

Abstract
The permeabilization of the mitochondrial outer membrane by Bax and Bak during apoptosis is considered a key step and a point of no return in the signalling pathway. It is always closely related to the reorganization of mitochondrial cristae that frees cytochrome c to the intermembrane space and to massive mitochondrial fragmentation mediated by the dynamin-like protein Drp1. Despite multiple evidence in favour of a functional link between these processes, the molecular mechanisms that connect them and their relevance for efficient apoptosis signalling remain obscure. In this review, we discuss recent progress on our understanding of how Bax forms pores in the context of Drp1-stabilized signalling platforms at apoptotic foci in mitochondria.
This article is part of the themed issue ‘Membrane pores: from structure and assembly, to medicine and technology’.
Keywords: Bcl-2, MOMP, mitochondrial fragmentation, cristae remodelling, membrane curvature
1. Introduction: role of mitochondria in apoptosis
Mitochondria are eukaryotic organelles that function as essential hubs in the coordination of a wide variety of metabolic reactions and signalling processes within the cell. For instance, they are considered the powerhouse of the cell due to their important role in respiration (i.e. ATP production). Among other things, they are also key players in lipid biosynthesis, calcium homeostasis and in the intrinsic pathway of a type of programmed cell death termed apoptosis.
Mitochondria are composed of a double [1–4] membrane system known as the mitochondrial outer and inner membranes (MOM and MIM, respectively). These membranes differ in protein and lipid composition as well as in morphology [5]. The transmembrane protein channels at the MOM make this membrane fluid and permeable to small polar molecules (up to 3–5 kDa). In contrast, the MIM shows restricted metabolite permeability and is folded into invaginations called cristae where specific proteins involved in relevant mitochondrial functions are located (e.g. the components of the electron chain transport, including cytochrome c, and the ATP synthase) [6]. The proton gradient that drives the ATP synthase during oxidative phosphorylation is indeed created and maintained across the MIM. The two spaces enclosed by the MOM and MIM are known as the mitochondrial matrix, which is inside the MIM and hosts the mitochondrial DNA, ribosomes and metabolic enzymes, and the intermembrane space, with a composition similar to the cytosol. Both membranes are connected at specific contact sites where the exchange of proteins and lipids between both membranes is enhanced [7]. Cardiolipin (CL) is an anionic phospholipid with four acyl chains that is specifically located at mitochondria of eukaryotic cells. It is mainly enriched in the MIM and found at lower levels at the MOM [8]. However, due to its special structure, it has been proposed that mitochondrial contact sites are enriched in CL and also phosphatidylethanolamine (PE), which share in common their tendency to form non-lamellar structures. This ability might allow them to stabilize the connections between both membranes, and they have also been proposed to play an important role as specific death foci during apoptosis [5,8–13].
It is widely assumed that mitochondrial shape and ultrastructure are closely linked to function. In this regard, mitochondria are able to change their morphology depending on the cell type and in response to cellular signalling and differentiation. They can vary from an interconnected filamentous network to separated spherical structures, where the cytoskeleton plays a crucial role in their morphological plasticity by controlling their distribution, motility and dynamics [14,15]. In particular, mitochondria undergo dramatic alterations in their structure and function under apoptotic stimuli [1,4,16,17].
Apoptosis at mitochondria is largely controlled by the members of the Bcl-2 family. They are mainly located at the MOM, where they form a complex interaction network whose outcome controls MOM permeabilization (MOMP). Concretely, the pro-apoptotic Bcl-2 proteins Bax and Bak are the direct executioners of this event [18–20]. Upon activation in apoptosis, they permeabilize the MOM and enable the release of the so-called apoptotic factors, including cytochrome c or SMAC/DIABLO, from the intermembrane space into the cytosol. This is considered the point of no return in the cell's commitment to death, and it leads to the activation of the caspase cascade, the dismantling of the cellular components and finally to cell death [21].
Besides MOMP, mitochondria also undergo a number of additional alterations during apoptosis, such as changes in lipid transfer between the MIM and the MOM [22], and between the mitochondria and other organelles [23], loss of mitochondrial function, including loss of transmembrane potential and incapability to maintain calcium homeostasis [24,25], mitochondrial fragmentation and cristae remodelling [1,4,16,17,26]. In this review, we focus on the current understanding of three mechanisms, mitochondrial fragmentation, cristae remodelling and MOMP, which occur close in time at specific mitochondrial foci during apoptosis. We pay special attention to new evidence related to Bax pore formation and the interconnection between these processes.
2. Structural changes in mitochondria during apoptosis
One of the first mitochondrial changes during apoptosis affects the lipid composition of its membranes. This alteration occurs at two different levels, involving the modification of the lipid species at mitochondria, as well as the lipid transfer between MIM and MOM, or between mitochondria and other cellular organelles (i.e. endoplasmic reticulum (ER)) [27]. At an early stage, there is an increased CL transfer from the MIM to the MOM [22,28]. This is accompanied by an early oxidation of CL mediated by the specific peroxidase activity of CL-bound cytochrome c, which plays a role in the release of the apoptotic factors [22,29]. Although it has been suggested that mitochondrial contact sites might be the specific foci for CL recruitment to the MOM, it is not clear yet what is the process that induces CL translocation [7]. One possible explanation for CL redistribution from the MIM to the MOM could be associated with the non-lamellar structures proposed for the membrane fusion sites between both membranes at these foci [7]. Other lipid alterations, like the accumulation at mitochondria of products of sphingolipid metabolism transferred from the ER, have also been shown to affect Bax/Bak activation and MOMP [30,31]. Indeed, the mitochondria/ER contact sites have been proposed to organize into lipid microdomains where proteins involved in mitochondrial fission are recruited [32,33]. At these sites, lipids and calcium are exchanged during apoptosis. For example, increased calcium flux from the ER to the mitochondria can easily derive in apoptosis by inducing the generation of CL microdomains, reactive oxygen species and CL peroxidation [29,34] (figure 1).
