

Abstract
Ataxia telangiectasia mutated (ATM) is a serine/threonine kinase critical to the cellular DNA-damage response, including DNA double-strand breaks (DSBs). ATM activation results in the initiation of a complex cascade of events facilitating DNA damage repair, cell cycle checkpoint control, and survival. Traditionally, protein kinases have been analyzed in vitro using biochemical methods (kinase assays using purified proteins or immunological assays) requiring a large number of cells and cell lysis. Genetically encoded biosensors based on optical molecular imaging such as fluorescence or bioluminescence have been developed to enable interrogation of kinase activities in live cells with a high signal to background. We have genetically engineered a hybrid protein whose bioluminescent activity is dependent on the ATM-mediated phosphorylation of a substrate. The engineered protein consists of the split luciferase-based protein complementation pair with a CHK2 (a substrate for ATM kinase activity) target sequence and a phospho-serine/threonine-binding domain, FHA2, derived from yeast Rad53. Phosphorylation of the serine residue within the target sequence by ATM would lead to its interaction with the phospho-serine-binding domain, thereby preventing complementation of the split luciferase pair and loss of reporter activity. Bioluminescence imaging of reporter expressing cells in cultured plates or as mouse xenografts provides a quantitative surrogate for ATM kinase activity and therefore the cellular DNA damage response in a noninvasive, dynamic fashion.
Keywords: ATM, Bioluminescence, Complementation, In vivo, Kinase activity, Live cell, Molecular imaging, Reporter, Split-luciferase
1 Introduction
Protein kinases constitute one of the largest gene families, comprising ∼2% of the human genome. It is estimated that approximately 30% of all cellular proteins are phosphorylated on at least one residue. Thus, protein kinases have key roles in many fundamental processes of cellular signaling in cancer as well as normal cells. Biochemical methods have been widely used to investigate whether or not a protein kinase of interest is active. Although biochemical methods are robust in vitro, they generally do not provide information about protein kinase activity in specific subcellular compartments; nor do they provide information about activity changes at the single-cell level. We and others have developed optical imaging reporters to measure the kinase activity of various oncologically important kinases ([1–11], Table 1) and have utilized these reporters in subsequent studies that lead to the identification of new inhibitors and discovery of novel signaling mechanisms [12, 13].
Table 1. Luciferase complementation-based reporters for imaging of kinase activities.
| Kinase | Substrate | Peptide sequence | Phospho peptide-binding domain | Reference |
|---|---|---|---|---|
| AKT | FOXO4/AFX1 | QSRPRSCTWPLPRPEKKK | FHA2 | [9] |
| ATM | CHK2 | LETVSTQELYSI | FHA2 | [12] |
| EGFR | EPS15 | KPANFSAYPSEEDMIE | SH2 | [2] |
| FADD KINASES | FADD | QNRSGAMSPMSWNSDASTSEAS | FHA2 | [3] |
| GSK3β/CKIα | β-CATENIN | SYLDSGIHSGATTTAPSLSG | FHA2 | [4] |
| c-MET | PYK2 | LSESCSIESDIYAEIPDETLR | SH2 | [10] |
| TGFβR | SMAD2 | LTQMGSPSVRCSSMS | FHA2 | [6] |
Bioluminescence is a chemical reaction where light is emitted by a living organism. Luciferases are a large family of light-generating enzymes that catalyze the oxidation of a substrate, generically called luciferin, to yield oxyluciferin with the concomitant production of light. For in vivo bioluminescence imaging of malignancy, tumor cells or cancer-related genes are tagged with a reporter gene that encodes a light-generating enzyme, luciferase [14–16]. When this reporter is in the presence of the substrate it emits a blue to yellow-green light with an emission spectra peaking at a wavelength between 490 and 620 nm [14]. An extremely sensitive cooled charged-coupled device (CCD) camera or a photomultiplier detects any low light that is emitted during the bioluminescence reaction. Due to its extreme sensitivity, broad dynamic range and exceptionally large signal-to-noise ratio, this type of noninvasive imaging permits a real-time analysis of an ample amount of various biological events [15]. Although there are more than 30 luciferase-luciferin systems that were derived independently of each other, the most frequently used luciferase for in vivo molecular imaging is the ATP-dependent firefly (Photinus pyralis) luciferase [17]. The reason for this is that 30% of the light produced by firefly luciferase has an emission spectra above 600 nm, a region in which the signal attenuation by the absorbing and scattering properties of live mammalian tissue is at a minimum [15, 17]. Recently, a very bright and smaller luciferase (NanoLuc; NLuc) from deep sea shrimp (Oplophorus gracilirostris) has been successfully used for dual luciferase imaging in a mouse model [18].
