

Abstract
Ewing’s sarcoma (EWS) is a primitive round cell sarcoma with a peak incidence in adolescence that is driven by a chimeric oncogene created from the fusion of the EWSR1 gene with a member of the ETS family of genes. Patients with metastatic and recurrent disease have dismal outcomes and need better therapeutic options. We screened a library of 309,989 chemical compounds for growth inhibition of EWS cells to provide the basis for the development of novel therapies, and to discover vulnerable pathways that might broaden our understanding of the pathobiology of this aggressive sarcoma. This screening campaign identified a class of benzyl-4-piperidone compounds which selectively inhibit growth of EWS cell lines by inducing apoptosis. These agents disrupt 19S proteasome function through inhibition of the deubiquitinating enzymes USP14 and UCHL5. Functional genomic data from a genome-wide shRNA screen in EWS cells also identified the proteasome as a node of vulnerability in EWS cells, providing orthologous confirmation of the chemical screen findings. Furthermore, shRNA-mediated silencing of USP14 or UCHL5 in EWS cells produced significant growth inhibition. Finally, treatment of a xenograft mouse model of EMS with VLX1570, a benzyl-4-piperidone compound derivative currently in clinical trials for relapsed multiple myeloma, significantly inhibited in vivo tumor growth. Overall, our results offer a preclinical proof of concept for the use of 19S proteasome inhibitors as a novel therapeutic strategy for EWS.
Introduction
Ewing sarcoma (EWS) is the second most common bone malignancy in children, with a peak incidence in adolescence and is characterized by specific translocations leading to the fusion of EWSR1 to a gene of the ETS family of transcription factors.(1,2) Although localized disease is curable with highly intensive chemotherapy combined with surgery or radiation therapy,(3,4) patients with metastatic, recurrent, or refractory disease, have dismal outcomes despite aggressive implementation of traditional chemotherapeutic agents.(5)
To identify novel active agents against EWS, several high-throughput compound screening strategies have been employed. Stegmaier et al. characterized a gene expression profile signature which could act as a surrogate signal for inhibition of EWSR1-FLI1.(6) They performed a screen of 1,040 small molecules against EWS cell lines and identified cytarabine arabinoside as inducing a gene signature consistent with EWSR1-FLI1 inhibition. Cytarabine therapy demonstrated significant efficacy in pre-clinical models, but disappointingly, a subsequent study in a limited number of patients with relapsed/refractory EWS showed no objective responses.(7) More recently, a chemical screen evaluating 50,000 compounds against EWS cell lines identified mithramycin as an agent which resulted in growth suppression as well as reduction of known targets of the EWSR1-FLI1 fusion protein.(8) A trial assessing the safety and efficacy of mithramycin (Clinical Trial Identifier: NCT01610570) for children with relapsed EWS was recently completed, but the results are yet to be published.
We performed a broad, unbiased screen of over 300,000 chemicals for growth-inhibitory activity against EWS using automated cell-based screening assays. The chemicals included synthetic compounds, as well as natural products from plants, micro-organisms, fungi, and deep sea algae. To broaden the biologic and therapeutic scope of the screen, we chose not to use EWSR1-FLI1 inhibition as the primary readout. Although the EWSR1-FLI1 fusion is widely recognized as the driving oncogenic feature in EWS, an understanding of its complex role is still evolving, as highlighted by the recent demonstration of both activating and repressive transcriptional effects of this chimeric protein.(9) Furthermore, effective disruption of critical EWSR1-FLI1 downstream targets may not lead to changes in EWSR1-FLI1 levels or function, and if used as a selection criterion for prioritization of compounds, could lead to dismissal of potentially relevant agents. In this report, we present the results of our broad chemical screen, which highlight a new class of inhibitors of the ubiquitin-proteasome system as having significant therapeutic potential in EWS. Proteasome inhibition was also defined as a specific vulnerability of EWS cells in a genome-wide shRNA screen.
Materials and Methods
Materials
A673, AK-PN-DW, SK-N-MC, and RD-ES were obtained from ATCC. CHP-100 and TC-71 were provided by Dr. Melinda Merchant (National Cancer Institute, Bethesda, Maryland). All cell lines were obtained in 2007, and re-authenticated within the past year by MSK-IMPACT sequencing, which includes 1,042 polymorphic SNPs.(10) Antibodies to GAPDH and S6 were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-UCHL5 antibody was purchased from Abcam (Cambridge, MA, USA). Anti-USP14 antibody was acquired from Bethyl Laboratories (Montgomery, TX, USA). Anti-ubiquitinylated proteins antibody (clone FK2) was purchased from EMD Millipore (Billerica, MA, USA). Anti-rabbit secondary antibodies conjugated to horseradish peroxidase, enhanced chemiluminescence kit, AlamarBlue and puromycin were obtained from Thermo Fisher Scientific (Pittsburg, PA. USA). ApoOne caspase assay and HIV p21 ELISA kits were obtained from Promega (Madison, WI). The 20S proteasome assay kit was purchased from Cayman Chemicals (Ann Arbor, MI, USA). Lentiviral shRNA plasmids (The RNAi Consortium 1.0 library) were obtained from the MSK RNAi Core Facility. MG262 was purchased from Calbiochem. Bortezomib and all 19S proteasome inhibitors used in conformation and animal studies were synthesized by the MSK Organic Synthesis Core Facility (Supplementary Methods). VLX1570 was kindly provided by Hans Rosen at Vivolux Inc. Animal care was conducted in accordance with institutional guidelines.
