

Abstract
AIM
To explore the effect of miR-382 on esophageal squamous cell carcinoma (ESCC) in vitro and its possible molecular mechanism.
METHODS
Eca109 cells derived from human ESCC and Het-1A cells derived from human normal esophageal epithelium were used. Lentivirus-mediated miR-382 was overexpressed in Eca109 cells. The effect of miR-382 on cell proliferation was evaluated by MTT and colony formation assay. For cell cycle analysis, cells were fixed and stained for 30 min with propidium iodide (PI) staining buffer containing 10 mg/mL PI and 100 mg/mL RNase A, and analyzed by BD FACSCalibur™ flow cytometer. For cell apoptosis assay, cells were stained with an Annexin V-FITC/PI Apoptosis Detection Kit according to the manufacturer’s instructions and analyzed by a dual-laser flow cytometer. Cell invasion and migration abilities were determined through use of transwell chambers, non-coated or pre-coated with matrigel. Levels of proteins related to cell growth and migration were examined by western blotting.
RESULTS
Endogenous miR-382 was down-regulated in Eca109 cells compared with Het-1A. Introduction of miR-382 not only significantly inhibited proliferation and colony formation, but also arrested cell cycle at the G2/M phase, as well as promoted apoptosis and autophagy in Eca109 cells. Migration, invasion and epithelial-mesenchymal transition of Eca109 cells were suppressed by overexpressing miR-382. Western blotting results showed that miR-382 inhibited the phosphorylation of mTOR and 4E-BP1.
CONCLUSION
miR-382 functions as a tumor suppressor against ESCC development and metastasis, and could be considered as a potential drug source for the treatment of ESCC patients.
Keywords: miR-382, Esophageal squamous cell carcinoma, Tumor suppressor, Proliferation, Migration, Invasion
Core tip: Our previous study revealed that miR-382 was significantly down-regulated in esophageal squamous cell carcinoma (ESCC) patients with short-term motility, implying that miR-382 may display antitumor function in ESCC development and metastasis. We present here that miR-382 functions as a tumor suppressor by inhibiting proliferation, migration, invasion and epithelial-mesenchymal transition, in addition to inducing cell cycle arrest, apoptosis and autophagy in Eca109 cells. Inhibitory influence on protein translation mediated by mTOR/4E-BP1 signaling might be involved in the antitumor activity of miR-382 against ESCC. Thus, manipulation of miR-382 level can be a potentially therapeutic intervention for ESCC.
INTRODUCTION
MicroRNAs (miRNAs) are classes of small non-coding, 17-25 nucleotides long, RNAs that bind to the 3’-untranslated region of target genes to induce post-transcription suppression or translational repression[1]. Evidence has demonstrated that miRNAs as either oncogenes or tumor suppressors are frequently dysregulated in many types of cancers and linked with its diagnosis and prognosis, implying that miRNAs play a substantial role in the pathogenesis of human cancers[2,3].
Esophageal cancer (EC) is one of the most common human malignancies[4] and can be pathologically classified into two major types, called esophageal squamous cell carcinoma (ESCC) and adenocarcinoma[5]. It is especially prevalent among Asian populations, with the most common type (90%) of EC in Asian countries being ESCC[6]. Despite improvement in diagnosis and treatment, ESCC has become one of the major diseases that negatively impact overall health and quality of life, as manifested by its rising incidence and poor 5-year survival rate (19%)[7]. ESCC is a complicated disease related to chromosomal instability, epigenetic variability, and expression abnormalities of genes, including both coding and noncoding genes[3].
miRNA as a noncoding gene has been demonstrated as having an expression signature associated with ESCC development and progression[8,9]. In our previous report, miRNA profiling results showed that 46 miRNAs were differentially expressed in ESCC tissues vs non-tumorous esophageal tissues, with further research demonstrating that four of these miRNAs affect the direction of patient outcomes[10]. These results imply that altered expression of these miRNAs may be potential predictive biomarkers for both prognosis and treatment of ESCC.
MicroRNA-382 (miR-382) is a member of the metastatic signature found in our previous study. Recent studies have demonstrated that miR-382 is dysregulated in multiple types of cancer, including breast, osteosarcoma, colorectal and ovarian cancers[11-14]. We found that miR-382 was significantly down-regulated in ESCC patients with short-term motility. Accordingly, in conjunction with relevant literature, our results indicate that low levels of miR-382 may contribute to the development and metastasis of ESCC[15]. However, the possible roles and mechanisms of miR-382 in human ESCC are still not well established.
In the present study, we found that miR-382 expression in the ESCC cell line was lower than that of the normal esophageal epithelial cell line. We determined a functional role of miR-382 in ESCC tumor progression using the in vitro cell model by lentivirus-mediated miR-382 overexpression. We found that overexpression of miR-382 inhibited ESCC cell proliferation by promoting cell cycle arrest at the G2/M phase as well as at apoptosis. Moreover, we observed that overexpression of miR-382 suppressed ESCC cell migration and invasion via the mechanism associated with blocking the epithelial-mesenchymal transition (EMT) process. The mammalian target of rapamycin (mTOR)/translation repressor 4E binding protein 1 (4E-BP1) signaling pathway and autophagy process might be involved in the antitumor activity of miR-382 on ESCC cells. Our study provides the evidence that miR-382 functions as a tumor suppressor against the development and metastasis of ESCC.
