

ABSTRACT
Bifidobacteria are commensals that colonize the orogastrointestinal tract and rarely cause invasive human infections. However, an increasing number of bifidobacterial blood culture isolates has lately been observed in Norway. In order to investigate the pathogenicity of the Bifidobacterium species responsible for bacteremia, we studied Bifidobacterium isolates from 15 patients for whom cultures of blood obtained from 2013 to 2015 were positive. We collected clinical data and analyzed phenotypic and genotypic antibiotic susceptibility. All isolates (11 Bifidobacterium longum, 2 B. breve, and 2 B. animalis isolates) were subjected to whole-genome sequencing. The 15 patients were predominantly in the extreme lower or upper age spectrum, many were severely immunocompromised, and 11 of 15 had gastrointestinal tract-related conditions. In two elderly patients, the Bifidobacterium bacteremia caused a sepsis-like picture, interpreted as the cause of death. Most bifidobacterial isolates had low MICs (≤0.5 mg/liter) to beta-lactam antibiotics, vancomycin, and clindamycin and relatively high MICs to ciprofloxacin and metronidazole. We performed a pangenomic comparison of invasive and noninvasive B. longum isolates based on 65 sequences available from GenBank and the sequences of 11 blood culture isolates from this study. Functional annotation identified unique genes among both invasive and noninvasive isolates of Bifidobacterium. Phylogenetic clusters of invasive isolates were identified for a subset of the B. longum subsp. longum isolates. However, there was no difference in the number of putative virulence genes between invasive and noninvasive isolates. In conclusion, Bifidobacterium has an invasive potential in the immunocompromised host and may cause a sepsis-like picture. Using comparative genomics, we could not delineate specific pathogenicity traits characterizing invasive isolates.
KEYWORDS: DNA sequencing, antibiotic resistance, bifidobacteria, blood culture, bloodstream infections, mass spectrometry, pangenome, probiotics, susceptibility testing, virulence factors
INTRODUCTION
Bifidobacteria are anaerobic, nonsporulating Gram-positive rods representing ubiquitous inhabitants of the human orogastrointestinal tract and vagina. The genus consists of more than 50 species, with only 10 species being found in humans. In breast-fed infants, bifidobacteria constitute more than 80% of the intestinal microbiota, whereas bifidobacteria comprise only 3 to 6% of the adult fecal flora (1, 2). Moreover, the species distribution is different in infants and adults; Bifidobacterium adolescentis and Bifidobacterium longum subsp. longum are the major bifidobacterial species in the adult intestinal flora, and Bifidobacterium longum subsp. infantis and Bifidobacterium breve are the predominant species in the intestinal tract of human infants (3–5). Selected members of the genus Bifidobacterium are believed to exert health benefits to the host, including competitive exclusion of pathogens (6, 7), modulation of the immune system (8, 9), and degradation of diet-derived carbohydrates (10). On the basis of these effects, bifidobacteria are often added to probiotic products in combination with other lactic acid bacteria to prevent or treat diseases (11, 12), although the evidence is inadequate. Nevertheless, a growing number of inpatients in U.S. hospitals often receive probiotics as part of their care (13).
The pathogenic potential of Bifidobacterium remains unclear. Data on the incidence of invasive infections are very limited, but Bifidobacterium species are estimated to represent 0.5 to 3% of anaerobic blood culture isolates (14, 15). Among adults, only 15 cases of Bifidobacterium bacteremias had been reported in the literature until 2015 (16), and these were predominantly among patients with underlying gastrointestinal disease and/or impaired immunity. There is a paucity of data on the clinical presentations, prognostic factors, and outcomes of patients with Bifidobacterium bacteremia.
Over the last few years, an increasing number of Bifidobacterium blood culture isolates have been reported to the Norwegian Organization for Surveillance of Antimicrobial Resistance (NORM) (17). The primary objective of this study was to describe the clinical characteristics, antimicrobial susceptibilities, treatments, and outcomes for 15 patients with Bifidobacterium bacteremia (11 with B. longum bacteremia, 2 with B. breve bacteremia, and 2 with B. animalis bacteremia). Furthermore, we analyzed the phylogeny, the resistome, and putative virulence factors by whole-genome sequencing (WGS). Finally, we performed a pangenome comparative analysis of all hitherto reported genome sequences of invasive versus noninvasive B. longum isolates of human origin in order to search for specific traits characterizing invasive B. longum isolates.
RESULTS
Patient characteristics, treatments, and clinical outcomes.
Demographic and clinical data are listed in Table 1. Six patients were above 80 years of age, and four patients were born prematurely, before 33 weeks of gestational age. The three extremely preterm infants (patients 13 to 15) had received a probiotic product containing B. longum, aiming to prevent necrotizing enterocolitis, as reported in a previous study (18). There was no information about probiotic supplementation in the medical records of the other 12 patients. The majority of the 15 patients were either immunocompromised or had signs of a severe underlying condition. Ten patients had gastrointestinal tract-related diseases, and nine of these patients had a compromised intestinal barrier or signs of a leaky gut. Four patients died before or during admission. Two patients (patients 2 and 9), both of whom were severely compromised and elderly, developed signs of sepsis/septic shock, and the blood culture showed monomicrobial growth of B. longum. On the basis of their clinical presentation, the blood culture results, and no other obvious infectious agent identified, we considered the deaths of these two patients to probably be attributable to B. longum sepsis. One patient (patient 4), an infant who died before admission to the hospital, had no fever or signs of infection immediately prior to death, no history of infections, and no signs of infection/inflammation on autopsy. We did not consider that there was enough evidence to define the death in this patient to be attributable to B. longum sepsis. The last patient who died (patient 12) was very old and frail. She died 14 h after admission to the hospital and only 3 h after the blood sample for culture was obtained. Due to her advanced age and clinical condition, no antibiotic therapy was started. There was polymicrobial growth in the blood culture (Table 1). On autopsy, there were signs of poor gut circulation (no perforation), and a dilated cardiomyopathy was confirmed. We did not consider that there was enough evidence to define the death in this patient to be attributable to B. longum sepsis. Thirteen patients received antibiotic treatment. Polymicrobial bloodstream infections, mainly caused by a combination of bifidobacteria and other organisms originating from the gastrointestinal tract, were observed in six patients.
TABLE 1.