Figure 1.

Scheme of structural changes in mitochondria during apoptosis: MOMP (a), cristae remodelling (b), mitochondrial fragmentation (bottom).
(a). Mitochondrial outer membrane permeabilization
The members of the Bcl-2 family are key mediators of the process that involves the permeabilization of the mitochondrial outer membrane. There are about 20 proteins in this family, which are further classified into three groups depending on their role in apoptosis: (i) anti-apoptotic proteins (e.g. Bcl-2, Bcl-xL and Mcl-1) that inhibit apoptosis by interacting with the pro-apoptotic proteins; (ii) the pro-apoptotic members Bax and Bak, which are considered direct executioners of MOMP by directly affecting the membrane integrity; and (iii) the pro-apoptotic BH3-only proteins (e.g. Bid, Bim, Bad and Bik), which have evolved to sense apoptotic stimuli and are able to induce Bax/Bak activation directly and/or by blocking the function of anti-apoptotic proteins during apoptosis [19,35,36].
Under physiological conditions, Bax and Bak are continuously translocating between cytosol and mitochondria in a steady state. Differences in the translocation rates lead to a major population of Bax being cytosolic in healthy cells, whereas most Bak molecules locate at the MOM [37,38]. In the presence of apoptotic stimuli, BH3-only proteins trigger the activation of Bax and Bak, leading to their recruitment and accumulation at discrete mitochondrial foci. There, Bax and Bak undergo conformational changes that allow extensive insertion into the membrane, oligomerization and pore formation. These sequential steps lead to MOMP and to the subsequent release of apoptotic factors [19,39]. The anti-apoptotic proteins of the Bcl-2 family block this process by acting at different levels, which include: (i) promoting the retrotranslocation of Bax and Bak from the MOM, (ii) sequestering the BH3-only activators, and (iii) directly binding and inhibiting the oligomerization of Bax and Bak at the MOM [40–44].
The Bcl-2 interaction network is very complex and many models have been postulated in order to explain how they orchestrate MOMP. The ‘direct activation’ model [45,46] suggests that Bax and Bak require direct interaction with activator BH3-only proteins in order to become activated. In contrast, the ‘neutralization’ model [47] assumes that Bax and Bak are constitutively active but kept in check by the pro-survival family members. Induction of the BH3-only proteins would then displace the executioners from these inhibitory complexes and promote MOMP. In addition, the ‘embedded together’ model [48,49] emphasizes the role of the membrane as the ‘locus of action’ for MOMP and Bcl-2 interactions, where CL also plays a role in Bax/Bak action [11,39,50,51]. Hence, the membrane environment is fully required to achieve the active conformation and stoichiometry of the complexes between Bcl-2 proteins, which will then determine whether apoptosis is induced or not [49,52]. More recently, the ‘unified’ and ‘hierarchical’ models [53,54] provide more sophisticated scenarios that account for the relative importance of the interactions between Bcl-2 proteins in the cytosol and the membrane environment.
(b). Mitochondrial fragmentation: a constant event in apoptosis
During apoptosis, and close in time with MOMP, mitochondria undergo massive fragmentation, which appears to be universally associated with this type of cell death [55,56]. In mammals, the main protein responsible for mitochondrial fission is a GTPase mechanoenzyme called dynamin-related protein 1 (Drp1) [57]. This protein shuttles between the cytosol and the MOM, where it binds when mitochondrial fission is required. Once at the MOM, it associates with specific adaptors and it oligomerizes into a spiral-like scaffold around the fission sites in mitochondria leading to organelle division [58]. From the literature currently available, it seems that different Drp1 adaptors can independently recruit Drp1 to the MOM, like mitochondrial fission factor (Mff) [59,60] and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51, respectively) [61–63]. Recent studies suggested that the role of each Drp1 adaptor in the recruitment of Drp1 to the MOM could represent distinct cellular pathways [64,65]. The complexity of this process is further increased with new evidence underscoring that fission sites are determined by interactions between mitochondria and ER [33]. These sites, which had already been established as sites for lipid and calcium transfer, are now considered as functional platforms where the coordinated polymerization of actin filaments could drive Drp1 assembly to mediate mitochondrial fission [66,67].
In the apoptotic context, Drp1 transitions from fast recycling to stable membrane association. This phenomenon seems to occur downstream of the recruitment of Bax and Bak but before cytochrome c release [68]. It is achieved thanks to the SUMOylation (small ubiquitin-like modifier) activity of MAPL, a mitochondrial anchored RING-finger containing protein with SUMO E3 ligase activity [69]. Thus, MAPL-induced covalent SUMOylation of Drp1 enables the stabilization of Drp1 scaffolds on ER-mitochondria contact sites [70,71]. Interestingly, Drp1 SUMOylation co-localizes with Bax/Bak at defined mitochondrial foci during cell death. However, the relevance of Drp1 recruitment to these foci for apoptosis progression remains undefined (see below). This notion has been challenged by some authors who reported little [72] or partial [73] resistance to apoptosis in Drp1-deficient cells, and Drp1-independent mitochondrial fission Drp1 knockout cells [73].
(c). Release of cytochrome c by cristae remodelling
The release of the apoptotic factors from the intramembrane space is dependent on their location within mitochondria [74]. For instance, SMAC/DIABLO is located at the inner boundary area of mitochondria, whereas cytochrome c is mainly sequestered at cristae structures. Consistent with this notion, Bax/Bak-induced MOMP can directly provide the release of SMAC/DIABLO, while the egress of cytochrome c, which is located within the cristae space, requires the remodelling of the highly stacked cristae structures [74–76].