MATERIALS AND METHODS
Reagents and antibodies
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), the Propidium Iodide (PI) Cell Cycle Assay Kit and the Annexin V-FITC/PI Apoptosis Detection Kit were purchased from Beyotime (Jiangsu, China). The All-in-One™ First-Strand cDNA Synthesis Kit, the All-in-One™ miRNA qRT-PCR Detection Kit and miRNA primers were purchased from Genecopoeia (Rockville, MD, United States). DMEM and fetal bovine serum were obtained from Thermo Fisher Scientific (Waltham, MA, United States). All primary antibodies including p21Cip1/Waf1, E-cadherin, β-catenin, vimentin and snail, mTOR, p-mTOR (Ser2448), p-4E-BP1 (Thr37/46), LC3 and β-actin were purchased from Cell Signaling Technologies (Danvers, MA, United States). All other common chemicals and buffers were from Boster (Wuhan, China).
Cell culture and lentivirus infection
Eca109 and Het-1A were obtained from Cobioer Biosciences (Nanjing, China). Both cell lines were cultured in DMEM medium containing 10% fetal bovine serum in a humidified atmosphere under 5% CO2 at 37 °C. Lentiviral vectors LV10-(U6/RFP & Puro) expressing a scrambled control (LV-Con) and mature miR-382 (MIMAT0000737, 5’GAAGUUGUUCGUGGUGGAUUCG3’, LV-miR-382) were generated by GenePharma (Shanghai, China). The virus infection was carried out according to GenePharma’s recommendations. Expression of mature miR-382 was confirmed by real-time reverse transcription (RT)-PCR.
RT and quantitative (q)PCR
Total RNA was isolated using TRIzol reagent from Ambion (Austin, TX, United States) according to the manufacturer’s protocol. The All-in-One™ First-Strand cDNA Synthesis Kit and the All-in-One™ miRNA qPCR Detection Kit were used for RT and qPCR respectively, and RT-qPCR was performed through Applied Biosystems QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA, United States). Expression of U6 was used to normalize the miR-382 level.
Cell proliferation and colony formation assay
MTT was used to measure cell proliferation. Eca109 cells (4 × 103 cells /well) were seeded in 96-well culture plates and incubated overnight at 37 °C in a humidified 5% CO2 incubator. At an indicated time, 10 μL of MTT dye was added to each well at a final concentration of 5 mg/mL. After 4 h incubation, the blue MTT formazan crystal was then dissolved in 100 μL/well of DMSO. The absorbance at 490 nm was measured on Multisken Spectrum (Thermo Fisher Scientific) microplate reader. The amount of viable cells after infection with LV-miR-382 and LV-Con was measured in triplicate. For the colony formation assay, cells were seeded into a 6-well plate with 1 × 103 cells/well. After 10 d of culturing, cells were washed twice with phosphate-buffered saline (PBS), then fixed with 4% paraformaldehyde for 15 min and stained with 0.5% crystal violet. The number of colonies was calculated by use of ImageJ software. The experiment was performed in triplicate.
Cell cycle and apoptosis analyses
Cells were seeded into a 35-mm dish with 1 × 105 cells/dish. After 72 h, harvested cells were washed with cold PBS for three times and fixed in 70% ethyl alcohol at 4 °C overnight. Cells were then treated with 10 μg/mL RNase and stained with 50 μg/mL PI for 30 min at room temperature in the dark. The cell cycle was then measured by BD FACSCalibur™ (BD Biosciences, San Jose, CA, United States) and the cell cycle distribution was analyzed by ModFit software (BD Biosciences). For the apoptosis analysis, cells were stained using the Annexin V-FITC/PI Apoptosis Detection Kit according to the manufacturer’s instructions. The apoptotic cells were detected by BD FACSCalibur™.
Transwell migration and invasion assay
A 24-well plate was used as a “feeder tray”. For the migration assay, 6.5-mm transwell chambers with 8 μm micro-pores (Corning Costar, Manassas, VA, United States) were used. For the invasion assay, the same filters were pre-coated with BD Matrigel (BD, Franklin Lakes, NJ, United States). A total of 60 μL of 1 mg/mL matrigel was briefly added on top of the filter into the upper chamber of each 24-well transwell and incubated at 37 °C for 8 h, followed by removal of the excess solution. In both migration and invasion tests, 200 μL of cell suspension (at 1 × 105 cells/mL) in serum-free medium was seeded on top of the filter, which was then immersed into the feeder tray and incubated at 37 °C in 600 μL of complete medium. After allowing the cells to migrate for 24 h, the non-migrated cells or non-invaded cells on the upper surface of each membrane were cleaned with a cotton swab. Cells adhering to the bottom surface of each membrane were fixed and stained with 0.1% crystal violet. Migrated or invaded cells in five different fields were counted through a phase contrast microscope. Both the migration assay and invasion assay were performed three times using triplicate wells.