Demographic and clinical data for 15 patients with Bifidobacterium bacteremia
| Patient no. | Hospital | Age | Gender | Underlying condition(s) | Antibiotic or immunosuppressive therapy(ies) prior to onset of bacteremia | Clinical presentation | Blood culture finding(s) | Antibiotic therapy(ies) | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A | 39 yr | Female | Diverticulitis | None | Fever, hypotension, back pain | B. longum and Clostridium paraputrificum at onset of disease, Escherichia coli 2 days later | Ampicillin, gentamicin, metronidazole, clindamycin | Recovered |
| 2 | A | 81 yr | Male | Parkinson's disease, abdominal surgery due to volvulus | Cephalothin, metronidazole, and doxycycline until 2 days before onset of bacteremia | Sepsis and respiratory failure 3 days after abdominal surgery | B. longum (monomicrobial) | Benzylpenicillin | Death |
| 3 | B | 81 yr | Male | Severe chronic obstructive pulmonary disease | Ampicillin, gentamicin, ciprofloxacin, cefuroxime, and meropenem until 4 days before onset of bacteremia | Fever and pneumonia | B. longum (monomicrobial), 2 of 4 blood culture bottles positive | Meropenem | Recovered |
| 4 | C | 3 wk | Male | Prematurity (32 wk of gestation) | None | Sudden infant death syndrome prior to hospital admission | B. longum (monomicrobial), obtained postmortem | No | Death |
| 5 | C | 40 yr | Male | Adenocarcinoma with perforated cecum | None (treated for acute leukemia at age 15 yr) | Peritonitis, septic shock | B. longum (monomicrobial) | Piperacillin-tazobactam, metronidazole | Recovered |
| 6 | D | 85 yr | Male | Metastatic colorectal cancer | None | Fever | B. longum, Streptococcus oralis, and Klebsiella pneumoniae at onset of disease | Ampicillin, gentamicin | Recovered |
| 7 | E | 49 yr | Female | Recurrent wound infections (over years) | Dicloxacillin at onset of bacteremia | Polymicrobial local bursitis, no signs of sepsis | B. breve (monomicrobial) | Benzylpenicillin, ciprofloxacin, clindamycin | Recovered |
| 8 | E | 69 yr | Male | Infected aortic graft with aortoenteric fistula | Prednisolone due to tendinitis 4 wk prior to onset of bacteremia | Chills | B. animalis, Fusobacterium nucleatum, and Veillonella parvula at onset of disease | Cefotaxime, ceftriaxone | Recovered |
| 9 | E | 71 yr | Male | Metastatic lung cancer | Methylprednisolone at onset bacteremia, chemotherapy and radiation therapy 4 mo prior to onset of bacteremia | Septic shock | B. longum (monomicrobial) | Cefotaxime, ciprofloxacin, metronidazole | Death |
| 10 | F | 84 yr | Female | Pyelonephritis, hydronephrosis caused by a kidney stone | Ciprofloxacin at onset of bacteremia | Fever, chills, abdominal pain | B. breve, Bacteroides spp., and Candida glabrata at onset of disease | Cefotaxime | Recovered |
| 11 | G | 84 yr | Male | Pancreatic cancer | Ciprofloxacin at onset of bacteremia | Fever, chills, abdominal pain, signs of sepsis | B. animalis and Lactobacillus johnsonii | Piperacillin-tazobactam | Recovered |
| 12 | H | 98 yr | Female | Congestive heart failure, systemic amyloidosis, chronic constipation | Prednisolone at onset of bacteremia | Afebrile on admission but developed fever and dyspnea, died 14 h after admission | B. longum and Bacteroides fragilis | No | Death |
| 13 | I | 2 wk | Male | Prematurity (23 wk of gestation), spontaneous gut perforation | Penicillin and gentamicin | Sepsis | B. longum (monomicrobial) | Cefotaxime, gentamicin | Recovered |
| 14 | I | 5 wk | Female | Prematurity (24 wk of gestation), leaky gut after necrotizing enterocolitis | Ampicillin and gentamicin 1 wk before onset of bacteremia | Sepsis | B. longum (monomicrobial) | Ampicillin, gentamicin, metronidazole | Recovered |
| 15 | B | 2 wk | Male | Prematurity (24 wk of gestation) | Ampicillin and gentamicin 1 wk before onset of bacteremia | Increasing apneas, bradycardia, temp instability | B. longum (monomicrobial) | Recovered |
Species identification and phylogenetic grouping.
Using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), the isolates were assigned to the following species: B. longum (n = 11), B. breve (n = 2), and B. animalis (n = 2). Whole-genome phylogenetics by comparison of the sequences of the isolate genomes to those of reference genomes further classified the 11 B. longum isolates to the subspecies level: B. longum subsp. infantis (n = 4) and B. longum subsp. longum (n = 7). Phylogenetic reconstruction grouped the 15 isolates into four clades (Fig. 1). B. breve and B. animalis grouped into clade I and clade IV, respectively. Clade II comprised only B. longum subsp. infantis isolates, while clade III comprised only B. longum subsp. longum isolates. There was no association between the different clades and the different hospitals in which the patients had received care.
FIG 1.

Dendrogram representing the arrangement of clusters between the 15 isolates and the prevalence of genes encoding resistance to antibiotic groups.
Phenotypic antimicrobial susceptibility.
All isolates showed low MIC values to vancomycin (0.25 to 1 mg/liter), meropenem (0.016 to 1 mg/liter), and piperacillin-tazobactam (0.064 to 1 mg/liter) (Table 2). One of the B. breve isolates and both B. animalis isolates displayed MICs of >16 mg/liter to tetracycline. Nine of 15 isolates displayed ciprofloxacin MICs of ≥32 mg/liter. High MIC values (MICs ≥ 256 mg/liter) for metronidazole were observed in six isolates.
TABLE 2.
Susceptibility to antimicrobial agents and putative resistance genes in bifidobacteriaa
| Clade and patient no. | Species | PEN |
MTZ |
CLI |
PIP-TAZ |
VAN |
CTX |
CIP |
MRP |
TET |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | MIC (mg/liter) | Res. genes | ||
| Clade I | |||||||||||||||||||
| 10 | B. breve | 0.25 | >256 | 0.064 | 0.25 | 0.50 | 1 | 1 | mfd, gyrA | 1 | 1 | ||||||||
| 7 | B. breve | 0.25 | >256 | 0.016 | 0.064 | 0.25 | 8 | >32 | mfd, gyrA | 1 | 32 | tet(T) | |||||||
| Clade II | |||||||||||||||||||
| 14 | B. longum subsp. infantis | 0.125 | 32 | 0.25 | 0.064 | 0.25 | 0.25 | 4 | mfd, gyrA | 0.016 | 4 | ||||||||
| 15 | B. longum subsp. infantis | 0.125 | 32 | 0.25 | 0.064 | 0.25 | 0.25 | 4 | mfd, gyrA | 0.016 | 4 | ||||||||
| 13 | B. longum subsp. infantis | 0.125 | 16 | 0.25 | 0.064 | 0.25 | 0.25 | 4 | mfd, gyrA | 0.016 | 4 | ||||||||
| 4 | B. longum subsp. infantis | 0.25 | >256 | 0.064 | lmrD | 0.125 | 0.25 | 0.50 | >32 | mfd, gyrA | 0.064 | 1 | |||||||
| Clade III | |||||||||||||||||||
| 12 | B. longum subsp. longum | 0.50 | >256 | 0.125 | 1 | 0.50 | 0.50 | 0.5 | mfd, gyrA | 1 | 32 | ||||||||
| 9 | B. longum subsp. longum | 0.50 | >256 | 0.064 | 0.5 | 0.50 | 1 | >32 | mfd, gyrA | 0.25 | 0.50 | ||||||||
| 3 | B. longum subsp. longum | 0.50 | AIE | 8 | 0.064 | 0.50 | 0.25 | 1 | AIE | >32 | mfd, gyrA | 0.50 | 1.0 | AIE | |||||
| 5 | B. longum subsp. longum | 0.25 | 8 | 0.032 | 0.25 | 0.50 | 0.50 | >32 | mfd, gyrA | 0.064 | 0.50 | ||||||||
| 6 | B. longum subsp. longum | 0.25 | 8 | 0.064 | 1 | 0.25 | 1 | >32 | mfd, gyrA | 0.125 | 0.50 | ||||||||
| 2 | B. longum subsp. longum | 0.50 | >256 | >256 | 0.5 | 0.25 | 1 | >32 | mfd, gyrA | 0.25 | 1.0 | ||||||||
| 1 | B. longum subsp. longum | 0.50 | 8 | 0.064 | 0.25 | 0.25 | 1 | >32 | mfd, gyrA | 0.125 | 0.50 | ||||||||
| Clade IV | |||||||||||||||||||
| 11 | B. animalis | 0.25 | 8 | 0.032 | 0.25 | 0.50 | 0.50 | 0.5 | mfd, gyrA-gyrB | 0.064 | 16 | tet(T) | |||||||
| 8 | B. animalis | 0.50 | 128 | 0.032 | 0.50 | 1 | 2 | >32 | mfd, gyr-gyrB | 0.125 | 16 | tet(T) | |||||||
PEN, penicillin; MTZ, metronidazole; CLI, clindamycin; PIP-TAZ, piperacillin-tazobactam; VAN, vancomycin; CTX, cefotaxime; CIP, ciprofloxacin; MRP, meropenem; TET, tetracycline; Res. genes, groups of genes presumed to predict resistance (Comprehensive Antibiotic Resistance Database [CARD]); AIE, antibiotic inactivation enzymes (encompasses several enzymes that catalyze the inactivation of an antibiotic); lmrD, lincomycin resistance gene; mfd, mutation frequency decline gene involved in strand-specific DNA repair (overexpression may lead to ciprofloxacin resistance); gyrA, Mycobacterium tuberculosis gyrA mutant; gyrB, Mycobacterium tuberculosis gyrB mutant; tet(T), tetracycline resistance gene.