Small molecule screen
Chemical screens were conducted as described previously.(11) In brief, chemicals were plated into clear-bottom white 384-well tissue culture plates and then cells added at a density of 2,000 cells per well and incubated for 72 h. During the last 24 h with compounds, AlamarBlue proliferation dye was added at a final concentration of 10% (vol/vol) and fluorescence measured (Ex: 555 nm, Em: 585). A primary screen was conducted using 10 μM compound in duplicates. Compounds that inhibited growth by ≥ 80% were considered “hits” and the growth inhibitory activity of these compounds were confirmed in secondary screens, using re-synthesized compounds. Dose-response studies were then carried out with candidate agents in a 12-point doubling dilution series. Data were processed and IC50 determined using Sigma Plot graphing software.
Cell growth and caspase 3/7 assays
For viability assays that were conducted outside of the chemical screen, cells were plated in clear-bottom, white 96-well plates at a density of 3,000 cells per well and incubated with compounds for 96 h. To measure caspase 3/7 activity cells were plated at density of 15,000 cells/well directly into inhibitors in white, clear-bottom 96-well plates and the activity of caspase 3/7 was determined using ApoOne caspase 3/7 activity assay kit (Promega). Fluorescence was measured using a SpectraMax M2 plate reader (Ex: 485 nm, Em: 530).
20S Proteasome Assay
20S proteasome activity was measured as per manufacturer instructions using a 20S proteasome assay kit (Cayman Chemicals). Briefly, the assay employs a substrate, SUC-LLVY-AMC, which upon cleavage by the 20S proteasome generates a highly fluorescent product (Ex: 380nm, Em: 480 nm). A Jurkat cell lysate supernatant with high level 20S activity was used as a positive control. Cells were treated with known 20S inhibitors bortezomib and MG-262, as well as compounds ES-P and ES-W in 5 concentrations in triplicate.
Microarray Analysis
CHP100 and TC-71 cell lines were treated with bortezomib (25nM), MG-262 (25nM), EWS-P (500nM), or EWS-W (25nM) for 6 h. All experiments were performed in triplicate including DMSO treated cells as a control. RNA was extracted using TRIzol Reagent (ThermoFisher Scientific) as per manufacturer instruction and analyzed on the Affymetrix Human Genome U133A 2.0 Array. The expression of probe sets was estimated with RMA.(12) The genes whose expression was differentially up- or down-regulated in treated versus control cells were determined with an empirical Bayes test using LIMMA.(13) Significant genes were those with an adjusted p-value (FDR) < 0.0001 and an absolute fold-change ≥2. Probe sets corresponding to significant genes were mapped to the U133A array annotation to ensure compatibility with, and query against, the Connective Map (CMAP) build 01 database.
Genome-wide shRNA library screen and data analysis
We performed a genome-wide functional genomic screen using the Sigma-Aldrich shRNA Human Genome Library consisting of 63,093 shRNAs targeting 11,748 human genes with a median of 5 shRNA/gene (range 1–58 shRNA/gene; 89.6% of genes with at least 5 shRNAs and 0.06% (7/11748) of genes were targeted by a single shRNA). shRNAs were introduced by lentivirus transduction using the pLKO.1 expression vector with puromycin selection. Specifically, lentivirus were generated using 293T cells. The titer of each preparation of virus was determined using a p21 ELISA assay (Promega). Cells were seeded at 350 cells/well in 35 ul medium and infected at 24 hours with MOI=5 using 6 μg/mL polybrene. Antibiotic selection was initiated 6 days after infection with 1.0 μg/mL puromycin. Cells were grown for an additional 7 days in the presence of puromycin. On day 13, cells were fixed with 4 % PFA (vol/vol) in PBS, permeabilized with 0.05% Triton-X 100 and nuclei stained with 10 μM Hoechst. Nuclear counts were used as the readout for cell number.
For analysis of the shRNA screen data, outliers of the nuclei counts for puromycin-treated control were removed. shRNAs for which the imaged nuclei count was less than or equal to the mean nuclei counts of the puromycin-treated controls were considered positive. For each gene, the percentage of positive shRNA was computed and ‘hits’ were defined as genes for which at least 80% of the shRNAs were positive. Pathway enrichment analysis of the ‘hits’ for networks, biological functions and canonical pathways was performed using Ingenuity Pathway Analysis (http://www.ingenuity.com/products/ipa).
Generation of xenograft tumors and drug administration
One million A673 or TC-71 cells were mixed with matrigel and injected subcutaneously into a single flank of female NOD scid gamma (NSG) or athymic mice (Harlan Labs). NSG mice were transduced with lentivirus expressing GFP-luciferase. NSG mice bearing A673 xenografts were treated with DMSO, bortezomib or b-AP15 after they reached 80–100mm3 and weekly tumor size measurements were taken. Athymic mice bearing A673 and TC-71 xenografts were employed for VLX1570 experiments. When tumors reached 80–100mm3, mice were randomized into 3 groups, 5 mice/group. Mice were treated with vehicle only, b-AP15 (25 mg/kg) or VLX1570 (4.4 mg/kg) daily. Drugs were administered via intraperitoneal injection. Tumors were measured twice weekly and tumor volume was calculated using the formula: length × width2 × 0.52. Body weight was also assessed twice weekly.
