

Abstract
Pore-forming toxins (PFTs) are proteins endowed with metamorphic properties that enable them to stably fold in water solutions as well as in cellular membranes. PFTs produce lytic pores on the plasma membranes of target cells conducive to lesions, playing key roles in the defensive and offensive molecular systems of living organisms. Actinoporins are a family of potent haemolytic toxins produced by sea anemones vigorously studied as a paradigm of α-helical PFTs, in the context of lipid–protein interactions, and in connection with nanopore technologies. We have recently reported that fragaceatoxin C (FraC), an actinoporin, engages biological membranes with a large adhesive motif allowing the simultaneous attachment of up to four lipid molecules prior to pore formation. Since actinoporins also interact with carbohydrates, we sought to understand the molecular and energetic basis of glycan recognition by FraC. By employing structural and biophysical methodologies, we show that FraC engages glycans with low affinity using its lipid-binding module. Contrary to other PFTs requiring separate domains for glycan and lipid recognition, the small single-domain actinoporins economize resources by achieving dual recognition with a single binding module. This mechanism could enhance the recruitment of actinoporins to the surface of target tissues in their marine environment.
This article is part of the themed issue ‘Membrane pores: from structure and assembly, to medicine and technology’.
Keywords: pore-forming protein, biomolecular recognition, lipid–protein interaction, carbohydrate–protein interaction, X-ray crystallography, sea anemone toxin
1. Introduction
Pore-forming toxins (PFTs) are water-soluble proteins that form transmembrane oligomeric pores on cellular membranes leading to cell damage [1–3]. PFTs comprise a heterogeneous family of proteins of various molecular sizes and number of domains, exhibiting a rich collection of target membranes and cell receptors, oligomerization states and mechanisms of action. For example, human perforin and the complement membrane attack complex play a key role in the immune system [4,5], whereas bacterial PFTs are potent virulence factors [1,6].
To effectively attack their targets, PFTs recognize specific receptors on the cell surface, such as lipids, carbohydrates and proteins. In addition to lipids, with which PFTs must interact before, during and after pore formation, some PFTs engage with secondary receptors on the membrane surface increasing their effective concentration and thus facilitating their oligomerization and assembly. For example, cholesterol-dependent bacterial cytolysins, such as pneumolysin from Streptococcus pneumoniae [7], streptolysin O from Streptococcus pyogenes [7] and lectinolysin from Streptococcus mitis [8,9], recognize glycans present on the membrane of target cells in addition to cholesterol [10]. Vibrio cholerae cytolysin (VCC) binds to glycans and lipids [11,12], and the translocation apparatus of the cholera toxin specifically recognizes the sugar moiety of ganglioside GM1 with high affinity [13]. Another PFT, aerolysin, is recruited to the cell membrane by the N-glycans of membrane-associated proteins, and by the glycan region of GPI-anchored proteins [14]. The structure of several of these glycan-binding sites has been determined for some PFTs, showing that these toxins use discrete domains to recognize their carbohydrate receptors [15,16].
Actinoporins are a group of small (approx. 20 kDa) and singe-domain α-helical PFTs secreted by sea anemones lethal to crustaceans, mollusks and fishes [17]. Actinoporins achieve their target specificity by using the lipid sphingomyelin (SM) as both a binding receptor [18] and an assembly cofactor [19]. The lytic activity of actinorins also depends on the physico-chemical properties of the membrane such as the lipid phase [20–22]. The most studied actinoporins are equinatoxin II [23], sticholysin II [24] and fragaceatoxin C (FraC) [25]. The mechanism of lipid-recognition and pore formation of FraC and other actinoporins has been recently described from both structural and energetic points of view [19,26–28] (figure 1), although some controversy remains about the nature of the assembled pore [29]. In addition, the structural features of the pores formed by actinoporins have potential biotechnological applications for the analysis of double- and single-stranded nucleic acids using nanopore technologies [30].
Figure 1.

Mechanism of actinoporins. In this study, we will address the hypothesis that carbohydrates act as weak secondary receptors, possibly helping to concentrate actinoporins on the surface of target cells. The dashed red circle represents a conformational change at the N-terminal region upon dimerization [19]. The existence of a prepore species bound to membranes was recently visualized [27]. The figure was adapted from Tanaka et al. [19].
Interestingly, previous studies have consistently shown that actinoporins experience a substantial delay in their elution profile in size-exclusion chromatography (SEC) columns due to specific interactions with the resin [31–33], which is largely composed of polymers of carbohydrate. However, the putative binding site for carbohydrates or their effect on the function and structure of actinoporins are still unknown.
We hypothesized that single-domain actinoporins may recognize lipids and carbohydrates by a mechanism different from that of multi-domain PFTs. Herein, we have investigated the ability of FraC to interact with carbohydrates from structural, functional and energetic viewpoints. We characterized the binding site of a sulfated monosaccharide at high resolution by X-ray crystallography. Surprisingly, the carbohydrate pocket overlaps with the lipid-binding module of actinoporins. Our study reveals a novel recognition mechanism by an ancient PFT that could enhance the activity of actinoporins in their natural coastal habitats.
2. Material and methods
(a). Materials
SM from porcine brain, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Glucose, galactose, mannose, fructose, N-acetyl glucosamine (GlcNAc), N-acetyl glucosamine 6-sulfate sodium salt (GlcNAc(6S)), raffinose and maltopentaose were obtained from Sigma-Aldrich (St Louis, MO, USA). Sucrose and occasionally glucose was from Merck. Chondroitin sulfate C sodium salt was obtained from Nacalai tesque (Kyoto, Japan). Defibrinated horse blood was purchased from Nippon Biotest Laboratories (Tokyo, Japan). Defibrinated sheep blood was from Pronadisa. Recombinant endoglycoceramidase II (EGCase II) with Activator II was obtained from TAKARA Bio (Siga, Japan). Biotin-hydrazide was purchased from Dojindo Laboratories (Kumamoto, Japan). Phosphocholine (POC) chloride sodium salt hydrate was obtained from Tokyo Chemical Industry (Tokyo, Japan). Reagents for the crystallization of proteins were purchased from Hampton Research (Aliso Viejo, CA, USA). Other chemicals were from Wako (Tokyo, Japan).