Western blot analysis
Cell lysates were prepared with RIPA lysis buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.1% SDS, 1% NP40, 0.5% sodium deoxycholate, 1 mmol/L phenylmethylsulfonyl fluoride, 100 μmmol/L leupeptin, and 2 μg/mL aprotinin, pH 8.0). A total of 30 μg protein extract was subjected to sodium dodecyl sulfate-polyacrylamide gel and transferred onto nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, United States). After blocking with 5% nonfat dry milk, membranes were incubated at 4 °C overnight with each of the following primary antibodies: p21Cip1/Waf1, E-cadherin, β-catenin, vimentin, snail, mTOR, p-mTOR (Ser2448), p-4E-BP1 (Thr37/46), LC3 (all 1:1000 dilution) and β-actin. Membranes were then washed with PBS-Tween and incubated with horseradish peroxidase-conjugated secondary antibodies. After incubation, the membranes were washed three times with Tris-buffered saline-Tween and processed with the SuperSignal West Pico Chemiluminescent Substrate Detection Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Chemiluminescent detection of western blots was performed using the AmershamTM Imager 600 System (GE Healthcare Bio-Sciences, Pittsburgh, PA, United States).
Statistical analysis
Data was analyzed using Student’s t-test, and all data was expressed as the mean ± SD. P < 0.05 was considered statistically significant.
RESULTS
Endogenous miR-382 was down-regulated in Eca109 cells
We have previously shown that miR382 level was explicitly down-regulated in ESCC patients with poor outcome compared to those with good outcome. However, the functional role of miR-382 in human ESCC remains elusive. In this study, we sought to confirm the miR-382 down-regulation in the Eca109 cell line derived from human ESCC tissue compared to the Het-1A cell line derived from human normal esophageal epithelium. As shown in Figure 1A, endogenous miR-382 in Eca109 was 7.69-fold lower than that in Het-1A cells (P < 0.01). This result is consistent with our previous clinical finding. In order to further explore the function of miR-382, Eca109 cells were infected with lentivirus-mediated miR-382 (LV-miR-382). As indicated in Figure 1B, miR-382 levels of LV-miR-382 infected Eca109 cells were 24153.06-fold higher than that of LV-Con control cells. Therefore, we utilized this in vitro cell model to investigate the functional role of miR-382 in ESCC progression in the following experiments.
Figure 1.
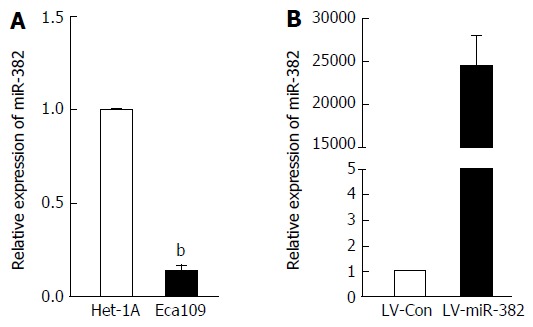
miR-382 was down-regulated in the esophageal squamous cell carcinoma cell line Eca109. A: The expression of miR-382 in Het-1A cells and Eca109 cells was examined by RT-qPCR; B: The relative expression level of miR-382 after introduction of either lentivirus-mediated miR-382 (LV-miR-382) or lentivirus-mediated scrambled control (LV-Con) into Eca109 cells was examined by RT-qPCR. The data in A and B is expressed as mean ± SD of three independent experiments. bP < 0.01, vs control. ESCC: Esophageal squamous cell carcinoma.
miR-382 inhibited Eca109 cell proliferation and colony formation
As the expression of endogenous miR-382 in Eca109 cells was down-regulated, we speculated that miR-382 might affect the proliferation rate of ESCC cells. Thus, we examined the effect of overexpression of miR-382 on the proliferation of Eca109 cells. As shown in Figure 2A, the cell proliferation rate of miR-382 overexpressed Eca109 cells was significantly inhibited when compared with that of the control cells at the 72-h culture time point (LV-miR-382 vs LV-Con, 312.01 ± 27.53 vs 176.76 ± 18.49, P < 0.01). Moreover, colony formation assay shown in Figure 2B and C showed that enforced expression of miR-382 resulted in a more than 69% decrease in colony numbers in miR-382 overexpressed Eca109 cells compared with control cells (LV-miR-382 vs LV-Con, 421.00 ± 41.24 vs 127.33 ± 27.46, P < 0.01). These results indicated that miR-382 inhibited Eca109 proliferation in vitro.
Figure 2.
