

Abstract
Poly(lactic-co-glycolic acid) (PLGA) microspheres have been widely examined for vaccine applications due to their attractive features of biocompatibility, biodegradability, ability to be internalized by antigen-presenting cells, and long-term antigen release. However, one of the major challenges for PLGA particle vaccines is the potential for antigen instability and loss of antigenicity and immunogenicity. To address this challenge, we have developed a new method of “self-healing” encapsulation in PLGA microspheres, where pre-made PLGA microspheres are loaded with protein antigens under aqueous conditions with minimal impact on their antigenicity and immunogenicity. In this report, we show that mice immunized with self-encapsulating PLGA microspheres in a prime-boost regimen generated significantly enhanced antigen-specific CD8α+ T cell and antibody responses, compared with mice immunized with free, soluble protein admixed with calcium phosphate gel, a widely used adjuvant. Furthermore, a single-dose of microspheres designed for >40 day sustained antigen release elicited robust cellular and humoral immune responses as efficiently as the prime-boost vaccinations with calcium phosphate gel. Overall, these results suggest excellent potential of our self-encapsulating PLGA microspheres as a vaccine platform for multiple-dose as well as single-dose vaccinations.
Keywords: antigen-presenting cells, calcium phosphate adjuvant, poly(lactic-co-glycolic acid) microspheres, self-encapsulation, vaccine delivery
Priming of adaptive immune responses requires internalization and processing of antigen (Ag) by antigen-presenting cells (APCs). Consequently, Ag delivery to APCs is a major focus of vaccine development that is crucial for eliciting strong protective immunity.[1–5] In particular, poly(lactic-co-glycolic acid) (PLGA) nano- and microspheres have been explored widely for vaccine delivery applications due to their biocompatibility, biodegradability, ability to be internalized by APCs, and potential for long-term Ag release and hence, single-dose vaccination.[6–12] Despite their promising attributes, one of the major challenges for PLGA particle vaccines is the potential for Ag instability, and loss of antigenicity and immunogenicity due to exposure of the Ag to harsh conditions during the production of polymeric particles.[13–15] To address this challenge, we have previously developed a novel method of self-healing encapsulation in PLGA microspheres, where large biomolecules are loaded into pre-made PLGA microspheres under aqueous conditions.[16,17] This “self-encapsulating” procedure exploits the passive healing of the polymer chains above the glass transition temperature (Tg) of PLGA polymer to close pores within the particles. By adjusting the environmental temperature, bio-macromolecules in aqueous conditions diffuse through open pores into the particle cores and then subsequently become trapped as pores close.[18,19] We have reported that the “self-encapsulating” procedure avoids exposure of Ags to harsh conditions, thus maintaining their antigenicity and immunogenicity.[17] In this work, we investigated the interaction between self-encapsulating PLGA microspheres and APCs and performed immunological analyses of Ag-specific T cell and humoral immune responses after subcutaneous immunization with these particles. Prime-boost immunizations with self-encapsulating PLGA microspheres significantly improved Ag-specific CD8α+ T cell and antibody responses, compared with immunizations with free, soluble protein admixed with calcium phosphate gel, a widely used adjuvant in Europe.[20–23] Importantly, a single-dose of microspheres designed for >40 d Ag release elicited Ag-specific CD8α+ T cell and antibody responses as efficiently as the prime-boost vaccinations with the equivalent total dose of Ag plus calcium phosphate gel. Our results suggest that self-encapsulating PLGA microspheres have great potential for further development as a versatile vaccine delivery system for multiple- as well as single-dose vaccinations, which may address a lack of patient compliance and poor medical infrastructure in resource-limited settings.
