathways of apoptosis. The proposed mechanism has been shown to involve the prevention of hyperglycemia-induced mitochondrial depolarization, which helps mitochondria to maintain their function[59].
In addition, other studies have suggested that treatment with anthocyanins increases mitochondrial fatty acid oxidation via activation of genes participating in this metabolic pathway (i.e., PPARα, PPARδ, UCP-2, UCP-3 and mitochondrial transcription factor A)[60].
Recent studies have suggested that these molecules can attenuate mitochondrial defects caused by inhibition of complex I in human and rat tissues[61,62]. In isolated mitochondria from ischemic hearts, anthocyanins were found to recover complex I activity, thereby enhancing the rate of ATP synthesis[63]. A normal functioning of respiratory complex I is necessary to ensuring mitochondrial ATP synthesis and its supply to cells. In fact, it is widely accepted that inhibition of complex I results in a decrease of oxidative phosphorylation[64]. The suggested mechanism of action of anthocyanins has been associated with their redox potential, since these molecules can act as electron acceptors in a similar way as endogenous coenzyme Q.
Epigallocatechin gallate
Epigallocatechin gallate is the ester of epigallocatechin and gallic acid. It is a flavonoid belonging to the chemical class of flavan-3-ols (catechins) and it is the most abundant catechin in green tea (Camellia sinensis L.), accounting for about 50% of its total polyphenols.
Some studies have suggested that epigallocatechin gallate can promote fat oxidation and energy expenditure[65]. At a cellular level, reduced energy uptake could activate the AMPK signalling pathway, which may further affect energy metabolism, inducing chronic alterations in mitochondrial architecture (morphology and mass). This may represent a crucial step for the generation of oxidative damage[66]. Such modifications also include increase in the levels of porin and mitochondrial respiratory proteins, and in mtDNA content. Therefore, epigallocatechin gallate is able to modulate systemic energy use by inducing mitochondrial changes[67]. On the other hand, this bioactive molecule has been reported to induce fatty acid β-oxidation through an increase in mRNA levels of CPT-1 and UCP2[68].
Curcumin
Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione] is a yellow polyphenol isolated from the rhizome of Curcuma longa, a characteristic plant of tropical and subtropical regions, which has been used as a spice and traditional medicine in Asia for centuries due to its antioxidant, antiinflammatory, antimutagenic, antimicrobial and anticancer properties.
The antioxidant properties of curcumin have been shown to protect against mitochondrial dysfunction in several experimental models[69]. In particular, curcumin has been reported to increase oxygen consumption and respiratory chain complexes’ activities, to prevent oxidative stress and to restore ATP content. Such an improvement in mitochondrial function was also shown to be associated with prevention against the decrease in the activity of aconitase[70], an oxidative stress marker. In addition, curcumin facilitated β-oxidation in in vitro experiments (by up-regulation of CPT-1), reducing lipogenesis at the same time[64].
CAROTENOIDS
Carotenoids are lipophilic pigments responsible for the yellow, orange or red colours of flowers and fruits. Their scavenging activity of ROS is well known, as well as the ability of β-carotenes to be the primary dietary source of provitamin A. The most prevalent carotenoids in the diet are α- and β-carotene, lutein, lycopene, zeaxanthin and cryptoxanthin.
Carotenoids accumulate mainly in the liver where they are incorporated into lipoproteins and released into the circulation in the form of lipoproteins. Dietary carotenoids may physiologically scavenge free radical species in the liver, thus inhibiting the development of hepatic dysfunction[71].
They are considered potent antioxidants and antiinflammatory micronutrients, and have been used in the prevention and treatment of NAFLD. Carotenoids are also critical regulators of macrophage polarization and thereby inhibit the development and the progression of NASH[72]. The molecular mechanism by which carotenoids exert their effects in NAFLD is not well defined, but it has been proposed that they may act through multiple mechanisms.
Lycopene
Lycopene is a carotenoid that lacks provitamin A activity and is responsible for the red to pink colours seen in tomatoes, red grapefruit, watermelon and apricots.
Epidemiological and experimental studies have suggested that lycopene may have chemopreventive properties against certain types of cancers, including NASH-promoted hepatocarcinogenesis, mainly as a consequence of oxidative stress decrease, which could be imparted through different mechanisms[73]. Moreover, lycopene also reduces the development of hepatic steatosis induced by an HF diet[74]. The key role of this carotenoid in protection against fatty liver was confirmed by the reduced plasma lycopene levels in subjects affected by NASH, suggesting a possible link between low lycopene levels and the development of liver diseases[75].