A significant advantage of cell-based bioluminescent kinase reporter is its adaptability for high-throughput screening. Bioluminescence generated in luciferase assays offers higher sensitivity than FRET-based systems due to amplification of the signal. In addition, luciferase is less susceptible to inference from nonspecific fluorescence of compounds. Thus, bioluminescence-based assays are highly suited for high-throughput screening. Furthermore, luciferase activity can be monitored dynamically and noninvasively, allowing bioluminescence-based cell assays to provide a unique method for identifying specific compounds that interact with the target in the correct cellular compartment and under normal cellular physiological conditions of that compartment (pH, concentrations of specific ions, etc). Reporters wherein the firefly luciferase enzyme has been divided into two halves (N-Luc and C-Luc) were originally developed to study protein-protein interaction [19]. These split- luciferase reporters were based on either the inter-molecular or intra-molecular complementation of the luciferase fragments to generate signal in response to cellular cues.
Ataxia Telangiectasia Mutated (ATM) is a member of the PI3- like family of serine/threonine kinases. It is a very large 370 KDa protein encoded by human chromosome 11q22-23. It plays a critical role in repair of DNA double-stranded breaks (DSBs) thereby maintaining genomic stability. These processes include, but are not limited to, DNA replication, DNA repair, cell cycle progression, apoptosis, and senescence. ATM exists in its inactive form as a noncovalently linked dimer where the kinase domain of one monomer is bound to the internal domain of another monomer covering the S1981 residue. In response to DSBs, the kinase domain of one monomer phosphorylates S1981 of the other interacting ATM resulting in subunit dissociation, ATM activation, and recruitment to DNA break sites [20]. Ionizing radiation-induced ATM activation results in the activation of a large number of ATM substrates [21-26] including P53, MDM2, SMC1, KAP1, BRCA1, γH2AX, and CHK2. The activated ATM triggers a sequence of events including cell cycle arrest, allowing time for the repair of the damaged DNA in sync with circadian rhythm [27]. If damaged DNA is left unrepaired it can lead to cell death, genomic instability, cancer, and/or other pathologies [28]. The 2015 award of the Nobel Prize in Chemistry for the discovery of DNA repair mechanisms highlights the importance of this pathway. Because of the important role ATM plays in cancer, therapeutics have been devised to target it [29].
In vitro kinase assays using purified substrate and kinase are routinely used to evaluate kinase activity. For traditional cell-based studies, immunohistological and biochemical techniques have been utilized for evaluating the kinase activity of ATM, such as counting pATM foci, γH2AX foci, immunofluorescence, or immunoprecipitation-western blotting [26, 30, 31]. Johnson, You, and Hunter [11] described a fluorescence resonance energy transfer (FRET)-based biosensor for monitoring ATM kinase activity in live cells. Although this reporter provides direct measurement of the ATM kinase activity, it has a limitation of usability in mouse model due to tissue penetration and autofluorescence in the CFP-YFP range. In this book chapter, we provide detailed methods of use for the recently developed split firefly-based bioluminescence reporter to noninvasively, dynamically, and sensitively measure ATM kinase activity in live cells and mouse models [7].
2 Materials
2.1 Molecular Biology
DNA encoding open reading frame for phospho-protein- binding domain (e.g., Rad53p FHA2 domain).
Full-length coding sequence for the Firefly luciferase (FLuc) or plasmids coding for the N-terminal luciferase (amino acids 1–416) and C-terminal luciferase (amino acids 398–550) fragments.
Expression vectors with constitutive promoters for expression in mammalian cells (e.g., pEF vector from Clontech).
Optional: Expression vectors and packaging plasmids for generating lentiviral particles.
High fidelity DNA polymerases (e.g., Pfu DNA polymerase), dNTPs, oligonucleotide primers for cloning and sequencing, PCR reaction buffers, PCR Thermocycler, restriction endonuclease, DNA ligase, site-directed mutagenesis kits, high efficiency competent cells, antibiotic, bacterial growth media (LB, SOC), agar plates, plasmid DNA extraction kits, DNA gel purification, and sequencing kits.
2.2 Cell Culture
HEK293T cells or other readily transfectable cell lines.
Desired cell line(s) for biologic question of interest (i.e., D54, U87).
Fetal bovine serum (FBS).
Complete growth medium with FBS: Growth media with 10% FBS, and 1% penicillin/streptomycin.
Serum-free medium: Growth media without FBS and penicillin/streptomycin.
Trypsinization medium: 0.05% Trypsin-EDTA.
1000× penicillin stock solutions: 10,000 Units/mL penicillin.
1000× streptomycin stock solution: 10 mg/mL streptomycin.
1000× geneticin/G418 stock solution: 50 mg/mL geneticin/G418.
Sterile 1× phosphate-buffered saline (PBS) solution.
