

Abstract
A variety of peptides induce pores in biological membranes; the most common ones are naturally produced antimicrobial peptides (AMPs), which are small, usually cationic, and defend diverse organisms against biological threats. Because it is not possible to observe these pores directly on a molecular scale, the structure of AMP-induced pores and the exact sequence of steps leading to their formation remain uncertain. Hence, these questions have been investigated via molecular modelling. In this article, we review computational studies of AMP pore formation using all-atom, coarse-grained, and implicit solvent models; evaluate the results obtained and suggest future research directions to further elucidate the pore formation mechanism of AMPs.
This article is part of the themed issue ‘Membrane pores: from structure and assembly, to medicine and technology’.
Keywords: antimicrobial peptides, pore formation, molecular dynamics, effective energy function 1, implicit membrane model 1
1. Introduction
Pore formation in lipid bilayers occurs in important biological processes, such as apoptosis [1–3], immunity [4,5], bacterial toxin function [6,7], viral infection [8] and protein translocation [9], some of which are described by other articles in this issue. In most of these processes, a soluble protein reconfigures and assembles into a membrane-embedded oligomer. Some available structures [10–12] show a completely proteinaceous pore. However, in other cases, lipids are speculated to participate in the pore's construction [13]. Pores can also be induced in pure lipid bilayers by applying electric fields (i.e. electroporation [14]).
The antibacterial defence mechanisms of a broad range of organisms also seem to involve membrane pore formation. Antimicrobial peptides (AMPs, or host defence peptides) are usually small, cationic peptides that provide a diverse array of immunological functions [15–18]. These amphipathic peptides, which assume various secondary structures, can permeabilize lipid bilayers in vivo [19–21] and in vitro [22–24]. This, together with the observed lack of dependence on amino acid chirality [25,26], led to the suggestion that they target the bacterial membrane, either by forming pores [27] or by dissolving the membrane in a detergent-like fashion (i.e. the carpet mechanism [28]). Their cationic charge is thought to impart selectivity for bacterial membranes, whose exterior lipid leaflet is negatively charged [29]. Whether membrane permeabilization is the actual lethal event is still actively debated [30,31]. Other proposed mechanisms include clustering of ionic lipids [32] and targeting intracellular components, such as DNA [33–35]. Nevertheless, the occurrence of AMP-induced membrane poration is unquestionable, and understanding peptide stabilization of membrane pores has fundamental value independent of its precise role in AMP action. In this article, we will focus on AMPs' membrane-permeabilizing function.
Extensive experimental effort has been invested in characterizing AMPs' membrane interactions and the nature of the pore state. For example, fluorescence measurements have been used to quantify membrane binding and leakage from vesicles [36,37]; fluorescence applied to giant unilamellar vesicles (GUVs) has allowed direct imaging of permeation [38–40]; and fluorescence imaging of live cells has elucidated the sequence of events [31,41]. Calorimetry has provided the thermodynamic properties of membrane binding [42]. Oriented circular dichroism has provided information on peptide orientation with respect to the bilayer normal [43,44]. X-ray diffraction has shown reduced membrane thickness upon peptide binding [45,46] and illustrated the shape of peptide-induced pores [47]. Neutron scattering has provided information on pore size [48]. Electrophysiology studies have described pore ion conductance and its voltage dependence [49–51]. Solution NMR in detergent micelles has provided structures and sometimes described oligomerization propensities [52]. Solid-state NMR (ssNMR) has provided structural and dynamic information in native environments [53,54]. Atomic force and electron microscopy have shown AMP-induced membrane damage [55–57]. However, these pores' lability and transience have prevented the acquisition of an experimental high-resolution structure of an AMP-stabilized pore.
A summary of experiments on the dozens of previously investigated AMPs would be beyond the scope of this review; therefore, we will mostly focus on a few well-studied peptides. Alamethicin is a 20-residue helical peptide of the peptaibol family with charge 0 or −1 [58]. Melittin is a 26-residue cytolytic peptide isolated from bee venom that has low target selectivity [59]. Magainin-2 (hereafter, magainin) is a 23-residue AMP isolated from frog skin that preferentially targets bacterial membranes [60]. Protegrin-1 (hereafter, protegrin) is an 18-residue β-hairpin derived from porcine leucocytes that is stabilized by two disulfide bonds [61]. The latter three peptides are cationic, and as expected, they bind more strongly to membranes containing anionic than zwitterionic lipids [62,63]. Alamethicin appears to form cylindrical barrel-stave pores, in which the pore lumen is completely lined by peptides [64], whereas melittin, magainin and protegrin appear to form toroidal pores, in which the two membrane leaflets curve together and the peptides are adjacent to lipid headgroups [48,65,66] (figure 1). Magainin exhibits synergy with another AMP from the same family, PGLa [67], which has also been the subject of ssNMR studies [68]. Dye leakage from vesicles usually does not proceed to completion in the presence of AMPs, suggesting that the pores are transient [69]. However, simple mutations to melittin generate peptides that form pores detectable long after equilibration [70]. Electrochemical impedance spectroscopy has shown the transience of melittin bilayer permeabilization [71], in sharp contrast with the behaviour of its MelP5 mutant [72]. There is ssNMR evidence that protegrin oligomerizes into a closed β-barrel composed of four or five dimers in anionic bacterial membrane mimetics [73]. Protegrin-1 dimers have been suggested to assume NCCN parallel topology [73] (figure 2a), although solution NMR in detergents yielded NCCN antiparallel topology (figure 2b) [74]. Further, protegrin-3 adopts NCCN antiparallel topology in DPC micelles [75].
Figure 1.

Schematics of (a) barrel-stave, (b) toroidal and (c) semitoroidal pores.
Figure 2.
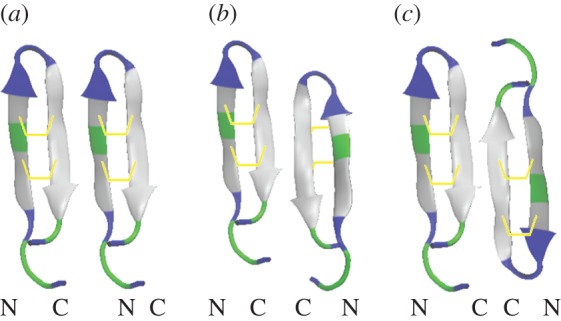
Three possible topologies of protegrin-1 dimerization. (a) NCNC parallel; (b) NCCN parallel; (c) NCCN antiparallel. The bridges represent disulfide bonds, which point inward only on the right side of (b). Arrows point N → C.
2. Key questions
Despite valuable information provided by over four decades of experimental investigations, our understanding of membrane pore formation, and therefore our predictive abilities, are severely limited. Presently, it is not possible to predict a peptide's pore-forming ability given its sequence. To generate such predictions, we would need to resolve the following key questions:
1. Pore versus carpet mechanisms. Do well-defined pores form [27], or is the membrane dissolved in a detergent-like fashion [28]? There is evidence supporting both models. In favour of the pore mechanism, many experiments using GUVs show permeation without membrane dissolution [39,76]. However, in other cases, the GUV bursts, supporting the carpet model [77]. Also in favour of the carpet model, some peptides seem unable to adopt transmembrane orientations [78], although whether such orientations are necessary for pore formation remains to be determined. The carpet model implies quite high peptide concentrations in the membrane, and membrane partitioning measurements suggest that such concentrations are attainable at typical solution concentrations [79,80].
2. Pore structure. When well-defined pores are formed, what is their detailed structure? The most common proposed models are the classical barrel-stave pore—where the pore is cylindrical and lined by peptides [81]—and the toroidal pore, where the two membrane leaflets bend and join together [48,82]. Although toroidal pores' overall lipidic shape has been visualized [27], the number, position and orientation of the peptide constituents with respect to the pore is unknown. Some computational studies have suggested highly disordered pores [83,84] (see §3a,b).
3. Pore lifetime. Are the pores long-lived or transient, and what determines their lifetime? What peptide features lead to pore stability or transience? Electrophysiological techniques are the primary sources of direct information on pore lifetime; however, AMP-induced membrane pores seem to change dynamically. For example, time-lapse atomic force microscopy has shown lateral pore expansion with time [85].
4. Aggregation. Do the peptides need to aggregate (i.e. directly contact each other) during the pore formation process? Numerous biophysical studies over the past decades have looked for signs of such aggregation (e.g. [86–89]). Direct contact is implied in the barrel-stave model but is not necessary in the toroidal model; indeed, most toroidal pore-forming peptides' high charge levels would discourage direct peptide–peptide contact [90]. However, dimerization has been detected for magainin [91], protegrin [74] and other AMPs in detergent micelles.
5. Pore formation pathway. Do the peptides adsorb to the membrane surface as monomers and then oligomerize? Is the reverse possible? At what point does pore opening occur? The exact sequence of steps and the factors that influence their interaction have not been completely elucidated for any peptide.
6. Selectivity and toxicity. What is the origin of most AMPs' selectivity towards bacterial membranes and other AMPs' toxicity towards mammalian cells? The prevalent explanation is that most AMPs' positive charge directs them towards negatively charged bacterial membranes (as opposed to the largely neutral outer leaflet of mammalian membranes). However, the situation is somewhat more complex. For example, both melittin and magainin are positively charged and bind anionic membranes more strongly than zwitterionic ones. However, melittin preferentially permeabilizes zwitterionic membranes [92], whereas magainin does so for anionic membranes [93]. This shows that there is no simple correlation between membrane affinity and permeabilization. Other contributions to selectivity include the effects of cholesterol (which is present in mammalian but not bacterial membranes) and the magnitude of the transmembrane voltage [29]. The dominant determinant of toxicity seems to be hydrophobicity, which is usually positively related to binding strength to neutral membranes and haemolysis (e.g. [94]).
3. Computational investigations
The lack of an experimental atomic-level picture of peptide-stabilized pores has prompted numerous molecular modelling studies. The standard computational technique in molecular biophysics is classical molecular dynamics (MD) simulations: all peptide, lipid and water atoms are represented by particles interacting according to an empirical energy function, and Newton's equations of motion are solved to provide an atomic-level view of the system on a time-scale dependent on the available resources, currently limited to the microsecond range. Because pore formation typically occurs on much longer time-scales, a single MD trajectory normally cannot show the entire sequence of events from water-soluble (usually unfolded) peptides to folded, membrane-inserted oligomeric pore. Hence, researchers have resorted to the following courses of action:
(a) Study putative initial steps of the pore formation process, such as monomer binding to a lipid bilayer [95] or association between two peptides in water, on the membrane surface or inside the membrane [96].