The structure of the cristae junctions that physically separate the cristae contents from the intermembrane space is stabilized by optic atrophy 1 (OPA1)-complex formation [77,78]. OPA1 is a GTPase located at the MIM, where it is responsible for MIM fusion and for the maintenance of cristae junction integrity [75,77,79]. During apoptosis, OPA1 oligomers are disassembled, cristae become open to the intermembrane space and cytochrome c is finally released from these structures to the cytosol [77,80]. It has been described that this process is closely linked to mitochondrial fragmentation [55,81,82] and downstream of Drp1 fission activity [83]. However, it is still unclear how cristae remodelling is regulated during apoptosis.
Previous investigations have speculated that some BH3-only proteins from the Bcl-2 family could also be involved in this process [16,77,84,85]. For instance, it has been described that the lipid transfer activity of tBid may help for CL translocation from MIM to MOM, favouring CL redistribution into microdomains and generating local alterations in the membrane curvature, somehow promoting cytochrome c release [12,86–88]. Similarly, another BH3-only protein termed Bik also seems to induce dynamic mitochondrial transformation, initiating the cristae remodelling regulated by Drp1 [85]. The latter study, together with recent reports, highlights the essential role that Drp1 may play in this process [71,74,76,89]. In fact, it has been proposed that the membrane remodelling function of Drp1 in this process could be different from its recognized role in regulating mitochondrial fission [85], although some authors suggest that both membrane constriction and lipid remodelling activities are also required for membrane fission [90,91]. Along these lines, current research focused on Drp1 adaptors has shed light on the specific contribution of MiD49/51 to this process [89]. Experiments performed in knockout cells for Drp1 or these adaptors confirmed that Drp1-mitochondrial fission through MiD49/51, but not Mff, facilitated cristae remodelling required for cytochrome c release during intrinsic apoptosis. Disassembly of OPA1 oligomers was not sufficient to enable cytochrome c release from the cristae space. Moreover, both the transmembrane domain and the Drp1-binding site of MiD51 were essential for cytochrome c release, suggesting that this adaptor may function as a spatial-temporal coupler to the cristae remodelling machinery in the presence of Drp1 [89].
3. Bax pore formation during apoptosis
The molecular mechanism by which Bax (and Bak) mediate MOMP has remained one of the key questions in the field. A lot has been learned from in vitro reconstituted systems, where Bax displays membrane-permeabilizing activity characterized by the formation of large and long-lived membrane pores, which are tunable in size and of toroidal nature [42,51,92,93]. Of note, the toroidal pore model implies that not only proteins, but also lipid molecules are exposed at the rim of the pore and thus also play a role in pore dimensions and stability [94]. Only recently Bax-mediated membrane pores could be directly visualized in the mitochondria of apoptotic cells [76,95].
(a). Assembly path of Bax
Bax/Bak activation is an essential step towards MOMP. This process involves the change from a monomeric, globular structure found in the cytosol or loosely associated to the MOM, into a partially unfolded, membrane-inserted, oligomeric conformation capable of disrupting the membrane integrity. Several lines of evidence support the role of the membrane to provide the right physical–chemical environment that stabilizes the active conformation of Bax [39,41,96]. It is therefore difficult to conceive that the active structure of Bax is long-lived in the absence of a lipid bilayer or a chemical environment that mimics it.
This also implies that Bax activation first starts with membrane association. Bax binds to the membrane as a monomer and rapidly self-assembles into higher oligomers [44]. The conformational changes that allow Bax activation are promoted by interaction with activator BH3-only proteins, which bind via their BH3 domains to a conserved hydrophobic groove or to a rear pocket situated between helices α1 and α6 [97,98]. Binding to an activator BH3-only protein is thought to induce a conformational change that displaces the hydrophobic C-terminal helix α9 from the hydrophobic groove, which inserts into the membrane and acts as an anchor to the MOM [99]. This is followed by exposure of the N-terminal helix α1 and rearrangement of the α2/BH3 domain. As a result, the BH3 domain of one Bax molecule is exposed and displaces the activator BH3-only from the groove in another Bax molecule, thus leading to the formation of Bax dimers. Structural data of active Bax in the membrane reveal a well-defined ‘dimerization’ or ‘core’ domain stabilized via BH3 domain/groove interactions, and a rather flexible ‘piercing’ or ‘latch’ domain, which unfolds from the rest of the protein via partial opening of the hairpin of helices α5 and α6 [98,100]. As a result, most of activated Bax lies on the membrane surface shallowly inserted, while only the C-terminal domain is believed to adopt a canonical transmembrane orientation [101].
After dimer formation, Bax continues to self-assemble in order to form higher order oligomers. Stoichiometry analysis at the single-molecule level revealed that Bax does not adopt a unique oligomeric form in the membrane, but rather organizes into a mixture of species that are based on dimer units [44]. While cBid was important for activation, it did not affect the final distribution of oligomers. In contrast, Bcl-xL was able to disassemble existing Bax complexes, suggesting that Bax oligomerization is a reversible process. The question remains whether there is a maximum or preferred oligomeric state for Bax. In addition, the role of Bak and other interacting proteins on Bax assembly needs further investigation (figure 2).
Figure 2.