Transfection reagents.
Filter paper discs and forceps for isolation of stable clones.
2.3 Cell Imaging
Black-walled or white-walled 96-well clear-bottom plates for tissue culture.
Sterile low adherence pipette tips with barrier filter.
Firefly luciferase substrate: 4 mg/mL D-luciferin 40× stock solution in PBS stored in dark colored vials at −80 °C. Alternatively, GloSensor c-AMP reagent (Promega) can be used to monitor firefly luciferase activity.
ATM inhibitor: 1000× stock solution 3 M caffeine.
ATM inhibitor: 1000× stock solution 20 mM KU-60019.
ATM inhibitor: 1000× stock solution 20 mM KU-55933.
ATM inhibitor: 1000× stock solution 20 mM CGK733.
Live cell bioluminescence imaging system with very high sensitivity and required software package for data generation and analysis (IVIS, Envision Xcite multi-label Plate Readers from Perkin Elmer; or similar system).
Liquid handling instrument (Biomek Nx from Beckman or similar).
Plate handling robot (Plate Handler II robot from Perkin Elmer or similar).
Optional: Cell culture incubator compatible with high- throughput instruments (LiCONiC StoreX STX44 IC precision incubator from Liconic Instruments or similar) for high-throughput assays.
2.4 Animal Imaging
Appropriate mouse strain for desired experimental system such as immunocompromised mice (nude, SCID, or NSG) for human tumor xenografts.
Optional: Small animal shaver such as Wahl trimmer.
40 mg/mL luciferin stock in PBS, store in tightly sealed dark tubes at −20 or −80 °C.
28–30 gauge insulin syringe for intra-peritoneal (IP) luciferin injection in mice.
Bioluminescence imaging instrument (IVIS or similar instrument) with a heated platform and isoflurane anesthesia injection and controller systems.
Additional accessories such as nose cones, animal partitions, black paper sheets, ear tags, markers, 70% alcohol, and 10% bleach or similar solution for disinfecting bench surfaces.
3. Methods
3.1 Construct Firefly Luciferase Complementation-Based ATM kinase Activity Reporter
Select a substrate such as CHK2 and determine the length of the substrate sequence that can be used for the construction of the reporter (see Note 1). We typically select 12–20 amino acid long substrate sequences with the target residue/s at the center of the sequence where possible (Table 1). For the construction of the ATM kinase reporter (ATMR), we selected a 12-residue sequence derived from CHK2 (Figs. 1 and 2).
Add a 5–7 amino acid long linker sequence at both the ends of the substrate sequence. We typically use GGSGG as the linker in our kinase reporters. For Ser/Thr kinases, attach a phosphopeptide-binding domain such as FHA2 (residues 420–582) [32]. For Tyr kinases, attach a SH2 domain (residue 374–465 of mouse shc2 [33, 34]). Use appropriate N-terminal (N-Luc) and C-terminal (C-Luc) firefly luciferase fragment pairs [19] at the flanks.
Generate chimeric reporter constructs using appropriate molecular biology procedures. We generally incorporate linkers, substrates, and restriction enzyme sites in primers. We typically confirm the clones by sequencing, construct expression by Western blotting and functional bioluminescence assays before proceeding to generate stable cell lines.
Generate relevant control constructs wherein the phospho-target residue/s within the substrate is mutated to neutral amino acids. We typically mutate Ser/Thr or Tyr to Ala in mutant reporters by site-directed mutagenesis (Fig. 2a).
Express complementation reporters in appropriate vectors for mammalian cells. Vectors should be selected with markers, such as antibiotic resistance genes or co-expressed fluorescent proteins that are suitable for generating stable cell lines. We typically use pEF vector in combination with the geneticin/G418 antibiotic for stable clone selection.
Fig. 1.

The DNA-coding sequence and translated amino acid sequence for all the domains of the ATM kinase activity reporter. In frame short linker sequences (linker) inserted between each functional domain provide flexibility for the intramolecular domain interaction in the chimeric reporter molecule. N-Luc denotes amino acids 1–416 of firefly luciferase and C-Luc denotes amino acids 398–550. The target peptide sequence was derived from the CHK2 coding sequence (amino acids 63–74). The Ser/Thr phospho-peptide-binding domain (FHA2) comprises amino acids 420–522 of Rad53P protein
Fig. 2.
