

Abstract
Mantle cell lymphoma (MCL) is an aggressive histotype of B-cell non-Hodgkin lymphoma that has increased in incidence over the past few decades and is incurable, usually poorly responsive to standard chemotherapy combinations, and associated with poor prognoses. Discovering new therapeutic agents with low toxicity that produce good outcomes in patients with MCL is an ongoing challenge. Recent studies showed that degrasyn, a novel small-molecule inhibitor of the Janus kinase (JAK)/signal transducer and activation of transcription (STAT) pathway, exerts antitumor activity in lymphoid tumors by inhibiting key growth and survival signaling pathways. In the present study, we found that treatment of both typical and blastoid-variant MCL cells with degrasyn in combination with bortezomib resulted in synergistic growth inhibition and apoptosis induction in vitro. The apoptosis in these cells was correlated with down-regulation of constitutive nuclear factor-κB and phosphorylated STAT3 activation, leading to inhibition of c-Myc, cyclin D1, and bcl-2 protein expression and upregulation of bax protein expression. In vivo, degrasyn and bortezomib interacted to synergistically prevent tumor development and prolong survival durations in a xeno-transplant severe combined immunodeficiency mouse (SCID) model of MCL. These findings suggest that agents such as degrasyn that can pharmacologically target constitutively expressed nuclear factor-κB and STAT3 in MCL cells, may be useful therapeutic agents for MCL when administered together with bortezomib.
Keywords: Bortezomib, degrasyn, NF-κB, STAT3, mantle cell lymphoma
Introduction
Mantle cell lymphoma (MCL) is an important histotype of aggressive B-cell non-Hodgkin lymphoma (NHL-B) with the poorest long-term outcome among all NHL-B subtypes, the incidence of which has increased over recent decades in the United States and worldwide (1). MCL is characterized by cell cycle abnormalities, including ATM, p53-mediated cell cycle anomalies, DNA repair, apoptosis, and in blastoid-variant MCL (BV-MCL), decreased expression of cyclin-dependent kinases such as p16, p21, and p27kip1 (2). Over-expression of the cyclin D1 oncogene and protein, which are associated with the characteristic non-random chromosomal translocation t(11:14)(q13;q32) in MCL as a result of enforced expression by the IgH enhancer on chromosome 14. MCL cases are classified pathologically into at least two subtypes: 1) typical (classic), the less aggressive, small- to medium-cell histotype; and 2) blastoid (BV-MCL), the more aggressive, large-cell histotype (3, 4). The most common clinical course of MCL consists of continual relapses with a median survival duration in most studies of 3-4 years (5). Both subtypes of MCL are currently incurable but some cases show sensitivity and partial (PR) or even complete (CR) response to dose-intensified regimens of combination chemotherapy (e.g., hyper-CVAD, R-CHOP-CVAD) with occasional extended periods of remission. Overall, however, both MCL and BV-MCL are associated with poor prognoses and relatively short survival (6, 7). Recent approaches to MCL therapy have included use of humanized monoclonal antibodies (rituximab), various strategies involving allogeneic and autologous bone marrow transplantation (8, 9), and molecular therapeutic targeting with proteasome inhibitors (bortezomib [Velcade]) (10, 11), mammalian target of rapamycin (mTOR) antagonists (12, 13), and newer immunomodulatory agents (lenalidomide [Revlimid]) (14, 15). However, these therapeutic approaches have failed to produce durable remissions in most patients with MCL. Therefore, the need for additional novel approaches to MCL therapy is clearly needed.
Bortezomib (also known as Velcade; PS-341) is one of the first therapeutic proteasome inhibitors (PSI) demonstrated to be effective in treating hematological disorders (16, 17). It is approved in the United States for treating relapsed multiple myeloma and MCL (10). Unfortunately, MCL frequently fails to respond to treatment with bortezomib or subsequently becomes refractory to bortezomib (11). Since single-agent therapy is rarely effective against diseases such as MCL, investigating whether the efficacy of bortezomib can be increased by combining the PSI with other novel type of pharmacologic agents may be necessary. Recent studies, both laboratory and clinical, have indicated that the addition of numerous agents, including thalidomide, lenalidomide, arsenic trioxide, and histone deacetylase inhibitors, to bortezomib may be beneficial in treating MCL (18-20).
Recently, WP1066, a novel small molecular weight compound, was synthesized, as a second-generation tyrphostin derivative of the classic Janus kinase 2 (JAK2) inhibitor AG490 (originally synthesized by Levitzky et al). (21). WP1066 was synthesized by screening a synthetic library for agents capable of suppressing cytokine (e.g., interleukin [IL]-6, IL-3) activation of signal transducer and activation of transcription 3 (STAT3) by down-regulating the expression of JAKs upstream from STAT, particularly JAK2 (22-27). Follow-up synthesis of degrasyn (WP1130), a more effective derivative of WP1066, showed that this analog was an even more potent inhibitor of both JAK2 and STAT3 than WP1066 and that it had additional activities that targeted degradation of the oncogene c-Myc (28). Specifically, degrasyn induces rapid degradation of c-Myc, an oncogene found highly-expressed in many tumor types that leads to tumor-growth inhibition in multiple myeloma and melanoma cells in vitro and in vivo. Degrasyn was also shown to target Bcr/Abl protein expression that leads to induction of cell death in chronic myelogenous leukemia cells through inhibition of the transcription factor phosphorylated Stat5 and the src kinase HcK (29).
The effect of the novel small-molecule inhibitor degrasyn and its molecular mechanism(s) targeting MCL has not been described. Because degrasyn and bortezomib target multiple pathways in different cancer cells, we hypothesized that the combination of bortezomib and degrasyn was likely to be an effective therapeutic strategy in treating MCL. In the present study, we evaluated the in vitro and in vivo activity of the novel small-molecule inhibitor degrasyn in combination with bortezomib in typical MCL and BV-MCL tumor cells.