A possible effect of lycopene on mitochondrial function has been proposed in cardiomyocytes, where the protective effects of this molecule have been attributed to its roles in improving mitochondrial function[76]. An important factor associated with mitochondrial dysfunction is activation of the mitochondrial permeability transition pore (mPTP). Activation of mPTP leads to functional breakdown, and subsequently morphological disintegration of the mitochondria progresses to cell death. Lycopene was shown to suppress the activation of mPTP by reducing intracellular ROS levels and inhibiting oxidative damage[76]. In this way, the impairment of mitochondrial membrane potential and decrease in ATP levels were alleviated, suggesting that the cell protection is, in part, due to the prevention of mitochondrial dysfunction.
In accordance with this hypothesis, some studies have shown the ability of lycopene to enhance the mitochondrial activity in HepG2 cells that were exposed to aflatoxin B1[77] and to improve the mitochondrial function in nervous system of rats that were exposed to 3-nitropropionic acid[78]. Therefore, the antioxidative potential of lycopene should account for its ability to preserve the activity of endogenous free radical scavengers and of respiratory chain complexes directly, thus preventing ROS production and secondary oxidative damage.
Astaxanthin
Astaxanthin is a xanthophyll carotenoid found in various marine organisms, such as yeast, salmon, shrimp, crayfish and green microalga Haematococcus pluvialis. It is well demonstrated that this carotenoid has strong antioxidant properties[79], since it is 100-fold to 500-fold more effective than vitamin E in preventing lipid peroxidation and in reducing lipid accumulation in liver[80]. In mice models it has also been reported that astaxanthin can down-regulate genes involved in lipogenesis and lipid-uptake, without influencing fatty acid oxidation-related genes in the liver[80]. Moreover, it has also been shown to reduce hepatic levels of TGs and cholesterol in mice[81].
A role has been suggested for astaxantin in improvement of mitochondrial function. This molecule appears to be able to increase mitochondrial membrane potential and respiratory control[82], which are important measures of mitochondrial health when investigating isolated mitochondria. We can hypothesize that the increase in mitochondrial respiration efficiency observed in astaxanthin-treated cells is due to the increase in mitochondrial membrane potential.
Interestingly, astaxanthin is also present in krill oil, a dietary source of n-3 long-chain PUFAs, the effects of which in reducing liver lipids were more pronounced than that found with fish oil-fed rats[83]. Moreover, astaxanthin induces fatty acid catabolism. Acyl-CoA oxidase (ACOX1) is a target gene of PPARα, a regulator of mitochondrial and peroxisomal β-oxidation, which can be activated by this carotenoid[84].
Fucoxanthin
Fucoxanthin is one of the most abundant marine carotenoids, accounting for more than 10% of the estimated total carotenoids in nature. It is a xanthophyll, the distinct structure of which includes an unusual allenic bond and some oxygenic functional groups in a polyene chain.
The hypolipidemic effects of fucoxanthin might be mediated by down-regulation of various lipogenic enzyme activities and up-regulation of fatty acid β-oxidation activity in fat tissues[85,86].
Recent literature[86] suggests that this carotenoid has also anti-obesity properties, mainly by stimulating UCP-1 expression in white adipose tissue. This mitochondrial protein is usually found in brown adipose tissue and is not expressed in white adipose tissue in the absence of any stimulation. The role of UCP-1 is the dissipation of the proton gradient created by respiration and this leads to heat generation (thermogenesis).
GLUCOSINOLATES
Glucosinolates are a family of sulphur-containing plant secondary metabolites usually found in cruciferous plants. They have a common structure containing a thioketal-linked glucose molecule and are hydrolysed by specific enzymes (i.e., myrosinases) to produce biologically active sulfureted aglycones, the isothiocyanates. Glucosinolates and their respective enzymatic hydrolysis scavange harmful radicals and modulate enzymes involved in detoxification processes, thus preventing oxidative damage.
Preliminary studies have suggested that these molecules can suppress the activation of hepatic macrophages and also contribute to decrease liver damage and to protect against the development of liver tumorigenesis[87].