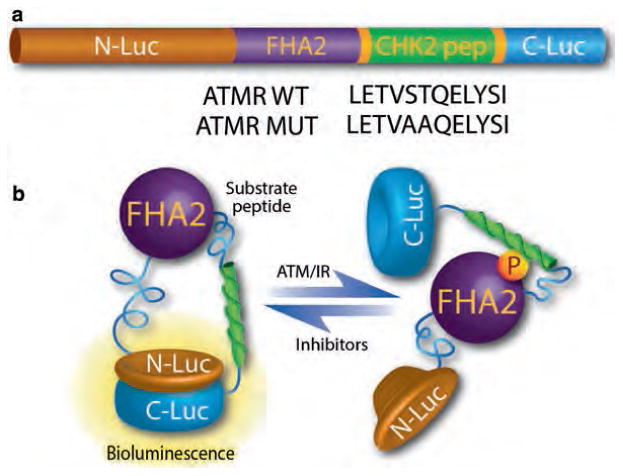
The components and functional basis of the ATM kinase activity reporter. (a) The ATM reporter consists of a phospho Ser/Thr-binding domain (FHA2), substrate peptide (CHK2), and split firefly luciferase. The substrate sequence is flanked by short linker sequences at either end. The functional basis of the reporter is demonstrated in part figure (b). In the presence of ATM kinase activity, the CHK2 target peptide is phosphorylated, resulting in interaction with the FHA2 domain, producing stearic constraints that inhibit functional reconstitution of the luciferase. In the absence of ATM kinase activity such as by small molecule inhibitors, siRNA-mediated knockdown (of ATM), or over-expression of phosphatases, the CHK2 consensus sequence is hypo-phosphorylated, allowing for lucif- erase enzyme reconstitution and increased bioluminescent activity
3.2 Cell-Based Bioluminescence Imaging of ATM Kinase Activity
We typically do all of our cell-based and in vivo bioluminescence assays using the reporter expressing stable cell lines. We fully select cell lines that represent an appropriate cellular and biological context for our studies (see Note 2). Cells are transfected with the reporter plasmids and allowed to grow under the antibiotic selection media. We typically pick 12–24 single-cell clones using sterilized filter paper discs and choose the best clones by measuring bioluminescence in response to specific kinase inhibitors (see Notes 3 and 4).
The three best reporter expressing stable cell lines (clones) are expanded and frozen at low passages for future use. Cells are maintained in 10 cm dishes with complete growth media containing serum and the appropriate amount of G418 (see Note 5).
Stable cell lines are plated overnight in black-walled or white-walled, clear-bottom 96-well plates for live cell assays. Cell density should be 2500–10,000 cells per well in 100 μL complete growth medium with serum (see Notes 6 and 7).
Cell culture media is removed and ATMR expressing cells are treated with different concentrations of ATM inhibitors such as caffeine, KU-60019, KU-55933, or CGK733 in serum-free media (100 μL per well).
After 5 min, 2.5 μL D-luciferin (black-walled plates) or cAMP- Glo reagent (white-walled plates) is added by multichannel pipette into each well for a working concentration of 100 μg/mL of firefly luciferase substrate.
Black-walled 96-well plates are imaged on the IVIS imaging system as soon as possible after adding luciferin. Typically, bioluminescence is acquired for 30–60 s at medium binning. For a time-course, the images are acquired with 3–10 min delay between the reads (Fig. 3a, b).
The white-walled plates are read on the Envision system right after the addition of the substrate. Generally, each well of the plate is read for 0.01–1.0 s. For a longer time-course activity measurement, a delay of 15–60 min between each read is set (Fig. 3c). For each read, the robot takes the plate out from the incubator, loads it on the reader where the plate is read, and is transferred back to the incubator until the next time point (see Note 8).
Quantify bioluminescence acquired on IVIS system by region- of-interest (ROI) analysis using Living Image software. The bioluminescence data from Envision system is automatically saved in quantitative form in tab-delimited file format.
Since radiation activates ATM within minutes, the bioluminescence activity of ATMR can be evaluated within 15 min after irradiation.
All the bioluminescence measurements should be validated by Western blotting in ATMR expressing cell lines in parallel experiments.
Fig. 3.