Materials and Methods
MCL Cell lines, Primary MCL Cells, and Normal Lymphocytes
The human typical MCL cell lines Mino, DBsp53, Jeko, and Granta and BV-MCL cell lines Z-138 and JMP-1, were described previously (30-34), and maintained in RPMI medium (Gibco, Rockville, MD) containing 15% fetal calf serum (FCS; Hyclone, Logan, UT). Fresh biopsy-or pheresis-derived MCL cells were obtained from patient samples stored in the Tissue Procurement and Banking Facility at The University of Texas M. D. Anderson Cancer Center. MCL cells were enriched using sheep red blood cell resetting followed by the RosetteSep (StemCell Technologies, Vancouver, British Columbia, Canada) and contained 98% CD20+ and less than 1% CD3+ T cells according to flow cytometry. These cells were also cultured in RPMI medium (Gibco) containing 15% FCS (Hyclone). Normal human B lymphocytes were purified from donors’ buffy coats using a human B-cell enrichment cocktail (StemCell Technologies, Vancouver, British Columbia, Canada). Purified B cells were activated via incubation for 48 h with an anti-IgM antibody (3.5 μg/mL; ICN, Aurora, OH) or with recombinant human CD154 (1 μg/mL; Alexis, San Diego, CA). Peripheral blood mononuclear cells (PBMCs) were purified from the donors’ buffy coats by Ficoll gradient.
This study was conducted in accordance with the Helsinki protocol and approved by the M. D. Anderson Cancer Center Institutional Review Board. Informed consent was obtained from all patients whose tumor samples were used.
Antibodies, Reagents, and Materials
The primary antibodies used in our study included STAT3 and phosphorylated STAT3 (pSTAT3; BD Biosciences Pharmingen, San Jose, CA), bcl-2, bax, cyclin D1, c-Myc, and Oct-1 (Santa Cruz Biotechnology, Santa Cruz, CA) and an anti-β-actin antibody (Sigma, St. Louis, MO). The secondary antibodies used were peroxidase-conjugated goat anti-mouse and anti-rabbit antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). WP1066, WP1129, and degrasyn were synthesized at M. D. Anderson; these compounds were solubilized in dimethyl sulfoxide (DMSO; 100 mM) that was further diluted in injectable saline for animal studies. Bortezomib was provided by Millennium (Cambridge, MA).
Gel Shift Assays and DNA-Binding Enzyme-Linked Immunosorbent Assay
Eletrophoretic mobility shift assays (EMSAs) for nuclear factor NF-κB DNA binding were performed according to procedures described previously (35). The DNA-binding activity of STAT3 subunits was analyzed using an enzyme-linked immunosorbent assay according to the manufacturer's instructions (TransAM STAT Family Transcription Factor Assay Kit; Active Motif, Carlsbad, CA). Briefly, nuclear extracts were placed in the wells of a 96-well plate that contained an immobilized oligonucleotide carrying a STAT consensus DNA-binding site. STAT3 proteins bound to this immobilized oligonucleotide were detected by incubating nuclear extracts with a primary antibody recognizing active STAT3 followed by a horseradish peroxidase-conjugated secondary antibody and were quantified using spectrophotometry at 450 nm with a reference wavelength of 650 nm.
NF-κB Reporter Plasmid Transfection and Luciferase Assays
Mino cells were transiently transfected with 5 μg of the 6xNF-κB-luc reporter plasmid according to a nucleofector protocol from Amaxa Biosystems (Cologne, Germany) as described previously (31). After transfection, cells were pooled and separated equally into a 12-well plate. Cells were then treated with specified drug concentrations for 6 and 24 h. At appropriate time point, cells were harvested and lysed. Whole cell lysates were used for luciferase assays using the BD Monolight Enhanced Luciferase Assay kit (BD Biosciences, San Jose, CA) that was normalized according to β-gal activity.
Cell Proliferation Assays and Synergy Calculation
In vitro thymidine incorporation proliferation assays were performed for cell growth as described previously (25). Briefly, cells were plated (in triplicate) at 4 × 104 cells/well in 200 μL of RPMI 1640 with 10% FCS and the indicated reagents in a 96-well plate and incubated in 5% CO2 at 37°C. After 24 h, each well was pulsed with 0.5 μCi/10 μL [3H]thymidine (Amersham, Arlington Heights, IL) for 16 h. Cells were harvested, and the radioactivity was measured. The CalcuSyn software program (Biosoft, Ferguson, MO) was used to analyze the result of non-constant ratio drug combination synergy studies. The combination index and isobologram plots for degrasyn and bortezomib were created using the Chou-Talalay method.
Immunoblot Analysis
Whole cell extracts were solubilized in 1% sodium dodecyl sulfate sample buffer and electrophoresed on a 4-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel (Bio-Rad, Richmond, CA). Proteins were transferred from the gel onto a polyvinylidene difluoride membrane and probed with various specific primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. Proteins were visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ).
Apoptosis and Caspase 3 Assays
MCL cells were washed and stained with annexin V-fluorescein isothiocyanate and propidium iodide in accordance with the manufacturer’s protocol recommendation (BD Pharmingen, San Diego, CA). Apoptosis in the cells was quantified using a fluorescence-activated cell sorter and the CellQuest software program (BD Biosciences, San Jose, CA). Caspase activity assays were performed using a colorimetric substrate and a protocol and materials supplied by the manufacturer (EMD Chemicals, Gibbstown, NJ). In brief, cells were lysed, and 50 μg of the resulting cell lysates was added to an assay buffer for a total volume of 90 μL and incubated at 37°C for 10 min. A colorimetric substrate for caspase 3 (final concentration, 200 μM) was added to the mixture. A405 values were recorded for each sample after incubation at 37°C for 2 h.
RNA isolation and Real-time PCR
Total RNA isolation was performed by using Trizol LS Reagent (Invitrogen) according to manufactures instructions. Reverse transcription of RNA was carried out with cDNA archive kit (ABI, Foster City, CA). Synthesized cDNA was subjected to real-time polymerase chain reaction for the detection of related genes transcripts (CYCLIN D1, C-MYC, 1-OCT, GAPDH). In brief, 2.5uL of cDNA was placed in 25uL of reaction volume containing 12.5uL of TaqMan Universal PCR Master Mix, No AmpErase UNG, 8.75uL water and 1.25uL of primers and probe sets. The primers and probes were purchased from Applied Biosystem (ABI, Foster City, CA) (CYCLIN D1: HS9999904, C-MYC: HS9999903, 1-OCT: HS00427552). Primers and Probes were designed to span exon-exon boundaries. Amplification was performed in ABI Fast 7500 Real Time PCR system (Applied Biosciences) using the cycling program: 95C for 10 min; 40 cycles of 95C for 15s, 60C for 60C. All samples were analyzed in triplicates. DNA contamination was evaluated by performing PCR on the non-reverse transcribed control of each sample. The relative expression levels of the genes of interest was normalized to endogenous reference GAPDH and relative to control sample as a calibrator using formula: 2−ΔΔCT. The Threshold Cycle (Ct) reflects the cycle number at which the fluorescence generated within a reaction crosses the threshold.