APCs, namely dendritic cells (DCs), are the main cell type that orchestrates the interaction between innate and adaptive immunity and thus are the major target for vaccine delivery.[1,2] DCs internalize and process exogenous Ags (e.g., bacterial products) and endogenous Ags (e.g., tumor and viral products). Processed Ag epitopes are then displayed on the cell surface to be recognized by Ag-specific T cells and contribute to cellular and humoral immune responses.[24] Therefore, vaccine strategies should address how to promote Ag delivery to DCs as well as subsequent Ag uptake and processing by DCs. PLGA particles have been extensively investigated for vaccine delivery applications because of their ability to target APCs, induce particulate phagocytosis, and serve as a sustained-release depot, thus prolonging Ag exposure to the immune system.[25] However, one of the remaining challenges for PLGA particle-based vaccine delivery systems is the potential for Ag instability, which can occur during particle production and Ag release.[13–15] Traditional methods for loading Ags into polymeric particles expose the Ags to detrimental conditions, including high shear stress and oil–water interfaces, that may lead to protein unfolding and aggregation and subsequent loss of antigenicity and immunogenicity.[8]
To address these issues, we have developed a method for protein loading into PLGA microspheres that bypasses many of the challenges of traditional techniques.[16,17] This method, which we termed “self-healing encapsulation” or more simply “self-encapsulation,” loads Ag by simple mixing of pre-made microspheres in an aqueous solution of protein (Figure 1a). These microspheres contain an interconnecting pore network and a protein-trapping agent (e.g., aluminum- or calcium-based adjuvant gel) that is accessible through the pores. During mixing, the Ag diffuses into the pores and binds to the trapping agent, allowing efficient remote loading. We have reported that addition of a protein-trapping agent strongly improves encapsulation efficiency, compared to microspheres without an internal trapping agent.[16,17] Subsequent heating of the system above the Tg of the polymer closes the pores, sealing Ag inside the microspheres (see Figure 1b for before and after images of pore healing). We have previously reported improved antigenicity of tetanus toxoid released from self-encapsulating microspheres over 28 d, compared with that loaded by traditional encapsulation techniques.[17] Furthermore, the use of Ag-binding aluminum or calcium-based trapping agents during the self-encapsulating procedure offers a unique and convenient strategy for incorporating one or more different types of Ags in PLGA microspheres produced in a single large batch.
Figure 1.
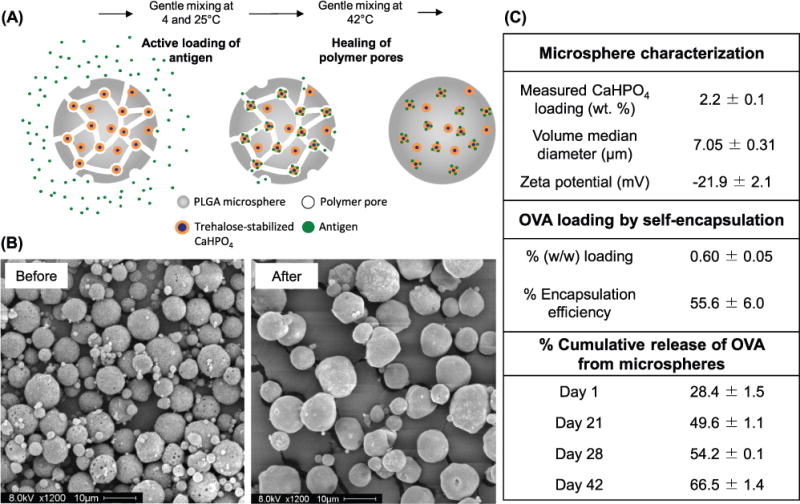
Formulation and characterization of self-encapsulating PLGA microspheres. A) Schematic illustration of the self-encapsulation of protein Ags into PLGA microspheres. B) SEM images of self-encapsulating PLGA microspheres containing CaHPO4 adjuvant gel as the protein-trapping agent. Microspheres are shown before (left) and after (right) self-encapsulation of OVA and pore healing. Scale bars represent 10 μm. C) Summary of key properties of the self-encapsulating microspheres and percentage cumulative OVA release from the microspheres. Data are shown as mean ± SEM (n = 3).