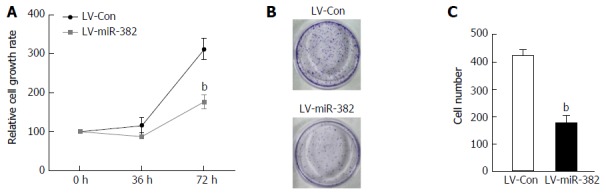
miR-382 inhibited Eca109 cell proliferation and colony formation. A: The proliferation of miR-382 overexpressed Eca109 cells and LV-Con control cells was measured by MTT. The Y-axis represents the relative number of viable cells presented as mean ± SD, bP < 0.01; B: Representative photomicrographs show the colony formation in cultured LV-Con Eca109 cells and LV-miR-382 Eca109; C: Data is presented as mean ± SD, bP < 0.01.
miR-382 induced cell cycle arrest at G2/M phase and apoptosis
To elucidate the possible mechanisms by which miR-382 inhibited the proliferation of ESCC cells, we studied the impact of miR-382 on Eca109 cell cycle progression and apoptosis. The cell cycle distribution analyzed by flow cytometry showed that the cell number at G2/M phase was increased in LV-miR-382 infected cells compared with LV-Con cells (LV-miR-382 vs LV-Con, 100% ± 10.14% vs 215% ± 16.60%, P < 0.05; Figure 3A-C). Western blot analysis was performed to determine the expression of p21Cip1/Waf1, which is an important negative cell cycle regulator. The result in Figure 3D clearly indicated that the expression level of p21Cip1/Waf1 was augmented by miR-382 overexpression. These data suggested that the miR-382-induced cell cycle arrest at G2/M phase in Eca109 cells may be mediated through p21Cip1/Waf1 down-regulation.
Figure 3.
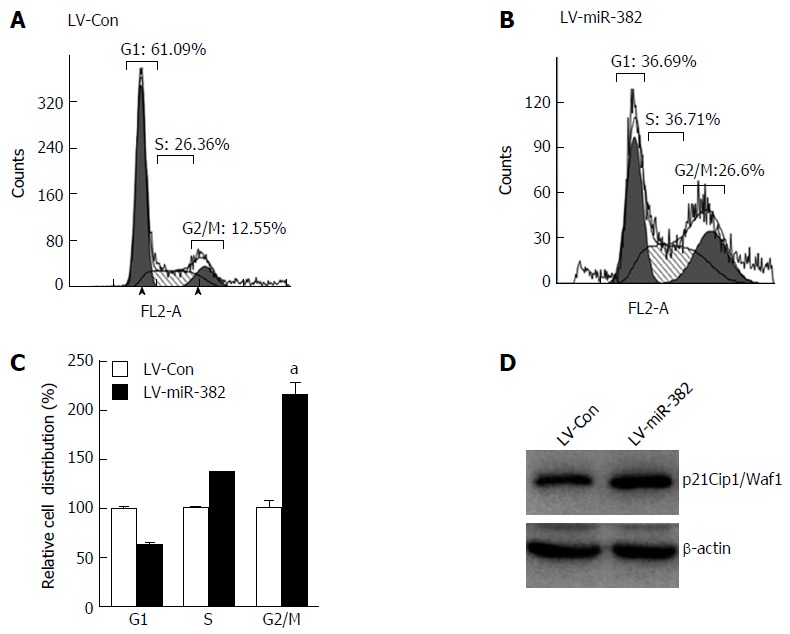
miR-382 induced cell cycle arrest at G2/M phase in Eca109 cells. A and B: Cell cycle distribution of miR-382 overexpressed Eca109 cells and control cells was analyzed by flow cytometry at post-culture 48 h; C: Relative cell distribution percentage of LV-miR-382 expressed Eca109 cells to LV-Con control cells in each cell cycle phase. The data presented is mean ± SD of three independent experiments. aP < 0.05, vs control; D: Protein expression level of p21Cip1/Waf1 was examined in LV-Con Eca109 cells and LV-miR-382 Eca109 cells. β-actin was as a loading control.
To investigate whether the anti-proliferative activity of miR-382 was related to an apoptotic effect, apoptosis was determined by flow cytometry after double staining with Annexin-V FITC/PI. As shown in Figure 4, either early or late apoptotic cell number of LV-miR-382 infected cells was clearly increased when compared to control cells (early: LV-miR-382 vs LV-Con, 1.00 ± 0.09 vs 5.50 ± 0.00, P < 0.05; late: LV-miR-382 vs LV-Con, 1.00 ± 0.72 vs 14.57 ± 1.84, P < 0.01). These results suggested that the miR-382 inhibitory effect of miR-382 on Eca109 proliferation may be related to increased G2/M cell cycle arrest and active apoptosis.
Figure 4.
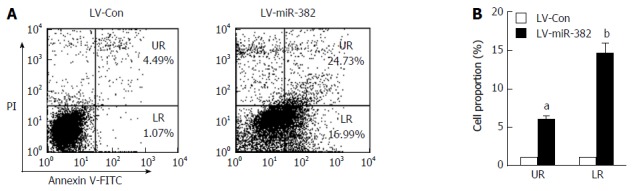
miR-382 promoted the apoptotic process in Eca109 cells. A: Apoptosis of LV-Con Eca109 cells and LV-miR-382 Eca109 cells was a negative cell cycle regulator, as measured using flow cytometry; B: Apoptotic cell values were expressed as mean ± SD of three experiments. aP < 0.05 and bP < 0.01, vs control.
