

Abstract
AIM
To improve anti-inflammatory activity while reducing drug doses, we developed a nanoformulation carrying dexamethasone and butyrate.
METHODS
Dexamethasone cholesteryl butyrate-solid lipid nanoparticles (DxCb-SLN) were obtained with the warm microemulsion method. The anti-inflammatory activity of this novel nanoformulation has been investigated in vitro (cell adhesion to human vascular endothelial cells and pro-inflammatory cytokine release by lipopolysaccharide-induced polymorphonuclear cells) and in vivo (disease activity index and cytokine plasma concentrations in a dextran sulfate sodium-induced mouse colitis) models. Each drug was also administered separately to compare its effects with those induced by their co-administration in SLN at the same concentrations.
RESULTS
DxCb-SLN at the lowest concentration tested (Dx 2.5 nmol/L and Cb 0.1 μmol/L) were able to exert a more than additive effect compared to the sum of the individual effects of each drug, inducing a significant in vitro inhibition of cell adhesion and a significant decrease of pro-inflammatory cytokine (IL-1β and TNF-α) in both in vitro and in vivo models. Notably, only the DxCb nanoformulation administration was able to achieve a significant cytokine decrease compared to the cytokine plasma concentration of the untreated mice with dextran sulfate sodium-induced colitis. Specifically, DxCb-SLN induced a IL-1β plasma concentration of 61.77% ± 3.19%, whereas Dx or Cb used separately induced a concentration of 90.0% ± 2.8% and 91.40% ± 7.5%, respectively; DxCb-SLN induced a TNF-α plasma concentration of 30.8% ± 8.9%, whereas Dx or Cb used separately induced ones of 99.5% ± 4.9% and 71.1% ± 10.9%, respectively.
CONCLUSION
Our results indicate that the co-administration of dexamethasone and butyrate by nanoparticles may be beneficial for inflammatory bowel disease treatment.
Keywords: Nanoparticles, Dexamethasone, Butyrate, Inflammatory bowel disease, Drug delivery systems
Core tip: The oral treatment with dexamethasone and butyrate co-loaded into nanoparticles was effective in achieving strong anti-inflammatory effects at doses significantly lower than those required for each single drug. This nanoformulation may open a new window on the treatment of chronic inflammatory conditions such as inflammatory bowel disease, where dose- and time-dependent side effects can limit the drug’s therapeutic usefulness. Notably, dexamethasone cholesteryl butyrate-solid lipid nanoparticles significantly relieved and repaired colon inflammation in a colitis mouse model thanks to the nanoformulation, which displayed an additive synergism among the corticosteroid, dexamethasone, and the short-chain fatty acid, butyrate.
INTRODUCTION
Inflammation is a physiological process that involves different cells, such as leukocytes and endothelial cells that establish adhesive interactions in order to transverse the vascular wall and migrate to the damaged tissue. Inflammatory bowel diseases (IBDs), including ulcerative colitis and Crohn’s disease, are comprised of chronic and deregulated inflammation of the intestinal mucosa characterized by active inflammation, tissue destruction and repeated attempts at tissue repair that lead to a waxing-waning course. This persistent inflammation is triggered by neutrophil and macrophage infiltration, with activated macrophages producing a potent mixture of broadly active inflammatory cytokines, including interleukin (IL)-1β and tumor necrosis factor (TNF)-α[1,2]. In response to such pro-inflammatory cytokines, endothelial cells undergo inflammatory activation, resulting in an increased surface expression of cell adhesion molecules (CAMs), such as ICAM-1, VCAM-1 and E-selectin[3]. These endothelial CAMs play a fundamental role in leukocyte recruitment from the blood for tissue infiltration. Chronic induction of these CAMs leads to abnormal leukocyte recruitment, like that observed in chronic inflammatory diseases characterized by profound tissue remodeling and loss of function[4].
Recently, the traditional therapeutic approach of IBD, with the introduction of biologic agents, has moved away from non-specific immunomodulators, including corticosteroids, thiopurines, and methotrexate toward a pathway-based anti-inflammatory approach. Even though the introduction of TNF inhibitors such as infliximab, as well as anti-integrins, has initiated a new therapeutic era, these biologics are clinically effective only in a subgroup of IBD patients[5,6]. Therefore, IBD treatment is still a difficult challenge, and efforts to facilitate effective drug treatment are still necessary. Corticosteroids exert their anti-inflammatory and immunosuppressive effects by reducing the expression of cytokines and adhesion molecules, inhibiting leukocyte traffic and access to the inflammation site. In particular, dexamethasone (Dx) has been used for decades in the treatment of IBD flares, even if a such life-long treatment might produce several adverse reactions that are mostly time- and dose-dependent, limiting its clinical usefulness[1,7,8]. Hence, attempts to maintain the IBD therapeutic effects of corticosteroids while minimizing their systemic side effects might provide a major therapeutic improvement.
With regard to corticosteroids and pharmaceutical technology, to date only the novel oral formulation of budesonide using multi-matrix (MMX) drug delivery technology has been introduced as a treatment option for patients with ulcerative colitis, allowing a wider colonic targeting with low systemic bioavailability. The MMX strategy is an extension of the pH-responsive polymer technique that allows the sustained release of a drug enclosed within a gastro-resistant, pH-dependent coating[9]. However, it seems likely that all such systems relying on pH-responsive polymers will not be truly colon site-specific[10].
Recent advances in nanotechnology have enabled the development of new corticosteroid formulations with a nanometric approach to ameliorate pharmacological properties, resulting in increased efficacy and reduction of side effects[11]. Different from the MMX strategy, the nanoparticle drug delivery strategy relies on the nanosize as the cardinal property for interaction with biological systems. Indeed, the nanosize determines the ability to penetrate cell membranes, thus facilitating the passage across biological barriers, interaction with the immune system, uptake, absorption and distribution[12]. For instance, the size of orally assumed nanoparticles may somehow determine their fate, addressing the kind of cell with which to interact (i.e., epithelial or phagocytic cells), or the depth level in the intestinal mucosa. Moreover, nanoparticles can directly enter into phagocytic cells populating the inflamed tissue, thus providing a wider distribution and an additional mechanism for drug targeting[13].
Furthermore, the potential inhibitory effect of nanoparticle formulations of Dx on cyclooxygenase-2 (COX-2) expression is interesting. Although the physiological activity of COX-2 may provide a benefit to the organism, its aberrant expression has been implicated in the pathogenesis of many diseases, such as chronic inflammation and carcinogenesis[14]. Moreover, the effective inhibitory activity displayed by nanoparticle formulations of Dx on CAM expression by “inflamed” endothelial cells may be beneficial in blunting detrimental inflammatory reactions[15,16]. In particular, the incorporation of Dx into solid lipid nanoparticles (SLN) showed a significant improvement of its anti-inflammatory activity in a human IBD whole-blood model. SLN loaded with Dx exerted earlier anti-inflammatory effects and at lower doses than free Dx, highlighting how this nanoparticle formulation may be of therapeutic interest[17]. It is well-known that nanoparticles are efficiently taken up by immunocompetent cells, so that nanoparticulate drug carriers may be useful in targeting the inflamed regions. Indeed, in the presence of IBD there is a strong cellular immunoresponse from the inflamed regions, and the nanoparticle passive targeting may allow for the accumulation of the drug loaded into the nanoparticulate carrier in the inflamed area[11].