UbVS labeling
EWS cells were treated with b-AP15 for 3 h followed by lysis (50 mM HEPES, 50 mM NaCl, 5 mM MgCl2, 0.25% Triton X-100, 2mM ATP, 1 mM DTT, 250 mM Sucrose). 25 μg of protein was labeled with 1 uM UbVS for 10 min, followed by quenching in loading buffer, fractionation on SDS-PAGE gels and immunoblotting with indicated antibodies.
CETSA assay
The CETSA assay was performed as described (14). In brief cells were collected, subject to freeze thaw, soluble fraction was extracted lysates were exposed to DMSO or drug for 30 min at room temp. Lysates were heated at 53°C, fractionated by SDS-PAGE. Thermostabilization was observed by immunoblotting with USP14 or UCHL5 antibodies.
Ub-AMC DUB assays
Recombinant UCHL1, UCHL3 and purified 26S proteasomes were obtained from Boston Biochem. 5nM 26S proteasomes in reaction buffer (50 mM HEPES, 50 mM NaCl, 5 mM MgCl2, 2mM ATP, 1 mM DTT, 250 mM Sucrose) were treated with b-AP15 fro 5 min prior to the addition of 500 nM Ub-AMC. Cleavage of AMC was monitored at 380 nm excitation and 460 nm emission wavelengths using Tecan plate reader.
Results
Identification of growth inhibitory compounds against EWS cell lines from a chemical screen
We performed a screen of a chemical library of 309,989 compounds to find agents with anti-proliferative activity in EWS cell lines. The primary screen was conducted against the TC-71 cell line, which contains the type 1 EWSR1-FLI1 fusion using 10 μM of each compound at the Memorial Sloan Kettering Cancer Center High-Throughput Drug Screening Facility (MSKCC HTSCF). Compounds that inhibited growth by ≥ 80% were moved forward for further analysis; 209 compounds met this criterion (Fig. 1). To identify highly active compounds against multiple EWS cell lines, we tested the 209 compounds against 5 EWS cell lines with the EWSR1-FLI1 fusion (TC-71, A673, RD-ES, SK-N-MC, SK-PN-DW). Dose-response studies were conducted and chemicals for which the IC50 for inhibition of growth was ≤ 1 μM were selected as potential growth inhibitors of EWS cell lines. This identified 23 compounds as inhibitors of the growth of EWS cells, based on IC50 concentrations of ≤ 1 μM in all 5 EWS cell lines.
Fig. 1. Summary of high-throughput compound screening strategy.

The TC-71 cell line was screened against 309,989 compounds. Two hundred and nine compounds were confirmed to result in ≥ 80% growth inhibition at 10 μM concentration following a 72 h incubation period. Four additional EWS cell lines (A673, RD-ES, SK-N-MC and SK-PN-DW) were screened against these 209 compounds. Twenty-three compounds were identified to achieve ≥50% growth inhibition at 1μM concentration against all five EWS cell lines. Eight of these 23 compounds were identified as having selective activity against the ES cell lines as compared to a panel of 11 other cell lines from various cancer types. The NCBI PubChem ID for each of the 8 compounds is listed; including the two lead benzyl-4-piperidone structures, which we designated as EWS-P and EWS-W.
Reasoning that pan-active compounds with low specificity for EWS would less likely be clinically novel or relevant, and would be less helpful in providing insight into the biology of EWS, we sought to identify which of these 23 compounds had relatively selective activity against EWS compared to other non-EWS cancer cell lines. We assembled a panel of 11 non-EWS cell lines from 10 different cancer types to perform a lateral screen with the 23 compounds identified to be active against the 5 EWS cell lines (Table S1). Cell lines for which the IC50 for inhibition of growth by any of the 23 compounds was < 1 μM were designated as sensitive. From the group of 23 compounds highly active against EWS cell lines, we identified 8 compounds to which at least 5/11 non-EWS cell lines were resistant in our comparative screening panel, suggesting selective or preferential activity against EWS. Two of the 8 compounds are analogs of daunorubicin and ellipticine (NCT608747, NCT176327), both of which are from chemotherapy families currently used as effective frontline chemotherapy for EWS (anthracyclines and topoisomerase-II inhibitors). Two of the remaining 6 compounds (NCT666038, NCT669441) were analogs that share a benzyl-4-piperidone scaffold, which prompted us to further explore this novel class of compounds. These compounds were designated EWS-P (NCT666038) and EWS-W (NCT669441).