Animal Studies
Three-week-old female severed combined immunodeficiency (SCID) mice were purchased from Taconic (Hudson, NY). The mice were housed five per cage and maintained under specific pathogen-free conditions at the SCID Mouse Barrier Facility at M. D. Anderson. The experimental protocol was reviewed and approved by the M. D. Anderson Institutional Animal Care and Use Committee. Mino cells (50 × 106) were injected intraperitoneally (IP) into the mice using a 27-gauge needle. One week after inoculation, mice were pooled and randomized into four treatment groups (n = 10/group). Group I received a vehicle control (DMSO) in injection saline (100 μL, twice weekly, IP), group II received bortezomib alone (0.25 mg/kg, 100 μL, twice weekly, IP), group III received degrasyn alone (20 mg/kg, 100 μL, twice weekly, IP), and group IV received a combination of bortezomib (0.25 mg/kg) and degrasyn (20 mg/kg) (100 μl, twice weekly, IP). Two additional groups (n = 5/group) that were not inoculated with the tumor cells but received treatment similar to that in groups III and IV were included. Treatment was continued for up to 8 weeks, at which time one mouse in each group was selected at random, euthanized, and subjected to necropsy. The remaining mice were monitored closely and euthanized based on the suggestion by veterinarians. Three different technicians on different schedule did treatment for these mice.
Statistical analysis
The IC50s were calculated by using the sigmoidal dose-response curves. Confidence intervals (CI) are shown in between parenthesis. Data from the in vitro assays were analyzed using a t test with a robust variance estimate. All significance testing was done at the P < 0.05 level. For the animal study, Kaplan-Meier survival curves were created for the mice using the GraphPad Prism software program (version 5.0b; GraphPad Software, La Jolla, CA) and pairwise comparison of survival curves and determination of p-values were done using the Log-rank (Mantel-Cox) Test.
Results
Growth inhibition of MCL cell lines with the AG490 congeners
Previous studies in non-lymphoid cells demonstrated that treatment with WP1066, a derivative of the classic JAK2-inhibiting compound AG490, inhibited both JAK2 and STAT3 activation, leading to inhibition of proliferation and induction of apoptosis in malignant glioma and melanoma cells (25, 26). Upon further screening and synthesis, we discovered that two more compounds, WP1129 and degrasyn (WP1130), had superior inhibitory activity in the JAK/STAT pathway when compared to the compound WP1066. We examined the antiproliferative effect of these compounds in MCL cell lines (two typical MCL and two BV-MCL lines). We exposed the four cell lines to each compound at increasing concentrations (0-5 μM) and then analyzed them for cell proliferation using thymidine incorporation assays. These studies showed that cell growth suppression was seen in all three compounds for all four cell lines (Fig. 1A), and the half maximal effective concentration (EC50) of degrasyn was lower than that of the other two compounds in all four MCL cell lines (Fig. 1B), suggesting that degrasyn is a more effective antitumor agent in MCL cells.
Figure 1.

Effect of treatment with AG490 congeners on MCL cell proliferation. Mino, DB, Z-138, and JMP-1 cells were treated with WP1066, WP1129, or degrasyn (WP1130) in a dose-dependent manner for 48 h and then analyzed for cell proliferation using thymidine incorporation assays. The data shown are the means and ranges of triplicate cultures of three independent experiments. Error bars represent standard deviation. (B) The EC50 of each compound was determined using the GraphPad Prism software program for all four cell lines, and the average EC50 for each compound was calculated. Statistical analysis was determined using the student t-test.
Degrasyn Inhibits Constitutively Activated pSTAT3 and NF-κB in MCL Cells
To determine the mechanism of growth suppression by degrasyn, we tested whether degrasyn can target STAT3 and NF-κB activation in MCL cells, as previous studies had shown that both STAT3 and NF-κB are constitutively activated in these cells (31, 36). First, we evaluated the expression of pSTAT3 in MCL cell lines and patient MCL cells by Western blotting and showed that pSTAT3 (serine 727) was constitutively expressed in all of the cell lines and patient MCL cells (Fig. 2A). Constitutive pSTAT3 expression in MCL cells can be down-regulated by treatment with degrasyn in a dose-and time-dependent manner in Mino MCL cells (Fig. 2B). Interestingly, degrasyn can also down-regulate constitutive cytoplasmic pIκBα protein level in MCL cells within 3 hours of treatment, suggesting that degrasyn targets upstream of the NF-kB pathway (Fig. 2C). Expression of transcription factor NF-kB was also assayed using a reporter gene assay, in which nuclear expression of NF-kB directed the induction of firefly luciferase activity. Degrasyn treatments resulted in reduced expression of luciferase activity in a dose-and time-dependent manner (Fig. 2D). These results suggest that degrasyn can directly or indirectly target both STAT3 and NF-κB pathways in MCL cells.
Figure 2.

Degrasyn inhibits constitutively activated pSTAT3 and NF-κB in MCL cells. (A) Cell extracts purified from six MCL cell lines (top panel), six primary MCL cells (middle panel) and control normal (unstimulated) and activated B cells (CD154 plus IgM) (bottom panel) were subjected to Western blotting for STAT3, pSTAT3 (ser-727), and actin (loading control). (B) Mino MCL cells were treated with dose-dependent (0-5 μM) of degrasyn for 24 h (top panel) or time-dependent (0-12 h) with 5 μM of degrasyn (bottom panel). Cells extracts from the cells were purified and analyzed by Western Blot for pSTAT3, STAT3 and Actin. (C) Western blots from part B (above) were stripped and reblotted for pIκBα and IκBα. (D) Mino cells were co-transfected with the 6xNF-κB-luc reporter plasmid and β-gal reporter plasmid and then treated with degrasyn or bortezomib (BZ), as a control, for the indicated times. Luciferase activity was measured and normalized according to β-gal activity. The data shown are the means and ranges of triplicate samples of three independent experiments. Error bars represent standard deviation.