Furthermore, our group investigated whether the association between Dx and another anti-inflammatory agent such as butyrate might be of therapeutic interest in IBD. Butyrate is a short-chain fatty acid (SCFA) normally released by intestinal epithelial cells, which exhibit several physiological and immunological functions[18]. Like other SCFA, such as acetate and propionate, butyrate has regulatory effects on the proliferation, differentiation, gene expression and immune regulation of colon epithelial and immune cells. In particular, in experimental models, butyrate has been demonstrated to stimulate mucus production by colon epithelial cells, to inhibit colon inflammation and oxidative stress, and to improve the colon defense barriers, inhibiting colon carcinogenesis as well[19-21]. Butyrate has emerged as a modulator of adaptive responses, owing to its multiple biofunctions, i.e., restoring transforming growth factor-β (TGF-β) and IL-10 production in the colonic mucosa, inducing T cell apoptosis and dampening interferon-γ (IFN-γ) secretion[22,23]. Clinical trials have shown the effectiveness of butyrate monotherapy and/or in combination with conventional treatment in patients with diversion colitis, acute radiation proctitis, as well as ulcerative colitis[24-27]. In this regard, in the 1990s non-controlled pilot clinical trials using oral administration or enemas of butyrate yielded promising results in ulcerative colitis patients[28]. However, extended confirmatory studies have not yet been performed. On the other hand, in a randomized, double-blind, placebo-controlled study on ulcerative colitis patients, the combined treatment of oral sodium butyrate tablets in combination with mesalazine significantly decreased the disease activity index score and improved disease outcomes with respect to mesalazine alone[26].
Therefore, owing to partial patient compliance or restricted indications, these treatments were not established as a standard of care. Recent studies have renewed the expectations in regard to strategies related to intestinal SCFA. The administration of probiotic bacteria with the capacity to produce butyrate has been shown to improve the symptoms in IBD models in vivo[29]. Moreover, the treatment with butyrate has been shown to increase apoptosis and differentiation, and to inhibit proliferation in colon, breast, gastric, lung, brain and pancreas cancer cells[30,31]. Butyrate is characterized by a short half-life, due to its rapid metabolism and excretion through the liver. Therefore, continuous administration of the drug is required in order to maintain therapeutic concentrations[32]. In addition, the use of butyrate in therapy is limited by its dose-dependent side effects, such as anemia, headache, nausea, diarrhea and abdominal cramps.
In order to overcome these limitations, SLN have been proposed for improving butyrate therapy, in that they constitute a drug delivery system able to ensure high drug loading, enhanced drug pharmacokinetic profile, good biocompatibility and scale-up feasibility[33-35]. The use of SLN has been under investigation in various preclinical and clinical trials, especially in cancer therapy, and their employment has been approved for clinical use in some cases[36]. Cholesteryl butyrate (Cb) as a butyrate SLN formulation has been evaluated in several in vitro and in vivo studies as an anticancer agent[37-41] and only in in vitro studies as anti-inflammatory agent[17,42].
Thus, our group sought to develop a new SLN formulation carrying dexamethasone and cholesteryl butyrate (DxCb) and investigated the efficacy of this strategy in strengthening the effect of each single drug in the treatment of inflammation. Specifically, investigations of this new anti-inflammatory SLN formulation were carried out in the following IBD models: (1) in vitro, evaluating the effects on cell adhesion to human vascular endothelial cells and on pro-inflammatory cytokine release by lipopolysaccharide (LPS)-induced polymorphonuclear cells; and (2) in vivo, evaluating the effects in dextran sulfate sodium-induced mouse colitis.
MATERIALS AND METHODS
Preparation and characterization of DxCb-SLN
Cb and DxCb-SLN were obtained with the warm microemulsion method (patent WO0030620). This process is based on mixing, in precise ratio, the melted lipid matrix loaded with hydrophobic drug with water phase (maintained at the same melting temperature as the lipid matrix) which contains surfactants, mainly phospholipids, and other co-surfactants, like SCFA, bile salts or short-chain fatty alcohols. When a clear warm microemulsion is obtained, it is dispersed in cold water (2 °C) to generate nanoparticles by solidifying the lipid matrix. The SLN dispersion obtained is then washed by tangential flow filtration (cut-off 30-100 kDa) to remove components and drug not incorporated into SLN, and the final product can then be filtered at 0.2 μm for sterility or can be subjected to freeze drying.
The Cb was prepared from cholesteryl butyrate (Asia Talent Chemical, Shenzen China), Epikuron 200 (Cargill, Milano, Italy) and sodium glycocholate (PCA, Basaluzzo, Italy). In this formulation, cholesteryl butyrate lipid matrix acts as a prodrug of butyrate. For preparation of the DxCb-SLN, Dx 21-acetate (hereafter referred to as Dx; Sigma-Aldrich, Milano, Italy) was previously added and dissolved into melted cholesteryl butyrate matrix before adding other excipients as by the preparation protocol of Cb. The full compositions of warm microemulsions used to prepare Cb and DxCb-SLN are reported in Table 1. The temperature of these warm microemulsions was 85 °C for both. After clear microemulsions had been obtained, they were dispersed in cold water (2 °C) under stirring, at a 1:5 volume ratio. The dispersions obtained were then washed by tangential flow filtration (Vivaflow50 membrane with cut-off of 100 kDa; Sartorius Stedim Biotech GmbH, Goettingen, Germany) by adding and removing the same volume of water 4 times (4 washings); the final concentrations of the main components are reported in Table 2. In both formulations, 2-phenylethanol was added to aid in microemulsion formation. In particular, it works mainly to reduce viscosity and further helps in the formation of an interface between the oil phase and the lipid phase. Due to the multiple washings applied to purify the final products - four washings in this case - the concentration of 2-phenylethanol was strongly reduced in the final dispersion, where it finally acted as a preservative. Dx (water:ethanol 9:1, 1 mmol/L) was also prepared as a free drug reference.
Table 1.
Composition of warm microemulsion for cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles formulations
| Molar composition of warm microemulsion | Cb, mmol/L | DxCb-SLN, mmol/L |
| Dexamethasone 21-acetate | - | 8.1 |
| Cholesteryl butyrate | 273.7 | 273.7 |
| Epikuron™ 200 (purified phosphatidylcholine 92%) | 335.5 | 335.5 |
| Sodium glycocholate | 194.8 | 194.8 |
Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles.
Table 2.
Concentration of main components of water dispersion of cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles formulations after 4 washing steps (HPLC method determination)
| Molar composition of final dispersion after washing | Cb, mmol/L | DxCb-SLN, mmol/L |
| Dexamethasone 21-acetate | - | 1.1 |
| Cholesteryl butyrate | 36.0 | 38.5 |
| Phosphatidylcholine | 48.4 | 49.2 |
| Sodium glycocholate | 12.5 | 11.2 |
Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles.
Physical characterization was performed by dynamic light scattering (DLS) (Malvern Zetasizer - Nano ZS; Malvern Instruments, Malvern, United Kingdom). The data are reported in Table 3. Finally, electron microscopy analysis by ZEISS Supra 40 Field Emission Scanning Electron Microscopy confirmed the regular shape and nanosize of the particles (Figure 1) (courtesy of Prof. Pirri, Laboratory FESEM Microscopy, DISAT, Politecnico of Torino).