Benzyl-4-piperidone compounds inhibition of the ubiquitin-proteasome pathway
To evaluate the mechanism of action of the EWS-P and EWS-W compounds, we studied the change in gene expression profiles of two EWS cell lines, CHP100 and TC-71, treated with these two agents compared to DMSO-treated controls (see Methods). Statistically significantly up- and down-regulated genes were queried against the Connectivity Map (CMAP) platform (http://www.broadinstitute.org/cmap/), a database of gene expression profiles generated from human cells treated with a wide range of bioactive small molecules.(15) To validate this approach, we also generated expression profiles of EWS cell lines treated with two compounds identified from the screen that were derivatives of camptothecin (EWS-A) and the topoisomerase II inhibitor ellipticine (EWS-S). The genes differentially expressed upon treatment with these compounds were highly similar to those expression profiles inferred from analogous compounds in CMAP (data not shown). The gene signatures inferred from both EWS cell lines treated with compounds EWS-P and EWS-W were highly congruent with expression signatures obtained for the proteasome inhibitor MG-262 (Fig. S1). Pairwise expression profile comparisons of EWS cell lines treated with MG-262 and its analog, bortezomib, demonstrated a statistically significant overlap with the expression profiles generated by compounds EWS-P and EWS-W. Furthermore, we identified 13 core genes which were up-regulated in both CHP100 and TC-71 following treatment with all 4 agents; bortezomib, MG-262, EWS-P, and EWS-W (Fig. S1A). The majority of these core genes are well established in the literature as being upregulated following inhibition of the proteasome (Fig. S1B).(16–19)
Activity of the proteasome is dependent upon the polyubiquitination of peptides destined for degradation and therefore, inhibition of proteasomal activity leads to intracellular accumulation of polyubiquitinated proteins.(20) To better define the effect of EWS-P and EWS-W on the proteasome, we treated TC-71 and CHP100 cell lines with these compounds and then looked at global protein ubiquitination by Western blot analysis. Treatment with compounds EWS-P and EWS-W resulted in significant accumulation of polyubiquitinated proteins providing further evidence that these agents are inhibitors of the proteasome (Fig. S1C). The human 26S proteasome consists of a central, barrel shaped 20S subunit and two outer structures designated as the 19S proteasome subunit. The 19S proteasome subunit is responsible for recognition of polyubiquitinated proteins, unfolding of the peptide structure, and passage of the peptide into the 20S subunit, the primary site of peptide degradation.(21–23) Agents such as bortezomib and MG-262 bind to the active site of the 20S proteasome leading to inhibition.(24). To assess activity against the 20S proteasome, we performed an assay that employs a specific 20S substrate which upon cleavage by the active proteasome generates a highly fluorescent product. As predicted, inhibition with bortezomib and MG-262 resulted in a marked decrease in fluorescence. Treatment with compounds EWS-P and EWS-W, however, did not result in a statistically significant reduction in fluorescence, indicating that these compounds inhibit proteasomal activity through a mechanism not dependent on the 20S subunit.
Growth reduction and apoptosis in EWS models in vitro and in vivo upon treatment with Benzyl-4-piperidone compounds
We generated analogs of EWS-P and EWS-W (MSK-EWS-4 and MSK-EWS-5) and tested their ability to inhibit growth of 4 EWS cell lines. Three of the benzyl-4-piperidone compounds inhibited the growth of all EWS lines tested, while one compound (MSK-EWS-4) was inactive. (Table S2). Compound MSK-EWS-5, which was previously published with the name b-AP15(25), was the most potent analog that we generated (Fig. 2A and Table S2). b-AP15 was previously shown to inhibit the deubiquitinating enzymes (DUBs) USP14 and UCHL5 (25). These enzymes associate with the 19S proteasome subunit and function to deubiquitinate proteins as they enter the 20S proteolytic core subunit (26–28). These results are consistent with our finding that these compounds, as a class, inhibit proteasomal activity through a 20S independent mechanism.
Fig. 2. Inhibition of DUBs USP14 and UCHL5 by benzyl-4-piperidone compounds.

(A) CHP100, A673, and TC-71 cells were treated with b-AP15 for 3 h followed by DUB activity labeling. DUB inhibition is indicated by loss of the higher molecular weight USP14-UbVS or UCHL5-UbVS band. (B) CETSA of USP14 following b-AP15 treatment. b-AP15 interacts with USP14 as indicated by increased stabilization at 53°C. Results represent the mean ± SD of 3 independent experiments.
To confirm DUB inhibition as a mechanism of action in EWS, we performed activity labeling using the suicide substrate Ub-vinylsulphone (UbVS) on cells following treatment with b-AP15 (1 μM) for 3 h. Immunoblotting following UbVS labeling showed the inhibition of USP14 and UCHL5 in all EWS cell lines tested (Fig 2A). As previously reported(25,29), USP14 appeared to be more effectively inhibited by b-AP15 compared to UCHL5. Building upon this we also examined the binding of b-AP15 to USP14 in EWS cells using a cellular thermo stability assay (CETSA)(14). We found stabilization of USP14 after EWS cell exposure to 1 μM b-AP15 (Fig 2B). Thermostabilization of UCHL5 was not observed at this drug concentration further supporting the idea that USP14 is the more sensitive DUB target for these compounds. Furthermore, b-AP15 did not demonstrate inhibition of other DUBs, including recombinant UCHL1 and UCHL3 (Fig S2).