miR-382 inhibited Eca109 cell migration and invasion
Cancer metastasis is greatly associated with cancer cell migratory and invasive abilities. To further investigate the influence of miR-382 over the migration and invasion of ESCC cells, migration transwell assay was initially performed to determine the migratory ability of the cells. As shown in Figure 5A and B, in the group infected with LV-miR-382 the rate of cell migration into the bottom chamber was significantly reduced by 32% compared with that of the scrambled control cells (LV-Con vs LV-miR-382, 433.50 ± 20.72 vs 292.25 ± 18.02, P < 0.05). Subsequently, matrigel invasion assay, a three-dimensional model, was utilized to investigate the inhibitory effect of miR-382 on the invasive potency of the cells. The result shown in Figure 5C and D demonstrates that miR-382 significantly suppressed invasion of Eca109 cells by 54% (LV-Con vs LV-miR-382, 603.80 ± 64.46 vs 273.80 ± 54.99, P < 0.01). These data indicated that miR-382 exerted an inhibitory effect on the migration and invasion of ESCC cells.
Figure 5.
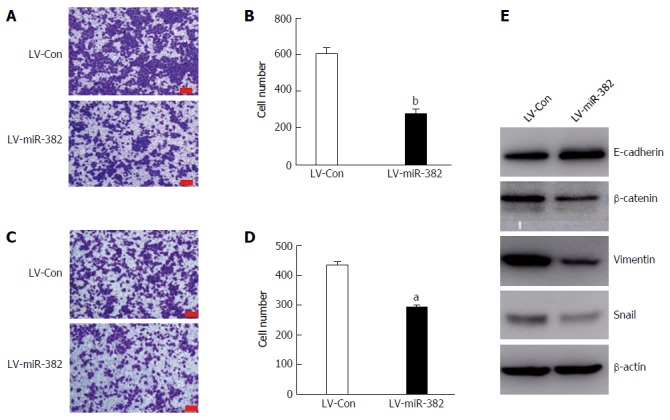
Overexpression miR-382 inhibited migration, invasion and epithelial-mesenchymal transition in Eca109 cells. A: Representative photomicrographs showed migrated cells stained with crystal violet after post-culture for 24 h in the transwell migration assay. The bars represent 200 μm; B: The migrated cells in five different fields were counted and the Y-axis represents the migrated cell number from three independent experiments. Data is presented as mean ± SD, bP < 0.01; C: Representative photomicrographs showing invaded cells stained with crystal violet after post-culture for 24 h in the transwell invasion assay. The bars are 200 μm; D: The invaded cells in five different fields were counted and the Y-axis represents the invaded cell number from three independent experiments. Data are presented as mean ± SD, aP < 0.05; E: Western blot analysis of the expression of epithelial marker E-cadherin, and mesenchymal markers β-catenin, vimentin and snail in Eca109 cells infected with LV-miR-382 or LV-Con(c). β-actin served as a loading control.
The role of miR-382 in regulating EMT was further studied by investigation of the protein expression of several key EMT effectors, including E-cadherin, β-catenin, vimentin and snail. Interestingly, miR-382 attenuated expressions of three mesenchymal markers (β-catenin, vimentin and snail). Moreover, we also observed that the expression of E-cadherin, which is one of the epithelial markers, was significantly augmented in LV-miR-382 cells, as shown in Figure 5E. Taken together, these results suggested that the inhibitory effect of miR-382 on the migration and invasion of ESCC cells may be mediated through restraining EMT processes in ESCC.
miR-382 inhibited mTOR signaling pathway and activated autophagy process
mTOR plays a key role in cell growth and homeostasis and may be abnormally regulated in tumors. To address the functional activation of mTOR regulated by miR-382, western blot analyses were performed to examine the phosphorylation level of both mTOR and 4E-BP1, a target of mTOR known to bind to translation initiation factor eIF4E to inhibit cap-dependent translation. Overexpression of miR-382 noticeably inhibited the phosphorylation of both mTOR (Ser2448) and 4E-BP1 (Thr37/46) (Figure 6). Previous studies have indicated that autophagy has roles in various cellular functions, including cell proliferation and apoptosis. During autophagy, LC3-I is converted to LC3-II and recruited into the membrane of the phagophore, a double membrane required for the recycling of protein aggregates and organelles. To determine whether miR-382 might induce autophagy in Eca109, we examined the level of LC3 protein in LV-miR-382 infected Eca109 cells and control cells. As shown in Figure 6, increased levels of either LC3-I or LC3-II were detected in LV-miR-382 cells compared to control cells, indicating that miR-382 activated the autophagy process. These data suggested that inhibition of the mTOR/4E-BP1 signaling pathway together with activation of the autophagy process may be important targets of miR-382 actions.
Figure 6.