Table 3.
Average value of hydrodynamic diameter (Zave) and polidispersity index of cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles formulations
| Physical characterization DLS analysis | Cb | DxCb-SLN |
| Zave in nm | 79.6 | 72.9 |
| Polydispersity index | 0.25 | 0.28 |
Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles.
Figure 1.
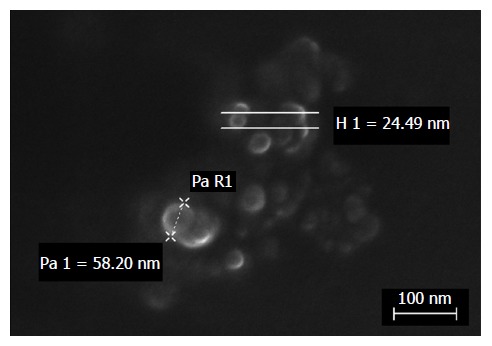
FeSEM micrograph of dexamethasone cholesteryl butyrate-solid lipid nanoparticles. Electron microscopy analysis showed regular shape and nanosize of particles by height (H) or radius (R) measurements.
Cell lines
Leukemic human T cells (Jurkat, clone E6-1) were obtained from American Type Culture Collection (ATCC) (Manassas, VA, United States), and were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) (v/v), 2 mmol/L L-glutamine and antibiotics (100 U/mL streptomycin and 200 U/mL penicillin) (Sigma-Aldrich).
Human vascular endothelial cells (HUVECs) were isolated from human umbilical veins from healthy parturients aged between 18-35 years undergoing a natural birth (informed consent was obtained from all donors). The umbilical cord was collected at birth and stored at 4 °C until the isolation procedure by trypsin treatment (1%). HUVECs were cultured in M199 medium with the addition of 20% FCS (v/v) and 100 U/mL penicillin, 100 mg/mL streptomycin, 5 UI/mL heparin, 12 mg/mL bovine brain extract and 200 mmol/L glutamine (Sigma-Aldrich). HUVECs were grown to confluence in flasks and used between the second and fifth passages; HUVEC viability was not affected by the drug treatment.
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized peripheral rat blood samples by density-gradient centrifugation over Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden) according to the method of Liu et al[43] (the study was approved by the Ethics Committee of the University of Torino). PBMCs were cultured in RPMI 1640 medium supplemented with 10% FCS (v/v), 2 mmol/L L-glutamine and antibiotics (100 U/mL streptomycin and 200 U/mL penicillin). All the cell lines were cultured at 37 °C in a humidified 5% CO2-95% air incubator.
In vitro cell adhesion assay
HUVECs were grown to confluence in 24-well culture plates, washed and rested for 1 d in M199 medium plus 10% FCS (v/v). Cells were pre-activated with IL-1β (0.01 μmol/L) for 1 h and then exposed or not exposed to increasing concentrations of Dx (2.5, 25 and 250 nmol/L), Cb (0.1, 1 and 10 μmol/L) and DxCb-SLN (2.5 nmol/L:0.1 μmol/L, 25 nmol/L:1 μmol/L and 250 nmol/L:10 μmol/L) for 24 h, washed with fresh medium twice and incubated for 1 h with Jurkat cells (1 × 105 per well). The 1 h incubation time was chosen to allow full sedimentation of the adhering cells, but similar results were obtained with shorter incubation times (10 and 20 min). After incubation, non-adherent cells were removed by being washed three times with M199 medium. The center of each well was analyzed by fluorescence imaging[42]. Adherent cells were counted by the ImagePro Plus Software for micro-imaging (version 5.0; Media Cybernetics, Bethesda, MD, United States). Single experimental points were assayed in triplicate, and the standard error of three replicates was always below 10%. Data are shown as the percentage of inhibition of treated cells vs the control adhesion measured on untreated cells (control adhesion was 65 ± 5 cells per microscope field; n = 5).
In vitro PBMC assay
PBMC viability was assayed by trypan blue dye exclusion, and 5 × 105/mL viable cells were cultured in 24-well culture plates with culture medium containing 1 μg/mL LPS (Sigma-Aldrich) for 24 h. PBMCs were then incubated with increasing concentration of Dx (2.5, 25 and 250 nmol/L), Cb (0.1, 1 and 10 μmol/L) and DxCb-SLN (2.5 nmol/L:0.1 μmol/L, 25 nmol/L:1 μmol/L and 250 nmol/L:10 μmol/L) for 24 h. In order to exclude the possibility that the drugs might affect cell viability, 24 h after drug incubation a trypan blue dye exclusion assay was performed for each condition.
The IL-1β and TNF-α protein concentrations in culture supernatants of PBMCs were determined at 24 h incubation by specific enzyme-linked immunosorbent assay (ELISA) (eBioscience, Thermo Fisher Scientific, Milano, Italy) according to the manufacturer’s instructions. Data are shown as the percentage of the cytokine secretion of LPS-treated PBMCs after each drug treatment vs the cytokine secretion of control cells, i.e., LPS-stimulated PBMCs.
Animals
Male, 8 wk-old BALB/c mice, with an average weight of 18 g, were obtained from Charles River (Milano, Italy). The mice were housed in a specific pathogen-free environment, and a 12 h light/dark cycle was maintained. The mice had access to water and rodent laboratory chow ad libitum; the weights of the mice as well as diarrhea were recorded daily. The procedures for the care and handling of the animals used in the study were approved by the local “Animal Use and Care Committee” (protocol number 12201), and they were in accordance with the European Directive 2010/63/EU on the protection of animals used for scientific purposes.
In vivo model of colitis
Colitis was induced in mice by adding 4% (w/v) dextran sulfate sodium salt (DSS, molecular weight 40000) (Sigma-Aldrich) to the drinking water and allowing ad libitum access, starting from day 0 for 5 d. Groups of mice (at least 5 mice per group) were then orally treated (by gavage) daily with Dx (0.0001 mg/g bw), Cb (0.004 mg/g bw) or DxCb-SLN (0.0001 mg/g bw:0.004 mg/g bw) starting from day 6 for 3 d. Moreover, in a group in which colitis was induced, as a sham treatment mice were administered orally with sterile phosphate-buffered saline solution (150 μL/mouse per day) starting from day 6 for 3 d (DSS group), whereas in another group colitis was not induced (control group). All groups were sacrificed on day 10. There were at least 5 mice per group, and two separate experiments were carried out. There was no significant difference in the water consumption and food intake of each group during all experimental periods.
Assessment of in vivo inflammation
The mice were weighed and inspected for diarrhea and rectal bleeding every day. The disease activity index (DAI) (i.e., the combined score of weight loss and bleeding) was determined according to a standard scoring system, as previously described by Rachmilewitz et al[44]. Specifically, the scores were defined as follows: (1) bodyweight (bw) loss (0: no bw loss; 1: 5%-10% bw loss; 2: 10%-15% bw loss; 3: 15%-20% bw loss; 4: > 20% bw loss); (2) fecal occult blood (0: no blood; 2: positive; 4: gross blood); and (3) diarrhea (0: no diarrhea; 1: mild diarrhea, 2: severe diarrhea). All groups were sacrificed on day 10.