Pangenome analysis and comparative genomics of B. longum species.
The genome sequences of 76 B. longum isolates were used to calculate the total gene repertoire of the B. longum taxon on the basis of clusters of orthologous groups (COGs). We identified a B. longum pangenome consisting of 7,876 COGs (Fig. 2). A total of 710 genes (COGs) shared by all 76 B. longum isolates represented the core genome. The functional classification of the genes in the core as well as the accessory genomes revealed that a large proportion had yet unknown functions. However, the most common functional classes represented genes involved in housekeeping functions, like carbohydrate and amino acid transport and metabolism, translation, ribosomal structure and biogenesis, transcription, and nucleotide transport and metabolism.
FIG 2.

Pangenome of B. longum showing the functional assignment of the core and accessory (soft core, shell, and cloud) genomes. The results are based on the analysis of 76 isolates.
The pangenome analysis of all invasive and noninvasive isolates of B. longum subsp. longum and B. longum subsp. infantis revealed unique clusters in both subspecies. For the 34 invasive and noninvasive B. longum subsp. longum isolates, there were 91 and 169 unique clusters, respectively. For the 13 invasive and noninvasive B. longum subsp. infantis isolates, there were 48 and 31 unique clusters, respectively. Functional classification of these clusters identified that unique genes involved in replication, recombination, repair, and transcription were more prevalent in the group of noninvasive isolates than invasive isolates. In contrast, unique genes involved in carbohydrate transport and metabolism and defense mechanisms were more prevalent in the group of invasive isolates than in noninvasive isolates (Fig. 3A and B).
FIG 3.

(A) Functional distribution (%) of unique genes from invasive and noninvasive isolates of B. longum subsp. infantis; (B) functional distribution (%) of unique genes from invasive and noninvasive isolates of B. longum subsp. longum.
To further discriminate clusters of invasive isolates from noninvasive isolates, phylogenetic trees based on the accessory genome were generated for all 34 isolates of B. longum subsp. longum and all 13 isolates of B. longum subsp. infantis. Interestingly, this showed that six of seven invasive B. longum subsp. longum isolates were positioned on subbranches of the same cluster (Fig. 4). However, a similar finding was not shown for invasive B. longum subsp. infantis isolates.
FIG 4.
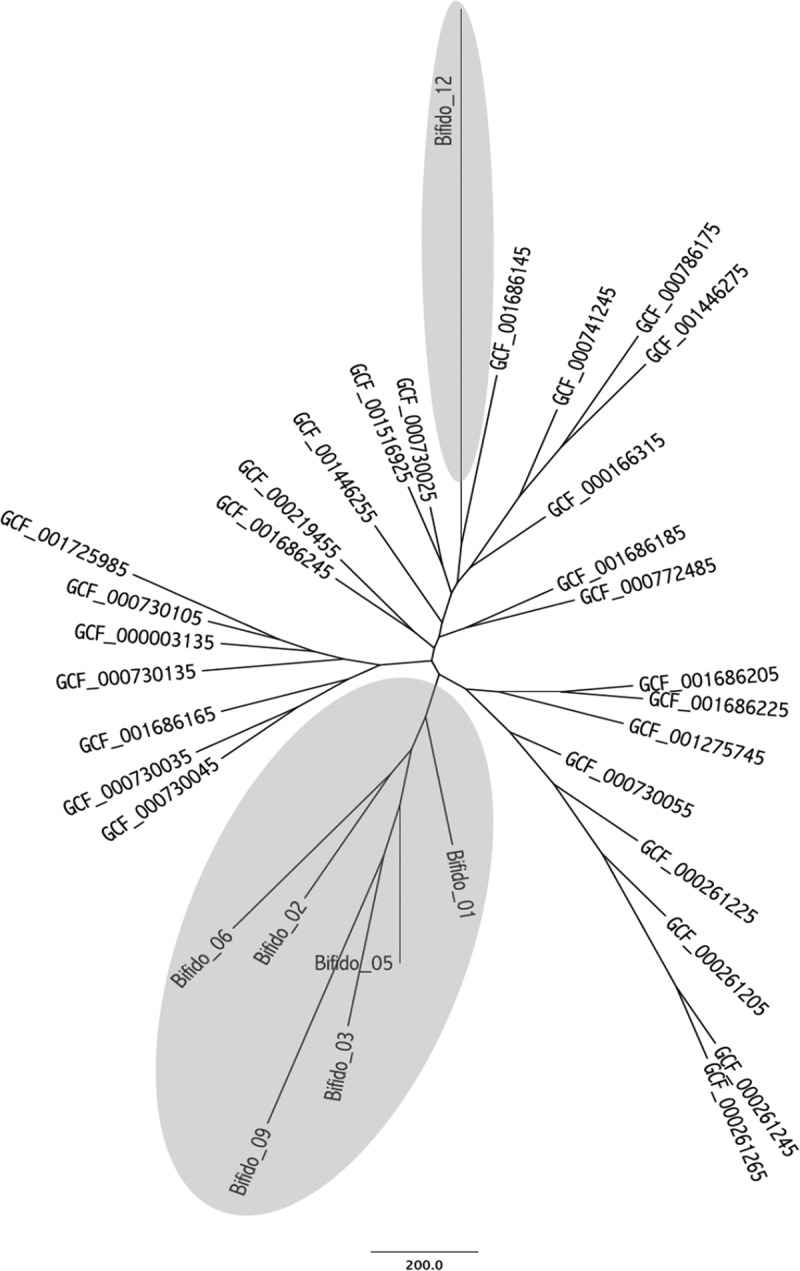
Genetic relationship between invasive and noninvasive B. longum subsp. longum isolates based on accessory genome analysis. Invasive isolates are presented on a gray background.