Having established a versatile procedure for Ag encapsulation, here we set out to investigate the interaction between APCs and self-encapsulating PLGA microspheres and to explore the potential of the sustained Ag release formulation for multiple-dose as well as single-dose vaccination in mice. Specifically, we studied the internalization of the microspheres by DCs and investigated the type and magnitude of the immune responses generated by the microspheres after subcutaneous immunization. We found that the microspheres were successfully internalized by murine DCs and produced balanced humoral and cellular immune responses. Importantly, a single dose of microspheres designed for >40 d Ag release elicited comparable, if not superior, CD8α+ T cell and antibody responses as the total equivalent dose of Ag administered in two separate doses with calcium phosphate gel, a routinely used adjuvant in human vaccines.[20–23] Our results presented here suggest that these self-encapsulating microspheres have great potential for further development as a vaccine delivery system for multiple-dose as well as single-dose vaccination.
To prepare self-encapsulating PLGA microspheres for vaccine delivery applications, we have employed the standard double emulsion-solvent evaporation technique and formulated antigen-free PLGA microspheres with trehalose and calcium phosphate (CaHPO4) adjuvant gel in the inner water phase (Figure 1a). We added trehalose as a porosigen to create the interconnecting pore network within the polymer spheres.[16] Calcium phosphate gel was used as the protein-trapping agent within the microsphere pores since it is a natural constituent of the body and is well-tolerated, readily resorbed, and has been widely used as an adjuvant in Europe.[20–23] We have recently reported the development of a “self-encapsulating” PLGA microsphere formulation and characterized its efficacy as an intranasal vaccine delivery system.[26] Briefly, CaHPO4 gel was incorporated within the PLGA microspheres at 2.2 ± 0.1% w/w loading as determined by inductively coupled plasma optical emission spectrometry (ICP-OES) (Figure 1c). Following the self-encapsulation procedure, ovalbumin (OVA), a prototypical model antigen, was loaded within the PLGA microspheres at 0.60 ± 0.05% w/w loading with 56% encapsulation efficiency (Figure 1c). A moderate burst release of 28% was observed on day 1 in phosphate buffered saline (PBS) at 37 °C. This was followed by steady, sustained release of OVA, achieving 67% total Ag release at the termination of the experiment on day 42 (Figure 1c). The size of the microspheres is an important factor that influences internalization by APCs. A number of studies have demonstrated that microspheres less than 10 μm in diameter are more efficiently internalized by APCs, compared with larger particles.[27–30] Therefore, we have refined the formulation parameters, including the polymer concentration and energy of agitation for emulsion, to produce microspheres of the desired size range with the median microsphere diameter of 7.0 ± 0.3 μm, with a negative zeta potential of −21.9 ± 2.1 mV (Figure 1c). Sonication was preferable to homogenization for forming the primary emulsion in our system since greater agitation generated smaller particles.[31]
Using microspheres in the target size range of less than 10 μm in diameter, we next studied the interaction between the vaccine microparticles and DCs, which are capable of internalizing PLGA particles via phagocytosis.[32] To evaluate this interaction, we used flow cytometry to measure particle uptake by JAWSII cells, a murine bone marrow-derived DC line. JAWSII cells were incubated with three different concentrations of rhodamine 6G-labeled microspheres for 6 or 24 h at 37 °C. Cells were then treated with phalloidin-iFluor 405 dye to stain the actin filaments. To quantify the percentage of cells with rhodamine fluorescence signal, we used an Amnis ImageStreamX Mark II imaging flow cytometer (Figure 2a). As we anticipated, the percentage of microsphere-positive cells increased with both the incubation time and microsphere concentration, with ∼60% of cells internalizing microspheres after 24 h of incubation with the initial particle concentration of 100 μg per well. Representative fluorescence images obtained from the flow cytometer showed the association of DCs with one or more PLGA particles (Figure 2b–e).
Figure 2.