The IL-1β and TNF-α plasma concentrations were determined on day 9, i.e., 24 h after the different treatments by specific sandwich enzyme immunoassay (eBioscience, Thermo Fisher Scientific) according to the manufacturer’s instructions. Data are shown as the percentage of the cytokine secretion of DSS-treated mice after each drug treatment vs the cytokine secretion of DSS-treated mice.
Statistical analysis
Results are expressed throughout as mean ± SD of three independent experiments for in vitro studies and of two independent experiments for in vivo studies. Statistical analyses were performed on GraphPad Prism 6.0 software (La Jolla, CA, United States). The two-way or one-way analysis of variance and Bonferroni’s test were used to determine statistical significance in the different treatment groups. The statistical significance threshold was set at P < 0.05.
RESULTS
Effects of DxCb-SLN on in vitro cell adhesion
First, we analyzed the effect of DxCb-SLN on the adhesion of Jurkat cells, a widely used continuous model of human T lymphocytes, to HUVECs comparing it with the effect of the drug separately, i.e., Dx and Cb. In order to reproduce an inflammatory environment, we pre-activated HUVECs with 0.01 μmol/L IL-1β for 1 h. The treatment with IL-1β increased Jurkat adhesion by 180%, and this value was used as control. The concentration used for each drug had been found not to be toxic for HUVECs.
HUVECs were treated with increasing concentrations of each single drug, i.e., Dx (2.5, 25 and 250 nmol/L) and Cb (0.1, 1 and 10 μmol/L), and of the DxCb nanoformulation (DxCb-SLN with a Dx:Cb concentration of 2.5 nmol/L:0.1 μmol/L, 25 nmol/L:1 μmol/L and 250 nmol/L:10 μmol/L) for 24 h, washed and used in the adhesion assay with Jurkat cells. Figure 2 shows that DxCb SLN inhibited cell adhesion to HUVEC in a concentration-dependent manner. A significant 43.1% ± 7.3% inhibition of cell adhesion was already determined at the lowest concentration tested of the DxCb nanoformulation (DxCb-SLN with a Dx:Cb concentration of 2.5 nmol/L M:0.1 μmol/L), reaching an 81.8% ± 11.7% inhibition of cell adhesion at the highest concentration tested (DxCb-SLN with a Dx:Cb concentration of 250 nmol/L:10 μmol/L). Considering the inhibition of cell adhesion determined by the single drugs, Dx produced a 4.2% ± 0.8% inhibition at the lowest concentration tested (2.5 nmol/L), reaching a 15.4% ± 0.9% inhibition at the highest concentration tested (250 nmol/L) and Cb determined a 14.9% ± 4.3% inhibition at the lowest concentration tested (0.1 μmol/L), reaching a 51.6% ± 7.8% inhibition at the highest concentration tested (10 μmol/L). Therefore, taking all the data together, the nanoformulation containing Dx 2.5 nmol/L and Cb 0.1 μmol/L was able to exert an inhibition of cell adhesion in a more than additive manner with respect to the sum of the individual effects of each drug if they had been used separately (Figure 2).
Figure 2.
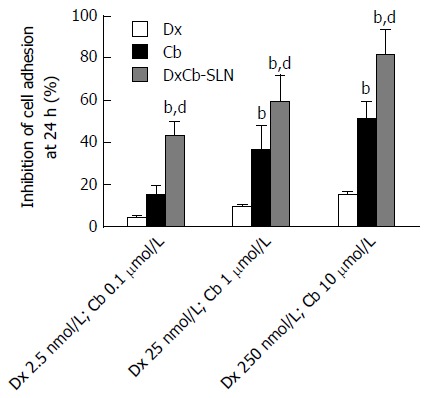
Effect of dexamethasone, cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles on human vascular endothelial cell adhesiveness to Jurkat cells. Human vascular endothelial cells were pre-activated with IL-1β (0.01 μmol/L) for 1 h and then exposed or not exposed to increasing concentrations of Dx (2.5, 25 and 250 nmol/L), Cb (0.1, 1 and 10 μmol/L) and DxCb-SLN (2.5 nmol/L:0.1 μmol/L, 25 nmol/L:1 μmol/L and 250 nmol/L:10 μmol/L) for 24 h and then incubated with Jurkat for 1 h. bP < 0.01, vs Dx; dP < 0.01, vs Cb. Dx: Dexamethasone; Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles.
Effects of DxCb-SLN on in vitro cytokine production
With respect to the effects on IL-1β production in PBMC culture supernatant, 24 h after the incubation a statistically significant (P < 0.05) higher decrease of IL-1β compared to the effect induced by each single drug was observed only with the nanoformulation containing the lowest concentrations tested (DxCb-SLN with a Dx:Cb concentration of 2.5 nmol/L:0.1 μmol/L; Figure 3A). Assuming as 100% the IL-1β production of untreated PBMCs, an IL-1β production of 74.3% ± 8.7% was observed with the nanoformulation, in contrast to a 98.7% ± 9.8% and a 89.1% ± 8.2% production with Dx (2.5 nmol/L) and with Cb (0.1 μmol/L), respectively. On increasing the concentrations, no significant differences on the IL-1β production were observed using either the DxCb nanoformulation or each single drug (Figure 3A).
Figure 3.
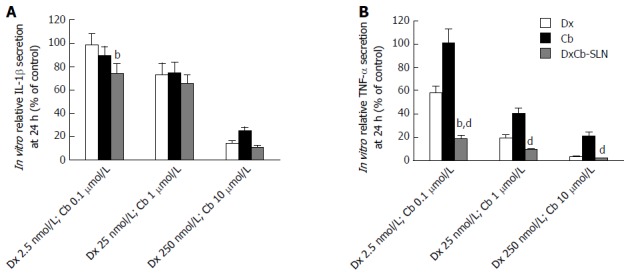
In vitro effect of dexamethasone, cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles on interleukin-1β and tumor necrosis factor-α secretion. Cells were treated with increasing concentrations of Dx (2.5, 25 and 250 nmol/L), Cb (0.1, 1 and 10 μmol/L) and DxCb-SLN (2.5 nmol/L:0.1 μmol/L, 25 nmol/L:1 μmol/L and 250 nmol/L:10 μmol/L) for 24 h. IL-1β (A) and TNF-α (B) secretion in culture supernatant of PBMCs stimulated with lipopolysaccharide (LPS; 1 μg/mL for 24 h) were analyzed by ELISA. bP < 0.01, vs Dx; dP < 0.01, vs Cb. Dx: Dexamethasone; Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles; IL: Interleukin; TNF: Tumor necrosis factor.
Regarding the effects on TNF-α production in the PBMC culture supernatant, 24 h after incubation a statistically significant (P < 0.001) higher decrease of TNF-α compared to the effect induced by single Dx was observed only with the nanoformulation at the lowest concentrations tested (DxCb-SLN with a Dx:Cb concentration of 2.5 nmol/L:0.1 μmol/L; Figure 3B). Assuming as 100% the TNF-α production of untreated PBMCs, a TNF-α production of 19.2% ± 2.8% was observed with the nanoformulation, compared to a 58.4% ± 5.3% and a 101.3% ± 11.3% production with Dx (2.5 nmol/L) and with Cb (0.1 μmol/L), respectively. On increasing the concentrations, no significant differences on TNF-α production were observed using either the DxCb nanoformulation or Dx single drug (Figure 3B). Therefore, in regard to all the data, the nanoformulation containing Dx 2.5 nmol/L and Cb 0.1 μmol/L was able to exert a strong decrease of TNF-α production in a more than additive manner with respect to the sum of the individual effects of each drug if they had been used separately (Figure 3B).