VLX1570 is an analog of b-AP15 with minor structural modifications improving its chemical properties for clinical use (29). Cell viability assays in 4 EWS cell lines demonstrate that VLX1570 is slightly more potent that b-AP15 at inhibiting growth in the four ES cell lines tested (Fig. 3A and Table S2). To determine if the reduction in growth of EWS cells by these benzyl-4-piperidone compounds is due in part to induction of apoptosis, we measured the enzymatic activity of caspase 3/7 following drug treatment. Four EWS cell lines were treated with b-AP15 or VLX1570 for 48 h followed by fluorescence-based measurement of caspase 3/7 activity. We observed a dose-dependent increase in caspase 3/7 activity, approximately 2–7-fold above DMSO-treated controls, in cells following treatment with b-AP15 or VLX1570 indicating activation of apoptosis (Fig. 3B).
Fig. 3. Effects of benzyl-4-piperidone compounds on growth and apoptosis in EWS pre-clinical models.
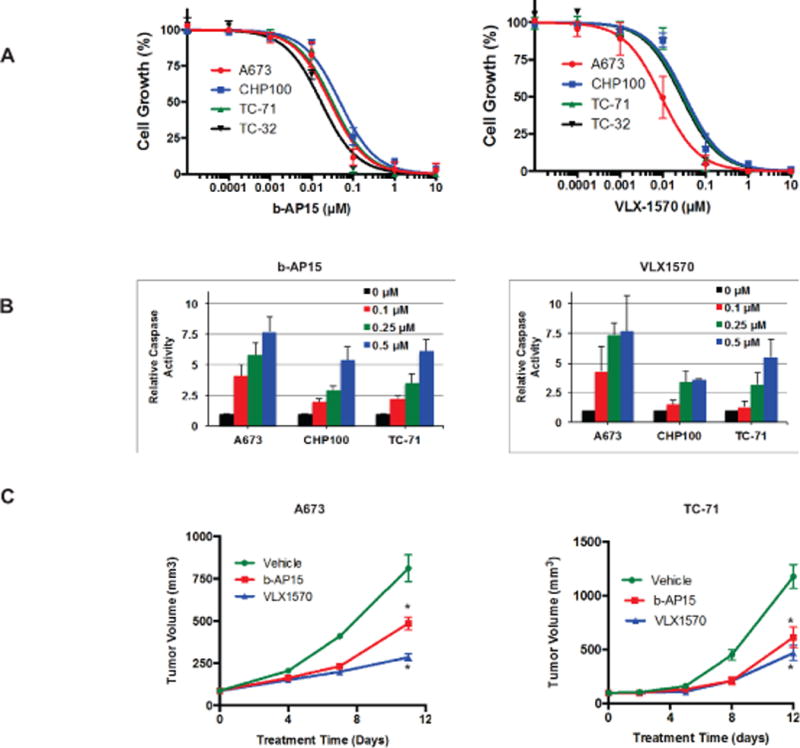
(A) Four EWS cell lines were treated with b-AP15 or VLX1570 for 96 h and the relative number of viable cells was then determined. Results represent the mean ± SE of 3–5 experiments in which each condition was assayed in triplicate. (B) Four EWS cell lines were treated with b-AP15 or VLX1570 for 48 h and then caspase 3/7 activity measured. Results are the mean ± SD of 2–4 experiments in which each condition was assayed in triplicate. (C) Athymic mice bearing A673 or TC-71 xenografts were treated daily with vehicle, b-AP15 or VLX1570. Tumors were measured twice per week. *Significantly different compared to vehicle-treated group (Two way ANOVA, p<0.001 compared to vehicle-treated group).
To evaluate the in vivo activity of benzyl-4-piperidone compounds, we first treated a GFP-luciferase expressing A673 xenograft model with b-AP15 following injection of cells to study the effect on tumor formation. Treatment with b-AP15 resulted in undetectable tumors by imaging at 4 weeks (Fig S3A). Next, we compared bortezomib and b-AP15 in an A673 xenograft model and demonstrated superior growth inhibition and improved survival with the former (Fig S3B and S3C). We then tested the ability of b-AP15 and VLX1570 to inhibit the growth of two EWS cell line xenografts, A673 and TC-71. Athymic mice were treated with b-AP15 (25mg/kg), VLX1570 (4.4mg/kg), or vehicle daily via intraperitoneal administration. We initiated treatment when tumors reached a volume of approximately 100 mm3. Growth of A673 and TC-71 xenograft tumors was significantly reduced by both compounds with VLX1570 being more potent (Fig. 3C). There was no weight loss in any treatment group (Fig. S3D).
Susceptibility of EWS to proteasome inhibition as supported by functional genomics screen and targeted RNAi
To provide orthogonal confirmation of EWS sensitivity to proteasome inhibition, we analyzed data from a functional genomic screen in EWS cells. Specifically, the CHP100 cell line was screened with the Sigma-Aldrich shRNA Human Genome Library by lentivirus transduction. The screen queried 11,748 genes with ≥ 5 shRNAs targeting 89.6% of the genes. Effects on proliferation and viability were measured by nuclear counts (Hoechst stain). The screen identified 627 genes with ≥ 80% shRNA scoring positive (Fig. 4A). Pathway analysis of these 627 genes revealed an enrichment of genes involved in the protein ubiquitination pathway (p=0.0004, Supplementary Table S3), including PSMB2/3/5/6/8, PSMC3, PSMD1/3/7, UBC and USP1/14/26. Furthermore, among these 627 “hits”, 26S proteasome component genes and associated DUBs were significantly over-represented (p < 0.0001, Fisher’s exact test) (Tables 1 and 2). Overall screen data and Ingenuity Pathway Analysis of the hits are provided in Supplementary Tables S3 and S4.