(b) Use free energy calculation techniques, such as umbrella sampling [97] or adaptive biasing force [98], to compute the free energy profiles (or potentials of mean force; PMFs) for these elementary steps. These calculations are time-consuming and expensive, with slow convergence reported [99].
(c) Start the simulation closer to the putative final pore state (e.g. with preformed pores [100] or several inserted transmembrane peptides [101]). Sufficiently long simulations should remove the initial bias and allow the system to relax to a local free energy minimum, which hopefully will be the desired pore state.
An alternative way to address the limitations of atomistic simulations is to simplify the model. One popular direction is to employ coarse-grained (CG) simulations, which represent the lipids, water and often the peptide itself with particles corresponding to more than one atom [102–104]. Several procedures to devise CG representations have been implemented: interactions between CG ‘beads’ can be calibrated to reproduce forces between corresponding groups of atoms in atomistic simulations [105], or the CG system's density, phase transitions or structure can be adjusted to match the physical system's characteristics [106]. CG modelling speeds up the computations considerably, but the added approximations can cause problems. For example, the representation of groups of four water molecules by one particle may not be appropriate for small pores [107], and peptide secondary structure is usually fixed, preventing conformational adaptation.
Even greater simplification is attained by implicit solvent simulations, which represent water and lipids implicitly through extra terms in the energy function. Implicit solvent models were first created for water-soluble proteins [108–110] and were extended to membranes soon afterwards [111–114]. Such models can easily account for surface charge using the Gouy–Chapman theory [115]. Implicit membrane models typically describe intact membranes, but one model permits dynamic changes in membrane thickness [116], and Implicit Membrane Model 1 (IMM1), developed in our laboratory, has been extended to pores [117,118]. In IMM1, pore shape is specified by making the radius R dependent on vertical position within the membrane, z′, according to a desired curvature k [118–120] (figure 3). Transmembrane voltage [121], membrane dipole potential [122] and lateral pressure effects [123] can also be included in IMM1. Because the Gouy–Chapman theory is restricted to modelling flat anionic membranes, the electrostatic potential in anionic membrane pores is found by numerical solution of the Poisson–Boltzmann equation, with the bilayer's dielectric properties represented by a five-slab model accounting for solvent, lipid headgroup and lipid tail regions [119,120].
Figure 3.

Dependence of solvation parameters on internal pore radius (R0), vertical position (z′) and pore curvature (R = R0 + k × z′2): (a) k = 0 in cylindrical pores; (b) k > 0 in toroidal pores.
Below (§3a–c), we review the available computational results on AMP pore formation obtained by all-atom, CG and implicit modelling. Earlier general reviews of this topic can be found in [124–128]. A review of computational studies of protegrins is also available [129].
(a). All-atom modelling
In addition to AMP studies, atomistic simulations have been used to study pore formation in pure lipid bilayers. Because this is not a spontaneous process, it has to be induced, either by application of an electric field (as in the experimental electroporation process [130–133]) or by constraining collective variables [134–136]. However, many of the latter approaches suffer from slow convergence and hysteresis [137]. In addition, pronounced force field dependence has been noted in the calculated free energy of pore formation [138], which impacts interpretation of the AMP simulations mentioned below. Strong force field dependence of results has also been reported in another study [139].
The simplest objective of AMP simulation research has been to examine monomer binding, usually parallel to the membrane surface and in a transmembrane orientation. This has been done for numerous peptides, such as melittin [140–143], alamethicin [144], magainin [145], protegrin [146] and others [147–150]. The peptide is usually placed on the membrane interface with the more hydrophobic side towards the membrane, but it can also be placed a small distance from the membrane, in which case it usually binds rapidly (e.g. [151]). Simulations have also been performed in micelles [152,153], because they are the standard medium in solution NMR. Simulations like these reveal details of AMPs' orientation, depth of membrane insertion and lipid interactions but give no information about the permeabilization mechanism.
Spontaneous pore formation starting from peptides dissolved in the water phase has rarely been observed in simulations. The only reported examples in AMPs are a study of a magainin derivative [83] and a similar subsequent study of melittin [84], which inspired the ‘disordered toroidal pore’ model. The exceedingly fast insertion observed in those studies may have been caused by the systems' lack of counterions. A similar issue was noted in a simulation of spontaneous translocation of an arginine-rich peptide [154]. Recent microsecond-scale atomistic MD simulations of multiple melittin–membrane systems showed bent U-shaped conformations of melittin without any transmembrane insertion or pore formation [155].
The difficulty of observing spontaneous pore formation has led to efforts to circumvent the time-scale problem by starting from preformed pores or preinserted peptides. An early example is simulations of transmembrane helical alamethicin bundles [156,157]. Similar studies have been done on other peptides; for example, a transmembrane melittin tetramer was found to decay to a trimer and generate a toroidal pore in 5.8 ns [158]. Increases in available computational power have gradually allowed much longer simulations. Work in our laboratory compared alamethicin with melittin in preformed pores and confirmed their preference for cylindrical and semitoroidal pores, respectively [159]. Further work examined the influence of intrapeptide charge position and imperfect amphipathicity (i.e. the presence of polar or charged sidechains within a peptide's hydrophobic part) on pore character [160].
Other groups have studied melittin tetramers, either half-inserted [161] or fully inserted [162] in preformed pores. The latter study found that antiparallel arrangement and reduced peptide folding yielded stable toroidal pores. Another group studied various oligomers on the surface or in transmembrane orientation [163]. Our group's much longer atomistic simulations of a melittin tetramer conducted on the Anton supercomputer revealed a classical toroidal pore characterized by dynamic orientation change of one or two peptides (figure 4a) [101]. Similar simulations of magainin and PGLa in lipid membranes showed a different picture: highly tilted peptides around a very small pore, with tilt angles in agreement with ssNMR experiments (figure 4b) [164]. This work also provided insights into the synergy between these two magainin-family peptides: more transmembrane orientations, stronger interactions, and a larger and more ordered pore were seen in the 1 : 1 heterotetramer with antiparallel helix arrangement than in homogeneous peptide mixtures [164]. Starkly different results were obtained for another AMP called piscidin: a 26 µs Anton simulation starting from 20 peptides in four barrel-stave pores resulted in almost all peptides moving to a surface-bound state, suggesting a non-pore mechanism for this peptide [165].
Figure 4.
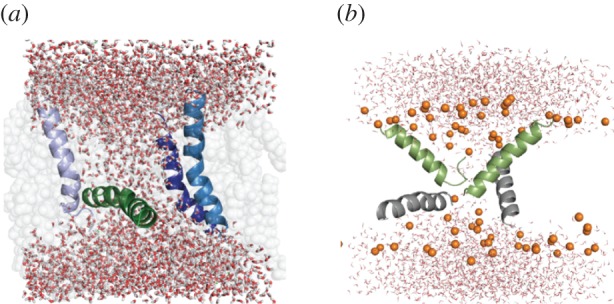
Snapshots from all-atom simulations of α-helical AMPs: (a) melittin tetramer at 8.3 µs [101]; (b) magainin–PGLa heterotetramer at 9 µs [164].
Complete β-barrel models of protegrin-1 based on the experimentally suggested NCCN parallel β-barrel structure have also been subjected to all-atom simulations [55,166–168]. Our laboratory has produced alternative models of protegrin barrels based on implicit modelling results (see §3c), which show the highest octameric β-barrel stability in NCNC parallel topology [169]. We also observed that the intrinsic tilt and twist values of NCNC parallel tetramers tend to form protegrin-1 into an arc shape, which form stable pores on a time-scale of 300 ns [100] (figure 5). However, longer explicit simulations (unpublished) show a lipid entering the pore lumen displacing water, implying that a single NCNC parallel tetrameric arc may be insufficient for a stable conductive pore.
Figure 5.

Snapshot from simulation of single NCNC parallel protegrin-1 tetrameric arc in implicit membrane [100].
In addition to standard MD simulations, attempts have been made to obtain thermodynamic information about putative steps in the pore formation pathway via free energy calculations. For example, PMFs have been calculated for protegrin dimerization [96], adsorption of monomers and dimers to membrane bilayer interfaces [170], and insertion into a lipid bilayer [171] in an effort to clarify the major steps in protegrin pore formation. Another study calculated PMFs for protegrin's orientation within membrane bilayers, finding a significant tilt angle at the optimal orientation [172]. The free energy of melittin binding and reorientation in phospholipid bilayers has also been calculated [95,173,174], as well as the free energy of membrane binding of a lipopeptide at the CG level [175] (see §3b).
(b). Coarse-grained modelling
CG modelling of AMP pore formation can offer unique insights, as it allows longer simulation times and larger systems than all-atom modelling. Most CG studies so far have used the MARTINI model [102,107], but models that employ coarser graining have also appeared [176]. MARTINI-based studies have included: a study of PAMAM dendrimers that showed unacetylated molecules forming pores [177]; a study of alamethicin that showed aggregation and some transitions to an interfacial state [178]; a study of the synthetic LS3 peptide that showed the spontaneous formation of mostly hexameric barrel-stave pores [179]; a study showing spontaneous pore formation by a hydrophobic magainin derivative [180]; a study of magainin and melittin that found U-shaped conformations for melittin and disordered toroidal pores for magainin [181,182]; studies of antimicrobial lipopeptides that found clustering of anionic lipids but no pore formation [183,184]; a dissipative particle dynamics study of eight AMPs employing a MARTINI-like model showing pore formation and translocation [185]; and a study of the AMP maculatin showing that an increased peptide-to-lipid ratio caused an interfacial–transmembrane orientation change and cooperative membrane insertion of peptide aggregates without a well-defined central pore lumen [186]. The last authors also observed maculatin-induced spontaneous membrane curvature and used that evidence to suggest that pore formation may not be responsible for maculatin's activity. In a larger-scale setting, beyond the practical limits of all-atom simulations, up to 1600 magainin peptides added to only one side of the bilayer induced spontaneous buckling and vesicle budding of the lipid bilayer [187].
While these results sound very promising, some issues raise questions about their reliability. The pores observed in CG simulations often contain no water [178,181,188], and coarse graining the water to a spherically symmetric particle corresponding to four water molecules limits reproduction of the pore's structural details. A detailed comparison of CG with atomistic simulations revealed deficiencies in the CG pore structures [189]. Models that employ polarizable CG water models provide improvements [107,190], although they still have difficulties forming aqueous pores under standard parametrization. Coarse graining of the abundant water molecules is generally a quite important factor in the computational efficiency gains offered by CG: one study estimated a speed-up factor of 50 from coarse graining the water molecules [191].