Effects of DxCb-SLN on in vivo mice colitis
In order to evaluate the effect of the DxCb nanoformulation on a mouse colitis model, the mice were divided into groups, and each was given drugs separately or as DxCb-SLN at the same concentrations. Specifically, doses of Dx (0.0001 mg/g bw), Cb (0.004 mg/g bw) or DxCb-SLN (0.0001 mg/g bw:0.004 mg/g bw) per day were administered orally from day 6 to day 8 after the colitis induction from day 0 to day 5. In addition, another group was composed of untreated mice (DSS group), and another of mice in which colitis was not induced (control group). Changes in mice bw were significantly different between the control group and the DSS group starting from day 7 and between groups treated with Dx or Cb and the control group starting from day 9 (Figure 4A). Instead, a slight but significant change in mice bw between DxCb-SLN treated-group and control group was recorded only at the last day of observation, i.e., day 10 (P < 0.05; Figure 4A). According to the DAI score determined for each treatment group, we observed that Dx alone was able to induce a significant decrease of the score compared to untreated mice (DSS group), with a 25% reduction of the disease symptoms (i.e., 6.0 vs 4.5, P < 0.05; Figure 4B). Notably, DxCb-SLN was able to induce a higher significant decrease of the disease score compared to untreated mice, with a 42% reduction of the disease symptoms (i.e., 6.0 vs 3.5, P < 0.01; Figure 4B).
Figure 4.
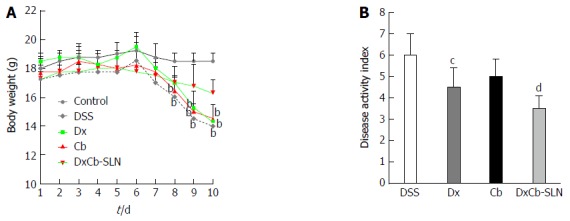
In vivo effect of dexamethasone, cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles on bodyweight and disease activity index. Animals received no treatment (control), DSS alone (DSS), or a combination of DSS and Dx (Dx, 0.0001 mg/g bw for 3 d), DSS and Cb (Cb, 0.004 mg/g bw for 3 d) and DSS and DxCb-SLN (DxCb-SLN, 0.0001 mg/g bw:0.004 mg/g bw for 3 d). After 7 d, DSS was replaced with a water cycle (ad libitum) for another 7 d. Body weight of the mice was recorded daily (A) and the disease activity rate at day 9 (B). bP < 0.01, vs control; cP < 0.05; dP < 0.01, vs DSS. Dx: Dexamethasone; Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles; DSS: Dextran sulfate sodium.
Considering the cytokine plasma concentration on day 9, i.e., 24 h after drug treatment, only the DxCb nanoformulation administration was able to achieve a significant cytokine decrease compared to the cytokine plasma concentration of the DSS group. Assuming as 100% the IL-1β or TNF-α production of mice with DSS-induced colitis on day 9, 24 h after the 3 d of oral treatments, only DxCb-SLN (0.0001 mg/g bw:0.004 mg/g bw) were able to induce a significant decrease (Figure 5). Specifically, DxCb-SLN induced a IL-1β plasma concentration of 61.77% ± 3.19%, whereas Dx or Cb used separately induced a concentration of 90.0% ± 2.8% and 91.40% ± 7.5%, respectively (Figure 5A); DxCb-SLN induced a TNF-α plasma concentration of 30.8% ± 8.9%, whereas Dx or Cb used separately induced ones of 99.5% ± 4.9% and 71.1% ± 10.9%, respectively (Figure 5B). Thus, DxCb-SLN significantly ameliorated DSS-induced colitis in the mice compared to the treatments with each drug separately, given that the observed anti-inflammatory effect was higher than what would be expected from a simple additive effect (Figures 4 and 5).
Figure 5.
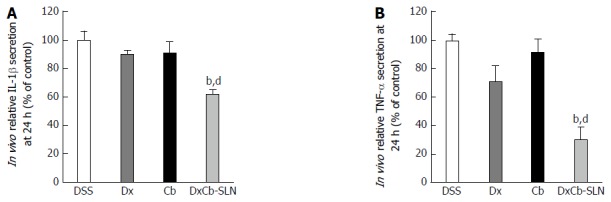
In vivo effect of dexamethasone, cholesteryl butyrate and dexamethasone cholesteryl butyrate-solid lipid nanoparticles on interleukin-1β and tumor necrosis factor-α secretion. Animals were treated with DSS alone (DSS), or a combination of DSS and Dx (Dx, 0.0001 mg/g bw for 3 d), DSS and Cb (Cb, 0.004 mg/g bw for 3 d) and DSS and DxCb-SLN (DxCb-SLN, 0.0001 mg/g bw:0.004 mg/g bw for 3 d). IL-1β and TNF-α secretion in mice plasma were analyzed by ELISA at day 9, 24 h after drug treatment. bP < 0.01, vs Dx; dP < 0.01, vs Cb. Dx: Dexamethasone; Cb: Cholesteryl butyrate; DxCb-SLN: Dexamethasone cholesteryl butyrate-solid lipid nanoparticles; DSS: Dextran sulfate sodium; IL: Interleukin; TNF: Tumor necrosis factor.
DISCUSSION
In a previous work, we observed that the incorporation of butyrate and Dx separately into SLN was effective in enhancing the anti-inflammatory activity of the drugs on PBMCs of IBD patients[17]. In the research presented herein, we observed that the combination therapy of Dx and butyrate co-loaded into an oral nanoformulation, namely DxCb-SLN, was effective in reducing the disease activity in a mouse model of DSS-induced colitis, as verified by its effect on macroscopic and biochemical parameters.
Before moving to an in vivo IBD model, we first tested DxCb-SLN on in vitro inflammation models. In a IL-1β-stimulated leukocyte-endothelial cell adhesion model, where the use of the cytokine allowed us to reproduce the initiation phase of IBD, the combination treatment with DxCb-SLN was able to significantly inhibit cell adhesion already at the lowest concentration tested, showing a significant inhibition at doses 10-fold lower than the dose required to achieve the same effects with each single drug. In a LPS-stimulated PBMC model, DxCb-SLN demonstrated a significant decrease of cytokine release that was higher for TNF-α rather than IL-1β secretion. Once again, the combination treatment was more effective at doses 10-fold lower than the dose required to achieve the same effects with the single drug treatment, i.e., 2.5 nmol/L:0.1 μmol/L Cb for DxCb-SLN with respect to 25 nmol/L for Dx and 1 μmol/L for Cb. We did not observe significant further decreases of both cytokine release at the highest concentration tested of DxCb-SLN because the free drugs, especially Dx, had a strong anti-inflammatory activity by themselves.