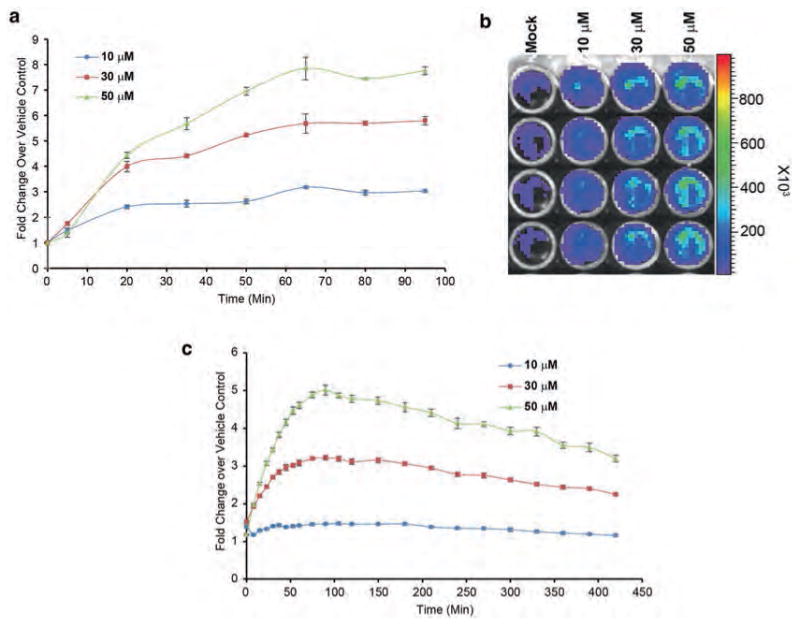
Firefly complementation-based live cell assay for noninvasive monitoring of ATM kinase activity. (a) D54 cells stably expressing the ATM kinase activity reporter (ATMR) were plated in black-walled 96-well plates (5000 cells/well). Cells were incubated with mock (DMSO) or an increasing concentration of ATM kinase inhibitor KU-55933, which increased the signal in a dose-dependent way. (b) A representative image of the bioluminescence acquired in response to various concentrations of KU-55933 is shown. ROI in a grid is created and overlaid in the pseudocolored image to quantitate the photons emitted. Scale bar shows photon flux in pseudocolor with blue as the lowest and red as the highest counts. (c) D54-ATMR cells plated in white-walled 96-well plates and treated as described above and read on Envision plate reader
3.3 In Vivo Imaging of ATM Kinase Activity
D54-ATMR cells are expanded, trypsinized, and suspended in serum-free media at 40 × 106 cells/mL. 50 μL of this suspension is injected into each flank (2 × 106 cells) in nude mice using a 22-gauge needle. We usually wait until the tumor reaches 60–100 mm3 size (3–4 weeks) before starting the experiments.
We acquire baseline bioluminescence measurements 3–6 h before starting the treatment (Fig. 4a). Each mouse is injected with 100 μL D-luciferin (4 mg/mL stock prepared in sterile PBS; 400 pg per mouse) anesthetized with 1–2% isoflurane for 5 min (see Note 9).
Transfer mice to the bioluminescence instrument, where they are maintained under anesthesia, and acquire bioluminescence. We typically acquire data on five mice at once isolated by a plastic separator. Generally, a 15–30 s acquisition at medium sensitivity is sufficient. We typically acquire data for 10–20 reads with a 1–5 min delay between the reads to cover the bioluminescence peak from all the tumors in each of the mice.
Treat the mice with appropriate inhibitors such as KU-55933 (both 25 mg/kg), or activators such as radiation (5 Gy) and monitor bioluminescence over time. Vehicle control (DMSO) or sham-irradiated mice should be used as control (Figs. 3c and 4b; see Note 10).
Remove mouse from imaging instrument and monitor for complete recovery from anesthesia.
Quantify imaging data by region-of-interest (ROI) analysis of bioluminescence produced by the tumor, using units of photon flux (Fig. 4a; see Notes 11–17).
Fig. 4.
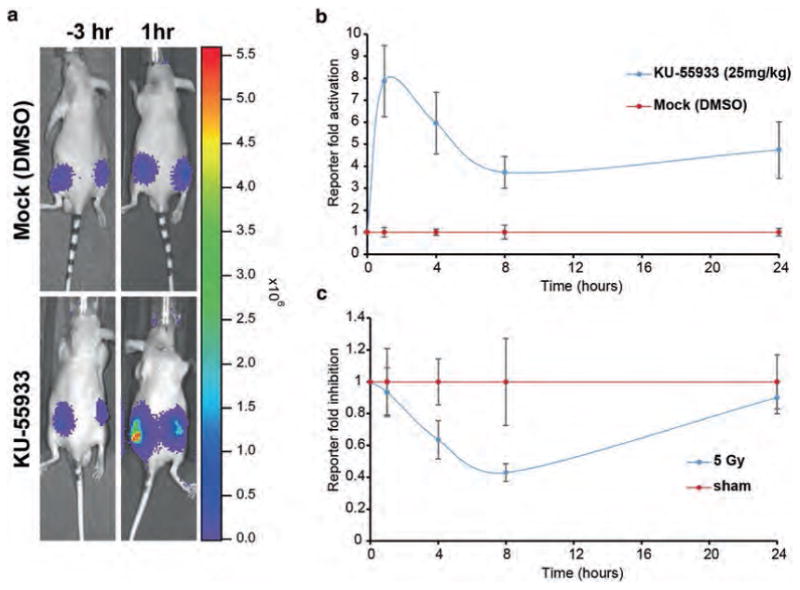
In vivo measurement of ATM kinase activity in mouse tumor xenograft model. (a) CD-1 nude mice harboring D54-ATMR WT tumor xenografts were injected with luciferin and bioluminescence was acquired as described 3 h before treatments. (b) The animals were injected with KU-55933 (25 mg/kg) or vehicle control (DMSO) and bioluminescence was acquired 1,4, 8, and 24 h posttreatment. The ATM reporter fold activation upon ATM inhibition is plotted over mock treatment. (c) Similarly, mouse harboring D54-ATMR WT tumor xenografts were whole body irradiated with 5Gy of radiation or sham irradiated and bioluminescence was measured for up to 24 h. About 70% decrease in the reporter activity was observed 8 h post irradiation
3.4 Conclusions
The method described herein is an adaptation of the traditional protein complementation assay for the detection of protein-protein interaction in live cells. Instead of monitoring the interaction of two proteins through the use of split reporter molecules, we have adapted the assay such that the interaction between the “bait” and the “prey” occurs in response to the activity of a specific kinase. The kinase can be a serine/threonine- or a tyrosine-kinase. The reporter has also been engineered such that increased complementation (and therefore reporter activity) occurs in response to decreased kinase activity. This approach is therefore very well suited for high-throughput screens for kinase inhibitor libraries since a positive hit would be detected as an increase in bioluminescence activity, thereby less likely to result in false positives. We have also used analogous reporters for whole genome siRNA screens. As an example, a reporter for TGF-β receptor serine/threonione kinase activity was used in a human kinome siRNA screen to yield a number of novel genes as regulators of the TGF- β receptor function [12]. Regulation of the molecular events that lead to the activation and/or inactivation of the ATM kinase activity is yet to be defined; therefore, it is anticipated that analogous whole genome siRNA screens against the ATMR will most likely yield new insights into the role of novel genes in the regulation of the cellular response to DNA damage.
