

Abstract
Kearns-Sayre syndrome (KSS) is a mitochondrial myopathy resulting from mitochondrial DNA deletion. This syndrome primarily involves the central nervous system, eyes, skeletal muscles and the heart. The most well-known cardiac complications involve the conduction system; however, there have been case reports describing cardiomyopathy. We describe a case of a child with KSS who presented with decompensated cardiac failure from dilated cardiomyopathy representing cardiomyocyte involvement of KSS. Our patient had a rapidly progressing course, despite maximal medical management, requiring emergent institution of extracorporeal membrane oxygenation and transition to a ventricular assist device. To the best of our knowledge, this is the youngest patient in the literature to have dilated cardiomyopathy in KSS.
Background
Kearns-Sayre syndrome (KSS), first described in 1958, is a mitochondrial myopathy characterised by the following features: age of onset prior to 20 years of age, chronic progressive ophthalmoplaegia and retinal pigmentary degeneration. It arises from large mitochondrial DNA deletions (mtDNA) in various tissues, causing impaired respiratory chain function, specifically in the metabolically active tissues such as of the heart, central nervous system and skeletal muscles. Clinical features of KSS include muscle weakness, central nervous system dysfunction such as deafness, ataxia, dementia, blindness resulting from retinal degeneration and endocrine and metabolic abnormalities such as diabetes mellitus. Cardiovascular complications primarily involve the conduction system, including progressive atrioventricular (AV) block requiring pacemaker implantation. Cardiomyopathy has been reported but is rare. This is the first report of a child with KSS who developed cardiomyopathy that progressed rapidly despite maximal medical management, requiring mechanical support and consideration of cardiac transplantation.
Case presentation
An 11-year-old patient with acute congestive cardiac failure and pulmonary oedema was transferred to our hospital, from an outside facility, for further management. This patient was known to have KSS and Fanconi's syndrome. The patient had, at the outside facility, presented in shock, with a 5-day history of diarrhoea and vomiting. There, the patient received fluid boluses of 80 mL/kg in the first few hours, and developed acute pulmonary oedema and signs of congestive heart failure requiring initiation of dobutamine. An echocardiogram performed at the time was suggestive of moderate left ventricular enlargement and poor left ventricular function with a shortening fraction of 8%. Owing to acute worsening of the clinical condition, the patient was transferred to our hospital for further management. On presentation to our hospital, the patient was afebrile, tachycardic and tachypnoeic, with mild subcostal and intercostal retractions. Blood pressure was 89/55 mm Hg and oxygen saturation was 100% on 0.5 L/min of oxygen by nasal cannula. On examination, the patient had facial oedema, pitting pedal oedema, weak peripheral pulses, capillary refill of 3 s, a 2/6 systolic murmur over the apex, no gallop and tender hepatomegaly of 5 cm below the costal margin. Neurological examination was positive for mild hypotonia in all extremities. The ECG on the day of the previous admission showed diffuse intraventricular conduction delay with a normal PR interval (figure 1). The echocardiogram on arrival at our hospital showed a dilated, poorly functioning left ventricle with moderate to severe mitral and tricuspid regurgitation (figure 2A, B). The working diagnosis was dilated cardiomyopathy secondary to mitochondrial disease with decompensated congestive heart failure.
Figure 1.
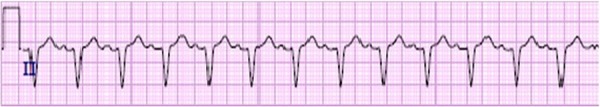
Rhythm strip on admission showing diffuse intraventricular conduction delay with normal PR interval.
Figure 2.