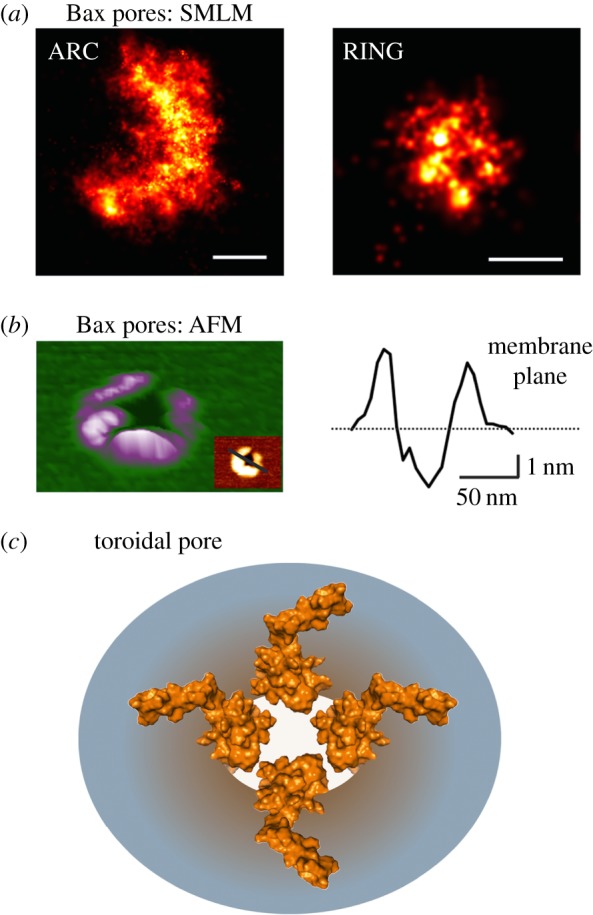
Bax pore architecture. (a) Magnified reconstructed super-resolution images of Bax pores in apoptotic HeLa cells corresponding to arc (left) and ring (right) structures (with permission from [95]). SMLM stands for Single Molecule Localization Microscopy. Scale bars, 100 nm. (b) Three-dimensional atomic force microscopy (AFM) topography of a Bax structure in a supported lipid bilayer (right) and its height profile (left) corresponding to the pore rim. (c) Representative scheme of Bax toroidal pore (top view) where both Bax dimers and lipids cooperate for pore formation. The structure of active, membrane-inserted Bax dimer is used based on the ‘clamp’ model [100].
(b). Bax pore architecture
Concomitant with oligomerization, Bax induces the formation of membrane pores that mediate MOMP. These pores were recently visualized in the mitochondria of apoptotic cells thanks to superresolution microscopy [76,95]. Permeabilized MOM contained large openings heterogeneous in size that, importantly, included Bax molecules enriched at the pore rim. These structures were very similar to membrane pores induced by Bax in liposomes and supported bilayers [95,102], suggesting that the properties of Bax pores discovered from in vitro studies also apply to the mitochondrial pores. For example, the heterogeneity of pore size in mitochondria is in line with in vitro experiments with Bax, where it was shown that Bax is capable of forming pores of tunable size. This fits well with a toroidal pore model where Bax oligomers reduce the stress caused by membrane distortion and curvature formation at the pore edge [51,92,93]. Nevertheless, it is likely that in the context of the cell, other proteins, like Bak or Drp1, and maybe lipids, also participate in MOM pores thereby modulating the features of Bax-mediated pores. Indeed, in the absence of Drp1, Bax continued to form large pores but cytochrome c release was hampered [76].
Bax clusters at the MOM exhibited a diverse mixture of distinct structures, including lines, arcs and full rings, which correlated well with Bax assemblies detected in vitro [95]. All these structures could represent different stages in the assembly of nascent evolving Bax pores. Bax molecules would initially organize into line and arc structures that further evolve into full rings. However, it cannot be discarded that they correspond to kinetically trapped intermediates during the assembly process. Interestingly, not only full rings but also a significant fraction of arc-shaped structures correlated with membrane pores. The architecture of the arc-shaped pores implies that Bax oligomers only cover partially the pore edge, which results in a fraction of the pore structure being formed only by lipids. This partial coverage of the pore edge by protein molecules constituted a demonstration of the toroidal pore model for Bax and confirmed the continuous membrane organization at the pore rim detected by X-ray for a peptide derived from helix α5 of Bax [103], which reproduces the pore activity of the full-length protein [104,105]. This reveals a scenario where Bax forms oligomers that are dynamic and heterogeneous, and that give rise to membrane pores of diverse sizes. Such diversity is in good agreement with the variety of oligomeric species detected at the membrane and with the flexible nature of the piercing domain in the structure of active Bax. Indeed, the recently proposed three-dimensional model for active, membrane-inserted Bax (i.e. the ‘clamp’ model based on double electron–electron resonance spectroscopy data) provides a physical–chemical basis for the stabilization of the pore edge by Bax assemblies at the membrane [100]. However, it is still unclear which is the minimum amount of Bax molecules able to stabilize a membrane pore capable of releasing the apoptotic factors. Studies with lipid nanodiscs suggest that it could be a number as small as monomers or dimers, although the exact assembly state of Bax in those systems was not conclusive [106].
4. Mechanistic link between mitochondrial structural changes during apoptosis
To date, several contradictory studies challenge the interplay between the alterations occurring at the mitochondria during intrinsic apoptosis. Among the open questions, it still remains unresolved whether the concentration of CL at the MOM under apoptotic conditions resembles the high concentration used in vitro to regulate the activity of Bcl-2 proteins. Similarly, there is an active debate regarding the link between MOMP and mitochondrial fragmentation. Although these two events occur very close in time and space, a number of reports in the literature suggest that they may not be strictly correlated [73,107–109].
However, a recent study brings together mitochondrial alterations with the efficient execution of apoptosis [71]. The authors claim that it is not necessarily mitochondrial fission that is required for Bax/Bak-mediated cytochrome c release, but rather the stabilization of specific interorganellar platforms that favour metabolite flux and membrane curvature. This is in line with previous work showing that hyperfragmented mitochondria could not support Bax-induced cytochrome c release because ER/mitochondria contact sites were abolished [110,111]. Similarly, Drp1 promotes tethering and hemifusion of membranes in vitro that might be linked to the stimulation of tBid-induced Bax oligomerization and cytochrome release during apoptosis [112,113]. Consistent with this notion, Bax/Bak-induced Drp1 SUMOylation at the ER/mitochondria interface stabilizes signalling platforms for calcium and lipid transfer between both organelles, as well as for cristae remodelling and cytochrome c release [30,71,114].