Bifidobacterium resistome.
The complete list of putative antibiotic resistance genes is reported in Table 2. Genes encoding efflux pumps were found in all isolates. One B. longum isolate harbored genes encoding antibiotic inactivation enzymes. The lmrD gene, conferring resistance to lincosamides in Streptomyces and Lactococcus species, was detected in one B. longum isolate. This isolate was susceptible to clindamycin. Three of the four isolates (two B. animalis isolates and one B. breve isolate) with decreased susceptibility to tetracycline (MICs, 16 to 32 mg/liter) harbored the tet(T) gene, known to confer tetracycline resistance. All isolates harbored the mfd gene and mutations in gyrA. Mutations in gyrB were found only in the two B. animalis isolates. Mutations in these genes are associated with resistance to fluoroquinolones, and 12 of 15 bifidobacterial isolates had MICs of ≥4 mg/liter to ciprofloxacin.
Putative virulence factors.
The number of putative virulence factors is summarized in Table 3. A comprehensive list of putative virulence genes is also included in Data Set S2 in the supplemental material. Ninety-eight putative virulence genes were detected among the 15 isolates, including genes associated with iron and magnesium transport, adhesion, stress proteins, proteins with immune-evasive properties, and toxin secretion. Twenty of the genes (clpC, clpP, bsh, mgtB, ppkA, msbA, phoP, hitC, relA, cylA, cylG, oatA, farB, pvdH, manB, ybtS, cpsA, bsc1, tagT, and essC) were present or partially present in the majority (>85%) of all isolates. Putative virulence genes supporting host cell invasion were detected only in the B. animalis isolates and were represented by the gene iap (cwhA), encoding the extracellular protein p60, a major virulence factor in Listeria monocytogenes (19). Two unique virulence genes, ureA and ureB, were detected in the four B. longum subsp. infantis isolates from neonates (clade II). These genes encode the urease alpha and beta subunits, respectively, which represent enzymes involved in the hydrolysis of urea to form ammonia and carbamate and increasing gastric pH, thereby providing a more permissive environment for colonization of the gastrointestinal tract (20). Forty-six putative virulence genes were shared among the three Bifidobacterium species, indicating a high level of relatedness (Fig. 5).
TABLE 3.
Number of putative virulence genes among different isolates of Bifidobacterium determined by BLAST analysis
| Virulence factora | No. of putative virulence genes in the indicated isolates in the following cladesb: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade I |
Clade II |
Clade III |
Clade IV |
||||||||||||
| B. breve (10) | B. breve (7) | B. longum subsp. infantis (14) | B. longum subsp. infantis (15) | B. longum subsp. infantis (13) | B. longum subsp. infantis (4) | B. longum subsp. longum (12) | B. longum subsp. longum (9) | B. longum subsp. longum (3) | B. longum subsp. longum (5) | B. longum subsp. longum (6) | B. longum subsp. longum (2) | B. longum subsp. longum (1) | B. animalis (11) | B. animalis (8) | |
| Target | |||||||||||||||
| Adherence | 7 | 7 | 7 | 4 | 4 | 3 | 5 | 7 | 5 | 6 | 4 | 4 | 4 | 8 | 9 |
| Invasion | 1 | 1 | 1 | ||||||||||||
| Toxin | 13 | 12 | 13 | 12 | 12 | 13 | 9 | 9 | 12 | 11 | 13 | 10 | 7 | 10 | 11 |
| Secretion | 8 | 8 | 6 | 5 | 4 | 6 | 10 | 4 | 5 | 6 | 5 | 7 | 2 | 7 | 8 |
| Defensive | |||||||||||||||
| Antiphagocytosis | 12 | 11 | 7 | 6 | 7 | 5 | 8 | 6 | 9 | 8 | 6 | 8 | 2 | 12 | 11 |
| Bile resistance | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Biofilm | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| Stress | 4 | 4 | 5 | 3 | 3 | 3 | 4 | 4 | 3 | 3 | 3 | 3 | 4 | 4 | 4 |
| Immune evasion | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 3 | 2 | |
| Nonspecific | |||||||||||||||
| Iron uptake | 9 | 7 | 8 | 8 | 9 | 4 | 6 | 5 | 6 | 7 | 7 | 8 | 5 | 8 | 8 |
| Mg uptake | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Enzyme | 2 | 1 | 1 | 1 | |||||||||||
| Exoenzyme | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| Other | |||||||||||||||
| Reg. of VAGc | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 2 |
| Regulation | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Efflux pumps | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Signaling | 1 | 1 | 1 | 1 | 1 | ||||||||||
| Unclassified | 1 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 1 | 2 | 2 |
| Total | 64 | 61 | 59 | 49 | 49 | 43 | 51 | 44 | 51 | 51 | 47 | 49 | 31 | 63 | 64 |
Target, virulence factors promoting colonization of the host, invasion into host cells by surface components, and production of endo- or exotoxins; defensive, virulence factors helping bacteria to evade host defense, including capsules that protect them from opsonization and phagocytosis; nonspecific, virulence factors promoting sophisticated adaptation to the host environment, including iron-binding factors that compete with the host for iron and factors involving altered magnesium uptake protecting the integrity of proteins or membranes, enzymatic activity altering the host environment to enhance bacterial survival, and colonization; other includes signaling molecules involved in regulation of cellular functions, such as motility and cell-cell aggregation, among others.
Patient numbers are given in parentheses.
Reg. of VAG, regulation of virulence-associated genes.
FIG 5.
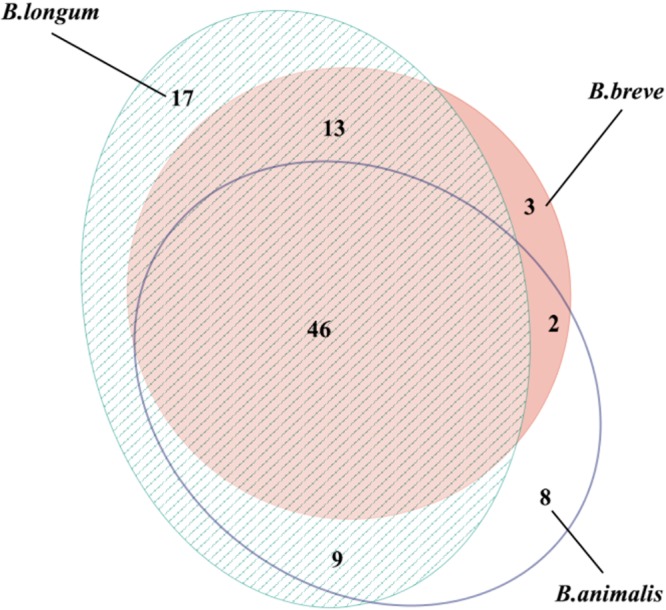
Area-proportional Venn diagram showing overlapping numbers of putative virulence factors between the three different species of bifidobacteria, B. longum, B. breve, and B. animalis.