4 Notes
Substrate sequence for the construction of the reporter should be decided based on literature searches. It is imperative to have good antibodies available to detect changes in the substrate phosphorylation as this will help in validating the kinase reporter bioluminescence data by biochemical techniques.
The selection of a specific cell line for creating a reporter-expressing stable line should be based on the specific questions being interrogated and the intrinsic activity and detectability of the kinase and the substrate in the cell line. This should be determined by experimentation as well as literature searches. We selected the D54 glioblastoma cell line because ATM inhibition in glioblastoma may sensitize them to IR and chemotherapy [35]. D54 cells express wild-type ATM protein and responds to ATM inhibitors and irradiation as seen by Western immunoblotting using antibodies against ATM and its substrate CHEK2.
We generally select for clones that exhibit low to moderate bioluminescence after adding luciferin. Since it is an activatable reporter that shows an increase in the light with inhibition of the ATM kinase, clones expressing the reporter in very high abundance may not yield high signal to background and thus may show a limited fold activation in response to inhibitors.
For picking up the clones, complete growth media with antibiotic is aspirated from the tissue culture dishes containing single-cell clones. Sterilized filter paper discs pre-wetted in trypsin are transferred to clones using sterilized forceps in a laminar flow hood. After 4–5 min, the discs are lifted from the tissue culture plate and swirled around in a 24-well plate containing complete growth media with half the concentration of antibiotic used for clone selection. Generally, 12–24 single-cell clones are picked. Forceps are sterilized either by heat or by dipping them in ethanol for 5–10 min. Make sure to let the ethanol evaporate before using the forceps to pick up the clones.
For expanding and maintaining of stable cell lines, we use half the concentration of G418 than that was used for selection. The G418 concentration for stable clone selection should be empirically decided.
The N-Luc and C-Luc fragments used in the construction of the complementation-based ATM reporter are derived from a firefly luciferase that has been optimized to work in mammalian cells at the physiological temperature of 37°C. Therefore, all the bioluminescence acquisitions should be performed at 37°C.
Since this reporter is based on the complementation of the light-generating enzyme luciferase, it works only in live cells under physiological conditions. Methods wherein cell lysate is used to measure the bioluminescence signals are incompatible with this reporter system and will not be able to yield any detectable change in signal to background (under different treatment conditions).
The Envision system is built with ultra-sensitive luminescence detection technology; thus 0.01–0.1 s measurement for each well is usually sufficient. The detection time can be increased if the signal is very weak and desired signal-to-noise is not reached with lower detection times. Furthermore, the bioluminescence signal from cAMP-Glo reagent is stable for a prolonged period of time, allowing us to measure the kinase activity of ATM for longer periods.
In our hands, we find that 400 μg luciferin/mouse gives us the best signal-to-background bioluminescence readings for split firefly luciferase-based kinase reporters. For smaller tumors or tumors generated from cell lines expressing very low levels of reporter, the amount of luciferin can be increased to 150 mg/kg body weight (i.e., 3.5–4 mg/mouse with 20–25 g average mouse weight).