(b). Protein expression and purification
FraC and muteins were expressed and purified as described previously [31], with minor modifications. Briefly, FraC was expressed in Escherichia coli BL21 (DE3) cells grown at 37°C. FraC was purified from the cell lysate with a Resource S cationic exchange column (GE Healthcare, Piscataway, NJ, USA) equilibrated with buffer A (50 mM Tris–HCl, pH 7.4). The protein was eluted with increasing concentrations of NaCl in buffer B (50 mM Tris–HCl, and 1 M NaCl, pH 7.4). The protein fractions were pooled and subjected to SEC in a HiLoad 16/60 Superdex 75 pg column (GE Healthcare) equilibrated with SEC buffer (50 mM Tris–HCl, 200 mM NaCl, pH 7.4).
(c). Haemolysis of deglycosylated red blood cells
Defibrinated horse blood was washed in 20 mM HEPES, 150 mM NaCl (pH 7.4). Washed red blood cells (RBC) (1.8 ml at OD600 of 1.0, corresponding to approx. 1.2 × 108 cells) were incubated with EGCase II [34] (50 mU) at 37°C overnight. RBC were then washed in 20 mM HEPES, 150 mM NaCl (pH 7.4). As a control, RBC were incubated without EGCase II. Aliquots of RBC treated with EGCase II or control RBC (260 µl, OD = 0.65) were incubated with FraC (19.5 nM) for 10 min followed by centrifugation at 3000g for 1 min. The haemolysis was monitored by the absorbance of haemoglobin released to the supernatant at a wavelength of 412 nm.
(d). Haemolytic assays
The time course of haemolysis was measured in a V-660 Spectrophotometer (Jasco, Japan) at 600 nm. Haemolytic activity was monitored by adding FraC to a suspension of horse RBC at an initial OD600 of approximately 0.5, in a total volume of 1 ml at room temperature. Proteins were added at final concentration 25 nM to a solution containing buffer supplemented with monosaccharides or PEG 200 at a concentration of 400 mM. High concentration of monosaccharides and PEG 200 produced no deleterious effects on the stability of RBC during the time course of the experiments as judged from OD600. When the effect of the number of units in the saccharide was examined, haemolysis of sheep RBC was carried out in the presence of glucose, sucrose, raffinose or maltopentaose at 37.5 mM in a PowerWaveTM XS microplate reader (Biotek, VT, USA) at 700 nm and 25°C with constant shaking and FraC at a final concentration of 2 nM.
The effect of the concentration of saccharide on haemolysis was determined by twofold serial dilutions of FraC in a 96-well plate with buffer supplemented with monosaccharides or PEG 200 in a cell of 200 µl. After an incubation period of 1 h at room temperature, samples were centrifuged at 6000g for 1 min. The absorbance of haemoglobin released to supernatant was monitored in a PHERAstar (BMG LABTECH, Ortenberg, Germany) at a wavelength of 412 nm.
(e). Micro-array glycan screening
FraC containing the additional residue Cys180 at the C-terminus was labelled using the fluorophore molecule Alexa Fluor 488 C5-maleimide (Thermo Fisher Scientific, MA, USA). The labelled protein was separated from unreacted dye in a NAP-5 column (GE Healthcare). The labelling efficiency was calculated to be approximately 100% from the ratio of absorbance at 495 and 280 nm. FraC retained its full haemolytic activity after labelling and lyophilization. Such labelled FraC was first lyophilized, subsequently dissolved in the appropriate volume and analysed at the Consortium for Functional Glycomics (CFG) using a mammalian printed array (Ver 5.2) containing 609 different glycans [35]. Labelled FraC was screened at 2, 20 and 200 µg ml−1. The results obtained from the screen are freely available on the public repository at the CFG website (Request ID: 2971).
(f). Surface plasmon resonance
Chondroitin sulfate C (2 mg ml−1) was oxidized using 2 mM sodium periodate in 20 mM sodium acetate (pH 5.5), and covalently attached to biotin using 10 µM Biotin-hydrazide in 100 mM MES (pH 6.5) at room temperature. The biotinylated molecule was reduced using 100 mM sodium cyanoborohydride in 100 mM MES, pH 7.0. The biotinylated carbohydrate was subsequently immobilized to a Series S Sensor Chip SA on a Biacore T200 instrument (GE Healthcare) equilibrated with HBS-P buffer (10 mM HEPES, 150 mM NaCl, 0.005% Surfactant P20, pH 7.4) at 25°C. Twofold serial dilutions of FraC were injected over immobilized chondroitin sulfate C for 60 s, and the dissociation was followed for an additional 600 s.
(g). Crystal structure of fragaceatoxin C and GlcNAc(6S) complex
A solution of microcrystals was first prepared like that described for unbound FraC [19]. Large rod-shaped single crystals of the complex of FraC and GlcNAc(6S) were obtained after mixing 1 µl of protein at 9 mg ml−1 in 10 mM Tris–HCl and 50 mM GlcNAc(6S) (pH 7.4), 0.2 µl solution of micro-seeds and 1 µl of crystallization solution composed of 19% PEG 3350, 200 mM ammonium chloride and 100 mM Bis–Tris (pH 6.3). Suitable crystals appeared after one month at 20°C. Crystals of an approximate size of 0.2 × 0.03 × 0.03 mm3 were transferred to a solution of mother liquor supplemented with 20% glycerol and subsequently frozen in liquid nitrogen. Data collection was carried out at beamline AR-NW12A of the Photon Factory (Tsukuba, Japan) under cryogenic conditions (100 K). The structure of FraC with GlcNAc(6S) was determined by the molecular replacement method using the coordinates of apo FraC (PDB entry code 3VWI) with the program PHASER [36]. The initial model was refined using the programs REFMAC5 [37] and COOT [38]. The quality of the final model was assessed using the programs COOT and PROCHECK [39]. Data collection and refinement statistics are shown in table 1.
Table 1.