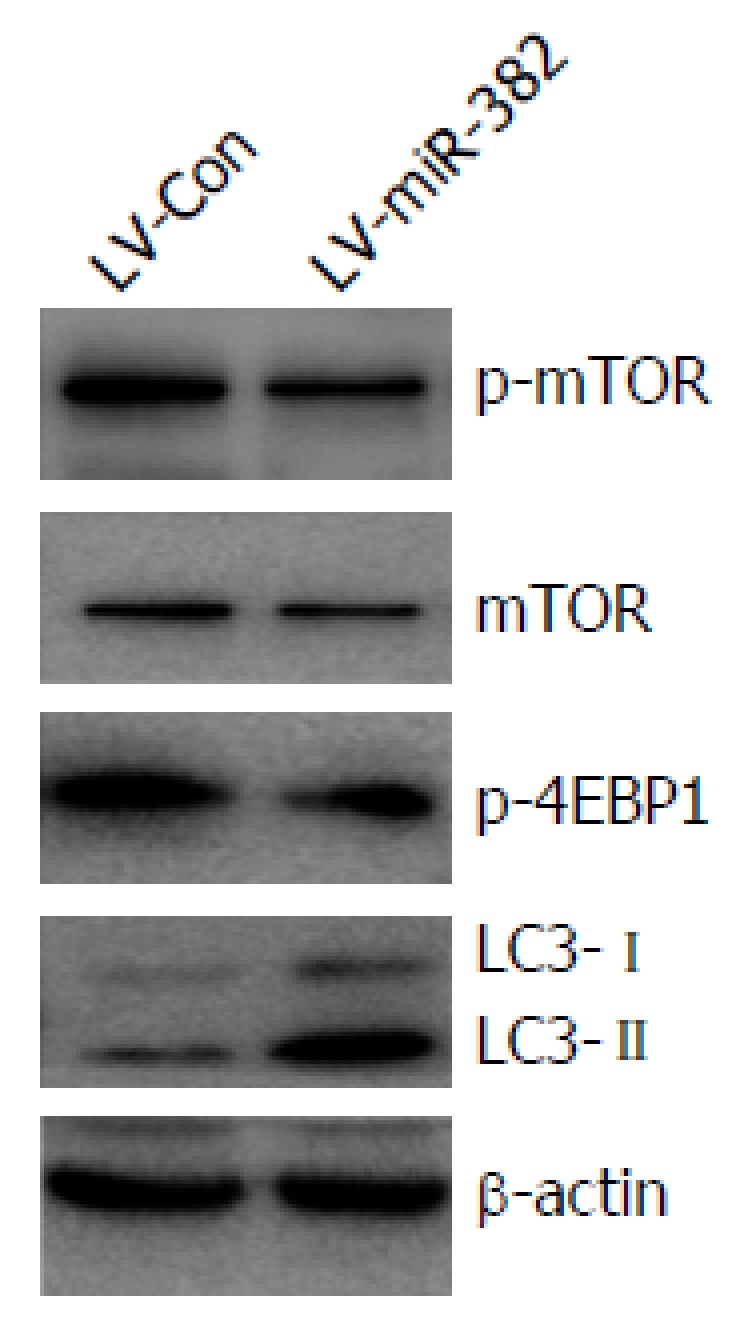
miR-382 inhibited mTOR and 4E-BP1 phosphorylation and activated autophagy. p-mTOR, mTOR, p-4E-BP1 and LC3 were evaluated by western blot. Experiments were performed in triplicate. Equal protein loading was confirmed by β-actin.
DISCUSSION
Accumulating evidence has revealed that miRNAs participate in the initiation and progression of human cancers. Thus, exploration of cancer-specific miRNAs contributes to the identification of novel biomarkers and therapeutic targets for human cancers. To date, correlation analyses between miRNAs and ESCC have shown that a number of dysregulated miRNAs levels correlate with ESCC patients’ outcomes.
Human miR-382 (has–mir-382, MIMAT0000737, 5’-GAAGUUGUUCGUGGUGGAUUCG-3’) resides in a miRNA cluster in the imprinted DLK1-DIO3 region on the 14q32 locus, which hosts one of the largest miRNA clusters in the genome. Many of these miRNAs are differentially expressed in several pathologic processes and various cancers, with recent studies reporting that miR-382 is aberrantly regulated in several types of human malignancies. miR-382 levels were found to be decreased in ovarian cancer[13] and osteosarcoma, but increased in breast cancer[16] and acute myeloid leukemia tumor tissue[17]. Based off these findings, the role of miR-382 in tumor development and metastasis is heterogeneous and it functions in a cell-driven and tissue-driven manner.
Our earlier study discovered that decreased miR382 expression in tumor tissues from ESCC patients was clinically correlated with short-term motility, and significantly linked to poor patient outcome. This indicates that miR-382 may play antitumor functions in esophageal carcinogenesis, and could be a potential predictive biomarker for ESCC prognosis. On the other hand, the mechanism by which miR-382 functionally affects ESCC phenotype has not been elucidated; therefore, in this study, we began by evaluating the expression of endogenous levels of miR-382 in Eca109 tumor cell line and Het-1A normal esophageal epithelia cell line. Consistent with our previous clinical results, we found that miR-382 was reduced in Eca109 cells compared with Het-1A. Moreover, we found that lentivirus-mediated overexpression of miR-382 significantly inhibited Eca109 cell proliferation, colony formation, migration and invasion. These findings together with our previous result suggest that miR-382 functions as a tumor suppressor in ESCC.