Degrasyn Interacts with Bortezomib to Synergize Growth Inhibition and Apoptosis Induction in MCL Cells
Bortezomib is a proteasome inhibitor approved for the conventional treatment of MCL. To examine the feasibility of combining degrasyn with bortezomib to treat MCL, we performed in vitro and in vivo experiments assessing the tolerability and antitumor efficacy of these agents in MCL. First, we evaluated the effects of degrasyn in combination with bortezomib in MCL cells in vitro. Fig. 3A shows the combined effect of degrasyn and bortezomib in the representative MCL and BV-MCL cell lines Mino and Z-138 on cell growth inhibition. Fig. 3B shows a significant decrease in cell growth of Mino and Z-138 cell line was observed in response to treatment with combined low doses of degrasyn (1 μM) and bortezomib (10 nM) than with either agent alone, in a time-dependent manner up to 72 hours. We then evaluated the synergistic effect of the two compounds in four representative MCL cell lines. The concentrations of bortezomib and degrasyn that produced the synergistic effects on cell-growth inhibition ranged from 2 to 12 nM and from 0.5 to 2.0 μM, respectively, as shown in a combination index for four representative MCL cell lines (Fig 3C). Fig. 3D shows representative isobologram analysis confirming the synergistic effect of degrasyn and bortezomib in Mino and Z-138 cells. These data demonstrate synergistic anti-MCL activity of degrasyn plus bortezomib.
Figure 3.


Degrasyn interacts with bortezomib to synergize growth inhibition in MCL cells. (A) Typical MCL (Mino) and BV-MCL (Z-138) cells were treated with a combination of bortezomib and degrasyn in a dose-dependent manner as indicated and analyzed for cell proliferation using thymidine incorporation assays. A significant cell growth inhibition was observed in degrasyn plus bortezomib treatment compared to single agent alone in both cell lines, Mino (degrasyn alone vs. BZ (15 nM); p=0.015) and Z-138 (degrasyn alone vs. BZ (12.5 nM); p=0.0018). The data shown are the means and ranges of triplicate cultures of three independent experiments. Error bars represent standard deviation. Statistical analysis was determined using the student t-test. (B) Typical MCL (Mino) and BV-MCL (Z-138) cells were left untreated or treated with low dose of degrasyn (DG, 1 uM), bortezomib (BZ, 10 nM), or the combination of both. Cell number was determined by tryphan blue exclusion. The data shown are the means and ranges of triplicate cultures of two independent experiments. Error bars represent standard deviation. (C) The drug combination index for bortezomib and degrasyn (CI) was calculated by using the CalcuSyn software program. A combination index value of 1 indicated an additive effect of the two drugs. Combination index values less than 1 indicated synergy; the lower the value, the stronger the synergy. In contrast, combination index values greater than 1 indicated antagonism. (D) Representative normalized isobolograms demonstrating the synergy between bortezomib and degrasyn in typical MCL and BV-MCL cells.
Next, we examined whether degrasyn and bortezomib can interact to synergize apoptosis induction using an annexin V-based method of early apoptosis detection. At a concentration of 10 nM, bortezomib induced apoptosis in approximately 15% of Mino cells after 48 h of treatment, whereas at a concentration of 1 μM, degrasyn induced apoptosis in 5% of Mino cells over the same period. However, the combination of bortezomib and degrasyn administered at similar concentrations induced apoptosis in more than 50% of MCL of Mino and Z-138 cells (Fig. 4A). These results were similar for both typical MCL and BV-MCL cell lines. In addition, we measured caspase 3 activation, a signature event during apoptosis, in both MCL cell lines and primary MCL tumor cells after 48 hr of treatment with degrasyn and bortezomib alone or in combination. The results showed that degrasyn interacted with bortezomib to activate caspase 3 in all four MCL cell lines (both typical MCL and BV-MCL) and four primary MCL tumor cells (two typical MCL [pat # 1 and 2] and two BV-MCL [pat # 3 and 4]) (Fig. 4B). Moreover, the apoptotic effect of degrasyn plus bortezomib was evidenced by the induction of bcl-2 protein cleavage and bax protein expression (Fig. 4C). These results suggested the existence of potential synergy between degrasyn and bortezomib in targeting MCL cells via cell-growth inhibition and apoptosis induction.
Figure 4.

Degrasyn interacts with bortezomib to synergize apoptosis induction. (A) Typical MCL (Mino) and BV-MCL (Z-138) cells were treated with bortezomib alone (10 nM), degrasyn alone (1 μM), or a combination of the two for 48 hours and then analyzed for apoptosis using annexin V assays. The data shown are the means and ranges of three independent experiments. Error bars represent standard deviation. (B) MCL cell lines and primary MCL cells were treated with bortezomib (BZ) alone, degrasyn (DG) alone, or a combination of the two for 48 h. Cell extracts were purified and subjected to caspase 3 assays. The data shown are the means and ranges of three independent experiments. Error bars represent standard deviation. (C) MCL cells were treated with bortezomib (BZ; 10 nM) alone, degrasyn (DG; 1 μM) alone, or a combination of the two. After 48 h, cell extracts were analyzed for bcl-2 and bax protein expression using Western blotting. Actin was used as a loading control. Cont, control. Numbers below each panel indicate the relative intensity of each protein band compared to the control sample for each cell line.