We then investigated this novel oral nanoformulation on a DSS-induced colitis in vivo model, which is one of the experimental models most frequently used in investigation of novel treatments for IBD[45]. Confirming the data observed in vitro, the in vivo pro-inflammatory cytokine release was significantly decreased by the DxCb-SLN oral administration, the decrease for TNF-α being more pronounced than IL-1β plasma concentration, 24 h after a daily treatment for 3 d. It is interesting that, on comparing the in vitro cytokine release at the lowest concentration of DxCb-SLN, we observed the same more pronounced decrease for TNF-α than IL-1β secretion.
This anti-inflammatory activity was consistent with the decreased DAI determined by the oral treatment with DxCb-SLN compared to the effects induced by the treatment with each drug separately. Notably, the bw loss induced by DSS was recovered significantly only after the oral treatment with DxCb-SLN. Therefore, thanks to this novel oral nanoformulation, the combination therapy of Dx and butyrate had better effects than any other single treatment, as specifically revealed by the significant decrease of plasma pro-inflammatory cytokines, i.e., IL-1β and TNF-α, and of the DAI. The efficacy of DxCb-SLN demonstrated in these in vitro and in vivo models may be explained by various mechanisms, such as particular abilities of nanoparticulate drug delivery systems and positive interaction mechanisms between Dx and butyrate.
However, further research is necessary to examine in depth the mechanism underpinning the enhanced anti-inflammatory effect determined by the simultaneous oral administration of Dx and butyrate as SLN formulation rather than as free drugs. For instance, a pharmacokinetic study comparing the simultaneous administration of the two drugs as free or loaded into the same SLN will be necessary to evaluate if the drug delivery system is effective in improving the bioavailability and inflamed tissue targeting. Also, molecular investigations will be necessary to evaluate differences in modulating inflammatory pathways by administering, at the same time, the two drugs as free or SLN formulation.
Thanks to pharmaceutical technology, we had the opportunity to develop an efficient drug delivery system able to improve the treatment of such a disease mediated by inflammation[11,46]. The use of a nanoparticulate drug carrier is useful to prevent early drug biological environmental degradation, to modulate drug pharmacokinetics, but also to enhance the treatment selectivity by targeting. Indeed, nanoparticles depending on their physico-chemical properties can preferentially accumulate in areas of intestinal inflammation when delivered orally[11,12]. They are particularly well-suited to the treatment of IBD through the local delivery of drugs to areas of inflammation, allowing site-specific delivery and minimizing side effects in other organs.
Targeting IBD sites is a challenging task to ensure the release of an intact and quantitatively clear amount of the administered drugs. Since drugs encounter a harmful environment after oral administration, high doses and/or frequent administration are usual to counter the degradation by stomach acidic pH or small intestine digestive enzymes; on the other hand, the occurrence of side effects are more likely[11,47]. In particular, SLN have been one of most studied carriers worldwide for drug delivery, since this nanosystem is mainly composed of solid lipid core and lecithin, has very low toxicity profile, good affinity for biological membrane, ability to facilitate up-taking/overcoming, and capacity to improve drug pharmacokinetics[48,49].
Therefore, the therapeutic potential of this novel anti-inflammatory drug nanoformulation in IBD is due to: (1) time protection of the loaded drug, especially for butyrate; (2) controlled release of the loaded drug, allowing a prolonged drug exposure; and (3) passive targeting of IBD sites, as a result of the abnormal permeability of inflamed colonic mucosa and the nanoparticle preferential uptake by immunocompetent cells.
Moreover, because some studies have reported the ability of butyrate to enhance the anti-inflammatory activity of corticosteroids or non-steroidal drugs[50,51], we decided to evaluate the effect of a combination therapy of Dx and butyrate. It is well-known that butyrate may play an important role in regulating intestinal inflammation[52,53]. As reported by Place et al[22], butyrate influences NF-κB activity by preventing the proteasome-dependent degradation of IκBα. This inhibition appears to arise from butyrate’s ability to inhibit histone deacetylase (HDAC)[54]. Specifically, the selective changes in gene expression induced by HDAC inhibitors, such as butyrate, arise from the enhanced acetylation of histone proteins and gene-regulatory transcription factors (e.g., p53, Sp1 and Sp3)[22].
NF-κB is a central mediator of the immune and inflammatory response and, upon activation, it rapidly enhances the expression of pro-inflammatory genes such as those encoding cytokines and cell adhesion molecules[55]. Dx effect on cytokine modulation in IBD is achieved through the translocation and activation of the glucocorticoid-receptor complex that can both bind to, and inactivate, key pro-inflammatory transcription factors, such as NF-κB[56]. Therefore, the greater effect observed in both in vitro and in vivo inflammation models by DxCb-SLN on TNF-α rather than IL-1β secretion might be modulated by a gene transcriptional regulation of NF-κB. Thus, according to our data we can speculate that an additive synergistic effect on NF-κB modulation due to the co-administration of Dx and butyrate might be responsible for the higher anti-inflammatory effect observed compared to the use of each drug separately, even if further molecular investigations are needed to confirm this hypothesis.
DxCb-SLN may provide a novel approach to treating IBD by taking advantage of a combination treatment achieved by co-loading Dx and butyrate into the same nanoparticle, which is able to exert a more than additive anti-inflammatory effect. Moreover, the pronounced anti-inflammatory activity of the DxCb-SLN oral treatment may be also due to passive targeting of the inflamed IBD sites, with the potential to reduce systemic side effects of each single drug in addition to the reduced amount of each drug required to achieve such an important anti-inflammatory activity.
ACKNOWLEDGMENTS
We are grateful to the Obstetrics and Gynecology Unit, Martini Hospital, Torino, Italy, for providing human umbilical cords.
COMMENTS
Background
Dexamethasone has been used for decades in the treatment of inflammatory bowel disease (IBD) flares, even if such a life-long treatment might produce several adverse reactions that are mostly time- and dose-dependent, limiting its clinical usefulness. Hence, attempts to maintain the IBD therapeutic effects of corticosteroids while minimizing their systemic side effects might provide a major therapeutic improvement.
Research frontiers
Nanotechnology can be used to improve the pharmacokinetic and pharmacodynamic properties of such a powerful drug. The authors have developed a nanoformulation carrying dexamethasone and the short-chain fatty acid, butyrate, to improve anti-inflammatory activity while reducing drug doses.
Innovations and breakthroughs
The authors developed a new solid lipid nanoparticle formulation carrying dexamethasone and butyrate highlighting the efficacy of this strategy in strengthening the effect of each single drug in the treatment of inflammation.
Applications
This nanoformulation may open a new window on the treatment of chronic inflammatory conditions such as inflammatory bowel disease, where dose- and time-dependent side effects can limit the drug’s therapeutic usefulness. Notably, dexamethasone cholesteryl butyrate-solid lipid nanoparticles significantly relieved and repaired colon inflammation in a colitis mouse model thanks to the nanoformulation, which displayed an additive synergism among the corticosteroid, dexamethasone, and the short-chain fatty acid, butyrate.
Terminology
Solid lipid nanoparticles are mainly composed of solid lipid core and lecithin, have very low toxicity profile, good affinity for biological membrane, ability to facilitate up-taking, and capacity to improve drug pharmacokinetics.
Peer-review
This is a very interesting article discussing a novel drug delivery using dexamethasone cholesteryl butyrate-solid lipid nanoparticles in in vitro and in vivo models.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Institutional Review Board of the University of Torino (Torino, Italy).