Overall, at the subspecies level, there were no differences in the number of putative virulence genes between invasive and noninvasive isolates. In B. longum subsp. infantis, 72 and 90 unique putative virulence genes were detected among the invasive and noninvasive isolates, respectively. Of these, 72 were shared among invasive and noninvasive isolates. In B. longum subsp. longum, 77 and 77 unique putative virulence genes were detected among the invasive and noninvasive isolates, respectively. Of these, 69 were shared among invasive and noninvasive isolates. However, among the B. longum subsp. longum isolates, one invasive isolate (from patient 12) accounted for most of the difference observed.
DISCUSSION
To our knowledge this is the largest case series of patients with Bifidobacterium bacteremia for which clinical, microbiological, and genome sequencing data have been described. There were three main clinical characteristics among patients with bacteremia. First, patients were predominantly in the extreme lower or upper age spectrum. Second, the majority of patients had some degree of immune impairment. Third, most (11/15) patients had gastrointestinal tract-related conditions or symptoms. Our clinical findings are in line with previous reports on patients with invasive Bifidobacterium infections indicating that they seem to be opportunistic infections in immunocompromised patients, probably secondary to bacterial translocation from the gut (16, 21). We found that in six patients with Bifidobacterium species bacteremia either there was polymicrobial growth in blood cultures or there were different bacteria isolated from the patients during the course of their acute disease. This made it difficult to interpret whether Bifidobacterium was the true cause of their acute infection episode or merely an innocent bystander in a sick patient (e.g., patients 3 and 7).
Bifidobacterium species are traditionally considered nonpathogenic commensals that rarely cause human infections. Indeed, a large cohort study focusing on bloodstream infections caused by probiotic bacteria in 3,500 hematopoietic transplant recipients did not find any cases of Bifidobacterium bacteremia (15). In Norway, 0 to 2 Bifidobacterium bacteremia cases were reported annually between 2007 and 2012. The apparent increase seen from 2013 to 2015 may have several reasons. In the recent past, diagnosis relied mostly on biochemical tests for species identification with known limitations. Thus, blood cultures with growth of Bifidobacterium may have been identified only as Gram-positive rods with no further specification of the species. This may have led to an underestimation of the incidence of Bifidobacterium bacteremia. New diagnostic tools, such as MALDI-TOF MS, improve detection to the species level. This technique was introduced between 2011 and 2014 in the hospitals from which the patients for our study were recruited, and its routine use may be one reason for the apparently recent increase in the number of cases of bacteremia caused by Bifidobacterium species observed in Norway.
B. longum and B. dentium are the species most frequently reported to cause bifidobacterial infections (16, 21). In our study, we recovered three different species: B. breve, B. animalis, and B. longum. Bacterial translocation from the gut to the bloodstream seems to be a likely mechanism since the majority of patients had gastrointestinal tract-related conditions with possible mucosal impairment and a leaky gut.
In Norway, B. animalis subsp. lactis and, to some extent, B. longum are the most common Bifidobacterium species included in functional food products. Despite their proposed health-promoting effects (22), antibiotic resistance determinants and virulence factors in commensals are of great concern, as commensals can serve as a reservoir of resistance genes for intestinal pathogens and have the ability to cause disease on their own (23). However, there is no experimental evidence for the transfer of antibiotic resistance genes from bifidobacteria to other pathogens (24). Most patients in our study had some degree of immune impairment. We did not have information about probiotic consumption in the adults, but we know that this is widespread both in Norway and in other countries (25). Although probiotic products generally are regarded as safe, vigilance regarding their potential virulence, antibacterial resistance, and adverse metabolic activity should be maintained, in particular, in patients with predisposing or underlying conditions, such as gastrointestinal surgery, malignancy, or immunodeficiency (26, 27).
The antibiotic susceptibility pattern was similar across all three Bifidobacterium species in this study, much in line with previous findings (28–30). All isolates had low MICs to vancomycin (28, 31). High MICs to clindamycin were rare. We detected one B. longum isolate with an MIC to clindamycin of >256 mg/liter. However, there were discrepancies between phenotypic and genotypic findings. In the clindamycin-resistant isolate, no macrolide, lincosamide, and/or streptogramin (MLS) resistance gene was identified, but other resistance mechanisms may have been involved. All Bifidobacterium isolates in our study harbored mutations in genes associated with resistance to fluoroquinolones, and in 12 of 15 isolates, the MIC to ciprofloxacin was ≥4 mg/liter. Previously, a variable and strain-specific susceptibility to ciprofloxacin among bifidobacteria has been described (32, 33). Resistance to tetracyclines is the most common resistance trait among bifidobacteria (32, 34, 35). We identified the presence of tet(T) in two B. animalis isolates and one B. breve isolate, which is in good concordance with the phenotypic findings. The tet genes are the most abundant genetic determinants responsible for tetracycline resistance among bifidobacteria, but the tet(W) gene has been the one most commonly found (30, 35, 36). To our knowledge, tet(T) has not previously been described in Bifidobacterium. MIC values were higher for cefotaxime than for penicillin G. Cell wall impermeability seems to be the main cause of cephalosporin resistance among the bifidobacteria (29, 37). Our finding suggests intrinsic resistance to metronidazole, much in line with previous reports (29, 37–39).
There was limited variation in the putative virulence gene content among the 15 Bifidobacterium isolates. In a classical risk assessment approach for pathogens, pathogenicity is demonstrated to be a consequence of several properties acting in concert, including colonization and virulence factors (40). We identified several genes playing an important role in bacterial virulence, including genes encoding proteins involved in adhesion, antiphagocytosis, immune evasion, iron uptake, and bile resistance, which presumably pose a risk of infection. However, our findings must be interpreted with caution, as these virulence factors also are essential features of most commensals. In fact, most of the mechanisms involved in adhesion of bifidobacteria to host tissue are similar or even identical to those employed by pathogens to cause disease (41). We therefore expanded our analysis with a pangenome approach comparing all published genome sequences from blood culture isolates and commensal strains of B. longum. Here we detected unique clusters among both invasive and noninvasive isolates. However, in the virulence prediction, we found limited variation in the putative virulence gene content, and most genes were present in both invasive and noninvasive isolates. Among the B. longum subsp. infantis isolates, we actually found a higher number of putative virulence genes among the noninvasive isolates than among the isolates causing invasive bacteremia. This was not observed for the B. longum subsp. longum isolates. However, the phylogenetic tree fr all B. longum subsp. longum isolates generated clusters of invasive isolates indicating possible common virulence determinants in their accessory genomes.
This study has limitations. First, the number of blood culture isolates was limited. Second, we were unable to track probiotic consumption via food or supplementation in 12 of the patients included. In addition, investigation of potential pathogenicity using a search for homologous genes in databases might be speculative in relation to their functional role in Bifidobacterium, as these online resources are based on other more well characterized bacteria, and sequence homology between different bacteria does not always predict function.