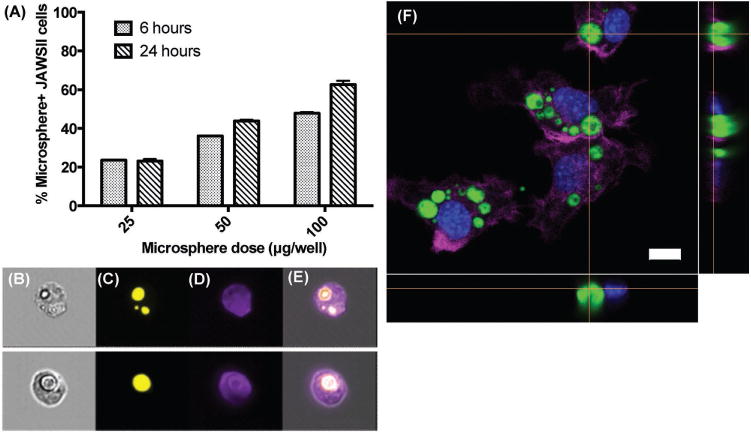
Internalization of self-healing PLGA microspheres by dendritic cells. A) Flow cytometry analysis of JAWSII dendritic cells treated with different doses of rhodamine 6G-labeled microspheres for 6 or 24 h. Columns show the percent of gated events containing cells with associated microspheres. Data represent mean ± SEM (n = 3). Representative ImageStreamX images showing B) bright-field image of JAWSII cells with associated microspheres; C, D) fluorescence images of the microspheres and JAWSII cells, respectively; and E) overlay of images. F) Confocal microscopy image showing JAWSII dendritic cells that internalized rhodamine-labeled self-encapsulating microspheres (green) after 24 h of incubation. Actin filaments were stained with Alexa Fluor 647-phalloidin (violet) and nuclei were stained with DAPI (blue). Scale bar represents 10 μm.
Next, to investigate the interaction between particles and JAWSII cells more closely, we used confocal microscopy. Cells were incubated with rhodamine-labeled microspheres for 24 h at 37 °C and stained with DAPI and Alexa Fluor 647-phalloidin to visualize cellular nuclei and actin filaments, respectively. As shown in Figure 2f, JAWSII cells successfully phagocytosed one or more microspheres over 24 h with a range of particle sizes taken up into the cells. Orthogonal confocal images confirmed that the microspheres were not merely attached to the cell surface but internalized within the cells, as demonstrated by actin filaments surrounding fluorophore-loaded particles. Taken together, results shown in Figure 2 indicated that self-encapsulating PLGA microparticles are readily taken up by DCs, allowing for intracellular Ag delivery to DCs.
Having characterized the particles and their cellular interaction with APCs in vitro, we performed vaccination studies in mice to examine the induction of Ag-specific cellular and humoral immune responses. The route of vaccine delivery plays a major role in shaping immune responses, perhaps due to local cell types (e.g., different subsets of APCs) and stability of the microspheres by a particular administration method (e.g., particle agglomeration at the site of injection).[33] We designed our immunization studies (1) to determine cellular and humoral immune responses after subcutaneous immunization with the microspheres; (2) to compare the immune responses elicited by the microspheres to those induced by commercial CaHPO4 adjuvant gel; and (3) to test a single-dose vaccination with the microspheres capable of long-term Ag release (as shown in Figure 1c). C57BL/6 mice were immunized subcutaneously at tail base with CaHPO4 adjuvant gel coadministered with free soluble OVA (100 μg gel + 10 μg OVA) or microspheres coloaded with CaHPO4 adjuvant gel and OVA (37 μg gel + 10 μg OVA, denoted as “10 μg OVA × 2”), with both groups treated on days 0 and 21 using the primeboost vaccine regimen shown in Figure 3a. Ag-specific T-cell responses were measured on day 28 (7 d after the booster dose) using tetramer staining on peripheral blood mononuclear cells, while anti-OVA serum IgG responses were measured on days 20 and 42 since subunit antigen vaccines generally induce peak cellular and humoral immune responses on week 1 and weeks 2–3 after immunization, respectively. In addition, to examine the potency of self-encapsulating PLGA microparticles as a platform for single-dose administration, we included a prime-only vaccination group with twice the dose of OVA-containing microspheres (74 μg gel + 20 μg OVA, denoted as “20 μg OVA × 1”) (Figure 3a). Subunit vaccination generally induces transient immune responses, thus necessitating multiple immunizations.[4,5,12] Therefore, we chose to use the prime-boost immunizations with CaHPO4 adjuvant gel with 10 μg OVA at the same total antigen dose as the control group rather than the prime-only CaHPO4 adjuvant gel with 20 μg OVA, which is expected to generate weaker immune responses.