The current data available support two alternative models for the role of Drp1 in apoptosis: (i) Drp1-mediated cristae remodelling, independent of Bax activity; and (ii) Drp1-induced Bax oligomerization, independent of cristae remodelling. Nevertheless, the fact that these proteins co-localize at the same mitochondrial foci during apoptosis suggests that there could be a direct interplay between them in order to regulate structural changes at mitochondria in apoptosis. Bcl-2 proteins could cooperate with the mitochondrial fission machinery to efficiently mediate cristae remodelling, fragmentation and MOMP during apoptosis. Further research will be needed to fully understand the molecular mechanisms involved in this interplay (figure 3).
Figure 3.
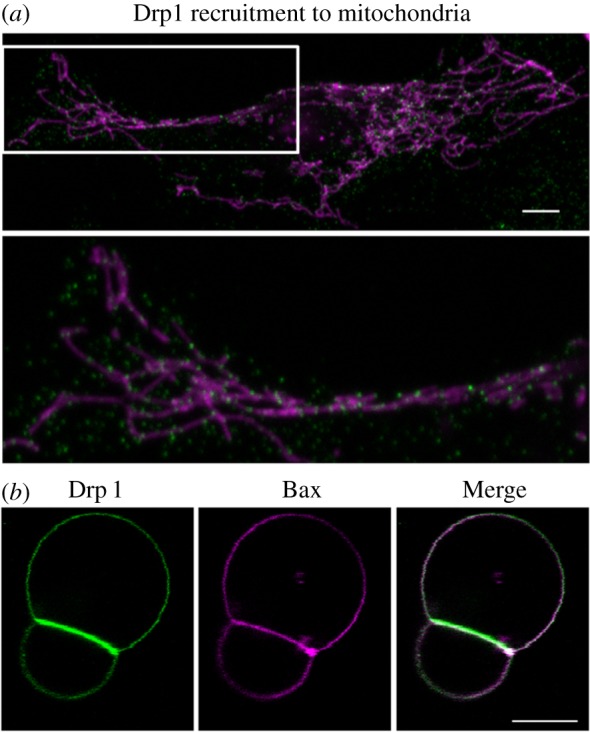
Role of Drp1 in mitochondrial fission and apoptosis. (a) Drp1 shuttles between cytosol and mitochondria for membrane division under physiological conditions. Overview (top panel) and zoomed image of the white box (bottom panel). Immunostaining of endogenous Drp1 (green) and mitochondrial staining with Mitotracker Red (magenta). Scale bar, 5 µm. (b) Drp1 (green) and Bax (magenta) binding to giant unilamellar vesicles composed of phosphatidylcholine: phosphatidylethanolamine: cardiolipin (54 : 20 : 26, mol : mol) at 42° for 30 min. Drp1 and Bax are concentrated at contact surfaces between two vesicles. Bax clusters at highly curved edges between vesicles. Scale bar, 10 µm.
5. Concluding remarks
The multiple alterations that mitochondria undergo during apoptosis include the remodelling of the cristae ultrastructure, the permeabilization of the MOM and dramatic fragmentation. Recent data reveal that during apoptosis Bax undergoes a conformational change and organizes into dimer units with a well-defined dimerization domain and a flexible piercing or latch domain. These dimers continue to self-assemble into multiple oligomeric species that organize into diverse structures at the MOM, including lines, arcs and rings, and concentrate at distinct foci. Both arcs and rings delineate membrane pores that mediate the release of the apoptotic factors. In parallel, Drp1 also becomes enriched in these foci where, together with MiD49/51, it stabilizes signalling platforms that modulate lipid and calcium exchange between the ER and mitochondria and play a role in mitochondrial fragmentation and in the reorganization of the MIM cristae. Exciting research lies ahead to uncover the molecular mechanisms that interconnect these processes and their functional relevance for efficient apoptosis induction.
Acknowledgements
We thank K. Cosentino and R. Salvador-Gallego for kindly providing images for the figures.
Authors' contributions
A.J.G.-S. and B.U.-U. conceived and wrote the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the DFG (FOR2036), the ERC (StG N°309966) and the Government of the Basque Country (fellowship for B.U.-U.).