Conflict-of-interest statement: The authors declare that they have no competing interests.
Data sharing statement: The technical appendix, statistical code, and dataset are available from the corresponding author at email address loredana.serpe@unito.it.
Peer-review started: February 10, 2017
First decision: March 21, 2017
Article in press: May 4, 2017
P- Reviewer: Battat R, Ozturk E, Vidal S S- Editor: Ma YJ L- Editor: Filipodia E- Editor: Zhang FF
References
- 1.Beattie RM, Croft NM, Fell JM, Afzal NA, Heuschkel RB. Inflammatory bowel disease. Arch Dis Child. 2006;91:426–432. doi: 10.1136/adc.2005.080481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 3.Sans M, Panés J, Ardite E, Elizalde JI, Arce Y, Elena M, Palacín A, Fernández-Checa JC, Anderson DC, Lobb R, et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology. 1999;116:874–883. doi: 10.1016/s0016-5085(99)70070-3. [DOI] [PubMed] [Google Scholar]
- 4.Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S. New markers of inflammation and endothelial cell activation: Part I. Circulation. 2003;108:1917–1923. doi: 10.1161/01.CIR.0000089190.95415.9F. [DOI] [PubMed] [Google Scholar]
- 5.Coskun M, Vermeire S, Nielsen OH. Novel Targeted Therapies for Inflammatory Bowel Disease. Trends Pharmacol Sci. 2017;38:127–142. doi: 10.1016/j.tips.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 6.van Deventer SJ. The future of inflammatory bowel disease therapy. Inflamm Bowel Dis. 2002;8:301–305; discussion 306. doi: 10.1097/00054725-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ, Adcock I. Anti-inflammatory actions of steroids: molecular mechanisms. Trends Pharmacol Sci. 1993;14:436–441. doi: 10.1016/0165-6147(93)90184-l. [DOI] [PubMed] [Google Scholar]
- 8.Sands BE. Therapy of inflammatory bowel disease. Gastroenterology. 2000;118:S68–S82. doi: 10.1016/s0016-5085(00)70007-2. [DOI] [PubMed] [Google Scholar]
- 9.Hoy SM. Budesonide MMX(®): a review of its use in patients with mild to moderate ulcerative colitis. Drugs. 2015;75:879–886. doi: 10.1007/s40265-015-0396-8. [DOI] [PubMed] [Google Scholar]
- 10.Amidon S, Brown JE, Dave VS. Colon-targeted oral drug delivery systems: design trends and approaches. AAPS PharmSciTech. 2015;16:731–741. doi: 10.1208/s12249-015-0350-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serpe L, Canaparo R, Foglietta F, Zara GP. Innovative formulations for the controlled and site-specific delivery of antiinflammatory drugs. Curr Pharm Des. 2013;19:7219–7236. doi: 10.2174/138161281941131219124517. [DOI] [PubMed] [Google Scholar]
- 12.Viscido A, Capannolo A, Latella G, Caprilli R, Frieri G. Nanotechnology in the treatment of inflammatory bowel diseases. J Crohns Colitis. 2014;8:903–918. doi: 10.1016/j.crohns.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 13.Collnot EM, Ali H, Lehr CM. Nano- and microparticulate drug carriers for targeting of the inflamed intestinal mucosa. J Control Release. 2012;161:235–246. doi: 10.1016/j.jconrel.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi M, Mutoh M, Kawamori T, Sugimura T, Wakabayashi K. Altered expression of beta-catenin, inducible nitric oxide synthase and cyclooxygenase-2 in azoxymethane-induced rat colon carcinogenesis. Carcinogenesis. 2000;21:1319–1327. [PubMed] [Google Scholar]
- 15.d'Alessio P. Endothelium as a pharmacological target. Curr Opin Investig Drugs. 2001;2:1720–1724. [PubMed] [Google Scholar]
- 16.Meager A. Cytokine regulation of cellular adhesion molecule expression in inflammation. Cytokine Growth Factor Rev. 1999;10:27–39. doi: 10.1016/s1359-6101(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 17.Serpe L, Canaparo R, Daperno M, Sostegni R, Martinasso G, Muntoni E, Ippolito L, Vivenza N, Pera A, Eandi M, et al. Solid lipid nanoparticles as anti-inflammatory drug delivery system in a human inflammatory bowel disease whole-blood model. Eur J Pharm Sci. 2010;39:428–436. doi: 10.1016/j.ejps.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Scheppach W, Weiler F. The butyrate story: old wine in new bottles? Curr Opin Clin Nutr Metab Care. 2004;7:563–567. doi: 10.1097/00075197-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3:858–876. doi: 10.3390/nu3100858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Shimoyama T, Suzuki K, Umeda T, Nakaji S, Sugawara K. Effect of sodium butyrate on reactive oxygen species generation by human neutrophils. Scand J Gastroenterol. 2001;36:744–750. doi: 10.1080/003655201300192012. [DOI] [PubMed] [Google Scholar]
- 21.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 22.Place RF, Noonan EJ, Giardina C. HDAC inhibition prevents NF-kappa B activation by suppressing proteasome activity: down-regulation of proteasome subunit expression stabilizes I kappa B alpha. Biochem Pharmacol. 2005;70:394–406. doi: 10.1016/j.bcp.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 23.Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011;22:849–855. doi: 10.1016/j.jnutbio.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Breuer RI, Soergel KH, Lashner BA, Christ ML, Hanauer SB, Vanagunas A, Harig JM, Keshavarzian A, Robinson M, Sellin JH, et al. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: a randomised, placebo controlled trial. Gut. 1997;40:485–491. doi: 10.1136/gut.40.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheppach W. Treatment of distal ulcerative colitis with short-chain fatty acid enemas. A placebo-controlled trial. German-Austrian SCFA Study Group. Dig Dis Sci. 1996;41:2254–2259. doi: 10.1007/BF02071409. [DOI] [PubMed] [Google Scholar]
- 26.Vernia P, Marcheggiano A, Caprilli R, Frieri G, Corrao G, Valpiani D, Di Paolo MC, Paoluzi P, Torsoli A. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment Pharmacol Ther. 1995;9:309–313. doi: 10.1111/j.1365-2036.1995.tb00386.x. [DOI] [PubMed] [Google Scholar]
- 27.Hamer HM, Jonkers DM, Vanhoutvin SA, Troost FJ, Rijkers G, de Bruïne A, Bast A, Venema K, Brummer RJ. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin Nutr. 2010;29:738–744. doi: 10.1016/j.clnu.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Wan P, Chen H, Guo Y, Bai AP. Advances in treatment of ulcerative colitis with herbs: from bench to bedside. World J Gastroenterol. 2014;20:14099–14104. doi: 10.3748/wjg.v20.i39.14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eeckhaut V, Ducatelle R, Sas B, Vermeire S, Van Immerseel F. Progress towards butyrate-producing pharmabiotics: Butyricicoccus pullicaecorum capsule and efficacy in TNBS models in comparison with therapeutics. Gut. 2014;63:367. doi: 10.1136/gutjnl-2013-305293. [DOI] [PubMed] [Google Scholar]