One may need to empirically determine the optimal inhibitor concentration for the best signal-to-noise bioluminescence detection in vivo. We usually test two to three different concentrations of the drugs in mouse tumor xenograft model to find the optimal concentration that gives highest fold change over vehicle control without being toxic to animals.
We create separate region-of-interest (ROI) for each tumor based on its size and shape. We also make sure that this ROI does not overlap with the ROI of any other tumors. We copy- paste the same ROI for each tumor for counting total photon flux for all the time points. We may move the position of the ROI so that it covers the tumor (because the position of the same animal with the same tumor might be slightly different between different time points) but do not change the overall shape or size of any ROI. This removes the chances of including background photon counts emanating from mouse skin.
We generally use four to five mice in an experimental group for bioluminescence data acquisition on the IVIS Spectrum system. Based on the number of animals in a group, we choose a stage level (distance of the CCD camera and the subject) and use the same stage level for the whole experiment. Changing the distance between the CCD camera and the subject height between different time points or reads would lead to different photon counts which will be difficult to analyze.
We usually use medium sensitivity settings with 15–60 s data collection time for each read. One should acquire photon flux without any saturated pixels. All the saturated pixels (above 108 counts) show the same value, and therefore, cannot be accurately analyzed.
We typically use the maximum photon flux emitted from each tumor separately for all the calculations. Each tumor may show the maximum emittance at different periods of time; therefore, we usually perform sequential reads with a delay between each read which allows us to collect the maximum photon flux for all the tumors.
We typically use the maximum photon count for each tumor for each time for the analysis. If a tumor does not respond at any time-point, we omit that reading from the analysis. We analyze and plot combined values of all the tumors from one treatment group. Photon counts from vehicle (DMSO) or sham treated animals are set as onefold and the ratios of other treatments are counted as fold change from vehicle/sham treatment.
Due to the sensitivity of the imaging system, one typically gets some background bioluminescence in nude and SCID mice. It is important to shave and NAIR® the mice if using a mouse strain with hair to reduce the background and determine the correct size, shape, and position of a ROI.
Bioluminescence data acquired in a mouse xenograft model should be validated by biochemical methods such as Western blotting or immunohistochemistry (IHC). For validation of the bioluminescence data for ATMR, tumor tissue should be analyzed with pATM and pCHK2 antibodies after control, KU-55933, KU-60019, or radiation treatments.
Acknowledgments
We would like to acknowledge members of the Center for Molecular Imaging at the University of Michigan for their valuable input and support for the studies. This work was supported by the National Institutes of Health grant R01CA193690 (AR), P01CA087634 (BDR, AR and SN) as well as a P30CA046592 award to the University of Michigan Cancer Center.
References
- 1.Bhojani MS, Nyati S, Rao HR, Rehemtulla A. Lung cancer metastasis. Springer; New York: 2010. Molecular imaging in lung cancer metastases; pp. 267–287. [Google Scholar]
- 2.Khan AP, Contessa JN, Nyati MK, Ross BD, Rehemtulla A. Molecular imaging of epidermal growth factor receptor kinase activity. Anal Biochem. 2011;417(1):57–64. doi: 10.1016/j.ab.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan AP, Schinske KA, Nyati S, Bhojani MS, Ross BD, Rehemtulla A. High- throughput molecular imaging for the identification of FADD kinase inhibitors. J Biomol Screen. 2010;15(9):1063–1070. doi: 10.1177/1087057110380570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyati S, Ranga R, Ross BD, Rehemtulla A, Bhojani MS. Molecular imaging of glycogen synthase kinase-3p and casein kinase-1a kinases. Anal Biochem. 2010;405(2):246–254. doi: 10.1016/j.ab.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyati S, Ross BD, Rehemtulla A, Bhojani MS. Novel molecular imaging platform for monitoring oncological kinases. Cancer Cell Int. 2010;10:23. doi: 10.1186/1475-2867-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nyati S, Schinske K, Ray D, Nyati M, Ross BD, Rehemtulla A. Molecular imaging of TGF beta-induced smad2/3 phosphorylation reveals a role for receptor tyrosine kinases in modulating TGF beta signaling. Clin Cancer Res. 2011;17(23):7424–7439. doi: 10.1158/1078-0432.Ccr-11-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams TM, Nyati S, Ross BD, Rehemtulla A. Molecular imaging of the ATM kinase activity. Int J Radiat Oncol Biol Phys. 2013;86(5):969–977. doi: 10.1016/j.ijrobp.2013.04.028. S0360-3016(13)00457-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Bhojani MS, Ross BD, Rehemtulla A. Molecular imaging of protein kinases. Cell Cycle. 