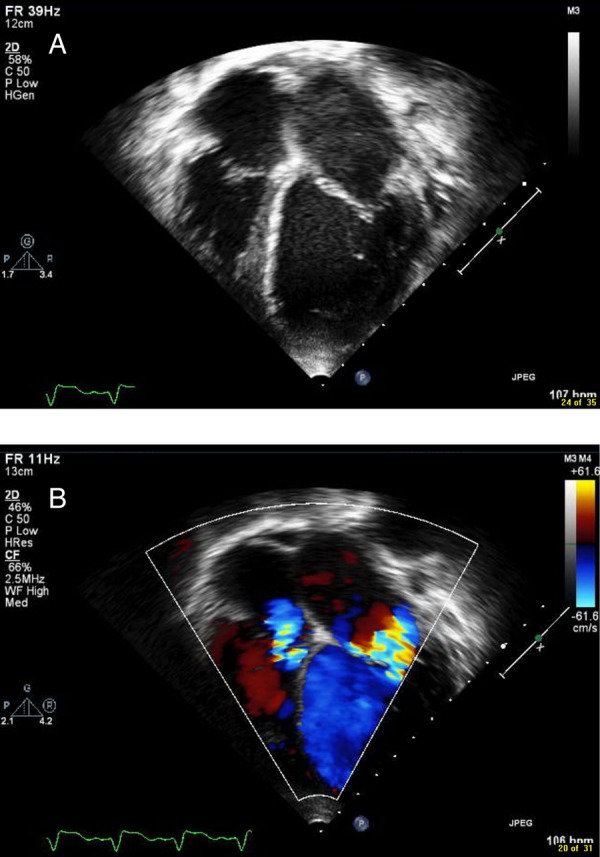
Still frame image from the echocardiogram on arrival showing a dilated left ventricle (A) on apical four-chamber view and (B) with moderate to severe mitral and tricuspid regurgitation on apical four-chamber view in colour.
On review of the medical history, we found that our patient was overall a healthy child, active in ballet, ice skating and playing piano. The patient had been diagnosed with Fanconi's syndrome 4 years prior to this presentation, with symptoms of decreased activity, fatigue and weight loss. Mitochondrial myopathy was subsequently diagnosed by muscle biopsy. Eventually, this child developed hearing loss and deterioration of vision, which led to the diagnosis of KSS. Genetic testing revealed a 5 kb mtDNA deletion with heteroplasmy of 65%, and treatment with coenzyme Q and l-carnitine was initiated. A cardiac evaluation had been performed within the year preceding hospitalisation. The ECG at that time showed normal AV conduction and mild intraventricular conduction delay (figure 3). The echocardiogram revealed normal left ventricular size and function.
Figure 3.

ECG 1 year prior to presentation.
Investigations
ECG showed second and intermittent third degree AV block.
Echocardiogram showed a dilated, poorly functioning left ventricle with moderate to severe mitral and tricuspid regurgitation, with fractional shortening of 8%.
Myocardial morphology on histopathology (figure 4) and light microscopy demonstrated myocardial fibrosis (lighter stained areas) and cardiomyocyte atrophy with cytoplasm vacuolisation, and accumulation of microgranules that corresponded to abnormal mitochondria (arrowheads), using Gomori-trichrome stain ×400; and electron microscopy showed a locus of vacuolated cytoplasm with myofibrils (left) pushed by numerous abnormally shaped polymorphous mitochondria (centre), using electron microscopy ×28 000.
Figure 4.

Myocardial morphology. (A) Light microscopy demonstrating myocardial fibrosis (lighter stained areas) and cardiomyocyte atrophy with cytoplasm vacuolisation, and accumulation of microgranules that correspond to abnormal mitochondria (arrowheads). Gomori-trichrome stain; ×400. (B) Electron microscopy showing a locus of vacuolated cytoplasm with myofibrils (left) pushed by numerous abnormally shaped polymorphous mitochondria (centre). Electron microscopy ×28 000.
Differential diagnosis
Dilated cardiomyopathy and myocarditis.
Treatment
Inotropic support with dopamine, dobutamine and milrinone was initiated in addition to diuretics, which resulted in significant improvement in the next 24–48 h. On day 4 of admission, the patient developed respiratory distress again, due to recurrence of pulmonary oedema, necessitating intubation. This deterioration was attributed to urosepsis imposing higher metabolic demands on her heart. In addition, the patient developed second and intermittent third degree AV block requiring temporary pacemaker placement in the next 48 h. Intravenous solumedrol and intravenous immunoglobulin were also administered, with a presumptive diagnosis of myocarditis as a contributing aetiology for the AV node dysfunction and heart failure. Despite maximal medical management, the patient's clinical condition continued to deteriorate and she developed end-organ dysfunction, including renal failure and lactic acidosis, necessitating the institution of extracorporeal membrane oxygenation on day 6 of admission, which was transitioned to a pulsatile flow left ventricular assist device on day 10 of admission. The patient was listed for transplant once her renal function improved on mechanical support.