Data collection and refinement statistics. Statistical values given in parentheses refer to the highest resolution bin.
| FraC + GlcNAc(6S) | |
|---|---|
| data collection | |
| space group | P 1 21 1 |
| unit cell | |
| a, b, c, Å | 77.16, 44.40, 114.36 |
| α, β, γ, ° | 90.0, 92.76, 90.0 |
| resolution, Å | 37.1–1.55 (1.63–1.55) |
| wavelength, Å | 1.000 |
| observations | 543 367 (59 662) |
| unique reflections | 103 405 (12 516) |
| Rmerge. (%)a | 0.069 (0.290) |
| CC(1/2) | 0.997 (0.937) |
| I/σ (I) | 14.8 (4.5) |
| multiplicity | 5.3 (4.8) |
| completeness (%) | 91.8 (76.8) |
| refinement statistics | |
| resolution, Å | 37.1–1.55 |
| Rwork/Rfree, %b | 11.8/16.3 (13.4/17.3) |
| no. of protein atoms | 5686 |
| no. of protein chains | 4 |
| no. of protein residues | 708 |
| no. of sugar molecules | 2 |
| no. of waters molecules | 775 |
| no. of other (not solvent) | 15 |
| protein B-factor, Å2 | 15.2 |
| sugar B-factor, Å2 | 23.3 |
| water B-factor, Å2 | 25.9 |
| other B-factor (not solvent), Å2 | 22.3 |
| Ramachandran plot | |
| preferred regions, % | 89.4 |
| allowed regions, % | 10.6 |
| outliers (%) | 0 |
| RMSD bond, Å | 0.012 |
| RMSD angle, ° | 1.5 |
| PDB identification code | 5GWF |
aRmerge = Σhkl Σi |(I(hkl)i − [I(hkl)]|/Σhkl Σi I(hkl).
bRwork = Σhkl |F(hkl)o − [F(hkl)c]|/Σhkl F(hkl)o; Rfree was calculated as Rwork, where F(hkl)o values were taken from 3% of data not included in the refinement.
(h). Affinity size-exclusion chromatography
A Superdex 200 10/300 GL column (GE Healthcare) was calibrated with Blue dextran 2000, acetone and marker proteins (aprotinin, ribonuclease A, carbonic anhydrase, ovalbumin, conalbumin, aldolase and ferritin) from a Gel Filtration Calibration Kit (GE Healthcare). The void volume (V0) and the column volume (Vtotal) were determined to be 7.6 and 21.2 ml, respectively. The elution profile of FraC (230 µg) was examined in the presence of SEC buffer supplemented with increasing concentrations of POC at 0, 2.5, 5, 10, 20, 40 and 80 mM. Assuming that a competition between the matrix of the column and POC for FraC is occurring, the relationship between the elution volume of the protein and the concentration of POC is the following [40]:
| 2.1 |
where V is the elution volume for the protein, Vnon the elution volume for the protein in the absence of interaction with the gel matrix, [POC] the concentration of ligand in solution, the constant KPOC is defined for binding the protein to POC and the constant A is a factor pertaining to the interaction between the protein and column matrix.
3. Results
(a). Haemolytic activity
Consistent with previous studies [19,31], we showed here that actinoporins experience a large delay in the SEC elution profile with chromatographic columns composed of carbohydrates (figure 2a). Because carbohydrates act as a weak receptor in the SEC column, we extrapolated that glycans present on biomembranes and tissues could play some role in the activity of FraC, possibly working as secondary receptors. To test this hypothesis, we first monitored the haemolysis of RBCs treated with an endoglycoceramidase (EGCase II), which cleaves the linkage between the glycans and the ceramide regions of glycosphingolipids. Other glycosylation linkages in proteins or in other lipids are not affected by EGCase II [34]. Although the fraction of glycolipids with respect to the total lipidome of the RBC is relatively small, as a result of the treatment with EGCase II, the RBC turned less susceptible to the haemolytic activity of FraC with respect to untreated cells. This observation suggested that the presence of glycans on the cell membrane enhances the potency of the toxin in a moderate fashion (figure 2b). Ceramides, the lipid product of the enzymatic reaction of EGCase II, have been shown to inhibit the activity of actinoporins in liposomes with little (5%) or no cholesterol present [41]. Although the effect of ceramides in the activity of actinoporins has not been studied in RBC yet, that study suggests an additional mechanism explaining the inhibition of haemolysis in figure 2b.
Figure 2.

Evidence of the interaction between FraC and carbohydrates. (a) Delayed elution profile of FraC on a column of SEC (HiLoad 16/600 Superdex 75) consisting of a composite base matrix of dextran and agarose. Anomalous elution profiles are also observed in other carbohydrate-based gel-filtration columns. V0, Vt and FraC indicate the void volume (45 ml), the column volume (120 ml) and the elution of the monomeric water-soluble form of FraC (198 ml), respectively. (b) Haemolysis of RBC treated with EGCase II, or untreated, in the presence of 19.5 nM FraC. The values corresponding to 100 and 0% haemolysis were obtained by addition of Triton X-100 (1% v : v) or buffer. (c) Inhibitory effect of monosaccharides on haemolysis. The protein was added at a final concentration of 25 µM. The compounds employed were PEG-200 (grey) and monosaccharides glucose (magenta), galactose (orange), mannose (blue), fructose (green) and GlcNAc (light brown).
To gain further insight, we tested the idea that monosaccharides could inhibit the haemolytic activity of FraC, as these small monosaccharides could compete with the glycans on the surface of RBC for the toxin without blocking the pore [42]. The extent of the inhibition was comparable among the few hexoses examined (glucose, galactose, mannose and fructose) (figure 2c). By contrast, the monosaccharide GlcNAc had a greater inhibitory potency than that of the other hexoses, suggesting some degree of specificity. PEG200, a small molecule of similar molecular weight to that of the monosaccharides, produced the lowest inhibitory effect. The extent of the inhibition increased with the concentration of the hexose in a nonlinear fashion, exhibiting midpoints at high concentration of monosaccharide or PEG200. The inhibitory effect is also observed in the kinetic profiles, and when exposing the RBC to saccharides composed of progressively more hexose rings (electronic supplementary material, figure S1). This latter observation suggests an avidity effect, although the inhibition caused by the large oligosaccharides is probably influenced also by an osmotic protection effect, as reported earlier [43,44]. As GlcNAc is also slightly larger than other monosaccharides, the presumably greater osmotic protective effect of this monosaccharide may also play some role in the differences reported in figure 2c; electronic supplementary material, figure S1a.
Because of the arguably low affinity of these sugars for the toxin, it was deemed necessary to examine other carbohydrates in order to find molecules with higher affinity for FraC. This search would be followed by a more direct characterization of the interaction between carbohydrates and FraC employing high-resolution structural and biophysical techniques.