Glucoraphanin and sulforaphane
Glucoraphanin (4-methylsulfinylbutyl glucosinolate) is the major glucosinolate found within broccoli and is the precursor of sulforaphane. Indeed, the biologically active sulforaphane is not present as such in Brassicaceae, but is produced by enzymatic hydrolysis upon tissue disruption, such as the chewing of raw vegetables, which brings the endogenous enzyme β-thioglucoside glucohydrolase (a myrosinase) into contact with the glucosinolate. Several factors determine the degree of conversion from glucoraphanin to sulforaphane and, therefore, its cellular activity and protective effects[88].
Studies on in vitro and animal models have demonstrated that broccoli sulforaphane can mitigate some of the effects produced by inducers of inflammation and cell damage, such as carcinogens, toxins and ROS. The proposed molecular mechanism involves the inhibition of enzymes responsible for reactions related to oxidation, reduction and hydrolysis (i.e., cytochrome P450 enzymes), and the induction of antioxidant and detoxification enzymes (i.e., quinone reductase, thioredoxin reductase 1, and heme oxygenase 1)[89,90]. These last enzymes, known as phase II antioxidant enzymes, plays an important protective role in the maintenance of redox state in mammalian cells by providing a precise balance between the level of ROS and endogenous thiol buffers, which protect cells from oxidative stress-induced damage. They also play a critical role in maintaining the NAD+/NADH and NADP+/NADPH ratios[91]. As a consequence, dietary supplementation with the sulforaphane precursor glucoraphanin represents a potent method for improving liver function and for maintaining good liver condition[92,93].
Recently, a role has been suggested for glucoraphanin in the modulation of mitochondrial function. Metabolomic studies have supported the proposal that such a modulation may be mediated by control of FAD levels[94]. FAD, a redox cofactor, takes part in several important reactions in energetic metabolism (i.e., fatty acid oxidation and Krebs cycle). Moreover, FAD acts as cofactor of lysine-specific demethylase-1, which, through epigenetic regulation, modulates the expression of genes involved in mitochondrial metabolism and energy expenditure[95]. Thus, when FAD levels are relatively high, expression of genes associated with mitochondrial respiration is down-regulated and export of citrate from the Krebs cycle is enhanced; this modulation results in elevated concentrations of fatty acids, lipids and steroids.
Sinigrin and allyl-isothiocyanate
Sinigrin is a major component of commonly consumed cruciferous vegetables, such as horseradish and wasabi. Its degradation product allyl-isothiocyanate is responsible for the characteristic pungent taste[96].
The potential of sinigrin to inhibit the growth of several types of cancer cells has been well investigated. In particular, sinigrin was shown to significantly prevent the proliferation of liver tumour cells, inducing apoptosis and gradually restoring liver functions[97].
A recent study suggested that allyl-isothiocyanate can improve mitochondrial respiration efficiency, reducing, at the same time, metabolic dysfunctions[98]. The effects of this molecule have been evaluated in muscle cells, which showed a higher mitochondrial activity after allyl-isothiocyanate treatment. In particular, increases in mitochondrial membrane potential, in mtDNA content and in respiratory capacity were observed.
CONCLUSION
Hepatic steatosis has become a very common cause of liver diseases in Western word. So far, there are no approved drugs for the treatment of this disease. Therefore, identification of the molecular mechanisms leading to the fat accumulation in liver may provide new avenues for the development of new diagnostic and therapeutic approaches.
Food bioactive compounds, which are important regulators of expression and activity of proteins associated with lipid homeostasis and metabolism, represent a novel therapeutic approach for fatty liver disease. In fact, there is evidence that several classes of antioxidants (i.e., polyphenols, carotenoids and glucosinolates) are able to reverse hepatic steatosis. Although the mechanism of action remains understood, an indirect effect of bioactive molecules on mitochondrial function is expected (Table 1).
Table 1.
Described effects of bioactive compounds on mitochondria
| Compound | Effect |
| Polyphenols | |
| Resveratrol | ↑Mitochondrial biogenesis |
| ↑Complex I activity | |
| Quercetin | ↑mtDNA |
| ↑Respiratory chain proteins | |
| Coumestrol | ↑Mitochondria number |
| ↑Respiratory chain proteins | |
| ↑Mitochondrial function | |
| Anthocyanins | ↑Fatty acid oxidation |
| ↑Compex I activity | |
| Epigallocatechin gallate | ↑Fatty acid oxidation |
| ↑Energy expenditure | |
| Curcumin | ↑Fatty acid oxidation |
| ↑Respiratory complexes activity | |
| Carotenoids | |
| Lycopene | ↓Activation of mPTP |
| Astaxanthin | ↑Fatty acid oxidation |
| ↑Mitochondrial membrane potential | |
| ↑Respiration efficiency | |
| Fucoxanthin | ↑Fatty acid oxidation |
| Glucosinolates | |
| Glucoraphanin | ↓FAD levels |
| Sinigrin | ↑Mitochondrial membrane potential |
| ↑Respiration efficiency | |
| ↑DNA content | |
mtDNA: Mitochondrial DNA; mPTP: Mitochondrial permeability transition pore.