Conclusion.
This study highlights the potential of Bifidobacterium as an opportunistic pathogen causing bacteremia in immunocompromised patients or patients with a compromised intestinal barrier. Our comparative genomic analysis indicated a possible phylogenetic separation between invasive and noninvasive B. longum subsp. longum isolates. Moreover, we found differences in genome content between the invasive and noninvasive isolates of both B. longum subspecies. However, invasive isolates were not associated with an increased number of putative virulence genes. Bifidobacterium bacteremia in infants and children is associated with impaired immunity (16). Our study indicates that similar risk factors apply to adults.
MATERIALS AND METHODS
Bacterial isolates and patients.
From 2013 to 2015, all Bifidobacterium bloodstream isolates identified in Norway (n = 15) were reported to NORM. Patients were eligible for inclusion in this study if there was one blood culture set with the presence of Bifidobacterium. We collected detailed clinical data from the medical records, including age, sex, underlying medical conditions, symptoms and signs prompting blood culture, use of antibiotics, and outcomes from all 15 Bifidobacterium bacteremia episodes. Patients received written information about this retrospective national study. Participation was voluntary with an opt-out option provided. The study was approved by the Norwegian Regional Ethical Committee (approval number 2016/1001).
Species identification and antimicrobial susceptibility testing.
The Bifidobacterium isolates were first isolated and species identification was obtained at nine different Norwegian hospital laboratories. Subsequently, all Bifidobacterium isolates were reanalyzed at a single laboratory. Species identification was confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) using a Microflex LT instrument (Bruker Daltonics, Bremen, Germany), Flex Control software, and MALDI Biotyper (v3.1) software (Bruker Daltonics, Bremen, Germany). Processing of samples was done according to the user's manual (42). In brief, one bacterial colony was placed on a target plate and 1 μl 70% formic acid was added for cell wall denaturation. Samples were then mixed with 1 μl matrix solution prior to mass spectrometry extraction. Samples with a log (score) value of ≥2 were considered to give a high probability of identification to the species level. Bifidobacteria were cultured on brucella blood agar plates supplemented with hemin and vitamin K1 (Becton Dickinson, Heidelberg, Germany). The plates were incubated in an anaerobic atmosphere (10% H2, 10% CO2, 80% N2) for 24 to 48 h, according to the instructions of the manufacturer. The quality control strain Bacteroides fragilis ATCC 25285 was used for growth control. The phenotypic susceptibility to nine antibiotics (penicillin G, metronidazole, clindamycin, tetracycline, meropenem, cefotaxime, ciprofloxacin, piperacillin-tazobactam, and vancomycin) was determined using MIC gradient strips (Liofilchem, Roseto degli Abbruzzi, Italy).
WGS, assembly, and annotation.
Bacterial DNA was extracted and prepared for whole-genome sequencing (WGS) using a Nextera XT kit (Illumina, San Diego, CA, USA), according to the manufacturer's instructions (43). The fragment size distribution (500 to 1,000 bp) was analyzed using an Agilent 2100 bioanalyzer system (Agilent Technologies, Waldbronn, Germany). The samples were multiplexed and sequenced by the Illumina MiSeq platform using v3 reagents with 2 sets of 300 cycles each according to the manufacturer's instructions. This yielded an average of 3.09 million reads per bacterial isolate. Each of the genomes was assembled de novo using SPAdes (v3.5.0) software with default parameters (44). Structural and functional annotations were performed using an in-house genome annotation pipeline (Department of Chemistry, University of Tromsø [https://arxiv.org/abs/1604.04103]).
Pangenome analysis of B. longum.
We performed a pangenome analysis of the genomes from 76 B. longum isolates. We included all 65 available B. longum genomes (complete and partial) of both human and animal origin deposited in GenBank (http://www.ncbi.nlm.nih.gov/GenBank/index.html) and the 11 B. longum genomes sequenced in the framework of this study (see Data Set S1 in the supplemental material). The genomes of B. animalis and B. breve were omitted from the pangenome analyses due to the limited number of published genomes of isolates of these species and the presence of only four isolates in our study. The amino acid sequences of the coding sequences (CDSs) for each of the 76 B. longum isolates and their subspecies were extracted and used as an input for the GET_HOMOLOGUES software package (45). Clustering of clusters of orthologous genes (COG) was performed using the OrthoMCL algorithm with default parameters (46). A gene cluster incorporating at least one representative from each isolate was defined as being part of the core genome, while gene clusters defying this definition were part of the accessory genome and could be further subdivided. Gene clusters represented in ≥72 isolates were regarded as the soft core, those represented in ≤2 isolates were regarded as the shell, and the rest of the accessory genome was regarded as the cloud. Each cluster was annotated, and functional grouping was made using the eggNOG (v4.5) database (47). The clusters with a functional classification within the core and subdivided accessory groups were counted individually.
We then excluded 29 of the B. longum genomes deposited in GenBank (from probiotic isolates, isolates of animal origin, isolates not further classified to the subspecies level, and isolates from subspecies other than B. longum subsp. longum and B. longum subsp. infantis) and performed separate pangenome analyses for B. longum subsp. longum (n = 34) and B. longum subsp. infantis (n = 13) isolates. In these pangenome analyses we compared invasive isolates of B. longum subsp. longum (n = 7) and B. longum subsp. infantis (n = 6) versus noninvasive isolates of B. longum subsp. longum (n = 27) and B. longum subsp. infantis (n = 7). Human blood culture isolates were defined as invasive isolates, whereas isolates from infant or adult feces or gut were defined as noninvasive isolates. Gene content trees from the binary pangenome cluster matrices (the presence or absence of genes in each isolate relative to the other isolates) were generated with the GET_HOMOLOGUES software package (45) using the discrete character parsimony algorithm. Clusters that were unique to the invasive isolates and/or to the noninvasive isolates from both subspecies were identified and functionally annotated with eggNOG classifications (47).
In silico analysis.
The subtyping of the 11 B. longum isolates compared to the reference strains B. longum subsp. infantis ATCC 15697, B. longum subsp. longum LMG 13197, and B. longum subsp. suis LMG 21814 was performed using the kSNP3 package (48) to identify single-nucleotide polymorphisms (SNPs) in the genomes and reconstruct a parsimony phylogenomic tree.
The resistance gene identifier in the comprehensive antibiotic resistance database (CARD; version 1.1.1; Department of Biochemistry and Biomedical Science, McMaster University, Canada [https://card.mcmaster.ca/home]) (49) was used to predict genes presumed to confer antibiotic resistance, and the findings were compared with the phenotypic susceptibility test results. The virulence factor database (VFDB; 2016, Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College, China [http://www.mgc.ac.cn/VFs/]) (50) was downloaded, and the CDSs from each isolate were searched against the sequences in the formatted database using the BLASTP program. Sequences that matched with E values of less than 1e−20 and sequence identities above 25% were considered homologs. The numbers of putative virulence genes in the three different Bifidobacterium species (B. longum, B. animalis, and B. breve) are presented in a Venn diagram (51). To further elucidate potential pathogenicity, putative virulence factors were identified in all 34 noninvasive B. longum isolates of human origin and matched to putative virulence factors in all 13 invasive B. longum isolates of human origin.