Figure 3.
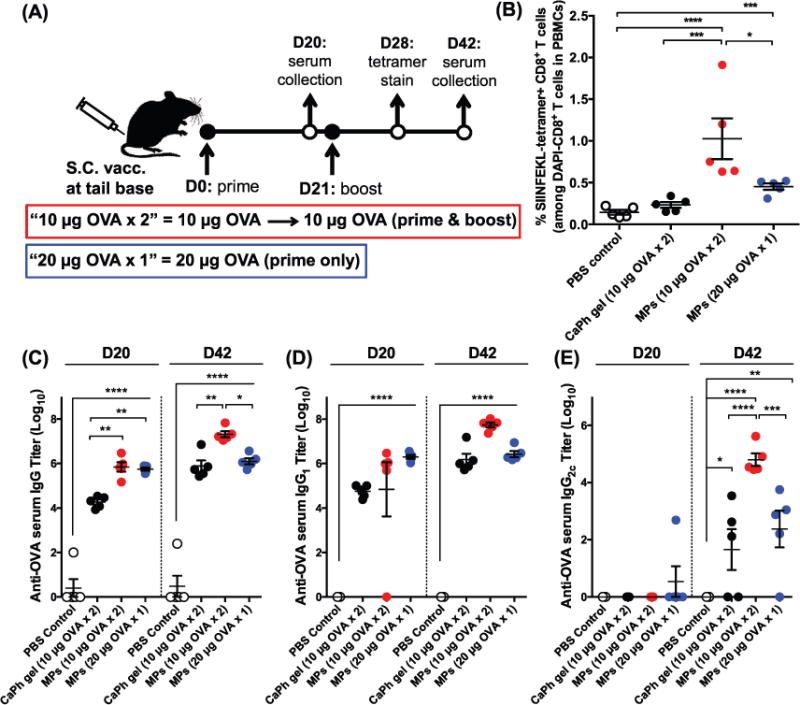
Cellular and humoral immune responses elicited by self-healing PLGA microspheres in vivo. A) Shown are the vaccine doses and regimen. Naïve C57BL/6 mice were administered subcutaneously at tail base on days 0 and 21 with 10 μg of OVA either admixed with CaHPO4 adjuvant gel or formulated with CaHPO4 adjuvant gel in self-healing PLGA microspheres (10 μg × 2). A group of mice was immunized on day 0 with a prime-only administration of 20 μg of OVA formulated with CaHPO4 adjuvant gel in self-healing PLGA microspheres (20 μg × 1). B) Shown are the percentages of SIINFEKL-tetramer + CD8α + T cells among total CD8α + T cells in PBMCs on day 28. C–E) Serum anti-OVA antibody titers were measured on day 20 (prime response) and day 42 (boost response). Shown are OVA-specific serum C) IgG, D) IgG1, and E) IgG2C titers. Data were fit using a 4-parameter curve, and titers were calculated by solving for the inverse dilution factor resulting in an absorbance value of 0.5. Data represent mean ± SEM (n = 5). All groups were compared using one-way ANOVA (B) or two-way ANOVA (C–E), followed by Bonferroni’s post-test (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).