We believe that gaining a more complete understanding of the biochemical mechanisms underlying the hepatoprotective roles of the bioactive compounds such as those ones included in this review will lead to more targeted and effective therapeutics for hepatic steatosis.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): A, A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare no conflict of interests for this article.
Peer-review started: January 22, 2017
First decision: February 9, 2017
Article in press: June 1, 2017
P- Reviewer: Gorrell MD, Kayadibi H, Lee HC S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Mikolasevic I, Milic S, Turk Wensveen T, Grgic I, Jakopcic I, Stimac D, Wensveen F, Orlic L. Nonalcoholic fatty liver disease - A multisystem disease? World J Gastroenterol. 2016;22:9488–9505. doi: 10.3748/wjg.v22.i43.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azzam H, Malnick S. Non-alcoholic fatty liver disease - the heart of the matter. World J Hepatol. 2015;7:1369–1376. doi: 10.4254/wjh.v7.i10.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCullough AJ. Update on nonalcoholic fatty liver disease. J Clin Gastroenterol. 2002;34:255–262. doi: 10.1097/00004836-200203000-00013. [DOI] [PubMed] [Google Scholar]
- 4.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 5.Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci. 2005;50:171–180. doi: 10.1007/s10620-005-1267-z. [DOI] [PubMed] [Google Scholar]
- 6.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Baran B, Akyüz F. Non-alcoholic fatty liver disease: what has changed in the treatment since the beginning? World J Gastroenterol. 2014;20:14219–14229. doi: 10.3748/wjg.v20.i39.14219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dyson J, Day C. Treatment of non-alcoholic fatty liver disease. Dig Dis. 2014;32:597–604. doi: 10.1159/000360511. [DOI] [PubMed] [Google Scholar]
- 9.Hossain N, Kanwar P, Mohanty SR. A Comprehensive Updated Review of Pharmaceutical and Nonpharmaceutical Treatment for NAFLD. Gastroenterol Res Pract. 2016;2016:7109270. doi: 10.1155/2016/7109270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferramosca A, Zara V. Modulation of hepatic steatosis by dietary fatty acids. World J Gastroenterol. 2014;20:1746–1755. doi: 10.3748/wjg.v20.i7.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferramosca A, Zara V. Dietary fat and hepatic lipogenesis: mitochondrial citrate carrier as a sensor of metabolic changes. Adv Nutr. 2014;5:217–225. doi: 10.3945/an.113.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O’Doherty RM. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol Endocrinol Metab. 2008;294:E969–E977. doi: 10.1152/ajpendo.00497.2007. [DOI] [PubMed] [Google Scholar]
- 16.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 17.Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism. 2016;65:1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.García-Ruiz C, Baulies A, Mari M, García-Rovés PM, Fernandez-Checa JC. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: cause or consequence? Free Radic Res. 2013;47:854–868. doi: 10.3109/10715762.2013.830717. [DOI] [PubMed] [Google Scholar]
- 20.Ferramosca A, Conte A, Damiano F, Siculella L, Zara V. Differential effects of high-carbohydrate and high-fat diets on hepatic lipogenesis in rats. Eur J Nutr. 2014;53:1103–1114. doi: 10.1007/s00394-013-0613-8. [DOI] [PubMed] [Google Scholar]
- 21.Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Auger C, Alhasawi A, Contavadoo M, Appanna VD. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol. 2015;3:40. doi: 10.3389/fcell.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flamment M, Rieusset J, Vidal H, Simard G, Malthièry Y, Fromenty B, Ducluzeau PH. Regulation of hepatic mitochondrial metabolism in response to a high fat diet: a longitudinal study in rats. J Physiol Biochem. 2012;68:335–344. doi: 10.1007/s13105-012-0145-3. [DOI] [PubMed] [Google Scholar]