(c). Implicit solvent modelling
In the 1980s and 1990s, when atomistic simulation of peptides in membranes was out of reach, researchers were forced to develop simple ways to account for the membrane environment [192–196]. The newer generation of implicit membrane models [112–114] are extensions of implicit aqueous solvation models [108–110] and thus account for the energetics of both membrane insertion and conformational changes. The Poisson–Boltzmann continuum model can also be used to study peptide–membrane interactions [197,198], but usually for static peptide configurations. Even with modern computational power, implicit membrane modelling allows studies that are impossible with all-atom and CG methods and facilitates unique insights because of its fast convergence and simple energetic interpretation.
Modern implicit membrane models are routinely and trivially used to determine the configurations and membrane binding energetics of AMP monomers (e.g. [118,199]). More sophisticated approaches have included the effects of transmembrane voltage, dipole potential and lateral pressure profile [121–123]. For example, using such simulations, we showed that voltage induces the interfacial–transmembrane orientation change in alamethicin [121], and the addition of a membrane dipole potential term reduced the membrane binding affinity of magainin [122]. Negative lateral pressure at the water–lipid interface stabilized interfacial binding and yielded an interfacial–transmembrane orientation shift with an increased peptide : lipid ratio, in accord with experiment. Further, with the inclusion of a lateral pressure term, an increased DOPE mole fraction in mixed DOPC : DOPE bilayers stabilized interfacial as opposed to transmembrane orientations [123].
Additional results of interest have been produced by implicit pore modelling [115,118,120]. When we compared peptide binding with pores of different shapes, we found that most AMPs bind more favourably to both zwitterionic and anionic toroidal pores than to flat membranes [118,120]. Alamethicin exhibited similar transfer energy to cylindrical and toroidal pores, whereas toroidal pore-forming peptides, such as melittin, exhibited a preference for toroidal pores. One factor driving these favourable transfer energies appears to be imperfect amphipathicity, which reduces binding strength to flat membranes but does not affect binding to membranes with positive curvature. Melittin and protegrin are examples of this type of amphipathic structure. (Note that the term ‘imperfect amphipathicity’ has a broader meaning in Wimley's independently developed interfacial activity model [200].)
A large-scale computational study using IMM1 analysed activity determinants of 53 α-helical AMPs [201], testing the hypothesis that antimicrobial and haemolytic activity correlate with binding affinity to anionic and zwitterionic membranes, respectively. Antibacterial and haemolytic activity were found to correlate best with transfer energy to membranes with anionic lipid fractions greater than 30% and 10%, respectively. Surface area occupation, insertion depth and structural fluctuation were significantly correlated with antibacterial, haemolytic and both activity levels, respectively. Peptides active at low membrane surface coverage ratios tended to be those identified in the literature as pore-forming. Further, the transfer energy to toroidal pores was negative in almost all cases, while that to cylindrical pores was more favourable in neutral than anionic pores, and pore transfer energy correlated with deviation from the predictions of the carpet model. There were significant correlations of hydrophobic quadrupole moment (a biophysical descriptor related to imperfect amphipathicity) with lethality against both Escherichia coli and red blood cells.
An investigation of putative protegrin β-barrels was an interesting application of this approach. ssNMR results had suggested NCCN parallel topology [73]; however, in that topology, the barrels were much less stable in implicit pores than those in NCNC parallel or NCCN antiparallel topologies, because the latter two topologies allow all monomers' hydrophobic sides to face the membrane [169]. Explicit simulations also showed the NCNC parallel topology to be more stable. Further work compared protegrin with a variety of similar β-hairpin AMPs (figure 6); the results showed that most lack stability when inserted into pores as β-barrels, casting doubt on the validity of the β-barrel pore formation mechanism for the entire β-hairpin AMP family [119]. These conclusions would have been very difficult to reach without decisive insights generated by implicit solvent simulations.
Figure 6.

Surface hydrophobicity levels of β-hairpin AMPs. Red: more hydrophobic; white: less hydrophobic according to a normalized consensus hydrophobicity scale [202]. Side views of each hairpin after 200 ps of MD simulation in water and 300 minimization steps: (a) protegrin-1; (b) θ-defensin; (c) tachyplesin; (d) polyphemusin; (e) gomesin; (f) androctonin; (g) YGKRGF; (h) AGGKGF. The latter two are from the combinatorial library in [203]. All peptides are oriented with the more hydrophobic side to the left and the turn region down. The black lines denote the borders of a 26-Å-thick membrane region with transmembrane insertion. The molecular graphics were produced using VMD 1.9.2 [204].
Implicit simulations in our laboratory that accounted for the free energy cost of acyl chain exposure showed that certain antiparallel alamethicin bundles have lower free energy than parallel ones and could be present under experimental conditions [205]. Multiple combinations of radius and oligomeric number were investigated, with all oligomeric numbers 5–9 forming open pores at their optimal radii. All oligomers 6–9 had comparable energies, consistent with multiple experimentally observed conductance levels [206] and the barrel-stave model. The N-terminal part was less tilted than the C-terminal part, resulting in hybrid funnel/hourglass pore shapes; this shape was not affected by transmembrane voltage, but pore stability was. The presence of a pore raised the total effective energy, suggesting that alamethicin pores may correspond to excited states stabilized by voltage and ion flux.
4. Conclusion and future directions
Computational studies have provided a wealth of insights on pore-forming peptides, some of which have been listed above. However, the limitations and uncertainties of the calculations have not allowed definitive conclusions. Moreover, many studies focus on certain stages of the pore formation process, such as early membrane binding and dimerization or the final pore state. Thus, a picture of the entire process is still lacking. In some cases, modelling results seem to conflict with each other: in the example case of melittin, one study found disordered pores with half-folded peptides [84], and another found mostly helical peptides around a classical-type toroidal pore [101]. The origin of these discrepancies—whether the force field or other technical details—needs to be clarified, and convergence needs to be achieved. As always, experimental validation will be necessary for reliable conclusions.
The ‘to-do’ list in this field is long and exciting. The ability to perform multi-microsecond MD simulations on Anton or other powerful computers has started to provide better-equilibrated pore structures and detailed views of peptide–lipid interactions. Extending the time-scale to a millisecond, which is now conceivable with Anton II [207], may bring into view processes like spontaneous aggregation, pore formation, expansion, contraction and annihilation. This will start to provide information on not only pore structure but also dynamics. Additional PMF calculations of higher-order oligomer insertion into lipid membranes, similar to those available for monomers and dimers, are also needed to obtain the complete energetic landscape for pore formation. It would also be useful to increase the diversity of lipid membrane compositions simulated, because activity and toxicity are clearly dependent on membrane lipid composition.
CG simulations face some challenges to reproduce the details of peptide-stabilized pores. One possible way forward could be mixed-resolution models that represent system components at different resolution levels according to importance (e.g. [208–210]), or adaptive resolution models whose resolution depends on location [211]. However, application to membrane pores will not be trivial. An additional challenge is that peptide conformation is usually restrained in CG simulations, which is not ideal, as considerable evidence indicates that conformational changes in AMPs are possible during the poration process [212]. A challenging solution to this problem could come from the development of CG peptide models that correctly reproduce the environment-dependent conformational distribution. Because of the above-mentioned difficulties, the investigation of large-scale membrane remodelling effects (e.g. membrane bilayer buckling and vesicular budding [187]) may be a more fruitful application of CG models. Significant work remains to be done, e.g. to understand the effects of the dependence of area per lipid on the peptide : lipid ratio and how this determines the properties of both quasi-spherical vesicular buds and their narrow necks.
Implicit membrane models could also be further developed. Extensions to curved membranes would allow a more systematic exploration of the effects of curvature, especially Gaussian curvature [213], on peptide binding. One significant limitation of most implicit models is the assumption that the membrane is fixed. Large-scale changes, like bilayer buckling or vesicular budding, are very difficult—and perhaps counterproductive—to pursue via implicit solvent methods. However, making pore radius or shape dynamic simulation variables is feasible. Some of these developments are in progress in our laboratory.
Eventually, to enable quantitative predictions, we need to develop a detailed molecular thermodynamic model of peptide-induced pore formation. That would require quantitative characterization of contributions to the pore state's free energy from peptide–peptide, peptide–lipid and lipid–lipid interactions. Implicit solvent models are best positioned to provide the first two terms; however, their quantitative accuracy needs to be ascertained by comparison with atomistic PMF calculations. Although we have previously done this by computing PMFs between ionizable side chains in solution [214] and at the bilayer–water interface [215], it is also necessary to consider free energy profiles for entire peptides, as has been done for glycophorin A [216] and model peptides [38]. A critical ingredient in this energetic landscape is the free energy of opening a pore in a pure bilayer (i.e. lipid–lipid interactions in the above scheme). This contribution has previously been estimated roughly from line tension measurements [217], but such values suffer from large experimental uncertainty and correspond to the lowest free energy state of a pore in a pure bilayer. Although we need to know the free energy of lipidic pore formation as a function of radius, shape and lipid headgroup distribution, existing approaches do not provide this level of detail [134–136].
Beyond thermodynamics, dynamic models of pore creation, growth and annihilation would also be useful. For example, the pore could represent a dynamic state constituted by a growing number of peptides: AMP pores may be able to expand laterally, even to the micrometre scale, until the membrane disintegrates [85]. Some elementary dynamic models have already appeared; e.g. one study estimated that 100 total protegrin pores are necessary to kill an E. coli cell [218] using a rough, static estimate of pore size. Future multi-scale approaches could integrate atomistic MD simulations, mesoscale models of single-pore electrodiffusion and models of transient bacterial cell ion transport [219].
Integrated multi-scale computational models could not only provide a general understanding of AMPs' pore formation mechanism but facilitate the development of efficient antimicrobial pharmaceutical products. Iseganan IB-367, a novel protegrin AMP designed for activity against microflora associated with oral mucositis [220], was granted fast-track status in both the USA and European Union for a Phase II/III trial for prevention of nosocomial pneumonia [221]. However, those trials were discontinued in 2004, as they failed to show improvements over existing treatments, and no AMP-based drugs have ultimately been given final approval, despite these compounds' promisingly low in vitro minimum inhibitory concentrations. A bottleneck has been most AMPs' high toxicity levels at therapeutic doses [129]. Several other AMPs are in clinical development, such as thanatin and heliomycin [222], but the lack of detailed understanding of AMPs' mechanisms of action has limited the rational design of AMP-based therapeutics. Future insights from computational studies could facilitate efficient drug design by identifying peptides with the desired pharmacological properties.