Accession number(s).
The sequences of the 15 Bifidobacterium isolates from this study have been deposited in the European Nucleotide Archive (www.ebi.ac.uk/ena) under study accession number PRJEB18553.
Supplementary Material
ACKNOWLEDGMENTS
We thank Runa Wolden for excellent technical assistance.
This work was supported by research grant from the Northern Norway Regional Heath Authority.
We have no potential conflicts of interest.
Eirin Esaiassen took part in all stages of the study and drafted the initial manuscript. Erik Hjerde performed and interpreted all bioinformatic analyses and revised the manuscript. Jorunn Pauline Cavanagh contributed to study design, took part in phenotypic analyses, and revised the manuscript. Gunnar Skov Simonsen conceptualized the study and revised the final manuscript. Claus Klingenberg conceptualized and designed the study and revised the final manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. Eirin Esaiassen, Erik Hjerde, and Claus Klingenberg have full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Original data are preserved and retrievable.
This study was performed as a collaborative project through the Norwegian Study Group on Invasive Bifidobacterial Infections. Members collected clinical data and identified the Bifidobacterium isolates analyzed in this study. The contributing members are Reidar Hjetland (Førde Hospital, Førde, Norway), Ingerid Skarstein (Haukeland University Hospital, Bergen, Norway), Aasmund Fostervold (Stavanger University Hospital, Stavanger, Norway), Karianne Wiger Gammelsrud (Oslo University Hospital, Ullevål, Oslo, Norway), Ståle Tofteland (Sørlandet Hospital, Kristiansand, Norway), Kjersti Wik Larssen (St. Olavs University Hospital, Trondheim, Norway), Ragnhild Støen (St. Olavs University Hospital, Trondheim, Norway), Nina Handal (Akershus University Hospital, Lørenskog, Norway), and Rolf Arne Sandnes (Innlandet Hospital, Lillehammer, Norway).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.00150-17.
REFERENCES
- 1.Saavedra JM. 2007. Use of probiotics in pediatrics: rationale, mechanisms of action, and practical aspects. Nutr Clin Pract 22:351–365. doi: 10.1177/0115426507022003351. [DOI] [PubMed] [Google Scholar]
- 2.Lewis ZT, Totten SM, Smilowitz JT, Popovic M, Parker E, Lemay DG, Van Tassell ML, Miller MJ, Jin YS, German JB, Lebrilla CB, Mills DA. 2015. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3:13. doi: 10.1186/s40168-015-0071-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milani C, Turroni F, Duranti S, Lugli GA, Mancabelli L, Ferrario C, van Sinderen D, Ventura M. 2015. Genomics of the genus Bifidobacterium reveals species-specific adaptation to the glycan-rich gut environment. Appl Environ Microbiol 82:980–991. doi: 10.1128/AEM.03500-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottacini F, Ventura M, van Sinderen D, O'Connell Motherway M. 2014. Diversity, ecology and intestinal function of bifidobacteria. Microb Cell Fact 13(Suppl 1):S4. doi: 10.1186/1475-2859-13-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arboleya S, Watkins C, Stanton C, Ross RP. 2016. Gut bifidobacteria populations in human health and aging. Front Microbiol 7:1204. doi: 10.3389/fmicb.2016.01204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collado MC, Gueimonde M, Hernandez M, Sanz Y, Salminen S. 2005. Adhesion of selected Bifidobacterium strains to human intestinal mucus and the role of adhesion in enteropathogen exclusion. J Food Prot 68:2672–2678. doi: 10.4315/0362-028X-68.12.2672. [DOI] [PubMed] [Google Scholar]
- 7.Serafini F, Strati F, Ruas-Madiedo P, Turroni F, Foroni E, Duranti S, Milano F, Perotti A, Viappiani A, Guglielmetti S, Buschini A, Margolles A, van Sinderen D, Ventura M. 2013. Evaluation of adhesion properties and antibacterial activities of the infant gut commensal Bifidobacterium bifidum PRL2010. Anaerobe 21:9–17. doi: 10.1016/j.anaerobe.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Round JL, Mazmanian SK. 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicola S, Amoruso A, Deidda F, Pane M, Allesina S, Mogna L, Del Piano M, Mogna G. 2016. Searching for the perfect homeostasis: five strains of Bifidobacterium longum from centenarians have a similar behavior in the production of cytokines. J Clin Gastroenterol 50(Suppl 2):S126–S130. [DOI] [PubMed] [Google Scholar]
- 10.Pokusaeva K, Fitzgerald GF, van Sinderen D. 2011. Carbohydrate metabolism in Bifidobacteria. Genes Nutr 6:285–306. doi: 10.1007/s12263-010-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Soud NH, Said RN, Mosallam DS, Barakat NA, Sabry MA. 2015. Bifidobacterium lactis in treatment of children with acute diarrhea. A randomized double blind controlled trial. Open Access Maced J Med Sci 3:403–407. doi: 10.3889/oamjms.2015.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singhi SC, Kumar S. 2016. Probiotics in critically ill children. F1000Res 5:407. doi: 10.12688/f1000research.7630.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi SH, Jernigan JA, McDonald LC. 2016. Prevalence of probiotic use among inpatients: a descriptive study of 145 U.S. hospitals. Am J Infect Control 44:548–553. doi: 10.1016/j.ajic.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brook I. 1996. Isolation of non-sporing anaerobic rods from infections in children. J Med Microbiol 45:21–26. doi: 10.1099/00222615-45-1-21. [DOI] [PubMed] [Google Scholar]
- 15.Cohen SA, Woodfield MC, Boyle N, Stednick Z, Boeckh M, Pergam SA. 2016. Incidence and outcomes of bloodstream infections among hematopoietic cell transplant recipients from species commonly reported to be in over-the-counter probiotic formulations. Transpl Infect Dis 18:699–705. doi: 10.1111/tid.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber E, Reynaud Q, Suy F, Gagneux-Brunon A, Carricajo A, Guillot A, Botelho-Nevers E. 2015. Bifidobacterium species bacteremia: risk factors in adults and infants. Clin Infect Dis 61:482–484. doi: 10.1093/cid/civ347. [DOI] [PubMed] [Google Scholar]
- 17.Norwegian Organization for Surveillance of Antimicrobial Resistance. 2016. Usage of antimicrobial agents and occurrence of antimicrobial resistance in Norway. Norwegian Organization for Surveillance of Antimicrobial Resistance, Tromsø/Oslo, Norway. [Google Scholar]
- 18.Esaiassen E, Cavanagh P, Hjerde E, Simonsen GS, Stoen R, Klingenberg C. 2016. Bifidobacterium longum subspecies infantis bacteremia in 3 extremely preterm infants receiving probiotics. Emerg Infect Dis 22:1664–1666. doi: 10.3201/eid2209.160033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu M, Zuo J, Gu H, Guo M, Yin Y. 2015. Domain function dissection and catalytic properties of Listeria monocytogenes p60 protein with bacteriolytic activity. Appl Microbiol Biotechnol 99:10527–10537. doi: 10.1007/s00253-015-6967-5. [DOI] [PubMed] [Google Scholar]