- 24.Ferramosca A, Conte A, Burri L, Berge K, De Nuccio F, Giudetti AM, Zara V. A krill oil supplemented diet suppresses hepatic steatosis in high-fat fed rats. PLoS One. 2012;7:e38797. doi: 10.1371/journal.pone.0038797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferramosca A, Conte A, Zara V. Krill Oil Ameliorates Mitochondrial Dysfunctions in Rats Treated with High-Fat Diet. Biomed Res Int. 2015;2015:645984. doi: 10.1155/2015/645984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y, Zhang H, Dong LY, Wang D, An W. Increased hepatic UCP2 expression in rats with nonalcoholic steatohepatitis is associated with upregulation of Sp1 binding to its motif within the proximal promoter region. J Cell Biochem. 2008;105:277–289. doi: 10.1002/jcb.21827. [DOI] [PubMed] [Google Scholar]
- 27.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–141. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- 28.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–14218. doi: 10.3748/wjg.v20.i39.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res. 2013;47:869–880. doi: 10.3109/10715762.2013.837577. [DOI] [PubMed] [Google Scholar]
- 31.Infante JP, Huszagh VA. Secondary carnitine deficiency and impaired docosahexaenoic (22: 6n-3) acid synthesis: a common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000;468:1–5. doi: 10.1016/s0014-5793(00)01083-8. [DOI] [PubMed] [Google Scholar]
- 32.Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J Clin Invest. 2004;113:1277–1287. doi: 10.1172/JCI19197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 34.Dongiovanni P, Lanti C, Riso P, Valenti L. Nutritional therapy for nonalcoholic fatty liver disease. J Nutr Biochem. 2016;29:1–11. doi: 10.1016/j.jnutbio.2015.08.024. [DOI] [PubMed] [Google Scholar]
- 35.Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, Fagà E, Silli B, Pagano G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–916. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 36.Chiu S, Sievenpiper JL, de Souza RJ, Cozma AI, Mirrahimi A, Carleton AJ, Ha V, Di Buono M, Jenkins AL, Leiter LA, et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of controlled feeding trials. Eur J Clin Nutr. 2014;68:416–423. doi: 10.1038/ejcn.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferramosca A, Savy V, Conte L, Zara V. Dietary combination of conjugated linoleic acid (CLA) and pine nut oil prevents CLA-induced fatty liver in mice. J Agric Food Chem. 2008;56:8148–8158. doi: 10.1021/jf8010728. [DOI] [PubMed] [Google Scholar]
- 38.Ferramosca A, Savy V, Zara V. Olive oil increases the hepatic triacylglycerol content in mice by a distinct influence on the synthesis and oxidation of fatty acids. Biosci Biotechnol Biochem. 2008;72:62–69. doi: 10.1271/bbb.70369. [DOI] [PubMed] [Google Scholar]
- 39.Del Rio D, Rodriguez-Mateos A, Spencer JP, Tognolini M, Borges G, Crozier A. Dietary (poly)phenolics in human health: structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid Redox Signal. 2013;18:1818–1892. doi: 10.1089/ars.2012.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Y, Chen X. Structures required of polyphenols for inhibiting advanced glycation end products formation. Curr Drug Metab. 2013;14:414–431. doi: 10.2174/1389200211314040005. [DOI] [PubMed] [Google Scholar]
- 41.Van De Wier B, Koek GH, Bast A, Haenen GR. The potential of flavonoids in the treatment of non-alcoholic fatty liver disease. Crit Rev Food Sci Nutr. 2017;57:834–855. doi: 10.1080/10408398.2014.952399. [DOI] [PubMed] [Google Scholar]
- 42.Serrano JC, Cassanye A, Martín-Gari M, Granado-Serrano AB, Portero-Otín M. Effect of dietary bioactive compounds on mitochondrial and metabolic flexibility. Diseases. 2016;4:14. doi: 10.3390/diseases4010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 44.Mercader J, Palou A, Bonet ML. Resveratrol enhances fatty acid oxidation capacity and reduces resistin and Retinol-Binding Protein 4 expression in white adipocytes. J Nutr Biochem. 2011;22:828–834. doi: 10.1016/j.jnutbio.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Kazgan N. Mammalian sirtuins and energy metabolism. Int J Biol Sci. 2011;7:575–587. doi: 10.7150/ijbs.7.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 2001;492:4–8. doi: 10.1016/s0014-5793(01)02198-6. [DOI] [PubMed] [Google Scholar]