Acknowledgements
The authors acknowledge Shanan Leeman of New York University's Tisch School of the Arts for generating figure 1 and all previous authors of papers from our laboratory for generating useful data and analysis.
Authors' contributions
R.L. designed and drafted the article, analysed literature and approved the final version of the manuscript; T.L. conceived the article, analysed and interpreted literature, critically revised the manuscript and approved the final version of the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the National Institutes of Health (GM117146-01) and the National Science Foundation (MCB 1244207). Infrastructure support was provided in part by an RCMI grant (5G12MD007603-30) from the National Institutes of Health.
References
- 1.Walensky LD, Gavathiotis E. 2011. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci. 36, 642–652. ( 10.1016/j.tibs.2011.08.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Saez AJ. 2012. The secrets of the Bcl-2 family. Cell Death Differ. 19, 1733–1740. ( 10.1038/cdd.2012.105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westphal D, Kluck RM, Dewson G. 2014. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 21, 196–205. ( 10.1038/cdd.2013.139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondos SC, Hatfaludi T, Voskoboinik I, Trapani JA, Law RH. P., Whisstock JC, Dunstone MA. 2010. The structure and function of mammalian membrane-attack complex/perforin-like proteins. Tissue Antigens 76, 341–351. ( 10.1111/j.1399-0039.2010.01566.x) [DOI] [PubMed] [Google Scholar]
- 5.Voskoboinik I, Whisstock JC, Trapani JA. 2015. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400. ( 10.1038/nri3839) [DOI] [PubMed] [Google Scholar]
- 6.Hotze EM, Tweten RK. 2012. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta Biomembr. 1818, 1028–1038. ( 10.1016/j.bbamem.2011.07.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ladokhin AS. 2013. pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 5, 1362–1380. ( 10.3390/toxins5081362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young R. 2013. Phage lysis: do we have the hole story yet? Curr. Opin. Microbiol. 16, 790–797. ( 10.1016/j.mib.2013.08.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walther TH, et al. 2013. Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism. Cell 152, 316–326. ( 10.1016/j.cell.2012.12.017) [DOI] [PubMed] [Google Scholar]
- 10.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. 1996. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274, 1859 ( 10.1126/science.274.5294.1859) [DOI] [PubMed] [Google Scholar]
- 11.Jiang J, Pentelute BL, Collier RJ, Zhou ZH. 2015. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 521, 545–549. ( 10.1038/nature14247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. 2009. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature 459, 726–730. ( 10.1038/nature08026) [DOI] [PubMed] [Google Scholar]
- 13.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. 2002. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342. ( 10.1016/S0092-8674(02)01036-X) [DOI] [PubMed] [Google Scholar]
- 14.Yarmush ML, Golberg A, Serša G, Kotnik T, Miklavčič D. 2014. Electroporation-based technologies for medicine: principles, applications, and challenges. Biomed. Eng. 16, 295 ( 10.1146/annurev-bioeng-071813-104622) [DOI] [PubMed] [Google Scholar]
- 15.Hancock RE. W. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1, 156–164. ( 10.1016/S1473-3099(01)00092-5) [DOI] [PubMed] [Google Scholar]
- 16.Papo N, Shai Y. 2005. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 62, 784–790. ( 10.1007/s00018-005-4560-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. ( 10.1038/415389a) [DOI] [PubMed] [Google Scholar]
- 18.Hancock RE. W., Haney EF, Gill EE. 2016. The immunology of host defence peptides: beyond antimicrobial activity. Nat. Rev. Immunol. 16, 321–334. ( 10.1038/nri.2016.29) [DOI] [PubMed] [Google Scholar]
- 19.Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, Selsted ME. 1989. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J. Clin. Invest. 84, 553–561. ( 10.1172/JCI114198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuzaki K, Sugishita K-I, Harada M, Fujii N, Miyajima K. 1997. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim. Biophys. Acta Biomembr. 1327, 119–130. ( 10.1016/S0005-2736(97)00051-5) [DOI] [PubMed] [Google Scholar]
- 21.da Silva A Jr, Teschke O. 2003. Effects of the antimicrobial peptide PGLa on live Escherichia coli. Biochim. Biophys. Acta Mol. Cell Res. 1643, 95–103. ( 10.1016/j.bbamcr.2003.10.001) [DOI] [PubMed] [Google Scholar]
- 22.Westerhoff HV, Juretić D, Hendler RW, Zasloff M. 1989. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl Acad. Sci. USA 86, 6597–6601. ( 10.1073/pnas.86.17.6597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falla TJ, Karunaratne DN, Hancock RE. W. 1996. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 271, 19 298–19 303. ( 10.1074/jbc.271.32.19298) [DOI] [PubMed] [Google Scholar]
- 24.Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y. 1999. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 341, 501 ( 10.1042/bj3410501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wade D, Boman A, Wåhlin B, Drain CM, Andreu D, Boman HG, Merrifield RB. 1990. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl Acad. Sci. USA 87, 4761–4765. ( 10.1073/pnas.87.12.4761) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oren Z, Shai Y. 1997. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: structure–function study. Biochemistry 36, 1826–1835. ( 10.1021/bi962507l) [DOI] [PubMed] [Google Scholar]
- 27.Huang HW. 2000. Action of antimicrobial peptides: two-state model. Biochemistry 39, 8347–8352. ( 10.1021/bi000946l) [DOI] [PubMed] [Google Scholar]
- 28.Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta Biomembr. 1462, 55–70. ( 10.1016/S0005-2736(99)00200-X) [DOI] [PubMed] [Google Scholar]
- 29.Matsuzaki K. 2009. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 1788, 1687–1692. ( 10.1016/j.bbamem.2008.09.013) [DOI] [PubMed] [Google Scholar]
- 30.Wenzel M, et al. 2014. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl Acad. Sci. USA 111, E1409–E1418. ( 10.1073/pnas.1319900111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sochacki KA, Barns KJ, Bucki R, Weisshaar JC. 2011. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc. Natl Acad. Sci. USA 108, E77–E81. ( 10.1073/pnas.1101130108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epand RM, Rotem S, Mor A, Berno B, Epand RF. 2008. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 130, 14 346–14 352. ( 10.1021/ja8062327) [DOI] [PubMed] [Google Scholar]
- 33.Hale JD. F, Hancock RE. W. 2007. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti-Infect. Ther. 5, 951–959. ( 10.1586/14787210.5.6.951) [DOI] [PubMed] [Google Scholar]
- 34.Hancock RE. W, Rozek A. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206, 143–149. ( 10.1111/j.1574-6968.2002.tb11000.x) [DOI] [PubMed] [Google Scholar]
- 35.Wu M, Maier E, Benz R, Hancock REW. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 38, 7235–7242. ( 10.1021/bi9826299) [DOI] [PubMed] [Google Scholar]
- 36.Matsuzaki K, Harada M, Handa T, Funakoshi S, Fujii N, Yajima H, Miyajima K. 1989. Magainin 1-induced leakage of entrapped calcein out of negatively-charged lipid vesicles. Biochim. Biophys. Acta Biomembr. 981, 130–134. ( 10.1016/0005-2736(89)90090-4) [DOI] [PubMed] [Google Scholar]
- 37.Ladokhin AS, Selsted ME, White SH. 1997. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys. J. 72, 1762 ( 10.1016/S0006-3495(97)78822-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J, Im W. 2008. Role of hydrogen bonding and helix−lipid interactions in transmembrane helix association. J. Am. Chem. Soc. 130, 6456–6462. ( 10.1021/ja711239h) [DOI] [PubMed] [Google Scholar]
- 39.Lee M-T, Sun T-L, Hung W-C, Huang HW. 2013. Process of inducing pores in membranes by melittin. Proc. Natl Acad. Sci. USA 110, 14 243–14 248. ( 10.1073/pnas.1307010110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Islam MZ, Alam JM, Tamba Y, Karal MA. S, Yamazaki M. 2014. The single GUV method for revealing the functions of antimicrobial, pore-forming toxin, and cell-penetrating peptides or proteins. Phys. Chem. Chem. Phys. 16, 15 752–15 767. ( 10.1039/C4CP00717D) [DOI] [PubMed] [Google Scholar]
- 41.Barns KJ, Weisshaar JC. 2016. Single-cell, time-resolved study of the effects of the antimicrobial peptide alamethicin on Bacillus subtilis. Biochim. Biophys. Acta Biomembr. 1858, 725–732. ( 10.1016/j.bbamem.2016.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seelig J. 2004. Thermodynamics of lipid–peptide interactions. Biochim. Biophys. Acta Biomembr. 1666, 40–50. ( 10.1016/j.bbamem.2004.08.004) [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Huang HW, Olah GA. 1990. Method of oriented circular dichroism. Biophys. J. 57, 797–806. ( 10.1016/S0006-3495(90)82599-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bürck J, Wadhwani P, Fanghänel S, Ulrich AS. 2016. Oriented circular dichroism: a method to characterize membrane-active peptides in oriented lipid bilayers. Acc. Chem. Res. 49, 184–192. ( 10.1021/acs.accounts.5b00346) [DOI] [PubMed] [Google Scholar]
- 45.Ludtke S, He K, Huang H. 1995. Membrane thinning caused by magainin 2. Biochemistry 34, 16 764–16 769. ( 10.1021/bi00051a026) [DOI] [PubMed] [Google Scholar]
- 46.Heller WT, Waring AJ, Lehrer RI, Harroun TA, Weiss TM, Yang L, Huang HW. 2000. Membrane thinning effect of the β-sheet antimicrobial protegrin. Biochemistry 39, 139–145. ( 10.1021/bi991892m) [DOI] [PubMed] [Google Scholar]