(b). Micro-array screening
To elucidate the preference of FraC for certain carbohydrates, a comprehensive screen using a glycan array composed of 609 different mammalian glycans was performed. The screen was carried out with FraC labelled at the C-terminus with the fluorescent dye Alexa Fluor 488 attached to an additional Cys residue appended at the C-terminus. The result of the screen failed to find a highly specific sugar, but instead it showed binding activity for a large subset of glycans (public data can be retrieved as indicated in Material and methods). Notably, sulfated glycans appeared most frequently among the highest-ranked glycans (figure 3a).
Figure 3.

Glycan screening and binding analysis by SPR. (a) The region corresponding to the top hits on a binding screen with 609 different glycans attached on a microarray is enriched on sulfated oligosaccharides. The sulfated glycans were classified according to the position of the sulfate group. Position 6, position 4 and position 3 are indicated by the purple, green and orange bars, respectively. (b) Kinetics of interaction between FraC and chondroitin sulfate C determined by SPR. The chondroitin sulfate C was immobilized on a chip and FraC was flowed at 0.5, 1.0, 2.0, 4.0, 8.0, 16.1, 32.2, 64.4 and 128.7 µM. The first and second arrow indicates the interval when FraC is accessing the sensor-chip where the polysaccharide was immobilized. Data were fit to single-site model to calculate kinetic parameters. (c) Determination of the equilibrium constant from the binding response in equilibrium (KD = 24 µM).
These data suggested that FraC possesses the ability to engage with a broad range of glycans, particularly with glycans modified with a sulfate group. Sulfated sugars are major components of glycosaminoglycans (GAGs), a class of carbohydrates rich in amino-sugars like GlcNAc. The stronger inhibitory potency of GlcNAc and the outcome of the experiment with the glycan micro-array suggested that GAGs are possibly low-affinity targets for FraC, at least in vitro. Because of the low binding affinity of small saccharides, we employed chondroitin sulfate C (a polysaccharide composed of alternating units of GlcNAc(6S) and glucuronic acid) as a representative member of this family of GAGs to quantify its binding affinity to FraC by surface plasmon resonance (SPR) (figure 3b,c). Binding of FraC to chondroitin sulfate C immobilized on the sensor chip was monitored at various concentrations of toxin. The sensorgram displayed a classical box shape [45], giving rise to a weak dissociation constant KD = 23.6 µM, and a fast dissociation rate (koff = 1.0 s−1). The association rate was too fast to be reliably determined. The fast dissociation rate, and the presence of negative charges in the polysaccharide and a positively charged surface in the lipid-binding region of FraC (electronic supplementary material, figure S2) [19], suggested an important role of the electrostatic forces in binding.
(c). Structural basis of interaction between FraC and GlcNAc(6S)
The high-resolution crystal structure of the complex of FraC with the sulfated monosaccharide GlcNAc(6S) was determined at 1.55 Å resolution (table 1). As the protein was crystallized from microcrystal seeds prepared like for the unbound protein (PDB entry code 3VWI), the crystals yielded the same space group and very similar cell-unit dimensions. In this particular three-dimensional arrangement, half of the protein chains are unavailable to engage the sugar because key binding residues are involved in crystal packing contacts, and therefore cannot interact with the monosaccharide.
The overall conformation of FraC was essentially identical to that of the unbound protein (RMSD = 0.19 ± 0.08 Å), or the lipid-bound species (PDB entry code 4TSQ; RMSD = 0.37 ± 0.04 Å) [19]. Interestingly, the binding site of GlcNAc(6S) overlaps with that of the fourth lipid (L4) described in the lipid-bound structure [19] (figure 4a,b). The position of all atoms of the monosaccharide in the crystal was unambiguously determined from the excellent features of the electron density maps (figure 4c). Close investigation of the GlcNAc(6S)-binding site reveals several hydrogen bonds between oxygens of the sulfate group and the hexose ring of the sugar, with residues Arg53 and Gln130 of the protein (figure 4c,d). The hydroxyl groups of aromatic residues Tyr51 and Tyr138 are members of hydrogen bond networks with the monosaccharide via interfacial water molecules. Notably, Arg53 and Tyr138 also formed hydrogen bonds with the phosphate oxygens of lipid L4 in the crystal structure with lipid bound [19] further demonstrating the overlap between the binding interfaces of lipid and carbohydrate (figure 4e).
Figure 4.

High-resolution crystal structure of FraC in complex with GlcNAc(6S). (a,b) Comparison of the crystal structure of Frac with GlcNAc(6S) (this work) or lipids bound (PDB entry code 4TQS). The position of GlcNAc(6S) (magenta) and DHPC (green) overlap. (c) Close-up view of the monosaccharide bound to FraC. The mesh corresponds to the sigma-A weighted 2Fo–Fc electron density map (σ = 1.0) of GlcNAc(6S). GlcNAc(6S) is shown in magenta. Protein–sugar and water-mediated hydrogen bonds are depicted with dashed lines, and their corresponding distances indicated in blue. (d,e) Residue environments around GlcNAc(6 s) (d) and DHPC L4 (e) as calculated with the program LigPlot+. (f) Conformational change in residues of the binding pocket in response to the presence of lipid (green) or carbohydrate (grey). GlcNAc(6S) is depicted in magenta. For clarity purposes, the lipid has been omitted from the figure. The arrows indicate the movement of the residues experiencing the greatest conformational changes.
Several residues involved in both binding GlcNAc(6S) and lipids underwent conformational changes in their side chains with respect to each other. These changes are clearly observed in residues Arg53, Glu130 and Gln134 (figure 4f). Changes restricted to the relative position of residues belonging to the aromatic cluster (Tyr 51, Tyr134, Tyr 136 and Tyr 138) are also visible. Although a limited number of residues contributed to the interaction with the monosaccharide GlnNAc(6S), it is reasonable to assume that the binding of oligo- and polysaccharides will require a more extensive interaction surface. Indeed, the lipid-binding module of actinoporins is composed of a cluster of aromatic residues accepting up to four lipid molecules before pore formation, and a region rich in basic residues surrounding it [46], being suitable for the engagement of carbohydrates bearing negative charges like GAGs [47,48] (electronic supplementary material, figure S2).