- 20.Marshall BJ, Barrett LJ, Prakash C, McCallum RW, Guerrant RL. 1990. Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology 99:697–702. doi: 10.1016/0016-5085(90)90957-3. [DOI] [PubMed] [Google Scholar]
- 21.Bourne KA, Beebe JL, Lue YA, Ellner PD. 1978. Bacteremia due to Bifidobacterium, Eubacterium or Lactobacillus; twenty-one cases and review of the literature. Yale J Biol Med 51:505–512. [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez B, Delgado S, Blanco-Miguez A, Lourenco A, Gueimonde M, Margolles A. 10 October 2016. Probiotics, gut microbiota, and their influence on host health and disease. Mol Nutr Food Res. doi: 10.1002/mnfr.201600240. [DOI] [PubMed] [Google Scholar]
- 23.Penders J, Stobberingh EE, Savelkoul PH, Wolffs PF. 2013. The human microbiome as a reservoir of antimicrobial resistance. Front Microbiol 4:87. doi: 10.3389/fmicb.2013.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gueimonde M, Sanchez B, de los Reyes-Gavilán CG, Margolles A. 2013. Antibiotic resistance in probiotic bacteria. Front Microbiol 4:202. doi: 10.3389/fmicb.2013.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varankovich NV, Nickerson MT, Korber DR. 2015. Probiotic-based strategies for therapeutic and prophylactic use against multiple gastrointestinal diseases. Front Microbiol 6:685. doi: 10.3389/fmicb.2015.00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Didari T, Solki S, Mozaffari S, Nikfar S, Abdollahi M. 2014. A systematic review of the safety of probiotics. Expert Opin Drug Saf 13:227–239. doi: 10.1517/14740338.2014.872627. [DOI] [PubMed] [Google Scholar]
- 27.Borriello SP, Hammes WP, Holzapfel W, Marteau P, Schrezenmeir J, Vaara M, Valtonen V. 2003. Safety of probiotics that contain lactobacilli or bifidobacteria. Clin Infect Dis 36:775–780. doi: 10.1086/368080. [DOI] [PubMed] [Google Scholar]
- 28.Moubareck C, Gavini F, Vaugien L, Butel MJ, Doucet-Populaire F. 2005. Antimicrobial susceptibility of bifidobacteria. J Antimicrob Chemother 55:38–44. [DOI] [PubMed] [Google Scholar]
- 29.Delgado S, Florez AB, Mayo B. 2005. Antibiotic susceptibility of Lactobacillus and Bifidobacterium species from the human gastrointestinal tract. Curr Microbiol 50:202–207. doi: 10.1007/s00284-004-4431-3. [DOI] [PubMed] [Google Scholar]
- 30.Mättö J, van Hoek AHAM, Domig KJ, Saarela M, Floréz AB, Brockmann E, Amtmann E, Mayo B, Aarts HJM, Danielsen M. 2007. Susceptibility of human and probiotic Bifidobacterium spp. to selected antibiotics as determined by the Etest method. Int Dairy J 17:1123–1131. doi: 10.1016/j.idairyj.2007.01.008. [DOI] [Google Scholar]
- 31.Lim KS, Huh CS, Baek YJ. 1993. Antimicrobial susceptibility of bifidobacteria. J Dairy Sci 76:2168–2174. doi: 10.3168/jds.S0022-0302(93)77553-0. [DOI] [PubMed] [Google Scholar]
- 32.Masco L, Van Hoorde K, De Brandt E, Swings J, Huys G. 2006. Antimicrobial susceptibility of Bifidobacterium strains from humans, animals and probiotic products. J Antimicrob Chemother 58:85–94. doi: 10.1093/jac/dkl197. [DOI] [PubMed] [Google Scholar]
- 33.Ouoba LI, Lei V, Jensen LB. 2008. Resistance of potential probiotic lactic acid bacteria and bifidobacteria of African and European origin to antimicrobials: determination and transferability of the resistance genes to other bacteria. Int J Food Microbiol 121:217–224. doi: 10.1016/j.ijfoodmicro.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Florez AB, Ammor MS, Alvarez-Martin P, Margolles A, Mayo B. 2006. Molecular analysis of tet(W) gene-mediated tetracycline resistance in dominant intestinal Bifidobacterium species from healthy humans. Appl Environ Microbiol 72:7377–7379. doi: 10.1128/AEM.00486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ammor MS, Florez AB, Alvarez-Martin P, Margolles A, Mayo B. 2008. Analysis of tetracycline resistance tet(W) genes and their flanking sequences in intestinal Bifidobacterium species. J Antimicrob Chemother 62:688–693. doi: 10.1093/jac/dkn280. [DOI] [PubMed] [Google Scholar]
- 36.Aires J, Doucet-Populaire F, Butel MJ. 2007. Tetracycline resistance mediated by tet(W), tet(M), and tet(O) genes of Bifidobacterium isolates from humans. Appl Environ Microbiol 73:2751–2754. doi: 10.1128/AEM.02459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charteris WP, Kelly PM, Morelli L, Collins JK. 1998. Antibiotic susceptibility of potentially probiotic Bifidobacterium isolates from the human gastrointestinal tract. Lett Appl Microbiol 26:333–337. doi: 10.1046/j.1472-765X.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- 38.Löfmark S, Edlund C, Nord CE. 2010. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis 50:S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]
- 39.Collado MC, Gonzalez A, Gonzalez R, Hernandez M, Ferrus MA, Sanz Y. 2005. Antimicrobial peptides are among the antagonistic metabolites produced by Bifidobacterium against Helicobacter pylori. Int J Antimicrob Agents 25:385–391. doi: 10.1016/j.ijantimicag.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Kitamoto S, Nagao-Kitamoto H, Kuffa P, Kamada N. 2016. Regulation of virulence: the rise and fall of gastrointestinal pathogens. J Gastroenterol 51:195–205. doi: 10.1007/s00535-015-1141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westermann C, Gleinser M, Corr SC, Riedel CU. 2016. A critical evaluation of bifidobacterial adhesion to the host tissue. Front Microbiol 7:1220. doi: 10.3389/fmicb.2016.01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruker Daltonics. 2012. MALDI Biotyper 3.1 user manual. Bruker Daltonics, Bremen, Germany. [Google Scholar]
- 43.Illumina. 2016. Nextera® DNA library prep reference guide. Illumina, San Diego, CA. [Google Scholar]
- 44.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Contreras-Moreira B, Vinuesa P. 2013. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, von Mering C, Bork P. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gardner SN, Hall BG. 2013. When whole-genome alignments just won't work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS One 8:e81760. doi: 10.1371/journal.pone.0081760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O'Brien JS, Pawlowski AC, Piddock LJ, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD. 2013. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. 2005. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33:D325–D328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Micallef LRP. 2013. eulerAPE: drawing area-proportional Euler and Venn diagrams using ellipses. University of Kent, Canterbury, United Kingdom: http://www.eulerdiagrams.org/eulerAPE/ Accessed April 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.