- 47.Qian S, Wang W, Yang L, Huang HW. 2008. Structure of the alamethicin pore reconstructed by X-ray diffraction analysis. Biophys. J. 94, 3512–3522. ( 10.1529/biophysj.107.126474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. 1996. Membrane pores induced by magainin. Biochemistry 35, 13 723–13 728. ( 10.1021/bi9620621) [DOI] [PubMed] [Google Scholar]
- 49.Cruciani RA, Barker JL, Durell SR, Raghunathan G, Guy HR, Zasloff M, Stanley EF. 1992. Magainin 2, a natural antibiotic from frog skin, forms ion channels in lipid bilayer membranes. Eur. J. Pharmacol. Mol. Pharmacol. Sect. 226, 287–296. ( 10.1016/0922-4106(92)90045-W) [DOI] [PubMed] [Google Scholar]
- 50.Tosteson MT, Tosteson DC. 1981. The sting. Melittin forms channels in lipid bilayers. Biophys. J. 36, 109 ( 10.1016/S0006-3495(81)84719-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordon LGM, Haydon DA. 1972. The unit conductance channel of alamethicin. Biochim. Biophys. Acta Biomembr. 255, 1014–1018. ( 10.1016/0005-2736(72)90415-4) [DOI] [PubMed] [Google Scholar]
- 52.Porcelli F, Buck-Koehntop BA, Thennarasu S, Ramamoorthy A, Veglia G. 2006. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 45, 5793–5799. ( 10.1021/bi0601813) [DOI] [PubMed] [Google Scholar]
- 53.Hong M. 2007. Structure, topology, and dynamics of membrane peptides and proteins from solid-state NMR spectroscopy. J. Phys. Chem. B 111, 10 340–10 351. ( 10.1021/jp073652j) [DOI] [PubMed] [Google Scholar]
- 54.Salnikov E, Aisenbrey C, Vidovic V, Bechinger B. 2010. Solid-state NMR approaches to measure topological equilibria and dynamics of membrane polypeptides. Biochim. Biophys. Acta Biomembr. 1798, 258–265. ( 10.1016/j.bbamem.2009.06.021) [DOI] [PubMed] [Google Scholar]
- 55.Capone R, Mustata M, Jang H, Arce FT, Nussinov R, Lal R. 2010. Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 98, 2644–2652. ( 10.1016/j.bpj.2010.02.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam KLH, Wang H, Siaw TA, Chapman MR, Waring AJ, Kindt JT, Lee KYC. 2012. Mechanism of structural transformations induced by antimicrobial peptides in lipid membranes. Biochim. Biophys. Acta Biomembr. 1818, 194–204. ( 10.1016/j.bbamem.2011.11.002) [DOI] [PubMed] [Google Scholar]
- 57.Hartmann M, Berditsch M, Hawecker J, Ardakani MF, Gerthsen D, Ulrich AS. 2010. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 54, 3132–3142. ( 10.1128/AAC.00124-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cafiso DS. 1994. Alamethicin: a peptide model for voltage gating and protein–membrane interactions. Annu. Rev. Biophys. Biomol. Struct. 23, 141–165. ( 10.1146/annurev.bb.23.060194.001041) [DOI] [PubMed] [Google Scholar]
- 59.Dempsey CE. 1990. The actions of melittin on membranes. Biochim. Biophys. Acta, Biomembr. 1031, 143–161. ( 10.1016/0304-4157(90)90006-X) [DOI] [PubMed] [Google Scholar]
- 60.Matsuzaki K. 1998. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta Biomembr. 1376, 391–400. ( 10.1016/S0304-4157(98)00014-8) [DOI] [PubMed] [Google Scholar]
- 61.Fahrner RL, Dieckmann T, Harwig SSL, Lehrer RI, Eisenberg D, Feigon J. 1996. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. (Oxford, UK) 3, 543–550. ( 10.1016/S1074-5521(96)90145-3) [DOI] [PubMed] [Google Scholar]
- 62.Batenburg AM, Van Esch JH, De Kruijff B. 1988. Melittin-induced changes of the macroscopic structure of phosphatidylethanolamines. Biochemistry 27, 2324–2331. ( 10.1021/bi00407a013) [DOI] [PubMed] [Google Scholar]
- 63.Matsuzaki K, Harada M, Funakoshi S, Fujii N, Miyajima K. 1991. Physicochemical determinants for the interactions of magainins 1 and 2 with acidic lipid bilayers. Biochim. Biophys. Acta Biomembr. 1063, 162–170. ( 10.1016/0005-2736(91)90366-G) [DOI] [PubMed] [Google Scholar]
- 64.He K, Ludtke SJ, Huang HW, Worcester DL. 1995. Antimicrobial peptide pores in membranes detected by neutron in-plane scattering. Biochemistry 34, 15 614–15 618. ( 10.1021/bi00048a002) [DOI] [PubMed] [Google Scholar]
- 65.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81, 1475–1485. ( 10.1016/S0006-3495(01)75802-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang L, Weiss TM, Lehrer RI, Huang HW. 2000. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 79, 2002–2009. ( 10.1016/S0006-3495(00)76448-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuzaki K, Mitani Y, Akada K-Y, Murase O, Yoneyama S, Zasloff M, Miyajima K. 1998. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry 37, 15 144–15 153. ( 10.1021/bi9811617) [DOI] [PubMed] [Google Scholar]
- 68.Strandberg E, Zerweck J, Wadhwani P, Ulrich AS. 2013. Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophys. J. 104, L9–L11. ( 10.1016/j.bpj.2013.01.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuzaki K, Murase O, Fujii N, Miyajima K. 1995. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 34, 6521–6526. ( 10.1021/bi00019a033) [DOI] [PubMed] [Google Scholar]
- 70.Krauson AJ, He J, Wimley WC. 2012. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 134, 12 732–12 741. ( 10.1021/ja3042004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wiedman G, Herman K, Searson P, Wimley WC, Hristova K. 2013. The electrical response of bilayers to the bee venom toxin melittin: evidence for transient bilayer permeabilization. Biochim. Biophys. Acta Biomembr. 1828, 1357–1364. ( 10.1016/j.bbamem.2013.01.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiedman G, Fuselier T, He J, Searson PC, Hristova K, Wimley WC. 2014. Highly efficient macromolecule-sized poration of lipid bilayers by a synthetically evolved peptide. J. Am. Chem. Soc. 136, 4724–4731. ( 10.1021/ja500462s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mani R, Cady SD, Tang M, Waring AJ, Lehrer RI, Hong M. 2006. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl Acad. Sci. USA 103, 16 242–16 247. ( 10.1073/pnas.0605079103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roumestand C, Louis V, Aumelas A, Grassy G, Calas B, Chavanieu A. 1998. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett. 421, 263–267. ( 10.1016/S0014-5793(97)01579-2) [DOI] [PubMed] [Google Scholar]
- 75.Usachev KS, Efimov SV, Kolosova OA, Klochkova EA, Aganov AV, Klochkov VV. 2015. Antimicrobial peptide protegrin-3 adopt an antiparallel dimer in the presence of DPC micelles: a high-resolution NMR study. J. Biomol. NMR 62, 71–79. ( 10.1007/s10858-015-9920-0) [DOI] [PubMed] [Google Scholar]
- 76.Tamba Y, Yamazaki M. 2005. Single giant unilamellar vesicle method reveals effect of antimicrobial peptide magainin 2 on membrane permeability. Biochemistry 44, 15 823–15 833. ( 10.1021/bi051684w) [DOI] [PubMed] [Google Scholar]
- 77.Domingues TM, Riske KA, Miranda A. 2010. Revealing the lytic mechanism of the antimicrobial peptide gomesin by observing giant unilamellar vesicles. Langmuir 26, 11 077–11 084. ( 10.1021/la100662a) [DOI] [PubMed] [Google Scholar]
- 78.Bechinger B. 2005. Detergent-like properties of magainin antibiotic peptides: a 31P solid-state NMR spectroscopy study. Biochim. Biophys. Acta, Biomembr. 1712, 101–108. ( 10.1016/j.bbamem.2005.03.003) [DOI] [PubMed] [Google Scholar]
- 79.Melo MN, Ferre R, Castanho MARB. 2009. Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 7, 245–250. ( 10.1038/nrmicro2095) [DOI] [PubMed] [Google Scholar]
- 80.Roversi D, Luca V, Aureli S, Park Y, Mangoni ML, Stella L. 2014. How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem. Biol. 9, 2003–2007. ( 10.1021/cb500426r) [DOI] [PubMed] [Google Scholar]
- 81.Baumann G, Mueller P. 1974. A molecular model of membrane excitability. J. Supramol. Struct. 2, 538–557. ( 10.1002/jss.400020504) [DOI] [PubMed] [Google Scholar]
- 82.Matsuzaki K, Murase O, Fujii N, Miyajima K. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 35, 11 361–11 368. ( 10.1021/bi960016v) [DOI] [PubMed] [Google Scholar]
- 83.Leontiadou H, Mark AE, Marrink SJ. 2006. Antimicrobial peptides in action. J. Am. Chem. Soc. 128, 12 156–12 161. ( 10.1021/ja062927q) [DOI] [PubMed] [Google Scholar]
- 84.Sengupta D, Leontiadou H, Mark AE, Marrink S-J. 2008. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta Biomembr. 1778, 2308–2317. ( 10.1016/j.bbamem.2008.06.007) [DOI] [PubMed] [Google Scholar]
- 85.Rakowska PD, et al. 2013. Nanoscale imaging reveals laterally expanding antimicrobial pores in lipid bilayers. Proc. Natl Acad. Sci. USA 110, 8918–8923. ( 10.1073/pnas.1222824110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buffy JJ, Waring AJ, Lehrer RI, Hong M. 2003. Immobilization and aggregation of the antimicrobial peptide protegrin-1 in lipid bilayers investigated by solid-state NMR. Biochemistry 42, 13 725–13 734. ( 10.1021/bi035187w) [DOI] [PubMed] [Google Scholar]
- 87.Chen F-Y, Lee M-T, Huang HW. 2002. Sigmoidal concentration dependence of antimicrobial peptide activities: a case study on alamethicin. Biophys. J. 82, 908–914. ( 10.1016/S0006-3495(02)75452-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.John E, Jähnig F. 1991. Aggregation state of melittin in lipid vesicle membranes. Biophys. J. 60, 319 ( 10.1016/S0006-3495(91)82056-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schwarz G, Stankowski S, Rizzo V. 1986. Thermodynamic analysis of incorporation and aggregation in a membrane: application to the pore-forming peptide alamethicin. Biochim. Biophys. Acta Biomembr. 861, 141–151. ( 10.1016/0005-2736(86)90412-8) [DOI] [PubMed] [Google Scholar]
- 90.Zemel A, Fattal DR, Ben-Shaul A. 2003. Energetics and self-assembly of amphipathic peptide pores in lipid membranes. Biophys. J. 84, 2242–2255. ( 10.1016/S0006-3495(03)75030-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wakamatsu K, Takeda A, Tachi T, Matsuzaki K. 2002. Dimer structure of magainin 2 bound to phospholipid vesicles. Biopolymers 64, 314–327. ( 10.1002/bip.10198) [DOI] [PubMed] [Google Scholar]