Degrasyn and Bortezomib Synergy is Associated with NF-κB and pSTAT3 Inhibition, Concurrent with Cyclin D1 and c-Myc Down-regulation in MCL Cells
Next, we sought to determine the molecular basis for the mechanism of degrasyn and bortezomib synergy in MCL cells by examining the expression of key growth and survival genes in MCL cells. In these experiments, we treated Mino cells with degrasyn and bortezomib alone or in combination for 24 and 48 h and then examined the STAT3 and NF-κB activity in the cells. The results showed that the combination of degrasyn and bortezomib effectively down-regulated constitutive STAT3 and NF-κB DNA-binding activity after 24 h; the combination significantly inhibited constitutive STAT3 (Fig. 5A) and NF-κB (Fig. 5B) DNA-binding activity after 48 h of treatment. We observed down-regulation of the protein expression (Fig. 5C) as well as mRNA expression (Fig. 5D) of c-Myc and cyclin D1, target genes of STAT3 and NF-κB, in a similar manner. These results suggested that the combination of degrasyn and bortezomib can target multiple growth and survival signaling pathways as well as targeting genes downstream from these pathways in MCL cells.
Figure 5.

Degrasyn and bortezomib synergy is associated with NF-κB and STAT3 inhibition, concurrent with down-regulation of the cell growth and survival genes cyclin D1 and c-Myc in MCL cells. (A) Mino cells were treated with bortezomib (BZ; 10 nM) alone, degrasyn (1 μM) alone, or a combination of the two. After 24 and 48 h, nuclear extracts from the cells were purified and subjected to STAT DNA-binding enzyme-linked immunosorbent assay. The data shown are the means and ranges of triplicate samples of three independent experiments. Error bars represent standard deviation. (B) Nuclear extracts from part (A) above were also examined by EMSA for NF-κB DNA binding. Cont, control; BZ, bortezomib; DG, degrasyn. (C) Nuclear extracts from part (A) were Western blotted for c-Myc, cyclin D1, and Oct-1 (loading control) protein expression. Cont, control; BZ, bortezomib; DG, degrasyn. (D) Mino cells were treated with bortezomib (BZ; 10 nM) alone, degrasyn (1 μM) alone, or a combination of the two. After 24 h, RNA from cells was purified and real-time PCR was performed for c-myc and cyclin D1. Relative mRNA expressions were normalized with GAPDH and Oct-1. The data shown are the means and ranges of triplicate samples of two independent experiments. Error bars represent standard deviation.
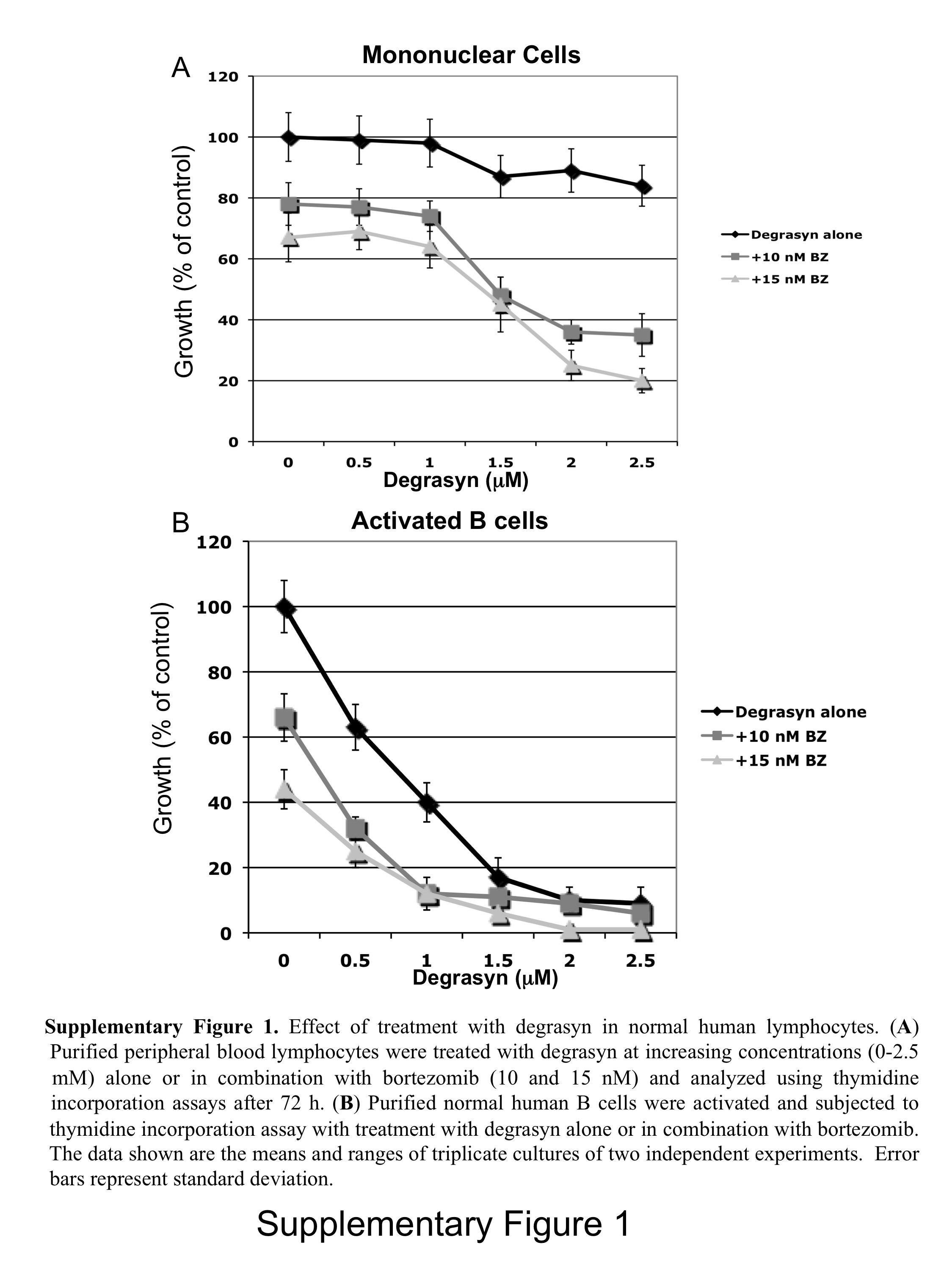
Effects of in Vitro Treatment of Normal Human Lymphocytes with Degrasyn and Bortezomib
To better understand the effects of degrasyn and bortezomib in the normal human B-cell lineage, we also evaluated the cytotoxicity of degrasyn alone and in combination with bortezomib in human PBMCs and activated normal B cells in vitro. We exposed purified PBMCs to degrasyn in dose response-studies at increasing concentrations (0-2.5 μM) alone and with 10 or 15 nM bortezomib. Degrasyn alone had a minimal cytotoxic effect on PBMCs, however, with exposure to the combination of degrasyn and bortezomib, the cytotoxicity of degrasyn and bortezomib in PBMCs increased as the concentration of degrasyn reached greater than 1 μM (Supplementary Figure 1A). We activated purified human B cells with recombinant CD40L, anti-IgM antibody, and IL-4 for 24 h and then exposed the proliferating B cell blasts to degrasyn alone or in combination with bortezomib in a manner similar to that utilized with PBMCs. Unlike PBMCs, the majority of the activated B cells responded to treatment with degrasyn preferentially alone as well as in combination with bortezomib (Supplementary Figure 1B), suggesting that degrasyn targets highly proliferative lymphocytes.