Cell cycle arrest and apoptosis are closely linked to cell proliferation in mammalian cells. In the present study, we found that overexpression of miR-382 significantly increased initiation of cell cycle arrest at G2/M phase and promoted the apoptotic process of Eca109 cells. Therefore, the inhibitory influence of miR-382 on Eca109 proliferation seems to be related with increased G2/M cell cycle arrest and active apoptosis. In addition, it is known that cell cycle progression and apoptosis are regulated by numerous proteins, with one of these proteins being the cyclin-dependent kinase inhibitor p21Cip1/Waf1, which facilitates cell cycle arrest in response to a variety of stimuli[18]. Our results showed that miR-382 clearly increased p21Cip1/Waf1 expression, indicating that the miR-382-induced cell cycle arrest in G2/M phase may occur through regulation of p21Cip1/Waf1.
EMT comprises the process by which epithelial cells are transited into mesenchymal cells, and display reduced intracellular adhesion and increased motility[19]. Most evidence supports EMT playing the integral roles through which epithelial cancers invade and metastasize. The EMT process is related to a number of cellular and molecular events and is accompanied by the loss of epithelial markers (e.g., E-cadherin) and the gain of mesenchymal markers (e.g., vimentin, β-catenin and snail). Our study showed that miR-382 inhibited Eca109 cell migration, as well as invasion in vitro. Overexpression of miR-382 enhanced the level of E-cadherin and reduced levels of vimentin, β-catenin, and snail. Accordingly, these data indicated that miR-382 might suppress the EMT process, so that Eca109 cell invasion and metastasis may be restrained.
Autophagy is a catabolic process for proteolytic degradation of damaged intracellular proteins and organelles in lysosomes. Autophagy dysfunction is associated with multiple human diseases, such as cancer. It has been reported that autophagy deficiency promotes cell proliferation, migration and invasion. Our data revealed that overexpression of miR-382 was able to up-regulate the expression of two distinctive hallmarks of autophagy, LC3-I and LC3-II[20], in Eca109 cells, implying that miR-382 could induce autophagy in Eca109 cells.
Regulation of translation is critical for controlling many major cellular processes, including cell proliferation, apoptosis and metastasis. The mTOR signaling pathway plays a pivotal role in regulation of the translation process, with one of the best-characterized downstream effectors of mTOR being 4E-BP1. mTOR phosphorylates 4E-BP1, which promotes the dissociation of eIF4E from 4E-BP1, consequently alleviating the inhibitory effect of 4E-BP1 on eIF4E-dependent translation initiation[21]. Therefore, overall translation levels are lowered when 4E-BP1 is active, which is thought to be regulated by mTOR-dependent phosphorylation. Our results showed that miR-382 dramatically inhibited the phosphorylation of mTOR as well as 4E-BP1. These results indicated that miR-382 might suppress the overall protein translation process through mTOR/4E-BP1, and could potentially function as a tumor suppressor against ESCC.
In conclusion, our results indicated that miR-382 was dramatically down-regulated in ESCC cells. Overexpression of miR-382 inhibited proliferation, migration and invasion of Eca109 cells. mTOR/4E-BP1 signaling mediated inhibitory influence on protein translation and may be involved in the antitumor activity of miR-382 against esophageal cancer. Our results could provide insights into a new therapeutic strategy for the treatment of ESCC through the up-regulation of miR-382 expression.
COMMENTS
Background
MicroRNAs (miRNAs) are classes of small non-coding RNAs involved in the process of silencing gene expression. Evidence has shown that miRNAs are frequently dysregulated in many types of cancers and play a substantial role in the pathogenesis of human cancers. They previously found that miRNA-382 (miR-382) was significantly down-regulated in esophageal squamous cell carcinoma (ESCC) patients with short-term motility, implying that miR-382 may contribute to the development and metastasis of ESCC. However, the possible roles and mechanisms of miR-382 in human ESCC are still not well established.
Innovations and breakthroughs
This study revealed that endogenous miR-382 was dramatically down-regulated in ESCC cells in comparison to normal esophageal epithelial cells. This is consistent with our previous clinical data. Overexpression of miR-382 inhibited cell proliferation, migration and invasion of Eca109 cells. The mTOR/4E-BP1 signaling-mediated inhibitory influence on protein translation might be involved in the antitumor activity of miR-382 against esophageal carcinoma.
Applications
This study suggests that the miR-382 is down-regulated in ESCC and plays an inhibitory role in the growth and invasion of ESCC. Because of this, miR-382 is considered a tumor suppressor of ESCC and provides new insights into a possible therapeutic strategy for ESCC through the up-regulation of miR-382 expression.
Terminology
Previous literature indicates that miR-382 was reduced and associated with poor survival in osteosarcoma patients. In contrast, increased miR-382 levels were found in acute myeloid leukemia tumor tissue. These findings illustrate the role of miR-382 in tumor development and metastasis as a heterogeneous one that is cell-driven and tissue-driven.
Peer-review
This is an interesting topic aiming to reveal more advanced and detailed functions of miR-382 on ESCC. The authors demonstrated that overexpression of miR-382 inhibited cell proliferation and invasion, and induced cell apoptosis and cell autophagy. The antitumor activity of miR-382 might be initiated by inhibition of an mTOR/4E-BP1-mediated protein translation process. Thus, manipulation of miR-382 level is a potential therapeutic strategy for ESCC.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was reviewed and approved by the First Affiliated Hospital of Xinxiang Medical University Institutional Review Board.