Outcome and follow-up
Despite stabilisation of the cardiac condition, the patient developed intracranial bleeding causing a devastating neurological injury that led to a decision to withdraw support. The examination of the heart on autopsy confirmed presence of abnormal mitochondria from involvement of heart muscle in KSS (figure 4).
Discussion
KSS is a rare mitochondrial encephalomyopathy first described in 1958 as a triad of chronic progressive ophthalmoplaegia, retinitis pigmentosa and complete heart block.1 Genetic studies have shown mitochondrial DNA deletion in affected tissues, usually with less extensive mitochondrial DNA deletion in myocardial tissue.2 Cardiac manifestations have been described in 57% of patients affected by KSS, typically including syncope, congestive cardiac failure and cardiac arrest.3 Cardiac involvement in this syndrome primarily consists of prolonged intraventricular conduction time, bundle-branch block and varying degrees of progressive AV block. Any degree of heart block may be seen with rapid and unpredictable progression to complete heart block.4 Patients with bifascicular block are more likely to progress to complete AV block.5 Electrophysiology studies in these patients clearly demonstrate abnormalities in the AV node. The His Purkinje system is evidenced by a short A-H (denoting impulse generation in the atria and conduction to the bundle of His) and prolonged H-V interval (conduction from bundle of His to the ventricles).6 Rarely, ventricular arrhythmias have been reported with normal or prolonged QT with sudden death from torsade.7 Progressive degeneration of the conduction system results in a mortality rate as high as 20%.4 A study on 67 patients over 10 years showed that 32% developed conduction defects, 12% required pacemaker implant and 5% suffered sudden death.8 Prophylactic pacemaker placement has been recommended even in asymptomatic patients with bifascicular block.6 Disappearance of spontaneous cardiac impulse formation has been described on long-term follow-up of a patient with a pacemaker.9
Cardiomyopathy is commonly seen in patients with respiratory chain disorders/mitochondrial myopathies, usually hypertrophic cardiomyopathy. Most studies involving patients with mitochondrial myopathy and KSS, reveal conduction disturbances and pacemaker dependence on long-term follow-up but no cardiomyopathy.2 10 There is one case series of five patients described in the literature, including subclinical left ventricular dysfunction in two patients with chronic progressive ophthalmoplaegia (presumably KSS). Only a few case reports of KSS and cardiomyopathy have been published, and these have been on adults who developed acute congestive cardiac failure and required heart transplant.11 12 All the patients were reportedly doing well 12–18 months after transplant. The management decision faced was whether our patient was suitable for cardiac transplant in the presence of renal dysfunction from Fanconi's syndrome, and the difficulty in prognosticating the progression of her KSS. Fanconi's syndrome is known to cause mild renal tubular dysfunction, but renal failure, interstitial nephritis and nephrotic syndrome have been rarely reported.13
From a neurological standpoint, there was no way of predicting this patient's clinical course. However, neurological involvement in KSS is typically manifested by oropharyngeal and oesophageal dysfunction, and proximal more than distal limb muscle weakness. Respiratory muscle weakness and ventilator dependency have been described, with seizures and strokes being very rare. Cardiac involvement has been found to be the most important prognostic factor, with conduction disturbances being the major source of morbidity and mortality in KSS. In one study, the mortality rate in KSS without heart disease was reported to be 26%, which increased to 71% when including heart disease.14 Death usually occur in the third or fourth decade from cardiac involvement. Our patient presented at 11 years of age with acute congestive heart failure without any conduction abnormality at baseline. To the best of our knowledge, this is, in the current literature, the youngest patient with KSS to develop acute cardiac decompensation requiring mechanical support and consideration for heart transplant. Although not common, patients with KSS can develop cardiomyopathy in addition to conduction abnormalities. This awareness is imperative in evaluation, appropriate referral and follow-up of these patients.