Antitumor Effects of Degrasyn in Combination with Bortezomib in a Xenotransplant SCID Mouse Model of MCL
To determine whether degrasyn also enhances the antitumor effects of bortezomib in MCL tumors cells in vivo, we utilized a xeno-transplant SCID mouse model of MCL. We intraperitoneally injected young SCID mice with Mino MCL cells and randomized the animals into four treatment groups as indicated in Fig. 6A. One week after tumor-cell inoculation, we began administering a low dose combination of degrasyn (20 mg/kg) and bortezomib (0.25 mg/kg) treatment to the mice (IP) twice weekly for approximately 8 weeks. The dose and schedule for both drugs were well tolerated and effective in other tumor models (29, 37). On day 65, when tumors began to appear, we randomly selected one mouse from each treatment group for sacrifice and necropsy. As shown in Fig. 6B, both bortezomib and degrasyn alone had some individual therapeutic effects in MCL tumor inhibition, whereas treatment with a combination of both drugs resulted in complete disappearance of tumors on gross pathological inspection. We closely monitored the remaining mice and sacrificed them at development of high tumor burden/morbidity. We created a survival curve showing that mice given both degrasyn and bortezomib survived longer than did mice given either agent alone (P<0.0001) (Fig. 6C). Bortezomib or degrasyn alone showed modest prolongation of survival compared to vehicle alone (p<0.0213 and p<0.105, respectively) (Fig. 6C). In addition, normal control mice (n = 5) given treatment with degrasyn alone or in combination with bortezomib remained alive and showed no signs of body-weight loss (data not shown) or organ (bone marrow, lungs, kidneys, and liver) toxicity 6 months later (Supplementary Figure 2). These results suggested that the combination of degrasyn and bortezomib has potential therapeutic efficacy against MCL and warrants further experimental and clinical investigation.
Figure 6.

Antitumor effect(s) of degrasyn in combination with bortezomib in a MCL xeno-transplant SCID mouse model. (A) Schematic representation of the experimental protocol for the animal study described in Materials and Methods. Mice received a vehicle control (DMSO; group I), bortezomib alone (0.25 mg/kg; group II), degrasyn alone (20 mg/kg; group III), or bortezomib plus degrasyn (group IV). (B) Images of representative mice in each group at necropsy (day 65). The arrows indicate tumors. BZ, bortezomib. (C) Kaplan-Meier survival curve showing survival for mice treated with bortezomib, degrasyn, or bortezomib plus degrasyn at the indicated concentrations in the xeno-transplant SCID mouse model of MCL (n = 10). BZ, bortezomib. Bortezomib plus degrasyn-treated mice show significantly increased survival (P<0.0001) compared to the untreated control group. The mean overall survival (OS) was 79 days (95% confidence interval, CI; 65-90) in the untreated or single agent-treated cohorts versus 104 days (95% CI; 90-120) in the group treated with combination of bortezomib and degrasyn.
Discussion
Intracellular signaling pathways that control cellular growth and survival mechanisms are complex, interactive and often crosstalk with each other in various types of human hematologic/lymphoid tumors (38, 39). Frequently however, similar or identical pathways (NF-κB, JAK/STAT, AKT/phosphatidylinositol 3-kinase, etc.) are utilized and constitutively activated in these tumors (31, 40, 41). Targeting these pathways with new therapeutic agents is likely to provide improved treatment outcomes if adequate agent targeting, molecular specificity, and drug delivery can be achieved. In the present study, we showed that the novel small-molecule compound degrasyn targets multiple signaling pathways in MCL cells, and that treating these cells with degrasyn in combination with the proteasome inhibitor bortezomib not only synergizes MCL cell-growth inhibition and apoptosis induction in vitro but also prolongs host survival in a SCID mouse model of MCL in vivo.
MCL, particularly BV-MCL, remains a poor responder to current combination chemotherapy among the various histotypes of NHL-B and represents a continuing therapeutic challenge (3). In some patients with MCL however, the disease does respond to treatment, sometimes for extended disease-free durations, with dose-intensification, most recently with combination chemotherapies such as hyper-CVAD and R-CHOP, which are often followed by autologous bone marrow transplantation (2, 7). Our goal in these studies was to continue to develop new, increasingly effective targeted approaches to MCL therapy, particularly for BV-MCL. These studies utilized experimental therapeutic methodologies, including in vitro techniques with representative MCL cell lines and patient tumor samples, as well as more translational in vivo studies with xeno-transplant SCID mouse models of MCL.