(d). Competition assay
Taking advantage of the specific interaction of FraC with SEC columns like Superdex 200 10/300, we sought to elucidate if fragments of lipids could compete with the column for the toxin (figure 5). As shown in figure 5a, the elution of FraC (26.9 ml) occurs at a significantly higher volume than the total volume of the column (21.2 ml) because FraC binds reversibly to the resin of the column, composed of dextrans and agarose. The expected elution volume estimated from the molecular weight of the protein in the absence of interactions with the resin is 17.2 ml. If lipid- and sugar-binding sites were overlapping, lipids would competitively inhibit the binding of FraC to the column, thus reducing the volume at which the elution would take place. We used POC as a water-soluble competitive inhibitor, as POC represents the headgroup of two major phospholipid molecules like phosphatidylcholine and sphygomyelin, to which FraC binds in liposomes [19,26,27].
Figure 5.

Elution profile of FraC on a Superdex column. (a) Chromatographic profile of WT FraC (black line) and mutein W112R/W116F (red line). The arrows point at the position of the void volume (V0), the volume of FraC in the absence of interactions (Vnon), the total volume (Vtotal) and the elution volume of FraC (V) as given in equation (2.1). (b) SEC profile of FraC in the presence of increasing amounts of POC (0, 2.5, 5, 10, 20, 40 and 80 mM). (c) Plot of 1/(V–Vnon) as a function of the concentration of POC. The black line corresponds to the fitted line from equation (2.1). The regression coefficient is also given in the figure (R2 = 0.986).
As the concentration of POC in the equilibration buffer increased, the elution volume of FraC (V) decreased (figure 5b). A linear correlation between the concentration of POC and the inverse of the mobility (V–Vnon)−1 is observed, as it would be expected for a phenomenon of competitive inhibition [40] (figure 5c). The fit with equation (2.1) plot yields a straight line with a good correlation coefficient (R2 = 0.986), corroborating the idea that the matrix of the column and POC are binding competitively to FraC.
The elution position of the mutein W112R/W116F, lacking binding to liposomes, appears at 19.2 ml, very close to the expected value for the non-interacting protein (17.2), and thus demonstrating that this mutein has a decreased ability to interact with the column. The residues Trp112 and Trp116 belonging to the attachment site for lipids 2 and 3 are in the vicinity of GlcNAc(6S), although we note that they are no completely adjacent (electronic supplementary material, figure S2). This observation suggests that the carbohydrate-binding module also comprises an extended surface that could facilitate the engagement of oligo- and polysaccharides by the incremental and favourable interactions of multiple residues with multiple hexose rings. These results thus reinforced the notion that the carbohydrate- and lipid-binding modules consist of overlapping regions of FraC.
(e). Structural similarity of fragaceatoxin C with fungal lectins
The top structures ranked by structural similarity to FraC as obtained by the Dali server [49] are shown in the electronic supplementary material, table S1. In addition to its close actinoporin relatives, FraC shows high structural homology with a variety of proteins from mushrooms, most of them lectins but also including the PFT pleurotolysin A, and two other PFT from bacteria. The similarities between actinoporins and fungal lectins were already noted in previous studies [2,17,46,50]; but not until now, has it been shown that both families of proteins shared the characteristic of binding carbohydrates. For example, both FraC and Boletus edulis lectin (BEL) display a core β-sandwich motif of quite similar topology (electronic supplementary material, figure S3). The β-sandwich of FraC is flanked by helices from both sides, whereas BEL display α-helices only one side of the β-sandwich. Importantly, the carbohydrate-binding site of FraC as observed in the crystal structure and the primary carbohydrate-binding site of BEL are in the same region between the β-sandwich and α helix (α2) (figure 6), suggesting common evolutionary traits.
Figure 6.
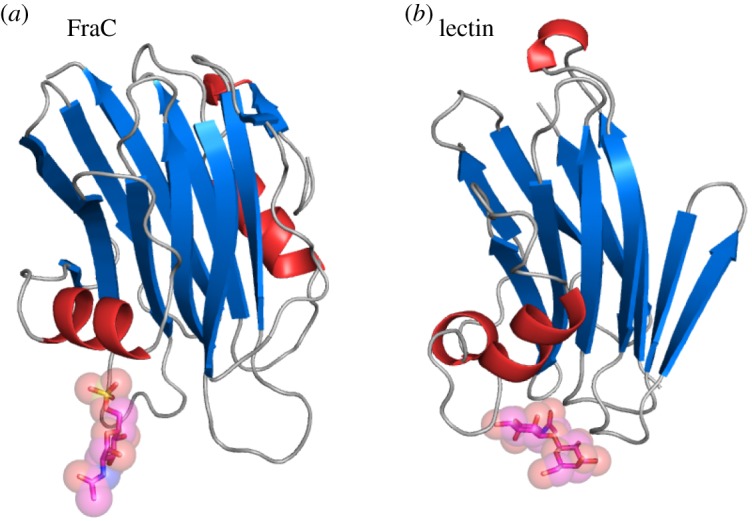
Structural comparison of FraC with a fungal lectin. Location of a carbohydrate bound to (a) FraC and (b) to the lectin from Boletus edulis (PDB ID: 3QDX). The carbohydrate bound to FraC is the monosaccharide GlcNAc(6S), whereas the carbohydrate bound to the lectin is di-GlcNAc.
4. Discussion
Herein, we have shown direct evidence that FraC of the family of actinoporins interacts with glycans using structural regions that overlap with the lipid-binding module. The efficient use of the interaction surface is therefore critical to recognize multiple receptors by a single-domain small protein. In contrast with FraC, large PFTs (greater than 40 kDa) often employ specialized domains to recognize multiple receptors on cell membranes [15]. Recently, it has been reported that some medium-sized PFTs (20–40 kDa) also have the ability to recognize multiple receptors [51]. However, the exact location of these receptor-binding sites has not yet been reported. Further research on this class of secondary binding sites in PFTs will shed light on the different strategies of recognition employed by toxins of different sizes and architectures. The ability of actinoporins to interact with carbohydrates is mirrored by the structurally homologue class of lectins from fungi (figure 6).