On day 28, four weeks after the prime vaccination (i.e., one week after the boost vaccination), mice were analyzed for the percentage of Ag-specific CD8α+ T cells using the tetramer staining assay with SIINFEKL-H-2Kb tetramer, as we described previously (Figure 3b).[34,35] As CaHPO4 adjuvant reportedly promotes humoral but not cellular immune responses,[20–23] we found that the control group, with soluble OVA admixed with CaHPO4 adjuvant, did not generate CD8α+ T cell responses above the PBS baseline even after prime-boost vaccinations (Figure 3b). By contrast, the microsphere group administered with prime-boost vaccinations (10 μg OVA × 2) produced ∼1% Ag-specific CD8α+ T cell responses, which is a sevenfold greater frequency of OVA-specific CD8α+ T cells than the PBS control group (P < 0.0001) and a fourfold greater frequency than the CaHPO4 adjuvant control group (P < 0.001, Figure 3b). Importantly, self-encapsulating PLGA microspheres administered in a prime-only vaccination (20 μg OVA × 1) also generated robust Ag-specific CD8α+ T cell responses with a threefold greater frequency of OVA-specific CD8α+ T cells among peripheral blood mononuclear cells (PBMCs) than the PBS control group (P < 0.001) and a twofold greater response than the dual prime-boost vaccination with soluble OVA plus CaHPO4 adjuvant (Figure 3b), although the latter difference was not statistically significant. Taken together, our results showed that the self-encapsulating PLGA microspheres elicited significantly enhanced Ag-specific CD8α+ T cell immune responses relative to the soluble vaccine formulated with the CaHPO4 adjuvant. In addition, we found that the self-encapsulating PLGA micro-spheres designed for long-term Ag release can serve as a single-dose vaccination platform for eliciting CD8α+ T cell immune responses.
We have performed parallel immunization trials and analyzed the induction of humoral immune responses (Figure 3c–e). We measured serum anti-OVA antibody titers for total IgG, IgG1, and IgG2C subclasses, as the subclass levels provide information on the polarization of Th responses, with IgG1 associated with a Th2-type response and IgG2C associated with a Th1-skewed response.[36] The PLGA microspheres (10 μg OVA × 2) elicited 38- and 27-fold increases in total anti-OVA serum IgG titers compared with soluble OVA plus CaHPO4 adjuvant after prime and boost immunizations, respectively (P < 0.01, Figure 3c). Similarly, compared with soluble OVA admixed with the CaHPO4 adjuvant, the PLGA microsphere group (10 μg OVA × 2) elicited 36- and 1400-fold higher serum titers for anti-OVA IgG1 and IgG2C subclasses, respectively, after boost immunizations (P < 0.0001 for IgG2C, Figure 3d, e). Importantly, the single prime-only vaccination group with PLGA microspheres (20 μg OVA × 1) also generated robust anti-OVA serum total IgG, IgG1, and IgG2C titers that were comparable to those observed only after two rounds of vaccinations with soluble OVA plus CaHPO4 adjuvant (Figure 3c–e). In addition, anti-OVA total IgG and IgG1 titers induced after a single vaccination with PLGA microspheres (20 μg OVA × 1) were maintained for the duration of the study, while anti-OVA IgG2C titers exhibited a 69-fold increase between day 20 and day 42 postvaccination (P < 0.01, Figure 3c–e). These results indicated that the sustained-release formulation of PLGA microspheres elicited robust and durable total IgG and IgG1 responses while simultaneously promoting maturation of Ag-specific IgG2C responses over time.
From these results we can conclude that two doses of OVA in microspheres administered in a prime-boost regimen (10 μg OVA × 2) elicited significantly greater Ag-specific CD8α+ T cell immune responses and generated stronger Th1/Th2-balanced humoral immune responses than the equivalent dose and regimen of protein vaccination with CaHPO4 adjuvant (Figure 3). Additionally, the single-dose vaccination with microspheres (20 μg OVA × 1) generated similar or stronger Ag-specific cellular and humoral immune responses, compared with the dual prime-boost vaccinations with soluble protein Ag plus CaHPO4 adjuvant (Figure 3).