- 49.Desquiret-Dumas V, Gueguen N, Leman G, Baron S, Nivet-Antoine V, Chupin S, Chevrollier A, Vessières E, Ayer A, Ferré M, et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J Biol Chem. 2013;288:36662–36675. doi: 10.1074/jbc.M113.466490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stefanatos R, Sanz A. Mitochondrial complex I: a central regulator of the aging process. Cell Cycle. 2011;10:1528–1532. doi: 10.4161/cc.10.10.15496. [DOI] [PubMed] [Google Scholar]
- 51.Davis JM, Murphy EA, Carmichael MD, Davis B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1071–R1077. doi: 10.1152/ajpregu.90925.2008. [DOI] [PubMed] [Google Scholar]
- 52.Rayamajhi N, Kim SK, Go H, Joe Y, Callaway Z, Kang JG, Ryter SW, Chung HT. Quercetin induces mitochondrial biogenesis through activation of HO-1 in HepG2 cells. Oxid Med Cell Longev. 2013;2013:154279. doi: 10.1155/2013/154279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruiz MJ, Fernández M, Picó Y, Mañes J, Asensi M, Carda C, Asensio G, Estrela JM. Dietary administration of high doses of pterostilbene and quercetin to mice is not toxic. J Agric Food Chem. 2009;57:3180–3186. doi: 10.1021/jf803579e. [DOI] [PubMed] [Google Scholar]
- 54.Wätjen W, Michels G, Steffan B, Niering P, Chovolou Y, Kampkötter A, Tran-Thi QH, Proksch P, Kahl R. Low concentrations of flavonoids are protective in rat H4IIE cells whereas high concentrations cause DNA damage and apoptosis. J Nutr. 2005;135:525–531. doi: 10.1093/jn/135.3.525. [DOI] [PubMed] [Google Scholar]
- 55.Robaszkiewicz A, Balcerczyk A, Bartosz G. Antioxidative and prooxidative effects of quercetin on A549 cells. Cell Biol Int. 2007;31:1245–1250. doi: 10.1016/j.cellbi.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 56.Seo DB, Jeong HW, Lee SJ, Lee SJ. Coumestrol induces mitochondrial biogenesis by activating Sirt1 in cultured skeletal muscle cells. J Agric Food Chem. 2014;62:4298–4305. doi: 10.1021/jf404882w. [DOI] [PubMed] [Google Scholar]
- 57.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang X, Tang X, Zhang P, Liu G, Guo H. Cyanidin-3-O-β-glucoside protects primary mouse hepatocytes against high glucose-induced apoptosis by modulating mitochondrial dysfunction and the PI3K/Akt pathway. Biochem Pharmacol. 2014;90:135–144. doi: 10.1016/j.bcp.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 60.Rupasinghe HP, Sekhon-Loodu S, Mantso T, Panayiotidis MI. Phytochemicals in regulating fatty acid β-oxidation: Potential underlying mechanisms and their involvement in obesity and weight loss. Pharmacol Ther. 2016;165:153–163. doi: 10.1016/j.pharmthera.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 61.Lagoa R, Graziani I, Lopez-Sanchez C, Garcia-Martinez V, Gutierrez-Merino C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim Biophys Acta. 2011;1807:1562–1572. doi: 10.1016/j.bbabio.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 62.Pajuelo D, Quesada H, Díaz S, Fernández-Iglesias A, Arola-Arnal A, Bladé C, Salvadó J, Arola L. Chronic dietary supplementation of proanthocyanidins corrects the mitochondrial dysfunction of brown adipose tissue caused by diet-induced obesity in Wistar rats. Br J Nutr. 2012;107:170–178. doi: 10.1017/S0007114511002728. [DOI] [PubMed] [Google Scholar]
- 63.Skemiene K, Liobikas J, Borutaite V. Anthocyanins as substrates for mitochondrial complex I - protective effect against heart ischemic injury. FEBS J. 2015;282:963–971. doi: 10.1111/febs.13195. [DOI] [PubMed] [Google Scholar]
- 64.Schapira AH. Complex I: inhibitors, inhibition and neurodegeneration. Exp Neurol. 2010;224:331–335. doi: 10.1016/j.expneurol.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 65.Rains TM, Agarwal S, Maki KC. Antiobesity effects of green tea catechins: a mechanistic review. J Nutr Biochem. 2011;22:1–7. doi: 10.1016/j.jnutbio.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 66.Serrano JC, Gonzalo-Benito H, Jové M, Fourcade S, Cassanyé A, Boada J, Delgado MA, Espinel AE, Pamplona R, Portero-Otín M. Dietary intake of green tea polyphenols regulates insulin sensitivity with an increase in AMP-activated protein kinase α content and changes in mitochondrial respiratory complexes. Mol Nutr Food Res. 2013;57:459–470. doi: 10.1002/mnfr.201200513. [DOI] [PubMed] [Google Scholar]