Utilization of lipid-binding pockets as weak interaction spots for glycans might be a strategy shared by other PFTs. Indeed, the type of residues that contribute to the engagement of lipids and carbohydrates like those shown in figure 4 appears often in both types of binding sites. First, aromatic residues are often observed at the recognition sites of lipids and glycans [47], and second, positively charged residues preferentially participate to attract the negatively charged GAGs and the headgroups of phospholipids [48]. Hydrogen bonds, considered a critical feature of glycan recognition [52] and binding to FraC (figure 4), are also an important aspect for the activity of actinoporins at the membrane level [19,53,54], thus reflecting an additional mechanistic similarity for the recognition of lipids and glycans by actinoporins in addition to aromatic interactions and electrostatic forces. In agreement with the idea of dual recognition sites, recent computational prediction of carbohydrate-binding sites for cholesterol-dependent cytolysins suggested that they could engage glycans using the lipid-binding regions [7].
The quantitative analysis indicated that the interaction of FraC with saccharides (KD ≈ 10−5 M) is significantly weaker than that of lipidic membranes (KD ≈ 10−7 M). Moreover, the dissociation rate constant of FraC–glycan interaction is fast (koff ≈ 100 s−1) compared with the nearly irreversible interaction of FraC (and the close homologue equinatoxin II) with liposomes [18,55]. This analysis suggests that FraC and glycans form transient complexes of short lifetime, helping to accumulate the toxin near on glycan-rich surfaces, but without compromising its ability to interact with membrane lipids. Notably, the mucus layer of fishes contains significant amounts of GAGs [56,57], which could act as a concentration platform for the accumulation of actinoporins on the surface of target preys. Further research to ascertain the exact glycan composition of mucus of the fish skin, and the ability of FraC to accommodate there, will be necessary to prove the full biological significance of this mechanism. In part, the failure of the glycan screen to find more specific and high-affinity glycans could be explained because this array employs only mammalian glycans, which are not among natural targets of sea anemones. Although the biological function of the carbohydrate–actinoporin interaction has not been fully established in our study, the recruitment of actinoporins on the surface of target cells and/or tissues by low-affinity interaction with carbohydrates could be an effective strategy to enhance their activity in conditions of high dilution such as the marine environment.
In summary, our data showed that FraC uses the glycans to enhance its haemolytic activity. FraC binds to saccharides without particular specificity, but it is enhanced by negative charges on the glycan. Notably, residues interacting with the carbohydrates also belong to the phospholipid binding module of the toxin. This dual usage of residues is an economic strategy that FraC may employ to achieve binding of multiple ligands with its limited molecular size.
Supplementary Material
Acknowledgements
We thank the staff of the Photon Factory (Tsukuba, Japan) for excellent technical support. Access to beamline AR-NE3A was granted by the Photon Factory Advisory Committee (Proposal no. 2013G738). We acknowledge the Protein-glycan Interaction Resource at the Consortium for Functional Glycomics (grant no. R24 GM098791) for the glycan screening. We thank Prof. Juan Manuel González-Mañas for valuable advice.
Data accessibility
The coordinates and structure factors for the structures of FraC in complex with GlcNAc(6S) have been deposited in the PDB under accession code 5GWF.
Authors' contributions
K.Ta., J.M.M.C. and K.Ts. designed research. K.Ta. performed experiments of haemolysis, crystallization, chromatography, SPR, protein labelling and site-directed mutagenesis. K.M. performed experiments of haemolysis. J.M.M.C. performed crystallization experiments, and processed and determined the crystal structure. All authors discussed the results. K.Ta. and J.M.M.C. wrote the manuscript, and received approval from K.M. and K.Ts.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by JSPS Grants-in-Aid for Scientific Research 25249115 (K.Ts.) and 15K06962 (J.M.M.C.) and a Grant-in-Aid for JSPS fellows (K.Ta.).
References
- 1.Iacovache I, Bischofberger M, van der Goot FG. 2010. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 20, 241–246. ( 10.1016/j.sbi.2010.01.013) [DOI] [PubMed] [Google Scholar]
- 2.Anderluh G, Lakey JH. 2008. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33, 482–490. ( 10.1016/J.Tibs.2008.07.004) [DOI] [PubMed] [Google Scholar]
- 3.Gilbert RJC, Serra MD, Froelich CJ, Wallace MI, Anderluh G. 2014. Membrane pore formation at protein–lipid interfaces. Trends Biochem. Sci. 39, 510–516. ( 10.1016/j.tibs.2014.09.002) [DOI] [PubMed] [Google Scholar]
- 4.Baran K, et al. 2009. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity 30, 684–695. ( 10.1016/j.immuni.2009.03.016) [DOI] [PubMed] [Google Scholar]
- 5.Serna M, Giles JL, Morgan BP, Bubeck D. 2016. Structural basis of complement membrane attack complex formation. Nat. Commun. 7, 10587 ( 10.1038/Ncomms10587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilley SJ, Saibil HR. 2006. The mechanism of pore formation by bacterial toxins. Curr. Opin. Struct. Biol. 16, 230–236. ( 10.1016/j.sbi.2006.03.008) [DOI] [PubMed] [Google Scholar]