In summary, our results presented in this communication showed that the self-encapsulating PLGA microspheres, with a median diameter of 7 μm, were successfully internalized by DCs and elicited potent Ag-specific cellular and humoral immune responses after subcutaneous immunizations in mice. Two-dose immunizations with the microspheres significantly improved CD8α+ T cell responses and Th1/Th2-balanced humoral responses, compared with two doses of protein vaccination with CaHPO4 adjuvant. Furthermore, a single dose of sustained-release formulation of microspheres, containing twice the amount of Ag, produced strong cellular and humoral immune responses, comparable to or stronger than those observed after two separate vaccinations with the equivalent total dose of Ag plus the CaHPO4 adjuvant. Overall, this report has provided proof-of-concept data showing excellent potential of our self-encapsulating PLGA microspheres as a vaccine platform for multiple-dose as well as single-dose vaccinations — an attractive and critical attribute of our vaccine technology that may address a lack of patient compliance and poor medical infrastructure in resource-limited settings.
Supplementary Material
Acknowledgments
The authors thank the following University of Michigan staff for their assistance: David Adams (Flow Cytometry Core), Linda Barthel (Microscopy and Image Analysis Laboratory), Dr. Gordon Moore (Electron Beam Analysis Laboratory), and Joel Whitfield (Cancer Center Immunology Core). Special thanks are given to Dr. Rajesh Gupta for his advice concerning the properties of calcium phosphate adjuvants. This work was supported in part by NIH (R21EB08873, S.P.S.; R01EB022563, J.J.M.; R01AI127070, J.J.M.; R01CA210273, J.J.M.). J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), NSF CAREER Award (1553831) and MTRAC for Life Sciences Hub. L.J.O. is supported by pre-doctoral fellowships from UM Rackham and the American Foundation for Pharmaceutical Education (AFPE). B.A.B. acknowledges financial support from the AFPE, the UM Pharmacological Sciences Training Program (PSTP) (GM007767 from NIGMS), and the UM Rackham Merit Fellowship. Opinions, interpretations, conclusions, and recommendations are those of the authors and are neither necessarily endorsed by the NIGMS nor by the Department of Defense. All animal experiments were handled according the University of Michigan Institutional Animal Care guidelines.
Footnotes
Experimental Section
Experimental protocols are reported in the Supporting Information available online.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Brittany A. Bailey, Department of Pharmaceutical Sciences, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA
Lukasz J. Ochyl, Department of Pharmaceutical Sciences, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA
Prof. Steven P. Schwendeman, Department of Pharmaceutical Sciences, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA Department of Biomedical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Prof. James J. Moon, Department of Pharmaceutical Sciences, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA Department of Biomedical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
References
- 1.O’Hagan DT, Valiante NM. Nat Rev. 2003;2:727. doi: 10.1038/nrd1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM. Immunity. 2008;29:319. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Reddy ST, Swartz MA, Hubbell JA. Trends Immunol. 2006;27:573. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Fan Y, Moon JJ. Wiley Interdiscip Rev.: Nanomed Nanobiotechnol. 2017;9:e1403. doi: 10.1002/wnan.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulendran B, Ahmed R. Nat Immunol. 2011;12:509. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta RK, Singh M, O’Hagan DT. Adv Drug Delivery Rev. 1998;32:225. [PubMed] [Google Scholar]
- 7.Katare YK, Panda AK, Lalwani K, Haque IU, Ali MM. Drug Delivery. 2003;10:231. doi: 10.1080/drd_10_4_231. [DOI] [PubMed] [Google Scholar]