Learning points.
Kearns-Sayre syndrome (KSS) is a mitochondrial myopathy that involves significant cardiac conduction abnormalities.
Although not reported often, cardiomyopathy in KSS can, specifically, be rapidly progressive.
Cardiac transplantation can be therapeutic in these patients and should be considered since the major source of mortality is cardiac dysfunction.
Footnotes
Competing interests: None declared.
Patient consent: Not obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmoplegia and complete heart block. Arch Ophthalmol 1958;60:280–9. 10.1001/archopht.1958.00940080296016 [DOI] [PubMed] [Google Scholar]
- 2.Anan R, Nakagawa M, Miyata M et al. Cardiac Involvement in Mitochondrial Diseases. A study of 17 patients with documented mitochondrial DNA defects. Circulation 1995;91:955–61. 10.1161/01.CIR.91.4.955 [DOI] [PubMed] [Google Scholar]
- 3.Berenberg RA, Pellock JM, DiMauro S et al. Lumping or splitting? “Ophthalmoplegia plus” or Kearns Sayre syndrome? Ann Neurol 1977;1:37–54. 10.1002/ana.410010104 [DOI] [PubMed] [Google Scholar]
- 4.Charles R, Holt S, Kay JM et al. Myocardial ultrastructure and the development of atrioventricular block in Kearns-Sayre syndrome. Circulation 1981;63:214–19. 10.1161/01.CIR.63.1.214 [DOI] [PubMed] [Google Scholar]
- 5.[No authors listed] How to diagnose diastolic heart failure. European Study Group on Diastolic Heart Failure. Eur Heart J 1998;19:990–1003. 10.1053/euhj.1998.1057 [DOI] [PubMed] [Google Scholar]
- 6.Polak PE, Zijlstra F, Roelandt JR. Indications for pacemaker implantation in the Kearns-Sayre syndrome. Eur Heart J 1989;10:281–2. [DOI] [PubMed] [Google Scholar]
- 7.Subbiah RN, Kuchar D, Baron D. Torsades de pointes in a patient with Kearns-Sayre syndrome: a fortunate finding. Pacing Clin Electrophysiol 2007;30:137–9. 10.1111/j.1540-8159.2007.00590.x [DOI] [PubMed] [Google Scholar]
- 8.Hayes DL, Hyberger LK, Hodge DO. Incidence of conduction system disease and need for permanent pacemaker in patients with Kearns Sayre syndrome. (abstract 149). PACE 2001;24(4 part II):576. [Google Scholar]
- 9.Yesil M, Bayata S, Postaci N et al. Progression of conduction system disease in a paced patient with Kearns-Sayre syndrome. Clin Cardiol 2007;32:65–7. 10.1002/clc.20242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limongelli G, Tome-Esteban M, Dejthevapron C et al. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail 2010;12:114–21. 10.1093/eurjhf/hfp186 [DOI] [PubMed] [Google Scholar]
- 11.Channer KS, Channer JL, Campbell MJ et al. Cardiomyopathy in the Kearns-Sayre syndrome. Br Heart J 1988;59:486–90. 10.1136/hrt.59.4.486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tranchant C, Moussan B, Mohr M et al. Cardiac transplantation in an incomplete Kearns–Sayre syndrome with mitochondrial DNA deletion. Neuromuscul Disord 1993;3:561–6. 10.1016/0960-8966(93)90116-2 [DOI] [PubMed] [Google Scholar]
- 13.Debray FG, Merouani A, Lambert M et al. Acute tubular dysfunction with Fanconi syndrome: a new manifestation of mitochondrial cytopathies. Am J Kidney Dis 2008;51:691–6. 10.1053/j.ajkd.2007.11.024 [DOI] [PubMed] [Google Scholar]
- 14.Bindoff LL. Mitochondria and the heart. Eur Heart J 2003;24:221–4. 10.1016/S0195-668X(02)00694-2 [DOI] [PubMed] [Google Scholar]