Bortezomib is a boronic acid-derived effective reversible inhibitor of the 26S proteasome that surprisingly, has had relatively minor toxic effects (e.g., minor platelet-count decreases, neuropathy) in initial clinical trials for MCL (17). These studies suggested that bortezomib likely would be more effective in combination with conventional chemotherapy and possibly even more effective with molecularly targeted novel therapeutic agents, particularly small-molecule inhibitors such as degrasyn. Although degrasyn is structurally related to the classic JAK2 inhibitor AG490, when tested against aggressive NHL-B-like MCL cells in vitro, we observed that degrasyn was also an effective inhibitor of constitutive NF-κB activation present in both typical MCL and BV-MCL cells. The molecular mechanism of how degrasyn down-regulates NF-κB activity in MCL cells remains uncertain. However, recent studies have indicated that degrasyn directly targets c-Myc for proteasomal degradation within hours after treatment (28), suggesting that NF-κB signaling may be a downstream target of c-Myc as previously shown in Drosophila flies (42). Another possible mechanism of NF-κB inhibition exerted by degrasyn occurs through the JAK/STAT signaling pathway, as previous studies have shown that STAT3 maintains constitutive NF-κB activity in tumor cells by prolonging NF-κB nuclear retention via acetyltransferase p300-mediated RelA acetylation (41). Therefore, inhibition of STAT3 activity by degrasyn may have affected the status of NF-κB activity in MCL cells, as seen in the present study. Constitutive STAT3 and NF-κB signaling and interaction are highly displayed in both murine and human cancers, and their roles are central to tumor-cell growth and survival (39, 41, 43). Studies have shown that concurrently targeting both constitutive STAT3 and NF-κB activity in lymphoid tumor cells is effective both in vivo and in vitro (38, 44). The synergy between degrasyn and bortezomib resulted in down-regulation of STAT3 and NF-κB activity in MCL cells may provide a molecular mechanism accounting for their effect in prolonging the survival of MCL xeno-transplanted SCID mice treated with these compounds. However, other still unidentified factors may also contribute to the observed synergistic effects of degrasyn and bortezomib on MCL cell growth and survival.
In terms of cell-type specificity, degrasyn has been shown to be more effective in myeloid and lymphoid tumor cells than normal CD34+ hematopoietic precursor cells, dermal fibroblasts, and endothelial cells (29). The present study also showed that degrasyn has minimal cytotoxicity in normal peripheral blood mononuclear lymphocytes but maximal cytotoxicity in targeted highly proliferative B lymphocytes, suggesting that degrasyn is a potential therapeutic drug candidate for aggressive or relapsed/refractory B-cell lymphomas, that very much need effective new therapies. When combined with bortezomib, degrasyn has antitumor activity in SCID mice with MCL but is not toxic in normal healthy mice, making degrasyn a candidate combinatorial therapeutic agent together with bortezomib in MCL therapy.
In summary, we have shown that MCL cells, both typical MCL and BV-MCL, treated with degrasyn in conjunction with bortezomib resulted in synergistic growth inhibition and apoptosis induction in vitro. Apoptosis in these cells was correlated with down-regulation of constitutive NF-κB and pSTAT3 activation, leading to inhibition of c-Myc and cyclin D, and upregulation of cleaved-bcl-2 and bax protein expression. In vivo, degrasyn and bortezomib combined to synergistically prevent MCL tumor development in a xeno-transplant SCID mouse model. Agents such as degrasyn that can pharmacologically target constitutively expressed key growth/survival signaling pathways and cell cycle regulators, including NF-κB, STAT3, c-Myc, and cyclin D1, in MCL cells may prove to be useful therapeutic agents for MCL when combined with effective proteasome inhibitors such as bortezomib.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Grant support: The Odyssey Program and Kimberly-Clark Foundation Award for Scientific Achievement at The University of Texas M. D. Anderson Cancer Center (to L.V.P.), National Cancer Institute grants CA-RO1-100836 (to R.J.F.) and CA-16672-26 (Cancer Center Support [Core] Grant), and a grant from The Leukemia & Lymphoma Society (to R.J.F.).
Abbreviations
- BV-MCL
blastoid-variant mantle cell lymphoma
- DMSO
dimethyl sulfoxide
- EC50
half maximal effective concentration
- EMSA
eletrophoretic mobility shift assay
- FCS
fetal calf serum
- IL
interleukin
- JAK
Janus kinase
- MCL
mantle cell lymphoma
- NF
nuclear factor
- NHL-B
B-cell non-Hodgkin lymphoma
- PBMC
peripheral blood mononuclear cell
- pSTAT3
phosphorylated signal transducer and activation of transcription 3
- SCID
severed combined immunodeficiency
- STAT
signal transducer and activation of transcription
References
- 1.Fisher RI. Mantle-cell lymphoma: classification and therapeutic implications. Ann Oncol. 1996;7(Suppl 6):S35–9. doi: 10.1093/annonc/7.suppl_6.s35. [DOI] [PubMed] [Google Scholar]
- 2.Obrador-Hevia A, Fernandez de Mattos S, Villalonga P, Rodriguez J. Molecular biology of mantle cell lymphoma: from profiling studies to new therapeutic strategies. Blood Rev. 2009;23:205–16. doi: 10.1016/j.blre.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Bernard M, Gressin R, Lefrere F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia. 2001;15:1785–91. doi: 10.1038/sj.leu.2402272. [DOI] [PubMed] [Google Scholar]
- 4.Jaffe ES, Harris NL, Diebold J, Muller-Hermelink HK. World Health Organization Classification of lymphomas: a work in progress. Ann Oncol. 1998;9(Suppl 5):S25–30. doi: 10.1093/annonc/9.suppl_5.s25. [DOI] [PubMed] [Google Scholar]
- 5.Bosch F, Lopez-Guillermo A, Campo E, et al. Mantle cell lymphoma: presenting features, response to therapy, and prognostic factors. Cancer. 1998;82:567–75. doi: 10.1002/(sici)1097-0142(19980201)82:3<567::aid-cncr20>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 6.Fisher RI. Mantle cell lymphoma: at last, some hope for successful innovative treatment strategies. J Clin Oncol. 2005;23:657–8. doi: 10.1200/JCO.2005.10.980. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt C, Dreyling M. Therapy of mantle cell lymphoma: current standards and future strategies. Hematol Oncol Clin North Am. 2008;22:953–63. doi: 10.1016/j.hoc.2008.07.001. ix. [DOI] [PubMed] [Google Scholar]
- 8.Khouri IF, Lee MS, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol. 2003;21:4407–12. doi: 10.1200/JCO.2003.05.501. [DOI] [PubMed] [Google Scholar]