- 8.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Adv Drug Delivery Rev. 2005;57:391. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Tamber H, Johansen P, Merkle HP, Gander B. Adv Drug Delivery Rev. 2005;57:357. doi: 10.1016/j.addr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Waeckerle-Men Y, Groettrup M. Adv Drug Delivery Rev. 2005;57:475. doi: 10.1016/j.addr.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Feng L, Qi XR, Zhou XJ, Maitani Y, Wang SC, Jiang Y, Nagai T. J Controlled Release. 2006;112:35. doi: 10.1016/j.jconrel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Sahdev P, Ochyl LJ, Moon JJ. Pharm Res. 2014;31:2563. doi: 10.1007/s11095-014-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso MJ, Gupta RK, Min C, Siber GR, Langer R. Vaccine. 1994;12:299. doi: 10.1016/0264-410x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 14.Kersten G, Hirschberg H. Expert Rev Vaccines. 2004;3:453. doi: 10.1586/14760584.3.4.453. [DOI] [PubMed] [Google Scholar]
- 15.Schwendeman SP. Crit Rev Ther Drug Carrier Syst. 2002;19:73. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 16.Reinhold SE, Desai KG, Zhang L, Olsen KF, Schwendeman SP. Angew Chem Int Ed. 2012;51:10800. doi: 10.1002/anie.201206387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai KG, Schwendeman SP. J Controlled Release. 2013;165:62. doi: 10.1016/j.jconrel.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzara JM, Balagna MA, Thouless MD, Schwendeman SP. J Controlled Release. 2013;171:172. doi: 10.1016/j.jconrel.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Mazzara JM, Schwendeman SP, Thouless MD. J Controlled Release. 2015;206:20. doi: 10.1016/j.jconrel.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 20.Gupta RK, Siber GR. Vaccine. 1995;13:1263. doi: 10.1016/0264-410x(95)00011-o. [DOI] [PubMed] [Google Scholar]
- 21.Gupta RK, Rost BE, Relyveld E, Siber GR. In: Vaccine Design: The Subunit and Adjuvant Approach. Powell MF, Newman MJ, editors. Vol. 6. Plenum Press; New York, USA: 1995. p. 229. [Google Scholar]
- 22.Dorozhkin SV, Epple M. Angew Chem Int Ed. 2002;41:3130. doi: 10.1002/1521-3773(20020902)41:17<3130::AID-ANIE3130>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Paneque-Quevedo AA. Biotecnol Apl. 2013;30:250. [Google Scholar]
- 24.Storni T, Kundig TM, Senti G, Johansen P. Adv Drug Delivery Rev. 2005;57:333. doi: 10.1016/j.addr.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Milacic V, Bailey BA, O’Hagan D, Schwendeman SP. In: Long Acting Injections and Implants. Wright JC, Burgess DJ, editors. Springer; USA: 2012. p. 429. [Google Scholar]
- 26.B. A. Bailey, K. H. Desai, L. J. Ochyl, S. M. Ciotti, J. J. Moon, S. P. Schwendeman, unpublished.
- 27.Champion JA, Walker A, Mitragotri S. Pharm Res. 2008;25:1815. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida M, Babensee JE. J Biomater Sci, Polym Ed. 2006;17:893. doi: 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- 29.Joshi VB, Geary SM, Salem AK. AAPS J. 2013;15:85. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Audran R, Peter K, Dannull J, Men Y, Scandella E, Groettrup M, Gander B, Corradin G. Vaccine. 2003;21:1250. doi: 10.1016/s0264-410x(02)00521-2. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Rouaud O, Poncelet D. Int J Pharm. 2008;363:26. doi: 10.1016/j.ijpharm.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 32.Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Adv Drug Delivery Rev. 2011;63:943. doi: 10.1016/j.addr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 33.Oyewumi MO, Kumar A, Cui Z. Expert Rev Vaccines. 2010;9:1095. doi: 10.1586/erv.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ochyl LJ, Moon JJ. J Vis Exp. 2015;98:e52771. doi: 10.3791/52771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan Y, Sahdev P, Ochyl LJ, Akerberg JJ, Moon JJ. J Controlled Release. 2015;208:121. doi: 10.1016/j.jconrel.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin RM, Brady JL, Lew AM. J Immun Methods. 1998;212:187. doi: 10.1016/s0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.