- 92.Hincha DK, Crowe JH. 1996. The lytic activity of the bee venom peptide melittin is strongly reduced by the presence of negatively charged phospholipids or chloroplast galactolipids in the membranes of phosphatidylcholine large unilamellar vesicles. Biochim. Biophys. Acta Biomembr. 1284, 162–170. ( 10.1016/S0005-2736(96)00122-8) [DOI] [PubMed] [Google Scholar]
- 93.Gregory SM, Pokorny A, Almeida PFF. 2009. Magainin 2 revisited: a test of the quantitative model for the all-or-none permeabilization of phospholipid vesicles. Biophys. J. 96, 116–131. ( 10.1016/j.bpj.2008.09.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Javadpour MM, Juban MM, Lo W-CJ, Bishop SM, Alberty JB, Cowell SM, Becker CL, McLaughlin ML. 1996. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 39, 3107–3113. ( 10.1021/jm9509410) [DOI] [PubMed] [Google Scholar]
- 95.Irudayam SJ, Berkowitz ML. 2012. Binding and reorientation of melittin in a POPC bilayer: computer simulations. Biochim. Biophys. Acta Biomembr. 1818, 2975–2981. ( 10.1016/j.bbamem.2012.07.026) [DOI] [PubMed] [Google Scholar]
- 96.Vivcharuk V, Kaznessis YN. 2010. Dimerization of protegrin-1 in different environments. Int. J. Mol. Sci. 11, 3177–3194. ( 10.3390/ijms11093177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Torrie GM, Valleau JP. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23, 187–199. ( 10.1016/0021-9991(77)90121-8) [DOI] [Google Scholar]
- 98.Darve E, Rodríguez-Gómez D, Pohorille A. 2008. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 128,144120 ( 10.1063/1.2829861) [DOI] [PubMed] [Google Scholar]
- 99.Neale C, Pomès R. 2016. Sampling errors in free energy simulations of small molecules in lipid bilayers. Biochim. Biophys. Acta Biomembr. 1858, 2539–2548. ( 10.1016/j.bbamem.2016.03.006) [DOI] [PubMed] [Google Scholar]
- 100.Prieto L, He Y, Lazaridis T. 2014. Protein arcs may form stable pores in lipid membranes. Biophys. J. 106, 154–161. ( 10.1016/j.bpj.2013.11.4490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.JM Leveritt III, Pino-Angeles A, Lazaridis T. 2015. The structure of a melittin-stabilized pore. Biophys. J. 108, 2424–2426. ( 10.1016/j.bpj.2015.04.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink S.-J. 2008. The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834. ( 10.1021/ct700324x) [DOI] [PubMed] [Google Scholar]
- 103.Ingólfsson HI, Lopez CA, Uusitalo JJ, de Jong DH, Gopal SM, Periole X, Marrink SJ. 2014. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 225–248. ( 10.1002/wcms.1169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saunders MG, Voth GA. 2013. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 42, 73–93. ( 10.1146/annurev-biophys-083012-130348) [DOI] [PubMed] [Google Scholar]
- 105.Izvekov S, Voth GA. 2006. Multiscale coarse-graining of mixed phospholipid/cholesterol bilayers. J. Chem. Theory Comput. 2, 637–648. ( 10.1021/ct050300c) [DOI] [PubMed] [Google Scholar]
- 106.Marrink SJ, De Vries AH, Mark AE. 2004. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 108, 750–760. ( 10.1021/jp036508g) [DOI] [Google Scholar]
- 107.Yesylevskyy SO, Schäfer LV, Sengupta D, Marrink SJ. 2010. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 6, e1000810 ( 10.1371/journal.pcbi.1000810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lazaridis T, Karplus M. 1999. Effective energy function for proteins in solution. Proteins Struct. Funct. Bioinf. 35, 133–152. ( 10.1002/(SICI)1097-0134(19990501)35:2%3C133::AID-PROT1%3E3.0.CO;2-N) [DOI] [PubMed] [Google Scholar]
- 109.Schaefer M, Karplus M. 1996. A comprehensive analytical treatment of continuum electrostatics. J. Phys. Chem. 100, 1578–1599. ( 10.1021/jp9521621) [DOI] [Google Scholar]
- 110.Still WC, Tempczyk A, Hawley RC, Hendrickson T. 1990. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112, 6127–6129. ( 10.1021/ja00172a038) [DOI] [Google Scholar]
- 111.Spassov VZ, Yan L, Szalma S. 2002. Introducing an implicit membrane in generalized Born/solvent accessibility continuum solvent models. J. Phys. Chem. B 106, 8726–8738. ( 10.1021/jp020674r) [DOI] [Google Scholar]
- 112.Lazaridis T. 2003. Effective energy function for proteins in lipid membranes. Proteins Struct. Funct. Bioinf. 52, 176–192. ( 10.1002/prot.10410) [DOI] [PubMed] [Google Scholar]
- 113.Im W, Lee MS, Brooks CL. 2003. Generalized Born model with a simple smoothing function. J. Comput. Chem. 24, 1691–1702. ( 10.1002/jcc.10321) [DOI] [PubMed] [Google Scholar]
- 114.Tanizaki S, Feig M. 2005. A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J. Chem. Phys. 122, 124706 ( 10.1063/1.1865992) [DOI] [PubMed] [Google Scholar]
- 115.Lazaridis T. 2005. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins Struct. Funct. Bioinf. 58, 518–527. ( 10.1002/prot.20358) [DOI] [PubMed] [Google Scholar]
- 116.Panahi A, Feig M. 2013. Dynamic heterogeneous dielectric generalized Born (DHDGB): an implicit membrane model with a dynamically varying bilayer thickness. J. Chem. Theory Comput. 9, 1709–1719. ( 10.1021/ct300975k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lazaridis T. 2005. Structural determinants of transmembrane β-barrels. J. Chem. Theory Comput. 1, 716–722. ( 10.1021/ct050055x) [DOI] [PubMed] [Google Scholar]
- 118.Mihajlovic M, Lazaridis T. 2010. Antimicrobial peptides bind more strongly to membrane pores. Biochim. Biophys. Acta Biomembr. 1798, 1494–1502. ( 10.1016/j.bbamem.2010.02.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lipkin R, Lazaridis T. 2015. Implicit membrane investigation of the stability of antimicrobial peptide β-barrels and arcs. J. Membr. Biol. 248, 469–486. ( 10.1007/s00232-014-9759-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He Y, Prieto L, Lazaridis T. 2013. Modeling peptide binding to anionic membrane pores. J. Comput. Chem. 34, 1463–1475. ( 10.1002/jcc.23282) [DOI] [PubMed] [Google Scholar]
- 121.Mottamal M, Lazaridis T. 2006. Voltage-dependent energetics of alamethicin monomers in the membrane. Biophys. Chem. 122, 50–57. ( 10.1016/j.bpc.2006.02.005) [DOI] [PubMed] [Google Scholar]
- 122.Zhan H, Lazaridis T. 2012. Influence of the membrane dipole potential on peptide binding to lipid bilayers. Biophys. Chem. 161, 1–7. ( 10.1016/j.bpc.2011.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhan H, Lazaridis T. 2013. Inclusion of lateral pressure/curvature stress effects in implicit membrane models. Biophys. J. 104, 643–654. ( 10.1016/j.bpj.2012.12.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bond PJ, Khalid S. 2010. Antimicrobial and cell-penetrating peptides: structure, assembly and mechanisms of membrane lysis via atomistic and coarse-grained molecular dynamic simulations. Protein Pept. Lett. 17, 1313–1327. ( 10.2174/0929866511009011313) [DOI] [PubMed] [Google Scholar]
- 125.Matyus E, Kandt C, Tieleman DP. 2007. Computer simulation of antimicrobial peptides. Curr. Med. Chem. 14, 2789–2798. ( 10.2174/092986707782360105) [DOI] [PubMed] [Google Scholar]
- 126.Tieleman DP, Sansom MSP. 2001. Molecular dynamics simulations of antimicrobial peptides: from membrane binding to trans-membrane channels. Int. J. Quantum Chem. 83, 166–179. ( 10.1002/qua.1208) [DOI] [Google Scholar]
- 127.La Rocca P, Biggin PC, Tieleman DP, Sansom MSP. 1999. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta Biomembr. 1462, 185–200. ( 10.1016/S0005-2736(99)00206-0) [DOI] [PubMed] [Google Scholar]
- 128.Kirsch SA, Böckmann RA. 2015. Membrane pore formation in atomistic and coarse-grained simulations. Biochim. Biophys. Acta Biomembr. 1858, 2266–2277. ( 10.1016/j.bbamem.2015.12.031) [DOI] [PubMed] [Google Scholar]
- 129.Bolintineanu DS, Kaznessis YN. 2011. Computational studies of protegrin antimicrobial peptides: a review. Peptides 32, 188–201. ( 10.1016/j.peptides.2010.10.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Piggot TJ, Holdbrook DA, Khalid S. 2011. Electroporation of the E. coli and S. aureus membranes: molecular dynamics simulations of complex bacterial membranes. J. Phys. Chem. B 115, 13 381–13 388. ( 10.1021/jp207013v) [DOI] [PubMed] [Google Scholar]
- 131.Böckmann RA, De Groot BL, Kakorin S, Neumann E, Grubmüller H. 2008. Kinetics, statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations. Biophys. J. 95, 1837–1850. ( 10.1529/biophysj.108.129437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tarek M. 2005. Membrane electroporation: a molecular dynamics simulation. Biophys. J. 88, 4045–4053. ( 10.1529/biophysj.104.050617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tieleman DP. 2004. The molecular basis of electroporation. BMC Biochem. 5, 1 ( 10.1186/1471-2091-5-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mirjalili V, Feig M. 2015. Interactions of amino acid side-chain analogs within membrane environments. J. Phys. Chem. B 119, 2877–2885. ( 10.1021/jp511712u) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bennett WFD, Sapay N, Tieleman DP. 2014. Atomistic simulations of pore formation and closure in lipid bilayers. Biophys. J. 106, 210–219. ( 10.1016/j.bpj.2013.11.4486) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wohlert J, Den Otter WK, Edholm O, Briels WJ. 2006. Free energy of a trans-membrane pore calculated from atomistic molecular dynamics simulations. J. Chem. Phys. 124,154 905 ( 10.1063/1.2171965) [DOI] [PubMed] [Google Scholar]
- 137.Awasthi N, Hub JS. 2016. Simulations of pore formation in lipid membranes: reaction coordinates, convergence, hysteresis, and finite-size effects. J. Chem. Theory Comput. 12, 3261–3269. ( 10.1021/acs.jctc.6b00369) [DOI] [PubMed] [Google Scholar]
- 138.Bennett WF. D., Hong CK, Wang Y, Tieleman DP. 2016. Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers. J. Chem. Theory Comput. 12, 4524–4533. ( 10.1021/acs.jctc.6b00265) [DOI] [PubMed] [Google Scholar]