Conflict-of-interest statement: The authors declared that no conflict of interest exists in this study.
Data sharing statement: No additional data are available.
Peer-review started: January 4, 2017
First decision: February 9, 2017
Article in press: March 31, 2017
P- Reviewer: Arigami T, Lin Q S- Editor: Yu J L- Editor: Filipodia E- Editor: Zhang FF
References
- 1.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 5.Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17:2–9. doi: 10.1016/j.semradonc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Su M, Liu M, Tian DP, Li XY, Zhang GH, Yang HL, Fan X, Huang HH, Gao YX. Temporal trends of esophageal cancer during 1995-2004 in Nanao Island, an extremely high-risk area in China. Eur J Epidemiol. 2007;22:43–48. doi: 10.1007/s10654-006-9086-x. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 8.Yang M, Liu R, Sheng J, Liao J, Wang Y, Pan E, Guo W, Pu Y, Yin L. Differential expression profiles of microRNAs as potential biomarkers for the early diagnosis of esophageal squamous cell carcinoma. Oncol Rep. 2013;29:169–176. doi: 10.3892/or.2012.2105. [DOI] [PubMed] [Google Scholar]
- 9.Yi Y, Lu X, Chen J, Jiao C, Zhong J, Song Z, Yu X, Lin B. Downregulated miR-486-5p acts as a tumor suppressor in esophageal squamous cell carcinoma. Exp Ther Med. 2016;12:3411–3416. doi: 10.3892/etm.2016.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao BS, Liu SG, Wang TY, Ji YH, Qi B, Tao YP, Li HC, Wu XN. Screening of microRNA in patients with esophageal cancer at same tumor node metastasis stage with different prognoses. Asian Pac J Cancer Prev. 2013;14:139–143. doi: 10.7314/apjcp.2013.14.1.139. [DOI] [PubMed] [Google Scholar]
- 11.Fu L, Li Z, Zhu J, Wang P, Fan G, Dai Y, Zheng Z, Liu Y. Serum expression levels of microRNA-382-3p, -598-3p, -1246 and -184 in breast cancer patients. Oncol Lett. 2016;12:269–274. doi: 10.3892/ol.2016.4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu M, Jin H, Xu CX, Sun B, Mao Z, Bi WZ, Wang Y. miR-382 inhibits tumor growth and enhance chemosensitivity in osteosarcoma. Oncotarget. 2014;5:9472–9483. doi: 10.18632/oncotarget.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B, Song J, Han T, Huang M, Jiang H, Qiao H, Shi J, Wang Y. MiR-382 inhibits cell growth and invasion by targeting NR2F2 in colorectal cancer. Mol Carcinog. 2016;55:2260–2267. doi: 10.1002/mc.22466. [DOI] [PubMed] [Google Scholar]
- 14.Tan H, He Q, Gong G, Wang Y, Li J, Wang J, Zhu D, Wu X. miR-382 inhibits migration and invasion by targeting ROR1 through regulating EMT in ovarian cancer. Int J Oncol. 2016;48:181–190. doi: 10.3892/ijo.2015.3241. [DOI] [PubMed] [Google Scholar]
- 15.Qi B, Lu JG, Yao WJ, Chang TM, Qin XG, Ji YH, Wang TY, Liu SG, Li HC, Liu YZ, et al. Downregulation of microRNA-382 is associated with poor outcome of esophageal squamous cell carcinoma. World J Gastroenterol. 2015;21:6884–6891. doi: 10.3748/wjg.v21.i22.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho JY, Hsu RJ, Liu JM, Chen SC, Liao GS, Gao HW, Yu CP. MicroRNA-382-5p aggravates breast cancer progression by regulating the RERG/Ras/ERK signaling axis. Oncotarget. 2017;8:22443–22459. doi: 10.18632/oncotarget.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang MR, Park KH, Yang JO, Lee CW, Oh SJ, Yun J, Lee MY, Han SB, Kang JS. miR-6734 Up-Regulates p21 Gene Expression and Induces Cell Cycle Arrest and Apoptosis in Colon Cancer Cells. PLoS One. 2016;11:e0160961. doi: 10.1371/journal.pone.0160961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang G, Yuan J, Li K. EMT transcription factors: implication in osteosarcoma. Med Oncol. 2013;30:697. doi: 10.1007/s12032-013-0697-2. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Jiang W, Hu Y, Da Z, Zeng C, Tu M, Deng Z, Xiao W. MicroRNA-199a-5p inhibits cisplatin-induced drug resistance via inhibition of autophagy in osteosarcoma cells. Oncol Lett. 2016;12:4203–4208. doi: 10.3892/ol.2016.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Chang J, Wang S, Liu X, Peng J, Huang D, Sun M, Chen Z, Zhang W, Guo W, et al. miRNA-99b-5p suppresses liver metastasis of colorectal cancer by down-regulating mTOR. Oncotarget. 2015;6:24448–24462. doi: 10.18632/oncotarget.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]