- 7.Shewell LK, et al. 2014. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl Acad. Sci. USA 111, E5312–E5320. ( 10.1073/pnas.1412703111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrand S, Hotze E, Friese P, Hollingshead SK, Smith DF, Cummings RD, Dale GL, Tweten RK. 2008. Characterization of a streptococcal cholesterol-dependent cytolysin with a Lewis y and b specific lectin domain. Biochemistry 47, 7097–7107. ( 10.1021/bi8005835) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feil SC, Lawrence S, Mulhern TD, Holien JK, Hotze EM, Farrand S, Tweten RK, Parker MW. 2012. Structure of the lectin regulatory domain of the cholesterol-dependent cytolysin lectinolysin reveals the basis for its Lewis antigen specificity. Structure 20, 248–258. ( 10.1016/j.str.2011.11.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouyain S, Geisbrecht BV. 2012. Host glycan recognition by a pore forming toxin. Structure 20, 197–198. ( 10.1016/j.str.2012.01.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De S, Bubnys A, Alonzo F, Hyun J, Lary JW, Cole JL, Torres VJ, Olson R. 2015. The relationship between glycan binding and direct membrane interactions in Vibrio cholerae cytolysin, a channel-forming toxin. J. Biol. Chem. 290, 28 402–28 415. ( 10.1074/jbc.M115.675967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levan S, De S, Olson R. 2013. Vibrio cholerae cytolysin recognizes the heptasaccharide core of complex N-glycans with nanomolar affinity. J. Mol. Biol. 425, 944–957. ( 10.1016/j.jmb.2012.12.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turnbull WB, Precious BL, Homans SW. 2004. Dissecting the cholera toxin–ganglioside GM1 interaction by isothermal titration calorimetry. J. Am. Chem. Soc. 126, 1047–1054. ( 10.1021/ja0378207) [DOI] [PubMed] [Google Scholar]
- 14.Abrami L, Fivaz M, van der Goot FG. 2000. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 8, 168–172. ( 10.1016/S0966-842x(00)01722-4) [DOI] [PubMed] [Google Scholar]
- 15.Dal Peraro M, van der Goot FG. 2016. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92. ( 10.1038/nrmicro.2015.3) [DOI] [PubMed] [Google Scholar]
- 16.Iacovache I, van der Goot FG, Pernot L. 2008. Pore formation: an ancient yet complex form of attack. Biochim. Biophys. Acta 1778, 1611–1623. ( 10.1016/j.bbamem.2008.01.026) [DOI] [PubMed] [Google Scholar]
- 17.Kristan KČ, Viero G, Dalla Serra M, Maček P, Anderluh G. 2009. Molecular mechanism of pore formation by actinoporins. Toxicon 54, 1125–1134. ( 10.1016/j.toxicon.2009.02.026) [DOI] [PubMed] [Google Scholar]
- 18.Bakrac B, Gutiérrez-Aguirre I, Podlesek Z, Sonnen AF-P, Gilbert RJC, Macek P, Lakey JH, Anderluh G. 2008. Molecular determinants of sphingomyelin specificity of a eukaryotic pore-forming toxin. J. Biol. Chem. 283, 18 665–18 677. ( 10.1074/jbc.M708747200) [DOI] [PubMed] [Google Scholar]
- 19.Tanaka K, Caaveiro JMM, Morante K, Gonzalez-Manas JM, Tsumoto K. 2015. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 6, 6337 ( 10.1038/ncomms7337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barlic A, Gutiérrez-Aguirre I, Caaveiro JMM, Cruz A, Ruíz-Arguello M-B, Pérez-Gil J, González-Mañas JM. 2004. Lipid phase coexistence favors membrane insertion of equinatoxin-II, a pore-forming toxin from Actinia equina. J. Biol. Chem. 279, 34 209–34 216. ( 10.1074/jbc.M313817200) [DOI] [PubMed] [Google Scholar]
- 21.Schön P, García-Sáez AJ, Malovrh P, Bacia K, Anderluh G, Schwille P. 2008. Equinatoxin II permeabilizing activity depends on the presence of sphingomyelin and lipid phase coexistence. Biophys. J. 95, 691–698. ( 10.1529/biophysj.108.129981) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caaveiro JMM, Echabe I, Gutiérrez-Aguirre I, Nieva JL, Arrondo JLR, González-Mañas JM. 2001. Differential interaction of equinatoxin II with model membranes in response to lipid composition. Biophys. J. 80, 1343–1353. ( 10.1016/S0006-3495(01)76107-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Athanasiadis A, Anderluh G, Maček P, Turk D. 2001. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure 9, 341–346. ( 10.1016/S0969-2126(01)00592-5) [DOI] [PubMed] [Google Scholar]
- 24.Mancheño JM, Martín-Benito J, Martínez-Ripoll M, Gavilanes JG, Hermoso JA. 2003. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 11, 1319–1328. ( 10.1016/j.str.2003.09.019) [DOI] [PubMed] [Google Scholar]
- 25.Mechaly AE, Bellomio A, Gil-Cartón D, Morante K, Valle M, González-Mañas JM, Guérin DMA. 2011. Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure 19, 181–191. ( 10.1016/j.str.2010.11.013) [DOI] [PubMed] [Google Scholar]
- 26.Morante K, Caaveiro JMM, Tanaka K, González-Mañas JM, Tsumoto K. 2015. A pore-forming toxin requires a specific residue for its activity in membranes with particular physicochemical properties. J. Biol. Chem. 290, 10 850–10 861. ( 10.1074/jbc.M114.615211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morante K, et al. 2016. Identification of a membrane-bound prepore species clarifies the lytic mechanism of actinoporins. J. Biol. Chem. 291, 19 210–19 219. ( 10.1074/jbc.M116.734053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alegre-Cebollada J, Cunietti M, Herrero-Galán E, Gavilanes JG, Martínez-del-Pozo Á. 2008. Calorimetric scrutiny of lipid binding by sticholysin II toxin mutants. J. Mol. Biol. 382, 920–930. ( 10.1016/j.jmb.2008.07.053) [DOI] [PubMed] [Google Scholar]
- 29.Rojko N, Dalla Serra M, Maček P, Anderluh G. 2016. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta 1858, 446–456. ( 10.1016/j.bbamem.2015.09.007) [DOI] [PubMed] [Google Scholar]
- 30.Wloka C, Mutter NL, Soskine M, Maglia G. 2016. Alpha-helical fragaceatoxin C nanopore engineered for double-stranded and single-stranded nucleic acid analysis. Angew. Chem. Int. Ed. Engl. 55, 12 494–12 498. ( 10.1002/anie.201606742) [DOI] [PubMed] [Google Scholar]
- 31.Bellomio A, Morante K, Barlič A, Gutiérrez-Aguirre I, Viguera AR, González-Mañas JM. 2009. Purification, cloning and characterization of fragaceatoxin C, a novel actinoporin from the sea anemone Actinia fragacea. Toxicon 54, 869–880. ( 10.1016/j.toxicon.2009.06.022) [DOI] [PubMed] [Google Scholar]