Fig. 4. Genome-wide and targeted RNAi in EWS cell lines.
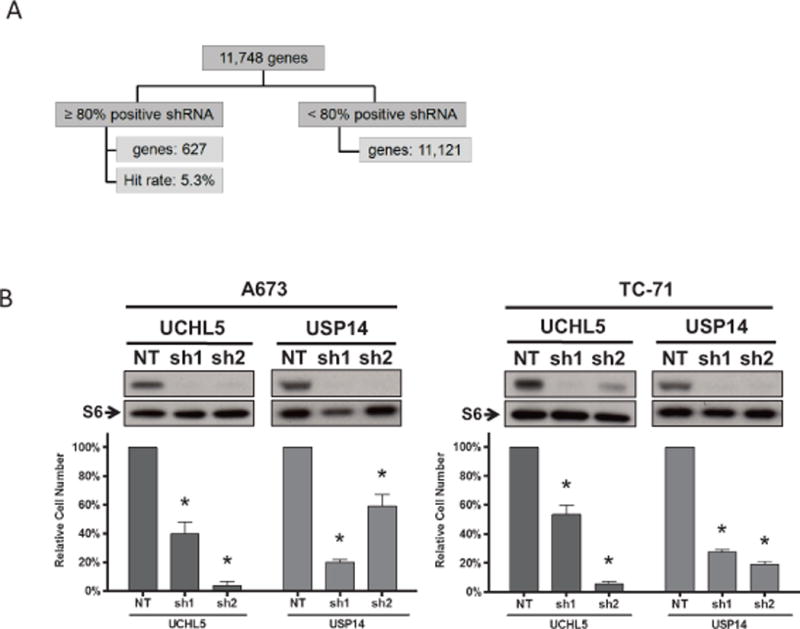
(A) A genome-wide functional screen was performed on the EWS cell line CHP100 using the Sigma Aldrich human genome library (~5 shRNAs per gene). 627 genes (5.3%) were scored as positive hits based on the criteria of ≥ 80% of shRNAs per gene resulting in threshold reduction of nuclei count. (B) Genes encoding 26S proteasome subunits or 19S-associated DUBs were significantly over-represented as positive hits.
Table 1.
Representation of 26S proteasome component genes found in functional genomic screen of EWS cells
| 26S Genes | Other Genes | Total | |
|---|---|---|---|
| shRNA pos | 10 | 617 | 627 |
| shRNA neg | 26 | 11095 | 11121 |
| Total | 36 | 11712 | 11748 |
p < 0.0001
Table 2.
Genes involved in the proteasome ubiquitination pathway identified as enriched in functional genomic screen of EWS cells
| 19S | 20S |
|---|---|
| PSMC3 | PSMB2 |
| PSMD1 | PSMB3 |
| PSMD3 | PSMB5 |
| PSMD7 | PSMB6 |
| USP14 | PSMB8 |
To further evaluate USP14 and UCHL5 as potential targets in EWS, we performed shRNA-mediated knockdown (2 shRNAs per gene) in A673 and TC-71 cells. RNA interference of these genes caused a significant reduction in expression level of the encoded proteins and a concomitant decrease in cell numbers (Fig. 4B).
Discussion
Given the current outcomes for patients with metastatic and recurrent EWS, there is a critical need for novel therapeutic agents. Proteasome inhibitors such as bortezomib have been evaluated in both pre-clinical and clinical studies of EWS. Bortezomib is a dipeptide boronic acid analogue which inhibits the chemotryptic activity of the 20S proteasome.(30) It is currently FDA-approved for the treatment of multiple myeloma and relapsed mantle cell lymphoma. In vitro cell viability assays performed on EWS cell lines have demonstrated drug susceptibility with IC50 levels as low as 20 nM (31) but subsequent xenograft studies performed by the Pediatric Preclinical Testing Program identified limited activity in EWS or any other pediatric solid tumor type.(32) Furthermore, in the COG ADVL0916 study, which evaluated the use of bortezomib in combination with escalating doses of vorinostat in 23 children with solid tumors, including 2 patients with EWS, no responses were observed.(33) More generally, numerous phase two studies have failed to demonstrate meaningful activity of bortezomib in adult solid tumors.(34–42) The poor activity of bortezomib in most solid tumor patients has been attributed to poor tumor penetration of the drug.(43) Clinical dosing schedules for bortezomib in multiple myeloma achieve only 70% proteasome inhibition in blood cells with complete recovery between doses, suggesting that the pharmacodynamic profile in solid tumors may be even less favorable.(44)
Novel inhibitors of the 20S proteasome with potentially advantageous pharmacologic properties are being investigated in solid tumors. Carfilzomib is a 20S inhibitor which, unlike bortezomib, irreversibly inhibits the chymotrypsin-like activity of the 20S proteasome.(45) A recent phase 1/2 study for adult patients with advanced solid tumors was conducted in which patients were treated twice weekly on consecutive days for 3 weeks per 28 day cycle. Among the 65 patients treated in the phase two portion of this trial, none achieved a partial response or better.(46) Although other studies evaluating the efficacy of carfilzomib in solid tumors are ongoing, these disappointing results highlight the need for the development and evaluation of alternative proteasome inhibition strategies.