- 9.Maris MB, Sandmaier BM, Storer BE, et al. Allogeneic hematopoietic cell transplantation after fludarabine and 2 Gy total body irradiation for relapsed and refractory mantle cell lymphoma. Blood. 2004;104:3535–42. doi: 10.1182/blood-2004-06-2275. [DOI] [PubMed] [Google Scholar]
- 10.Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–74. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 11.Goy A, Bernstein SH, Kahl BS, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20:520–5. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rizzieri DA, Feldman E, Dipersio JF, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008;14:2756–62. doi: 10.1158/1078-0432.CCR-07-1372. [DOI] [PubMed] [Google Scholar]
- 13.Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 14.Habermann TM, Lossos IS, Justice G, et al. Lenalidomide oral monotherapy produces a high response rate in patients with relapsed or refractory mantle cell lymphoma. Br J Haematol. 2009;145:344–9. doi: 10.1111/j.1365-2141.2009.07626.x. [DOI] [PubMed] [Google Scholar]
- 15.Tempescul A, Ianotto JC, Morel F, Marion V, De Braekeleer M, Berthou C. Lenalidomide, as a single agent, induces complete remission in a refractory mantle cell lymphoma. Ann Hematol. 2009;88:921–2. doi: 10.1007/s00277-008-0692-4. [DOI] [PubMed] [Google Scholar]
- 16.Goy A. New directions in the treatment of mantle cell lymphoma: an overview. Clin Lymphoma Myeloma. 2006;7(Suppl 1):S24–32. doi: 10.3816/clm.2006.s.005. [DOI] [PubMed] [Google Scholar]
- 17.Suh KS, Goy A. Bortezomib in mantle cell lymphoma. Future Oncol. 2008;4:149–68. doi: 10.2217/14796694.4.2.149. [DOI] [PubMed] [Google Scholar]
- 18.O'Connor OA, Czuczman MS. Novel approaches for the treatment of NHL: Proteasome inhibition and immune modulation. Leuk Lymphoma. 2008;49(Suppl 1):59–66. doi: 10.1080/10428190802365033. [DOI] [PubMed] [Google Scholar]
- 19.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 20.Wen J, Feng Y, Huang W, et al. Enhanced antimyeloma cytotoxicity by the combination of arsenic trioxide and bortezomib is further potentiated by p38 MAPK inhibition. Leuk Res. 2009 doi: 10.1016/j.leukres.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 21.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–8. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 22.Ferrajoli A, Faderl S, Van Q, et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007;67:11291–9. doi: 10.1158/0008-5472.CAN-07-0593. [DOI] [PubMed] [Google Scholar]
- 23.Hussain SF, Kong LY, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–6. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 24.Iwamaru A, Szymanski S, Iwado E, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–44. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 25.Kong LY, Abou-Ghazal MK, Wei J, et al. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759–68. doi: 10.1158/1078-0432.CCR-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong LY, Wei J, Sharma AK, et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol Immunother. 2009;58:1023–32. doi: 10.1007/s00262-008-0618-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verstovsek S, Manshouri T, Quintas-Cardama A, et al. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin Cancer Res. 2008;14:788–96. doi: 10.1158/1078-0432.CCR-07-0524. [DOI] [PubMed] [Google Scholar]
- 28.Bartholomeusz G, Talpaz M, Bornmann W, Kong LY, Donato NJ. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 2007;67:3912–8. doi: 10.1158/0008-5472.CAN-06-4464. [DOI] [PubMed] [Google Scholar]
- 29.Bartholomeusz GA, Talpaz M, Kapuria V, et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109:3470–8. doi: 10.1182/blood-2006-02-005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu L, Lin-Lee YC, Pham LV, Tamayo A, Yoshimura L, Ford RJ. Constitutive NF-kappaB and NFAT activation leads to stimulation of the BLyS survival pathway in aggressive B-cell lymphomas. Blood. 2006;107:4540–8. doi: 10.1182/blood-2005-10-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003;171:88–95. doi: 10.4049/jimmunol.171.1.88. [DOI] [PubMed] [Google Scholar]
- 32.Amin HM, McDonnell TJ, Medeiros LJ, et al. Characterization of 4 mantle cell lymphoma cell lines. Arch Pathol Lab Med. 2003;127:424–31. doi: 10.5858/2003-127-0424-COMCLC. [DOI] [PubMed] [Google Scholar]
- 33.Goy A, Remache YK, Gu J, et al. Establishment and characterization of a new mantle cell lymphoma cell line M-1. Leuk Lymphoma. 2004;45:1255–60. doi: 10.1080/10428190310001642729. [DOI] [PubMed] [Google Scholar]
- 34.Medeiros LJ, Estrov Z, Rassidakis GZ. Z-138 cell line was derived from a patient with blastoid variant mantle cell lymphoma. Leuk Res. 2006;30:497–501. doi: 10.1016/j.leukres.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 35.Pham LV, Tamayo AT, Yoshimura LC, et al. A CD40 Signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in B cell lymphomas. Immunity. 2002;16:37–50. doi: 10.1016/s1074-7613(01)00258-8. [DOI] [PubMed] [Google Scholar]
- 36.Lai R, Rassidakis GZ, Medeiros LJ, Leventaki V, Keating M, McDonnell TJ. Expression of STAT3 and its phosphorylated forms in mantle cell lymphoma cell lines and tumours. J Pathol. 2003;199:84–9. doi: 10.1002/path.1253. [DOI] [PubMed] [Google Scholar]
- 37.LeBlanc R, Catley LP, Hideshima T, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 38.Lam LT, Wright G, Davis RE, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–13. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee TL, Yeh J, Friedman J, et al. A signal network involving coactivated NF-kappaB and STAT3 and altered p53 modulates BAX/BCL-XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer. 2008;122:1987–98. doi: 10.1002/ijc.23324. [DOI] [PubMed] [Google Scholar]
- 40.Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O. Regulation and Importance of the PI3K/Akt/mTOR Signaling Pathway in Hematologic Malignancies. Anticancer Agents Med Chem. 2009 doi: 10.2174/187152009789377772. [DOI] [PubMed] [Google Scholar]
- 41.Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–93. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orian A, van Steensel B, Delrow J, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–14. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bharti AC, Shishodia S, Reuben JM, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–84. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie GG, Queisser N, Wolfson ML, Fraga CG, Adamo AM, Oteiza PI. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin's lymphoma cells. Int J Cancer. 2008;123:56–65. doi: 10.1002/ijc.23477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.