- 32.de los Ríos V, Mancheño JM, del Pozo AM, Alfonso C, Rivas G, Oñaderra M, Gavilanes JG. 1999. Sticholysin II, a cytolysin from the sea anemone Stichodactyla helianthus, is a monomer–tetramer associating protein. FEBS Lett. 455, 27–30. ( 10.1016/S0014-5793(99)00846-7) [DOI] [PubMed] [Google Scholar]
- 33.Morante K, Caaveiro JMM, Viguera AR, Tsumoto K, González-Mañas JM. 2015. Functional characterization of Val60, a key residue involved in the membrane-oligomerization of fragaceatoxin C, an actinoporin from Actinia fragacea. FEBS Lett. 589, 1840–1846. ( 10.1016/j.febslet.2015.06.012) [DOI] [PubMed] [Google Scholar]
- 34.Ito M, Ikegami Y, Yamagata T. 1991. Activator proteins for glycosphingolipid hydrolysis by endoglycoceramidases—elucidation of biological functions of cell-surface glycosphingolipids in situ by endoglycoceramidases made possible using these activator proteins. J. Biol. Chem. 266, 7919–7926. [PubMed] [Google Scholar]
- 35.Drickamer K, Taylor ME. 2002. Glycan arrays for functional glycomics. Genome Biol. 3, 1–4. ( 10.1186/gb-2002-3-12-reviews1034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. ( 10.1107/S0021889807021206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Struct. Biol. 53, 240–255. ( 10.1107/S0907444996012255) [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Struct. Biol. 66, 486–501. ( 10.1107/S0907444910007493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. ( 10.1107/S0021889892009944) [DOI] [Google Scholar]
- 40.Dunn BM, Chaiken IM. 1974. Quantitative affinity chromatography. Determination of binding constants by elution with competitive inhibitors. Proc. Natl Acad. Sci. USA 71, 2382–2385. ( 10.1073/Pnas.71.6.2382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alm I, García-Linares S, Gavilanes JG, Martínez-del-Pozo Á, Slotte JP. 2015. Cholesterol stimulates and ceramide inhibits Sticholysin II-induced pore formation in complex bilayer membranes. Biochim. Biophys. Acta 1848, 925–931. ( 10.1016/j.bbamem.2014.12.017) [DOI] [PubMed] [Google Scholar]
- 42.Bernheimer AW, Avigad LS. 1976. Properties of a toxin from the sea anemone Stoichacis helianthus, including specific binding to sphingomyelin. Proc. Natl Acad. Sci. USA 73, 467–471. ( 10.1073/Pnas.73.2.467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tejuca M, Dalla Serra M, Potrich C, Alvarez C, Menestrina G. 2001. Sizing the radius of the pore formed in erythrocytes and lipid vesicles by the toxin sticholysin I from the sea anemone Stichodactyla helianthus. J. Membr. Biol. 183, 125–135. ( 10.1007/S00232-001-0060-Y) [DOI] [PubMed] [Google Scholar]
- 44.Belmonte G, Pederzolli C, Maček P, Menestrina G. 1993. Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J. Membr. Biol. 131, 11–22. ( 10.1007/Bf02258530) [DOI] [PubMed] [Google Scholar]
- 45.Kobe A, Caaveiro JMM, Tashiro S, Kajihara D, Kikkawa M, Mitani T, Tsumoto K. 2013. Incorporation of rapid thermodynamic data in fragment-based drug discovery. J. Med. Chem. 56, 2155–2159. ( 10.1021/jm301603n) [DOI] [PubMed] [Google Scholar]
- 46.García-Ortega L, Alegre-Cebollada J, García-Linares S, Bruix M, Martínez-del-Pozo Á, Gavilanes JG. 2011. The behavior of sea anemone actinoporins at the water–membrane interface. Biochim. Biophys. Acta 1808, 2275–2288. ( 10.1016/j.bbamem.2011.05.012) [DOI] [PubMed] [Google Scholar]
- 47.Asensio JL, Ardá A, Cañada FJ, Jiménez-Barbero J. 2013. Carbohydrate–aromatic interactions. Acc. Chem. Res. 46, 946–954. ( 10.1021/ar300024d) [DOI] [PubMed] [Google Scholar]
- 48.Esko JD, Linhardt RJ. 2009. Proteins that bind sulfated glycosaminoglycans. In Essentials of glycobiology (eds Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME), 2nd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- 49.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549. ( 10.1093/nar/gkq366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutiérrez-Aguirre I, Trontelj P, Maček P, Lakey JH, Anderluh G. 2006. Membrane binding of zebrafish actinoporin-like protein: AF domains, a novel superfamily of cell membrane binding domains. Biochem. J. 398, 381–392. ( 10.1042/BJ20060206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DuMont AL, Torres VJ. 2014. Cell targeting by the Staphylococcus aureus pore-forming toxins: it's not just about lipids. Trends Microbiol. 22, 21–27. ( 10.1016/j.tim.2013.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weis WI, Drickamer K. 1996. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 65, 441–473. ( 10.1146/annurev.bi.65.070196.002301) [DOI] [PubMed] [Google Scholar]
- 53.García-Linares S, Palacios-Ortega J, Yasuda T, Åstrand M, Gavilanes JG, Martínez-del-Pozo Á, Slotte JP. 2016. Toxin-induced pore formation is hindered by intermolecular hydrogen bonding in sphingomyelin bilayers. Biochim. Biophys. Acta 1858, 1189–1195. ( 10.1016/j.bbamem.2016.03.013) [DOI] [PubMed] [Google Scholar]
- 54.Maula T, et al. 2013. 2NH and 3OH are crucial structural requirements in sphingomyelin for sticholysin II binding and pore formation in bilayer membranes. Biochim. Biophys. Acta 1828, 1390–1395. ( 10.1016/j.bbamem.2013.01.018) [DOI] [PubMed] [Google Scholar]
- 55.Tanaka K, Caaveiro JMM, Tsumoto K. 2015. Bidirectional transformation of a metamorphic protein between the water-soluble and transmembrane native states. Biochemistry 54, 6863–6866. ( 10.1021/acs.biochem.5b01112) [DOI] [PubMed] [Google Scholar]
- 56.Shephard KL. 1994. Functions for fish mucus. Rev. Fish Biol. Fish. 4, 401–429. ( 10.1007/Bf00042888) [DOI] [Google Scholar]
- 57.Van de Winkel JGJ, Van Kuppevelt THMSM, Janssen HMJ, Lock RAC. 1986. Glycosaminoglycans in the skin mucus of rainbow trout (Salmo gairdneri). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 85, 473–475. ( 10.1016/0305-0491(86)90030-1) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors for the structures of FraC in complex with GlcNAc(6S) have been deposited in the PDB under accession code 5GWF.