Thus, while in vitro data suggest that proteasome function may be an effective therapeutic target in some solid tumors, clinical experience with 20S inhibitors in patients with solid tumors has been disappointing, suggesting that alternate strategies for proteasome inhibition should be pursued.
Here, we have provided strong pre-clinical evidence that benzyl-4-piperidone compounds are highly active in EWS. Two analogs were identified as lead compounds from our high-throughput chemical screen based on stringent requirements for both potency and selectivity. We have demonstrated that treatment of EWS cell lines with this family of compounds leads to accumulation of polyubiquitinated proteins, without inhibition of the 20S proteasome, thereby acting at a site within the ubiquitin-proteasome system distinct from that targeted by proteasome inhibitors currently employed in clinical practice. Our findings are supported by recent publications that have identified the 19S proteasome component as the target for this group of compounds.(25,47) We also demonstrate that this class of compounds act synergistically with bortezomib, and can overcome bortezomib resistance mutations. This is an important observation for the potential evaluation of dual 19S and 20S inhibition in future trials for both liquid and solid tumor types. Finally, we have also established the susceptibility of EWS to proteasomal disruption through an unbiased functional genomics screen, and through specific knockdown experiments targeting the DUBs USP14 and UCHL5. Our data supports USP14 as a primary target given its identification as a lead hit on our genome-wide shRNA screen as well as preferential b-AP15 inhibitory activity against USP14 seen in our in vitro binding studies.
We have identified a highly active novel class of proteasome inhibitors from a screen of an exceptionally broad library of chemicals against EWS, and have validated these functionally and pre-clinically. Importantly, a clinical compound is now available for this novel class of proteasome inhibitors; VLX1570 was recently developed in an effort to improve on the chemical properties of b-AP15 for use in a clinical setting. This novel derivative was generated by substituting the piperidine ring in b-AP15 with an azepane ring. VLX1570 has a superior clinical profile compared to b-AP15 with significantly improved solubility as well as an increase in biologic activity.(29) A phase 1/2 trial assessing the safety and efficacy of VLX1570 in patients with relapsed/refractory multiple myeloma is ongoing (NCT02372240). Our findings now provide a compelling rationale for a clinical trial evaluating VLX1570 as a novel therapeutic strategy in patients with relapsed/refractory EWS.
Supplementary Material
Acknowledgments
We thank Joseph Olechnowicz for editorial assistance.
Financial Support: This study was funded by the Ewing Research Foundation, the Sarcoma Foundation of America, the Hyundai Hope on Wheels Program, and the MSKCC Experimental Therapeutics Center. Research infrastructure support was provided by the National Cancer Institute under grant no. P30CA008748.
Footnotes
Conflicts of interest: P.D. and S.L are shareholders of Vivolux AB.
References
- 1.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359(6391):162–5. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 2.Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annual review of pathology. 2012;7:145–59. doi: 10.1146/annurev-pathol-011110-130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(33):4148–54. doi: 10.1200/JCO.2011.41.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolb EA, Kushner BH, Gorlick R, Laverdiere C, Healey JH, LaQuaglia MP, et al. Long-term event-free survival after intensive chemotherapy for Ewing’s family of tumors in children and young adults. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21(18):3423–30. doi: 10.1200/JCO.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 5.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. The New England journal of medicine. 2003;348(8):694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 6.Stegmaier K, Wong JS, Ross KN, Chow KT, Peck D, Wright RD, et al. Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS medicine. 2007;4(4):e122. doi: 10.1371/journal.pmed.0040122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DuBois SG, Krailo MD, Lessnick SL, Smith R, Chen Z, Marina N, et al. Phase II study of intermediate-dose cytarabine in patients with relapsed or refractory Ewing sarcoma: a report from the Children’s Oncology Group. Pediatric blood & cancer. 2009;52(3):324–7. doi: 10.1002/pbc.21822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grohar PJ, Woldemichael GM, Griffin LB, Mendoza A, Chen QR, Yeung C, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. Journal of the National Cancer Institute. 2011;103(12):962–78. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sankar S, Bell R, Stephens B, Zhuo R, Sharma S, Bearss DJ, et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 2013;32(42):5089–100. doi: 10.1038/onc.2012.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD. 2015;17(3):251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Somwar R, Shum D, Djaballah H, Varmus H. Identification and preliminary characterization of novel small molecules that inhibit growth of human lung adenocarcinoma cells. Journal of biomolecular screening. 2009;14(10):1176–84. doi: 10.1177/1087057109350919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 13.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 14.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–7. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 15.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 16.Tang ZY, Wu YL, Gao SL, Shen HW. Effects of the proteasome inhibitor bortezomib on gene expression profiles of pancreatic cancer cells. The Journal of surgical research. 2008;145(1):111–23. doi: 10.1016/j.jss.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 17.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14374–9. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimmermann J, Erdmann D, Lalande I, Grossenbacher R, Noorani M, Furst P. Proteasome inhibitor induced gene expression profiles reveal overexpression of transcriptional regulators ATF3, GADD153 and MAD1. Oncogene. 2000;19(25):2913–20. doi: 10.1038/sj.onc.1203606. [DOI] [PubMed] [Google Scholar]