References
- 1.Martinou JC, Youle RJ. 2011. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 21, 92–101. ( 10.1016/j.devcel.2011.06.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osman C, Voelker DR, Langer T. 2011. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16. ( 10.1083/jcb.201006159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glancy B, Balaban RS. 2012. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. ( 10.1021/bi2018909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ugarte-Uribe B, García-Sáez AJ. 2014. Membranes in motion: mitochondrial dynamics and their role in apoptosis. Biol. Chem. 395, 297–311. ( 10.1515/hsz-2013-0234) [DOI] [PubMed] [Google Scholar]
- 5.Horvath SE, Daum G. 2013. Lipids of mitochondria. Prog. Lipid Res. 52, 590–614. ( 10.1016/j.plipres.2013.07.002) [DOI] [PubMed] [Google Scholar]
- 6.Ow Y-LP, Green DR, Hao Z, Mak TW. 2008. Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542. ( 10.1038/nrm2434) [DOI] [PubMed] [Google Scholar]
- 7.Reichert AS, Neupert W. 2002. Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim. Biophys. Acta 1592, 41–49. ( 10.1016/S0167-4889(02)00263-X) [DOI] [PubMed] [Google Scholar]
- 8.Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. 1990. Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265, 18 797–18 802. [PubMed] [Google Scholar]
- 9.Van Venetië R, Verkleij AJ. 1982. Possible role of non-bilayer lipids in the structure of mitochondria. A freeze-fracture electron microscopy study. Biochim. Biophys. Acta 692, 397–405. ( 10.1016/0005-2736(82)90390-X) [DOI] [PubMed] [Google Scholar]
- 10.Aguilar L, et al. 1999. Phospholipid membranes form specific nonbilayer molecular arrangements that are antigenic. J. Biol. Chem. 274, 25 193–25 196. ( 10.1074/jbc.274.36.25193) [DOI] [PubMed] [Google Scholar]
- 11.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. 2000. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. 2, 754–761. ( 10.1038/35036395) [DOI] [PubMed] [Google Scholar]
- 12.Gonzalvez F, et al. 2005. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 12, 614–626. ( 10.1038/sj.cdd.4401571) [DOI] [PubMed] [Google Scholar]
- 13.Unsay JD, Cosentino K, Subburaj Y, García-Sáez AJ. 2013. Cardiolipin effects on membrane structure and dynamics. Langmuir 29, 15 878–15 887. ( 10.1021/la402669z) [DOI] [PubMed] [Google Scholar]
- 14.Anesti V, Scorrano L. 2006. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 1757, 692–699. ( 10.1016/j.bbabio.2006.04.013) [DOI] [PubMed] [Google Scholar]
- 15.Boldogh IR, Pon LA. 2007. Mitochondria on the move. Trends Cell Biol. 17, 502–510. ( 10.1016/j.tcb.2007.07.008) [DOI] [PubMed] [Google Scholar]
- 16.Suen DF, Norris KL, Youle RJ. 2008. Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. ( 10.1101/gad.1658508) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cosentino K, García-Sáez AJ. 2014. Mitochondrial alterations in apoptosis. Chem. Phys. Lipids 181, 62–75. ( 10.1016/j.chemphyslip.2014.04.001) [DOI] [PubMed] [Google Scholar]
- 18.García-Sáez AJ, Fuertes G, Suckale J, Salgado J. 2010. Permeabilization of the outer mitochondrial membrane by Bcl-2 proteins. Adv. Exp. Med. Biol. 677, 91–105. ( 10.1007/978-1-4419-6327-7_8) [DOI] [PubMed] [Google Scholar]
- 19.García-Sáez AJ. 2012. The secrets of the Bcl-2 family. Cell Death Differ. 19, 1733–1740. ( 10.1038/cdd.2012.105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamas-Din A, Bindner S, Zhu W, Zaltsman Y, Campbell C, Gross A, Leber B, Andrews DW, Fradin C. 2013. tBid undergoes multiple conformational changes at the membrane required for Bax activation. J. Biol. Chem. 288, 22 111–22 127. ( 10.1074/jbc.M113.482109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei MC, et al. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. ( 10.1126/science.1059108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagan VE, et al. 2005. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. ( 10.1038/nchembio727) [DOI] [PubMed] [Google Scholar]
- 23.Hoppins S, Nunnari J. 2012. Cell biology. Mitochondrial dynamics and apoptosis—the ER connection. Science 337, 1052–1054. ( 10.1126/science.1224709) [DOI] [PubMed] [Google Scholar]
- 24.Wang X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15, 2922–2933. [PubMed] [Google Scholar]
- 25.Green DR, Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305, 626–629. ( 10.1126/science.1099320) [DOI] [PubMed] [Google Scholar]
- 26.Wasilewski M, Scorrano L. 2009. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Metab. 20, 287–294. ( 10.1016/j.tem.2009.03.007) [DOI] [PubMed] [Google Scholar]
- 27.Crimi M, Esposti MD. 2011. Apoptosis-induced changes in mitochondrial lipids. Biochim. Biophys. Acta 1813, 551–557. ( 10.1016/j.bbamcr.2010.09.014) [DOI] [PubMed] [Google Scholar]
- 28.Fernandez GM, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. 2002. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 13, 449–455. [PubMed] [Google Scholar]
- 29.Grijalba MT, Vercesi AE, Schreier S. 1999. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 38, 13 279–13 287. ( 10.1021/bi9828674) [DOI] [PubMed] [Google Scholar]
- 30.Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. 2012. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. ( 10.1016/j.cell.2012.01.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tafesse FG, Vacaru AM, Bosma EF, Hermansson M, Jain A, Hilderink A, Somerharju P, Holthuis MJC. 2014. Sphingomyelin synthase-related protein SMSr is a suppressor of ceramide-induced mitochondrial apoptosis. J. Cell Sci. 127, 445–454. ( 10.1242/jcs.138933) [DOI] [PubMed] [Google Scholar]
- 32.Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, Hajnóczky G. 2010. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. ( 10.1016/j.molcel.2010.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. 2011. ER tubules mark sites of mitochondrial division. Science 334, 358–362. ( 10.1126/science.1207385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naon D, Scorrano L. 2014. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 1843, 2184–2194. ( 10.1016/j.bbamcr.2014.05.011) [DOI] [PubMed] [Google Scholar]
- 35.Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. ( 10.1038/nrm2308) [DOI] [PubMed] [Google Scholar]
- 36.Kim H, Tu H-C, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ-D, Cheng EH-Y. 2009. Stepwise activation of BAX and BAK by tBID, BIM, PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499. ( 10.1016/j.molcel.2009.09.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ. 2011. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116. ( 10.1016/j.cell.2011.02.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Todt F, et al. 2015. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 34, 67–80. ( 10.15252/embj.201488806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. 2002. Bid, Bax, lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342. ( 10.1016/S0092-8674(02)01036-X) [DOI] [PubMed] [Google Scholar]