2008;7(3):314–317. doi: 10.4161/cc.7.3.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Lee KC, Bhojani MS, Khan AP, Shilman A, Holland EC, Ross BD, Rehemtulla A. Molecular imaging of Akt kinase activity. Nat Med. 2007;13(9):1114–1119. doi: 10.1038/nm1608. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Virani S, Zhang Y, Bhojani MS, Burgess TL, Coxon A, Galban CJ, Ross BD, Rehemtulla A. Molecular imaging of c-Met tyrosine kinase activity. Anal Biochem. 2011;412(1):1–8. doi: 10.1016/j.ab.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson SA, You Z, Hunter T. Monitoring ATM kinase activity in living cells. DNA Repair. 2007;6(9):1277–1284. doi: 10.1016/j.dnarep.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 12.Nyati S, Schinske-Sebolt K, Pitchiaya S, Chekhovskiy K, Chator A, Chaudhry N, Dosch J, Van Dort ME, Varambally S, Kumar-Sinha C, Nyati MK, Ray D, Walter NG, Yu H, Ross BD, Rehemtulla A. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF- beta signaling. Sci Signal. 2015;8(358) doi: 10.1126/scisignal.2005379. ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schinske KA, Nyati S, Khan AP, Williams TM, Johnson TD, Ross BD, Tomás RP, Rehemtulla A. A novel kinase inhibitor of F ADD phosphorylation chemosensitizes through the inhibition of nf-κb. Mol Cancer Ther. 2011;10(10):1807–1817. doi: 10.1158/1535-7163.MCT-11-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCaffrey A, Kay MA, Contag CH. Advancing molecular therapies through in vivo bioluminescent imaging. Mol Imaging. 2003;2(2):75–86. doi: 10.1162/15353500200303124. [DOI] [PubMed] [Google Scholar]
- 15.Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002;4:235–260. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 16.Choy G, Choyke P, Libutti SK. Current advances in molecular imaging: noninvasive in vivo bioluminescent and fluorescent optical imaging in cancer research. Mol Imaging. 2003;2(4):303–312. doi: 10.1162/15353500200303142. [DOI] [PubMed] [Google Scholar]
- 17.Greer LF, 3rd, Szalay AA. Imaging of light emission from the expression of luciferases in living cells and organisms: a review. Luminescence. 2002;17(1):43–74. doi: 10.1002/bio.676. [DOI] [PubMed] [Google Scholar]
- 18.Stacer AC, Nyati S, Moudgil P, Iyengar R, Luker KE, Rehemtulla A, Luker GD. NanoLuc reporter for dual luciferase imaging in living animals. Mol Imaging. 2013;12(7):1–13. [PMC free article] [PubMed] [Google Scholar]
- 19.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA. 2004;101(33):12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 21.Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, Aebersold R, Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3(151) doi: 10.1126/scisignal.2001034. rs3. [DOI] [PubMed] [Google Scholar]
- 22.Bhatti S, Kozlov S, Farooqi AA, Naqi A, Lavin M, Khanna KK. ATM protein kinase: the linchpin of cellular defenses to stress. Cell Mol Life Sci. 2011;68(18):2977–3006. doi: 10.1007/s00018-011-0683-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi S, Srivas R, Fu KY, Hood BL, Dost B, Gibson GA, Watkins SC, Van Houten B, Bandeira N, Conrads TP, Ideker T, Bakkenist CJ. Quantitative proteomics reveal ATM kinase-dependent exchange in DNA damage response complexes. J Proteome Res. 2012;11(10):4983–4991. doi: 10.1021/pr3005524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6(8):931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 25.Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J. A proteomic analysis of ataxia telangiectasia- mutated (ATM)/ATM-Rad 3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282(24):17330–17334. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 26.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274(53):37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 27.Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010;584(12):2618–2625. doi: 10.1016/j.febslet.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Front Genet. 2013;4:37. doi: 10.3389/fgene.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33(5):547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozlov S, Gueven N, Keating K, Ramsay J, Lavin MF. ATP activates ataxia- telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. J Biol Chem. 2003;278(11):9309–9317. doi: 10.1074/jbc.m300003200. [DOI] [PubMed] [Google Scholar]
- 32.Durocher D, Jackson SP. The FHA domain. FEBS Lett. 2002;513(1):58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- 33.Filippakopoulos P, Muller S, Knapp S. SH2 domains: modulators of nonreceptor tyrosine kinase activity. Curr Opin Struct Biol. 2009;19(6):643–649. doi: 10.1016/j.sbi.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlessinger J. SH2/SH3 signaling proteins. Curr Opin Genet Dev. 1994;4(1):25–30. doi: 10.1016/0959-437x(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 35.Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7(7):989–999. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]