- 67.Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM. Epigallocatechin gallate and mitochondria-A story of life and death. Pharmacol Res. 2016;104:70–85. doi: 10.1016/j.phrs.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 68.Lee MS, Kim CT, Kim Y. Green tea (-)-epigallocatechin-3-gallate reduces body weight with regulation of multiple genes expression in adipose tissue of diet-induced obese mice. Ann Nutr Metab. 2009;54:151–157. doi: 10.1159/000214834. [DOI] [PubMed] [Google Scholar]
- 69.Trujillo J, Granados-Castro LF, Zazueta C, Andérica-Romero AC, Chirino YI, Pedraza-Chaverrí J. Mitochondria as a target in the therapeutic properties of curcumin. Arch Pharm (Weinheim) 2014;347:873–884. doi: 10.1002/ardp.201400266. [DOI] [PubMed] [Google Scholar]
- 70.Granados-Castro LF, Rodríguez-Rangel DS, Fernández-Rojas B, León-Contreras JC, Hernández-Pando R, Medina-Campos ON, Eugenio-Pérez D, Pinzón E, Pedraza-Chaverri J. Curcumin prevents paracetamol-induced liver mitochondrial alterations. J Pharm Pharmacol. 2016;68:245–256. doi: 10.1111/jphp.12501. [DOI] [PubMed] [Google Scholar]
- 71.Yilmaz B, Sahin K, Bilen H, Bahcecioglu IH, Bilir B, Ashraf S, Halazun KJ, Kucuk O. Carotenoids and non-alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:161–171. doi: 10.3978/j.issn.2304-3881.2015.01.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ni Y, Zhuge F, Nagashimada M, Ota T. Novel Action of Carotenoids on Non-Alcoholic Fatty Liver Disease: Macrophage Polarization and Liver Homeostasis. Nutrients. 2016;8:pii: E391. doi: 10.3390/nu8070391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Ausman LM, Greenberg AS, Russell RM, Wang XD. Dietary lycopene and tomato extract supplementations inhibit nonalcoholic steatohepatitis-promoted hepatocarcinogenesis in rats. Int J Cancer. 2010;126:1788–1796. doi: 10.1002/ijc.24689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bahcecioglu IH, Kuzu N, Metin K, Ozercan IH, Ustündag B, Sahin K, Kucuk O. Lycopene prevents development of steatohepatitis in experimental nonalcoholic steatohepatitis model induced by high-fat diet. Vet Med Int. 2010;2010:pii: 262179. doi: 10.4061/2010/262179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Erhardt A, Stahl W, Sies H, Lirussi F, Donner A, Häussinger D. Plasma levels of vitamin E and carotenoids are decreased in patients with Nonalcoholic Steatohepatitis (NASH) Eur J Med Res. 2011;16:76–78. doi: 10.1186/2047-783X-16-2-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L, Zhang S, Li K, Yu Z. Lycopene protects against hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial dysfunction in primary neonatal mouse cardiomyocytes. PLoS One. 2012;7:e50778. doi: 10.1371/journal.pone.0050778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reddy L, Odhav B, Bhoola K. Aflatoxin B1-induced toxicity in HepG2 cells inhibited by carotenoids: morphology, apoptosis and DNA damage. Biol Chem. 2006;387:87–93. doi: 10.1515/BC.2006.012. [DOI] [PubMed] [Google Scholar]
- 78.Kumar P, Kalonia H, Kumar A. Lycopene modulates nitric oxide pathways against 3-nitropropionic acid-induced neurotoxicity. Life Sci. 2009;85:711–718. doi: 10.1016/j.lfs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 79.Guerin M, Huntley ME, Olaizola M. Haematococcus astaxanthin: applications for human health and nutrition. Trends Biotechnol. 2003;21:210–216. doi: 10.1016/S0167-7799(03)00078-7. [DOI] [PubMed] [Google Scholar]
- 80.Ni Y, Nagashimada M, Zhuge F, Zhan L, Nagata N, Tsutsui A, Nakanuma Y, Kaneko S, Ota T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci Rep. 2015;5:17192. doi: 10.1038/srep17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ikeuchi M, Koyama T, Takahashi J, Yazawa K. Effects of astaxanthin in obese mice fed a high-fat diet. Biosci Biotechnol Biochem. 2007;71:893–899. doi: 10.1271/bbb.60521. [DOI] [PubMed] [Google Scholar]