- 40.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. 2008. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 6, e0060147 ( 10.1371/journal.pbio.0060147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084. ( 10.1016/j.cell.2008.11.010) [DOI] [PubMed] [Google Scholar]
- 42.Bleicken S, Wagner C, Garcia-Saez AJ. 2013. Mechanistic differences in the membrane activity of Bax and Bcl-xL correlate with their opposing roles in apoptosis. Biophys. J. 104, 421–431. ( 10.1016/j.bpj.2012.12.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber K, Harper N, Schwabe J, Cohen GM. 2013. BIM-mediated membrane insertion of the BAK pore domain is an essential requirement for apoptosis. Cell Rep. 5, 409–420. ( 10.1016/j.celrep.2013.09.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subburaj Y, Cosentino K, Axmann M, Pedrueza-Villalmanzo E., Hermann E, Bleicken S, Spatz J, García-Sáez AJ. 2015. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 6, 8042 ( 10.1038/ncomms9042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. ( 10.1016/S1535-6108(02)00127-7) [DOI] [PubMed] [Google Scholar]
- 46.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535. ( 10.1016/j.molcel.2005.02.003) [DOI] [PubMed] [Google Scholar]
- 47.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DCS. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305. ( 10.1101/gad.1304105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leber B, Lin J, Andrews DW. 2007. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12, 897–911. ( 10.1007/s10495-007-0746-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leber B, Lin J, Andrews DW. 2010. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene 29, 5221–5230. ( 10.1038/onc.2010.283) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landeta O, Landajuela A, Gil D, Taneva S, Di Primo C, Sot B, Valle M, Frolov VA, Basañez G. 2011. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J. Biol. Chem. 286, 8213–8230. ( 10.1074/jbc.M110.165852) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bleicken S, Landeta O, Landajuela A, Basanez G, Garcia–Saez AJ. 2013. Proapoptotic Bax and Bak proteins form stable protein-permeable pores of tunable size. J. Biol. Chem. 288, 33 241–33 252. ( 10.1074/jbc.M113.512087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Saez AJ, Ries J, Orzaez M, Perez-Paya E, Schwille P. 2009. Membrane promotes tBID interaction with BCL(XL). Nat. Struct. Mol. Biol. 16, 1178–1185. ( 10.1038/nsmb.1671) [DOI] [PubMed] [Google Scholar]
- 53.Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 44, 517–531. ( 10.1016/j.molcel.2011.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H-C, et al. 2015. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 17, 1270–1281. ( 10.1038/ncb3236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frank S, Gaume B, Bergmann-Leitner ES., Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. ( 10.1016/S1534-5807(01)00055-7) [DOI] [PubMed] [Google Scholar]
- 56.Martinou J-C, Youle RJ. 2006. Which came first, the cytochrome c release or the mitochondrial fission?. Cell Death Differ. 13, 1291–1295. ( 10.1038/sj.cdd.4401985) [DOI] [PubMed] [Google Scholar]
- 57.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. ( 10.1091/mbc.12.8.2245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strack S, Cribbs JT. 2012. Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J. Biol. Chem. 287, 10 990–11 001. ( 10.1074/jbc.M112.342105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gandre-Babbe S, van der Bliek AM. 2008. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 19, 2402–2412. ( 10.1091/mbc.E07-12-1287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. 2010. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. ( 10.1083/jcb.201007152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. 2011. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. ( 10.1038/embor.2011.54) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM. 2013. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl Acad. Sci. USA 110, E1342–E1351. ( 10.1073/pnas.1300855110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loson OC, Song Z, Chen H, Chan DC. 2013. Fis1, Mff, MiD49, MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. ( 10.1091/mbc.E12-10-0721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. 2013. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 288, 27 584–27 593. ( 10.1074/jbc.M113.479873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT. 2016. Cooperative and independent roles of Drp1 adaptors Mff and MiD49/51 in mitochondrial fission. J. Cell Sci. 129, 2170–2181. ( 10.1242/jcs.185165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hatch AL, Gurel PS, Higgs HN. 2014. Novel roles for actin in mitochondrial fission. J. Cell Sci. 127, 4549–4560. ( 10.1242/jcs.153791) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ji W, Hatch AL, Merrill RA, Strack S, Higgs HN. 2015. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4, e11553 ( 10.7554/eLife.11553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wasiak S, Zunino R, McBride HM. 2007. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177, 439–450. ( 10.1083/jcb.200610042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neuspiel M, et al. 2008. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. ( 10.1016/j.cub.2007.12.038) [DOI] [PubMed] [Google Scholar]
- 70.Braschi E, Zunino R, McBride HM. 2009. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 10, 748–754. ( 10.1038/embor.2009.86) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. 2015. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 59, 941–955. ( 10.1016/j.molcel.2015.08.001) [DOI] [PubMed] [Google Scholar]
- 72.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. 2009. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816. ( 10.1083/jcb.200903065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishihara N, et al. 2009. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. ( 10.1038/ncb1907) [DOI] [PubMed] [Google Scholar]
- 74.Ban-Ishihara R., Ishihara T, Sasaki N, Mihara K, Ishihara N. 2013. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl Acad. Sci. USA 110, 11 863–11 868. ( 10.1073/pnas.1301951110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67. ( 10.1016/S1534-5807(01)00116-2) [DOI] [PubMed] [Google Scholar]
- 76.Große L, Wurm CA, Brüser C, Neumann D, Jans DC, Jakobs S. 2016. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35, 402–413. ( 10.15252/embj.201592789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frezza C, et al. 2006. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. ( 10.1016/j.cell.2006.06.025) [DOI] [PubMed] [Google Scholar]
- 78.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. 2008. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, independent of Bak oligomerization. Mol. Cell 31, 557–569. ( 10.1016/j.molcel.2008.07.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. 2004. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl Acad. Sci. USA 101, 15 927–15 932. ( 10.1073/pnas.0407043101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cipolat S, et al. 2006. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175. ( 10.1016/j.cell.2006.06.021) [DOI] [PubMed] [Google Scholar]