- 139.Wang Y, Zhao T, Wei D, Strandberg E, Ulrich AS, Ulmschneider JP. 2014. How reliable are molecular dynamics simulations of membrane active antimicrobial peptides? Biochim. Biophys. Acta Biomembr. 1838, 2280–2288. ( 10.1016/j.bbamem.2014.04.009) [DOI] [PubMed] [Google Scholar]
- 140.Bernèche S, Nina M, Roux B. 1998. Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophys. J. 75, 1603–1618. ( 10.1016/S0006-3495(98)77604-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bachar M, Becker OM. 2000. Protein-induced membrane disorder: a molecular dynamics study of melittin in a dipalmitoylphosphatidylcholine bilayer. Biophys. J. 78, 1359–1375. ( 10.1016/S0006-3495(00)76690-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Andersson M, Ulmschneider JP, Ulmschneider MB, White SH. 2013. Conformational states of melittin at a bilayer interface. Biophys. J. 104, L12–L14. ( 10.1016/j.bpj.2013.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen CH, Wiedman G, Khan A, Ulmschneider MB. 2014. Absorption and folding of melittin onto lipid bilayer membranes via unbiased atomic detail microsecond molecular dynamics simulation. Biochim. Biophys. Acta Biomembr. 1838, 2243–2249. ( 10.1016/j.bbamem.2014.04.012) [DOI] [PubMed] [Google Scholar]
- 144.Tieleman DP, Berendsen HJ. C, Sansom MS. P. 1999. Surface binding of alamethicin stabilizes its helical structure: molecular dynamics simulations. Biophys. J. 76, 3186–3191. ( 10.1016/S0006-3495(99)77470-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kandasamy SK, Larson RG. 2006. Effect of salt on the interactions of antimicrobial peptides with zwitterionic lipid bilayers. Biochim. Biophys. Acta Biomembr. 1758, 1274–1284. ( 10.1016/j.bbamem.2006.02.030) [DOI] [PubMed] [Google Scholar]
- 146.Jang H, Ma B, Nussinov R. 2007. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 7, 1–15. ( 10.1186/1472-6807-7-21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shepherd CM, Vogel HJ, Tieleman DP. 2003. Interactions of the designed antimicrobial peptide MB21 and truncated dermaseptin S3 with lipid bilayers: molecular-dynamics simulations. Biochem. J. 370, 233–243. ( 10.1042/bj20021255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang Y, Schlamadinger DE, Kim JE, McCammon JA. 2012. Comparative molecular dynamics simulations of the antimicrobial peptide CM15 in model lipid bilayers. Biochim. Biophys. Acta Biomembr. 1818, 1402–1409. (doi:j.bbamem.2012.02.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Neale C, Hsu JCY, Yip CM, Pomes R. 2014. Indolicidin binding induces thinning of a lipid bilayer. Biophys. J. 106, L29–L31. ( 10.1016/j.bpj.2014.02.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Perrin BS, Jr, et al. 2014. High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J. Am. Chem. Soc. 136, 3491–3504. ( 10.1021/ja411119m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mika JT, Moiset G, Cirac AD, Feliu L, Bardají E., Planas M, Sengupta D, Marrink SJ, Poolman B. 2011. Structural basis for the enhanced activity of cyclic antimicrobial peptides: the case of BPC194. Biochim. Biophys. Acta Biomembr. 1808, 2197–2205. ( 10.1016/j.bbamem.2011.05.001) [DOI] [PubMed] [Google Scholar]
- 152.Langham AA, Khandelia H, Kaznessis YN. 2006. How can a β-sheet peptide be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of protegrin-1 in micelles. Pept. Sci. 84, 219–231. ( 10.1002/bip.20397) [DOI] [PubMed] [Google Scholar]
- 153.Khandelia H, Kaznessis YN. 2007. Cation-π interactions stabilize the structure of the antimicrobial peptide indolicidin near membranes: molecular dynamics simulations. J. Phys. Chem. B 111, 242–250. ( 10.1021/jp064776j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Herce HD, Garcia AE. 2007. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl Acad. Sci. USA 104, 20 805–20 810. ( 10.1073/pnas.0706574105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Upadhyay SK, Wang Y, Zhao T, Ulmschneider JP. 2015. Insights from micro-second atomistic simulations of melittin in thin lipid bilayers. J. Membr. Biol. 248, 497–503. ( 10.1007/s00232-015-9807-8) [DOI] [PubMed] [Google Scholar]
- 156.Tieleman DP, Shrivastava IH, Ulmschneider MR, Sansom MS. P. 2001. Proline-induced hinges in transmembrane helices: possible roles in ion channel gating. Proteins Struct. Funct. Bioinf. 44, 63–72. ( 10.1002/prot.1073) [DOI] [PubMed] [Google Scholar]
- 157.Tieleman DP, Sansom MS. P, Berendsen HJ. C. 1999. Alamethicin helices in a bilayer and in solution: molecular dynamics simulations. Biophys. J. 76, 40–49. ( 10.1016/S0006-3495(99)77176-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lin J.-H, Baumgaertner A. 2000. Stability of a melittin pore in a lipid bilayer: a molecular dynamics study. Biophys. J. 78, 1714–1724. ( 10.1016/S0006-3495(00)76723-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mihajlovic M, Lazaridis T. 2010. Antimicrobial peptides in toroidal and cylindrical pores. Biochim. Biophys. Acta Biomembr. 1798, 1485–1493. ( 10.1016/j.bbamem.2010.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mihajlovic M, Lazaridis T. 2012. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 1818, 1274–1283. ( 10.1016/j.bbamem.2012.01.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Manna M, Mukhopadhyay C. 2009. Cause and effect of melittin-induced pore formation: a computational approach. Langmuir 25, 12 235–12 242. ( 10.1021/la902660q) [DOI] [PubMed] [Google Scholar]
- 162.Irudayam SJ, Berkowitz ML. 2011. Influence of the arrangement and secondary structure of melittin peptides on the formation and stability of toroidal pores. Biochim. Biophys. Acta Biomembr. 1808, 2258–2266. ( 10.1016/j.bbamem.2011.04.021) [DOI] [PubMed] [Google Scholar]
- 163.Lyu Y, Zhu X, Xiang N, Narsimhan G. 2015. Molecular dynamics study of pore formation by melittin in a 1,2-dioleoyl-sn-glycero-3-phosphocholine and 1, 2-di(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) mixed lipid bilayer. Ind. Eng. Chem. Res. 54, 10 275–10 283. ( 10.1021/acs.iecr.5b01217) [DOI] [Google Scholar]
- 164.Pino-Angeles A, Leveritt JM III, Lazaridis T. 2016. Pore structure and synergy in antimicrobial peptides of the magainin family. PLoS Comput. Biol. 12, e1004570 ( 10.1371/journal.pcbi.1004570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Perrin BS Jr, Fu RC, Myriam LP, Richard W. 2016. Simulations of membrane-disrupting peptides II: AMP piscidin 1 favors surface defects over pores. Biophys. J. 111, 1258–1266. ( 10.1016/j.bpj.2016.08.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Langham AA, Ahmad AS, Kaznessis YN. 2008. On the nature of antimicrobial activity: a model for protegrin-1 pores. J. Am. Chem. Soc. 130, 4338–4346. ( 10.1021/ja0780380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jang H, Ma B, Lal R, Nussinov R. 2008. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic Alzheimer β-amyloid ion channels. Biophys. J. 95, 4631–4642. ( 10.1529/biophysj.108.134551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jang H, Teran Arce F, Ramachandran S, Capone R, Lal R, Nussinov R. 2010. Structural convergence among diverse, toxic β-sheet ion channels. J. Phys. Chem. B 114, 9445–9451. ( 10.1021/jp104073k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lazaridis T, He Y, Prieto L. 2013. Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys. J. 104, 633–642. ( 10.1016/j.bpj.2012.12.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vivcharuk V, Kaznessis Y. 2010. Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer. J. Phys. Chem. B 114, 2790–2797. ( 10.1021/jp909640g) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vivcharuk V, Kaznessis YN. 2011. Thermodynamic analysis of protegrin-1 insertion and permeation through a lipid bilayer. J. Phys. Chem. B 115, 14 704–14 712. ( 10.1021/jp205153y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rui H, Im W. 2010. Protegrin-1 orientation and physicochemical properties in membrane bilayers studied by potential of mean force calculations. J. Comput. Chem. 31, 2859–2867. ( 10.1002/jcc.21580) [DOI] [PubMed] [Google Scholar]
- 173.Irudayam SJ, Pobandt T, Berkowitz ML. 2013. Free energy barrier for melittin reorientation from a membrane-bound state to a transmembrane state. J. Phys. Chem. B 117, 13 457–13 463. ( 10.1021/jp406328d) [DOI] [PubMed] [Google Scholar]
- 174.Sun D, Forsman J, Woodward CE. 2015. Multistep molecular dynamics simulations identify the highly cooperative activity of melittin in recognizing and stabilizing membrane pores. Langmuir 31, 9388–9401. ( 10.1021/acs.langmuir.5b01995) [DOI] [PubMed] [Google Scholar]
- 175.Lin D, Grossfield A. 2014. Thermodynamics of antimicrobial lipopeptide binding to membranes: origins of affinity and selectivity. Biophys. J. 107, 1862–1872. ( 10.1016/j.bpj.2014.08.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Illya G, Deserno M. 2008. Coarse-grained simulation studies of peptide-induced pore formation. Biophys. J. 95, 4163–4173. ( 10.1529/biophysj.108.131300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee H, Larson RG. 2006. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. J. Phys. Chem. B 110, 18 204–18 211. ( 10.1021/jp0630830) [DOI] [PubMed] [Google Scholar]
- 178.Thøgersen L, Schiøtt B, Vosegaard T, Nielsen NC, Tajkhorshid E. 2008. Peptide aggregation and pore formation in a lipid bilayer: a combined coarse-grained and all atom molecular dynamics study. Biophys. J. 95, 4337–4347. (doi:0.1529/biophysj.108.133330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gkeka P, Sarkisov L. 2009. Interactions of phospholipid bilayers with several classes of amphiphilic α-helical peptides: insights from coarse-grained molecular dynamics simulations. J. Phys. Chem. B 114, 826–839. ( 10.1021/jp908320b) [DOI] [PubMed] [Google Scholar]