- 30.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 31.Pajak B, Orzechowski A, Gajkowska B. Molecular basis of sodium butyrate-dependent proapoptotic activity in cancer cells. Adv Med Sci. 2007;52:83–88. [PubMed] [Google Scholar]
- 32.Daniel P, Brazier M, Cerutti I, Pieri F, Tardivel I, Desmet G, Baillet J, Chany C. Pharmacokinetic study of butyric acid administered in vivo as sodium and arginine butyrate salts. Clin Chim Acta. 1989;181:255–263. doi: 10.1016/0009-8981(89)90231-3. [DOI] [PubMed] [Google Scholar]
- 33.Brioschi A, Zara GP, Calderoni S, Gasco MR, Mauro A. Cholesterylbutyrate solid lipid nanoparticles as a butyric acid prodrug. Molecules. 2008;13:230–254. doi: 10.3390/molecules13020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellizzaro C, Coradini D, Morel S, Ugazio E, Gasco MR, Daidone MG. Cholesteryl butyrate in solid lipid nanospheres as an alternative approach for butyric acid delivery. Anticancer Res. 1999;19:3921–3925. [PubMed] [Google Scholar]
- 35.Ugazio E, Marengo E, Pellizzaro C, Coradini D, Peira E, Daidone MG, Gasco MR. The effect of formulation and concentration of cholesteryl butyrate solid lipid nanospheres (SLN) on NIH-H460 cell proliferation. Eur J Pharm Biopharm. 2001;52:197–202. doi: 10.1016/s0939-6411(01)00176-x. [DOI] [PubMed] [Google Scholar]
- 36.Manjunath K, Reddy JS, Venkateswarlu V. Solid lipid nanoparticles as drug delivery systems. Methods Find Exp Clin Pharmacol. 2005;27:127–144. doi: 10.1358/mf.2005.27.2.876286. [DOI] [PubMed] [Google Scholar]
- 37.Foglietta F, Serpe L, Canaparo R, Vivenza N, Riccio G, Imbalzano E, Gasco P, Zara GP. Modulation of butyrate anticancer activity by solid lipid nanoparticle delivery: an in vitro investigation on human breast cancer and leukemia cell lines. J Pharm Pharm Sci. 2014;17:231–247. doi: 10.18433/j3xp4r. [DOI] [PubMed] [Google Scholar]
- 38.Minelli R, Occhipinti S, Gigliotti CL, Barrera G, Gasco P, Conti L, Chiocchetti A, Zara GP, Fantozzi R, Giovarelli M, et al. Solid lipid nanoparticles of cholesteryl butyrate inhibit the proliferation of cancer cells in vitro and in vivo models. Br J Pharmacol. 2013;170:233–244. doi: 10.1111/bph.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minelli R, Serpe L, Pettazzoni P, Minero V, Barrera G, Gigliotti C, Mesturini R, Rosa AC, Gasco P, Vivenza N, et al. Cholesteryl butyrate solid lipid nanoparticles inhibit the adhesion and migration of colon cancer cells. Br J Pharmacol. 2012;166:587–601. doi: 10.1111/j.1476-5381.2011.01768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serpe L, Catalano MG, Cavalli R, Ugazio E, Bosco O, Canaparo R, Muntoni E, Frairia R, Gasco MR, Eandi M, et al. Cytotoxicity of anticancer drugs incorporated in solid lipid nanoparticles on HT-29 colorectal cancer cell line. Eur J Pharm Biopharm. 2004;58:673–680. doi: 10.1016/j.ejpb.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 41.Serpe L, Laurora S, Pizzimenti S, Ugazio E, Ponti R, Canaparo R, Briatore F, Barrera G, Gasco MR, Bernengo MG, et al. Cholesteryl butyrate solid lipid nanoparticles as a butyric acid pro-drug: effects on cell proliferation, cell-cycle distribution and c-myc expression in human leukemic cells. Anticancer Drugs. 2004;15:525–536. doi: 10.1097/01.cad.0000127329.83568.15. [DOI] [PubMed] [Google Scholar]
- 42.Dianzani C, Cavalli R, Zara GP, Gallicchio M, Lombardi G, Gasco MR, Panzanelli P, Fantozzi R. Cholesteryl butyrate solid lipid nanoparticles inhibit adhesion of human neutrophils to endothelial cells. Br J Pharmacol. 2006;148:648–656. doi: 10.1038/sj.bjp.0706761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu FC, Hoyt DB, Coimbra R, Junger WG. Proliferation assays with human, rabbit, rat, and mouse lymphocytes. In Vitro Cell Dev Biol Anim. 1996;32:520–523. doi: 10.1007/BF02722976. [DOI] [PubMed] [Google Scholar]
- 44.Rachmilewitz D, Karmeli F, Takabayashi K, Hayashi T, Leider-Trejo L, Lee J, Leoni LM, Raz E. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122:1428–1441. doi: 10.1053/gast.2002.32994. [DOI] [PubMed] [Google Scholar]
- 45.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takedatsu H, Mitsuyama K, Torimura T. Nanomedicine and drug delivery strategies for treatment of inflammatory bowel disease. World J Gastroenterol. 2015;21:11343–11352. doi: 10.3748/wjg.v21.i40.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakase H, Okazaki K, Tabata Y, Uose S, Ohana M, Uchida K, Matsushima Y, Kawanami C, Oshima C, Ikada Y, et al. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J Pharmacol Exp Ther. 2000;292:15–21. [PubMed] [Google Scholar]
- 48.Geszke-Moritz M, Moritz M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater Sci Eng C Mater Biol Appl. 2016;68:982–994. doi: 10.1016/j.msec.2016.05.119. [DOI] [PubMed] [Google Scholar]
- 49.Suchaoin W, Bernkop-Schnürch A. Nanocarriers protecting toward an intestinal pre-uptake metabolism. Nanomedicine (Lond) 2017;12:255–269. doi: 10.2217/nnm-2016-0331. [DOI] [PubMed] [Google Scholar]
- 50.Song M, Xia B, Li J. Effects of topical treatment of sodium butyrate and 5-aminosalicylic acid on expression of trefoil factor 3, interleukin 1beta, and nuclear factor kappaB in trinitrobenzene sulphonic acid induced colitis in rats. Postgrad Med J. 2006;82:130–135. doi: 10.1136/pgmj.2005.037945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guerron AD, Rawat R, Sali A, Spurney CF, Pistilli E, Cha HJ, Pandey GS, Gernapudi R, Francia D, Farajian V, et al. Functional and molecular effects of arginine butyrate and prednisone on muscle and heart in the mdx mouse model of Duchenne Muscular Dystrophy. PLoS One. 2010;5:e11220. doi: 10.1371/journal.pone.0011220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics. 2012;4:4. doi: 10.1186/1868-7083-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rieder F, Karrasch T, Ben-Horin S, Schirbel A, Ehehalt R, Wehkamp J, de Haar C, Velin D, Latella G, Scaldaferri F, et al. Results of the 2nd scientific workshop of the ECCO (III): basic mechanisms of intestinal healing. J Crohns Colitis. 2012;6:373–385. doi: 10.1016/j.crohns.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 54.Felice C, Lewis A, Armuzzi A, Lindsay JO, Silver A. Review article: selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;41:26–38. doi: 10.1111/apt.13008. [DOI] [PubMed] [Google Scholar]
- 55.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]