- 82.Wolf AM, Asoh S, Hiranuma H, Ohsawa I, Iio K, Satou A, Ishikura M, Ohta S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J Nutr Biochem. 2010;21:381–389. doi: 10.1016/j.jnutbio.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 83.Ferramosca A, Conte L, Zara V. A krill oil supplemented diet reduces the activities of the mitochondrial tricarboxylate carrier and of the cytosolic lipogenic enzymes in rats. J Anim Physiol Anim Nutr (Berl) 2012;96:295–306. doi: 10.1111/j.1439-0396.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 84.Jia Y, Wu C, Kim J, Kim B, Lee SJ. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J Nutr Biochem. 2016;28:9–18. doi: 10.1016/j.jnutbio.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 85.Ha AW, Kim WK. The effect of fucoxanthin rich power on the lipid metabolism in rats with a high fat diet. Nutr Res Pract. 2013;7:287–293. doi: 10.4162/nrp.2013.7.4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gammone MA, D’Orazio N. Anti-obesity activity of the marine carotenoid fucoxanthin. Mar Drugs. 2015;13:2196–2214. doi: 10.3390/md13042196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen YJ, Wallig MA, Jeffery EH. Dietary Broccoli Lessens Development of Fatty Liver and Liver Cancer in Mice Given Diethylnitrosamine and Fed a Western or Control Diet. J Nutr. 2016;146:542–550. doi: 10.3945/jn.115.228148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.James D, Devaraj S, Bellur P, Lakkanna S, Vicini J, Boddupalli S. Novel concepts of broccoli sulforaphanes and disease: induction of phase II antioxidant and detoxification enzymes by enhanced-glucoraphanin broccoli. Nutr Rev. 2012;70:654–665. doi: 10.1111/j.1753-4887.2012.00532.x. [DOI] [PubMed] [Google Scholar]
- 89.Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 2007;64:1105–1127. doi: 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y, Tang L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol Sin. 2007;28:1343–1354. doi: 10.1111/j.1745-7254.2007.00679.x. [DOI] [PubMed] [Google Scholar]
- 91.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52 Suppl 1:S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 92.Kikuchi M, Ushida Y, Shiozawa H, Umeda R, Tsuruya K, Aoki Y, Suganuma H, Nishizaki Y. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J Gastroenterol. 2015;21:12457–12467. doi: 10.3748/wjg.v21.i43.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoshida K, Ushida Y, Ishijima T, Suganuma H, Inakuma T, Yajima N, Abe K, Nakai Y. Broccoli sprout extract induces detoxification-related gene expression and attenuates acute liver injury. World J Gastroenterol. 2015;21:10091–10103. doi: 10.3748/wjg.v21.i35.10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Armah CN, Traka MH, Dainty JR, Defernez M, Janssens A, Leung W, Doleman JF, Potter JF, Mithen RF. A diet rich in high-glucoraphanin broccoli interacts with genotype to reduce discordance in plasma metabolite profiles by modulating mitochondrial function. Am J Clin Nutr. 2013;98:712–722. doi: 10.3945/ajcn.113.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai K, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. 2012;3:758. doi: 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kushad MM, Brown AF, Kurilich AC, Juvik JA, Klein BP, Wallig MA, Jeffery EH. Variation of glucosinolates in vegetable crops of Brassica oleracea. J Agric Food Chem. 1999;47:1541–1548. doi: 10.1021/jf980985s. [DOI] [PubMed] [Google Scholar]
- 97.Jie M, Cheung WM, Yu V, Zhou Y, Tong PH, Ho JW. Anti-proliferative activities of sinigrin on carcinogen-induced hepatotoxicity in rats. PLoS One. 2014;9:e110145. doi: 10.1371/journal.pone.0110145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahn J, Lee H, Im SW, Jung CH, Ha TY. Allyl isothiocyanate ameliorates insulin resistance through the regulation of mitochondrial function. J Nutr Biochem. 2014;25:1026–1034. doi: 10.1016/j.jnutbio.2014.05.006. [DOI] [PubMed] [Google Scholar]