- 180.Rzepiela AJ, Sengupta D, Goga N, Marrink SJ. 2010. Membrane poration by antimicrobial peptides combining atomistic and coarse-grained descriptions. Faraday Disc. 144, 431–443. ( 10.1039/B901615E) [DOI] [PubMed] [Google Scholar]
- 181.Santo KP, Berkowitz ML. 2012. Difference between magainin-2 and melittin assemblies in phosphatidylcholine bilayers: results from coarse-grained simulations. J. Phys. Chem. B 116, 3021–3030. ( 10.1021/jp212018f) [DOI] [PubMed] [Google Scholar]
- 182.Santo KP, Irudayam SJ, Berkowitz ML. 2013. Melittin creates transient pores in a lipid bilayer: results from computer simulations. J. Phys. Chem. B 117, 5031–5042. ( 10.1021/jp312328n) [DOI] [PubMed] [Google Scholar]
- 183.Horn JN, Cravens A, Grossfield A. 2013. Interactions between fengycin and model bilayers quantified by coarse-grained molecular dynamics. Biophys. J. 105, 1612–1623. ( 10.1016/j.bpj.2013.08.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Horn JN, Sengillo JD, Lin D, Romo TD, Grossfield A. 2012. Characterization of a potent antimicrobial lipopeptide via coarse-grained molecular dynamics. Biochim. Biophys. Acta Biomembr. 1818, 212–218. ( 10.1016/j.bbamem.2011.07.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen L, Li X, Gao L, Fang W. 2014. Theoretical insight into the relationship between the structures of antimicrobial peptides and their actions on bacterial membranes. J. Phys. Chem. B 119, 850–860. ( 10.1021/jp505497k) [DOI] [PubMed] [Google Scholar]
- 186.Bond PJ, Parton DL, Clark JF, Sansom MS. P. 2008. Coarse-grained simulations of the membrane-active antimicrobial peptide maculatin 1.1. Biophys. J. 95, 3802–3815. ( 10.1529/biophysj.108.128686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Woo H.-J, Wallqvist A. 2011. Spontaneous buckling of lipid bilayer and vesicle budding induced by antimicrobial peptide magainin 2: a coarse-grained simulation study. J. Phys. Chem. B 115, 8122–8129. ( 10.1021/jp2023023) [DOI] [PubMed] [Google Scholar]
- 188.Bond PJ, Wee CL, Sansom MS. P. 2008. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry 47, 11 321–11 331. ( 10.1021/bi800642m) [DOI] [PubMed] [Google Scholar]
- 189.Bennett WFD, Tieleman DP. 2011. Water defect and pore formation in atomistic and coarse-grained lipid membranes: pushing the limits of coarse graining. J. Chem. Theory Comput. 7, 2981–2988. ( 10.1021/ct200291v) [DOI] [PubMed] [Google Scholar]
- 190.Wu Z, Cui Q, Yethiraj A. 2010. A new coarse-grained model for water: the importance of electrostatic interactions. J. Phys. Chem. B 114, 10 524–10 529. ( 10.1021/jp1019763) [DOI] [PubMed] [Google Scholar]
- 191.Wang H, Junghans C, Kremer K. 2009. Comparative atomistic and coarse-grained study of water: what do we lose by coarse-graining? Eur. Phys. J. E Soft Matter 28, 221–229. ( 10.1140/epje/i2008-10413-5) [DOI] [PubMed] [Google Scholar]
- 192.Biggin PC, Breed J, Son HS, Sansom MS. P. 1997. Simulation studies of alamethicin–bilayer interactions. Biophys. J. 72, 627 ( 10.1016/S0006-3495(97)78701-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Milik M, Skolnick J. 1992. Spontaneous insertion of polypeptide chains into membranes: a Monte Carlo model. Proc. Natl Acad. Sci. USA 89, 9391–9395. ( 10.1073/pnas.89.20.9391) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ram P, Kim E, Thomson DS, Howard KP, Prestegard JH. 1992. Computer modelling of glycolipids at membrane surfaces. Biophys. J. 63, 1530 ( 10.1016/S0006-3495(92)81729-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Edholm O, Jähnig F. 1988. The structure of a membrane-spanning polypeptide studied by molecular dynamics. Biophys. Chem. 30, 279–292. ( 10.1016/0301-4622(88)85023-3) [DOI] [PubMed] [Google Scholar]
- 196.Brasseur R, Deleers M, Malaisse WJ, Ruysschaert J-M. 1982. Conformational analysis of the calcium–A23187 complex at a lipid–water interface. Proc. Natl Acad. Sci. USA 79, 2895–2897. ( 10.1073/pnas.79.9.2895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Diraviyam K, Stahelin RV, Cho W, Murray D. 2003. Computer modeling of the membrane interaction of FYVE domains. J. Mol. Biol. 328, 721–736. ( 10.1016/S0022-2836(03)00325-5) [DOI] [PubMed] [Google Scholar]
- 198.Ben-Tal N, Ben-Shaul A, Nicholls A, Honig B. 1996. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys. J. 70, 1803 ( 10.1016/S0006-3495(96)79744-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kessel A, Cafiso DS, Ben-Tal N. 2000. Continuum solvent model calculations of alamethicin-membrane interactions: thermodynamic aspects. Biophys. J. 78, 571–583. ( 10.1016/S0006-3495(00)76617-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wimley WC. 2010. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 5, 905–917. ( 10.1021/cb1001558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.He Y, Lazaridis T. 2013. Activity determinants of helical antimicrobial peptides: a large-scale computational study. PLoS ONE 8, e66440 ( 10.1371/journal.pone.0066440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eisenberg D, Schwarz E, Komarony M, Wall R. 1984. Amino acid scale: normalized consensus hydrophobicity scale. J. Mol. Biol. 179, 125–142. ( 10.1016/0022-2836(84)90309-7) [DOI] [PubMed] [Google Scholar]
- 203.Rausch JM, Marks JR, Wimley WC. 2005. Rational combinatorial design of pore-forming β-sheet peptides. Proc. Natl Acad. Sci. USA 102, 10 511–10 515. ( 10.1073/pnas.0502013102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. ( 10.1016/0263-7855(96)00018-5) [DOI] [PubMed] [Google Scholar]
- 205.Rahaman A, Lazaridis T. 2014. A thermodynamic approach to alamethicin pore formation. Biochim. Biophys. Acta Biomembr. 1838, 98–105. ( 10.1016/j.bbamem.2013.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bezrukov SM, Vodyanoy I. 1993. Probing alamethicin channels with water-soluble polymers. Effect on conductance of channel states. Biophys. J. 64, 16 ( 10.1016/S0006-3495(93)81336-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shaw DE, et al. 2014. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer In Proc. of the Int. Conf. for High Performance Computing, Networking, Storage and Analysis, pp. 41–53. New Orleans, LA: IEEE Press. [Google Scholar]
- 208.Wassenaar TA, Ingólfsson HI, Prieß M, Marrink SJ, Schäfer LV. 2013. Mixing MARTINI: electrostatic coupling in hybrid atomistic–coarse-grained biomolecular simulations. J. Phys. Chem. B 117, 3516–3530. ( 10.1021/jp311533p) [DOI] [PubMed] [Google Scholar]
- 209.Rzepiela AJ, Louhivuori M, Peter C, Marrink SJ. 2011. Hybrid simulations: combining atomistic and coarse-grained force fields using virtual sites. Phys. Chem. Chem. Phys. 13, 10 437–10 448. ( 10.1039/C0CP02981E) [DOI] [PubMed] [Google Scholar]
- 210.Han W, Schulten K. 2012. Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: improved backbone hydration and interactions between charged side chains. J. Chem. Theory Comput. 8, 4413–4424. ( 10.1021/ct300696c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Praprotnik M, Delle Site L, Kremer K. 2005. Adaptive resolution molecular-dynamics simulation: changing the degrees of freedom on the fly. J. Chem. Phys. 123, 224106 ( 10.1063/1.2132286) [DOI] [PubMed] [Google Scholar]
- 212.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. ( 10.1038/nrmicro1098) [DOI] [PubMed] [Google Scholar]
- 213.Schmidt NW, Wong GC. L. 2013. Antimicrobial peptides and induced membrane curvature: geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mater. Sci. 17, 151–163. ( 10.1016/j.cossms.2013.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Masunov A, Lazaridis T. 2003. Potentials of mean force between ionizable amino acid side chains in water. J. Am. Chem. Soc. 125, 1722–1730. ( 10.1021/ja025521w) [DOI] [PubMed] [Google Scholar]
- 215.Yuzlenko O, Lazaridis T. 2011. Interactions between ionizable amino acid side chains at a lipid bilayer–water interface. J. Phys. Chem. B 115, 13 674–13 684. ( 10.1021/jp2052213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hénin J, Pohorille A, Chipot C. 2005. Insights into the recognition and association of transmembrane α-helices. The free energy of α-helix dimerization in glycophorin A. J. Am. Chem. Soc. 127, 8478–8484. ( 10.1021/ja050581y) [DOI] [PubMed] [Google Scholar]
- 217.Verkleij AJ, Zwaal RFA, Roelofsen B, Comfurius P, Kastelijn D, van Deenen LLM. 1973. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta Biomembr. 323, 178–193. ( 10.1016/0005-2736(73)90143-0) [DOI] [PubMed] [Google Scholar]
- 218.Bolintineanu D, Hazrati E, Davis HT, Lehrer RI, Kaznessis YN. 2010. Antimicrobial mechanism of pore-forming protegrin peptides: 100 pores to kill E. coli. Peptides 31, 1–8. ( 10.1016/j.peptides.2009.11.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bolintineanu DS, Vivcharuk V, Kaznessis YN. 2012. Multiscale models of the antimicrobial peptide protegrin-1 on Gram-negative bacteria membranes. Int. J. Mol. Sci. 13, 11 000–11 011. ( 10.3390/ijms130911000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mosca DA, Hurst MA, So W, Viajar BSC, Fujii CA, Falla TJ. 2000. IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob. Agents Chemother. 44, 1803–1808. ( 10.1128/AAC.44.7.1803-1808.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Elad S, Epstein JB, Raber-Durlacher J, Donnelly P, Strahilevitz J. 2012. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 41, 229–234. ( 10.1111/j.1600-0714.2011.01094.x) [DOI] [PubMed] [Google Scholar]
- 222.Andres E. 2012. Cationic antimicrobial peptides in clinical development, with special focus on thanatin and heliomicin. Eur. J. Clin. Microbiol. Infect. Dis. 31, 881–888. ( 10.1007/s10096-011-1430-8) [DOI] [PubMed] [Google Scholar]