- 81.Lee Y, Jeong S-Y, Karbowski M, Smith CL, Youle RJ. 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. ( 10.1091/mbc.E04-04-0294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cassidy-Stone A, et al. 2008. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. ( 10.1016/j.devcel.2007.11.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Estaquier J, Arnoult D. 2007. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 14, 1086–1094. ( 10.1038/sj.cdd.4402107) [DOI] [PubMed] [Google Scholar]
- 84.Arnoult D, Grodet A, Lee Y-J, Estaquier J, Blackstone C. 2005. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J. Biol. Chem. 280, 35 742–35 750. ( 10.1074/jbc.M505970200) [DOI] [PubMed] [Google Scholar]
- 85.Germain M, Mathai JP, McBride HM, Shore GC. 2005. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 24, 1546–1556. ( 10.1038/sj.emboj.7600592) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Esposti MD, Erler JT, Hickman JA, Dive C. 2001. Bid, a widely expressed proapoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol. Cell. Biol. 21, 7268–7276. ( 10.1128/MCB.21.21.7268-7276.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esposti MD, Cristea IM, Gaskell SJ, Nakao Y, Dive C. 2003. Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 10, 1300–1309. ( 10.1038/sj.cdd.4401306) [DOI] [PubMed] [Google Scholar]
- 88.Lutter M, Perkins GA, Wang X. 2001. The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol. 2 ( 10.1186/1471-2121-2-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Otera H, Miyata N, Kuge O, Mihara K. 2016. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 212, 531–544. ( 10.1083/jcb.201508099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bashkirov PV, Akimov SA, Evseev AI, Schmid SL, Zimmerberg J, Frolov VA. 2008. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell 135, 1276–1286. ( 10.1016/j.cell.2008.11.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pucadyil TJ, Schmid SL. 2008. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell 135, 1263–1275. ( 10.1016/j.cell.2008.11.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Basañez G, et al. 1999. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl Acad. Sci. USA 96, 5492–5497. ( 10.1073/pnas.96.10.5492) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Terrones O, Antonsson B, Yamaguchi H, Wang H.-G., Liu J, Lee RM, Herrmann A, Basañez G. 2004. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 279, 30 081–30 091. ( 10.1074/jbc.M313420200) [DOI] [PubMed] [Google Scholar]
- 94.Gilbert RJC, Dalla Serra M, Froelich CJ, Wallace MI, Anderluh G. 2014. Membrane pore formation at protein–lipid interfaces. Trends Biochem. Sci. 39, 510–516. ( 10.1016/j.tibs.2014.09.002) [DOI] [PubMed] [Google Scholar]
- 95.Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, García-Sáez AJ. 2016. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 35, 389–401. ( 10.15252/embj.201593384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O'Neill KL, Huang K, Zhang J, Chen Y, Luo X. 2016. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 30, 973–988. ( 10.1101/gad.276725.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gavathiotis E, et al. 2008. BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081. ( 10.1038/nature07396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Czabotar PE, et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531. ( 10.1016/j.cell.2012.12.031) [DOI] [PubMed] [Google Scholar]
- 99.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. 2010. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell 40, 481–492. ( 10.1016/j.molcel.2010.10.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, García-Sáez AJ, Bordignon E. 2014. Structural model of active Bax at the membrane. Mol. Cell 56, 496–505. ( 10.1016/j.molcel.2014.09.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Westphal D, et al. 2014. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl Acad. Sci. USA 111, E4076–E4085. ( 10.1073/pnas.1415142111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuwana T, Olson NH, Kiosses WB, Peters B, Newmeyer DD. 2016. Pro-apoptotic Bax molecules densely populate the edges of membrane pores. Sci. Rep. 6, 27299 ( 10.1038/srep27299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Qian S, Wang W, Yang L, Huang HW. 2008. Structure of transmembrane pore induced by Bax-derived peptide: evidence for lipidic pores. Proc. Natl Acad. Sci. USA 105, 17 379–17 383. ( 10.1073/pnas.0807764105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.García-Sáez AJ, Coraiola M, Dalla Serra M, Mingarro I, Menestrina G, Salgado J. 2005. Peptides derived from apoptotic Bax and Bid reproduce the poration activity of the parent full-length proteins. Biophys. J. 88, 3976–3990. ( 10.1529/biophysj.104.058008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.García-Sáez AJ, Coraiola M, Serra MD, Mingarro I, Müller P, Salgado J. 2006. Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. FEBS J. 273, 971–981. ( 10.1111/j.1742-4658.2006.05123.x) [DOI] [PubMed] [Google Scholar]
- 106.Xu X-P, Zhai D, Kim E, Swift M, Reed JC, Volkmann N, Hanein D. 2013. Three-dimensional structure of Bax-mediated pores in membrane bilayers. Cell Death Dis. 4, e683 ( 10.1038/cddis.2013.210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parone PA, James DI, Da Cruz S, Mattenberger Y, Donzé O, Barja F, J-C. 2006. Martinou. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol. Cell. Biol. 26, 7397–7408. ( 10.1128/MCB.02282-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Breckenridge DG, Kang B-H, Kokel D, Mitani S, Staehelin LA, Xue D. 2008. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol. Cell 31, 586–597. ( 10.1016/j.molcel.2008.07.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sheridan C, Delivani P, Cullen SP, Martin SJ. 2008. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome c release. Mol. Cell 31, 570–585. ( 10.1016/j.molcel.2008.08.002) [DOI] [PubMed] [Google Scholar]
- 110.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. 2004. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 16, 59–68. ( 10.1016/j.molcel.2004.09.026) [DOI] [PubMed] [Google Scholar]
- 111.Renault TT, et al. 2015. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol. Cell 57, 69–82. ( 10.1016/j.molcel.2014.10.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Montessuit S, et al. 2010. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. ( 10.1016/j.cell.2010.08.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ugarte-Uribe B, Müller H-M, Otsuki M, Nickel W, García-Sáez AJ. 2014. Dynamin-related protein 1 (Drp1) promotes structural intermediates of membrane division. J. Biol. Chem. 289, 30 645–30 656. ( 10.1074/jbc.M114.575779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Colombini M. 2010. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 1797, 1239–1244. ( 10.1016/j.bbabio.2010.01.021) [DOI] [PubMed] [Google Scholar]