- 19.Wu WT, Chi KH, Ho FM, Tsao WC, Lin WW. Proteasome inhibitors up-regulate haem oxygenase-1 gene expression: requirement of p38 MAPK (mitogen-activated protein kinase) activation but not of NF-kappaB (nuclear factor kappaB) inhibition. The Biochemical journal. 2004;379(Pt 3):587–93. doi: 10.1042/BJ20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10(1):29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nature reviews Molecular cell biology. 2009;10(2):104–15. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- 22.Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS biology. 2006;4(8):e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(21):4776–89. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 24.Guedat P, Colland F. Patented small molecule inhibitors in the ubiquitin proteasome system. BMC biochemistry. 2007;8(Suppl 1):S14. doi: 10.1186/1471-2091-8-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nature medicine. 2011;17(12):1636–40. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 26.Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Molecular & cellular proteomics : MCP. 2011;10(5):R110 003871. doi: 10.1074/mcp.R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. The EMBO journal. 2001;20(18):5187–96. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nature cell biology. 2006;8(9):994–1002. doi: 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, et al. Synthesis and Evaluation of Derivatives of the Proteasome Deubiquitinase Inhibitor b-AP15. Chemical biology & drug design. 2015 doi: 10.1111/cbdd.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer research. 1999;59(11):2615–22. [PubMed] [Google Scholar]
- 31.Lu G, Punj V, Chaudhary PM. Proteasome inhibitor Bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing’s sarcoma family of tumors and synergizes with TRAIL. Cancer Biol Ther. 2008;7(4):603–8. doi: 10.4161/cbt.7.4.5564. [DOI] [PubMed] [Google Scholar]
- 32.Houghton PJ, Morton CL, Kolb EA, Lock R, Carol H, Reynolds CP, et al. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatric blood & cancer. 2008;50(1):37–45. doi: 10.1002/pbc.21214. [DOI] [PubMed] [Google Scholar]
- 33.Muscal JA, Thompson PA, Horton TM, Ingle AM, Ahern CH, McGovern RM, et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: a Children’s Oncology Group phase I consortium study (ADVL0916) Pediatric blood & cancer. 2013;60(3):390–5. doi: 10.1002/pbc.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah MH, Young D, Kindler HL, Webb I, Kleiber B, Wright J, et al. Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(18 Pt 1):6111–8. doi: 10.1158/1078-0432.CCR-04-0422. [DOI] [PubMed] [Google Scholar]
- 35.Engel RH, Brown JA, Von Roenn JH, O’Regan RM, Bergan R, Badve S, et al. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer investigation. 2007;25(8):733–7. doi: 10.1080/07357900701506573. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg JE, Halabi S, Sanford BL, Himelstein AL, Atkins JN, Hohl RJ, et al. Phase II study of bortezomib in patients with previously treated advanced urothelial tract transitional cell carcinoma: CALGB 90207. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2008;19(5):946–50. doi: 10.1093/annonc/mdm600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch TJ, Fenton D, Hirsh V, Bodkin D, Middleman EL, Chiappori A, et al. A randomized phase 2 study of erlotinib alone and in combination with bortezomib in previously treated advanced non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2009;4(8):1002–9. doi: 10.1097/JTO.0b013e3181aba89f. [DOI] [PubMed] [Google Scholar]
- 38.Scagliotti GV, Germonpre P, Bosquee L, Vansteenkiste J, Gervais R, Planchard D, et al. A randomized phase II study of bortezomib and pemetrexed, in combination or alone, in patients with previously treated advanced non-small-cell lung cancer. Lung cancer. 2010;68(3):420–6. doi: 10.1016/j.lungcan.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Aghajanian C, Blessing JA, Darcy KM, Reid G, DeGeest K, Rubin SC, et al. A phase II evaluation of bortezomib in the treatment of recurrent platinum-sensitive ovarian or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecologic oncology. 2009;115(2):215–20. doi: 10.1016/j.ygyno.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 40.Trinh XB, Sas L, Van Laere SJ, Prove A, Deleu I, Rasschaert M, et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncology reports. 2012;27(3):657–63. doi: 10.3892/or.2011.1562. [DOI] [PubMed] [Google Scholar]
- 41.Parma G, Mancari R, Del Conte G, Scambia G, Gadducci A, Hess D, et al. An open-label phase 2 study of twice-weekly bortezomib and intermittent pegylated liposomal doxorubicin in patients with ovarian cancer failing platinum-containing regimens. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2012;22(5):792–800. doi: 10.1097/IGC.0b013e318251051a. [DOI] [PubMed] [Google Scholar]
- 42.Maki RG, Kraft AS, Scheu K, Yamada J, Wadler S, Antonescu CR, et al. A multicenter Phase II study of bortezomib in recurrent or metastatic sarcomas. Cancer. 2005;103(7):1431–8. doi: 10.1002/cncr.20968. [DOI] [PubMed] [Google Scholar]
- 43.Williamson MJ, Silva MD, Terkelsen J, Robertson R, Yu L, Xia C, et al. The relationship among tumor architecture, pharmacokinetics, pharmacodynamics, and efficacy of bortezomib in mouse xenograft models. Mol Cancer Ther. 2009;8(12):3234–43. doi: 10.1158/1535-7163.MCT-09-0239. [DOI] [PubMed] [Google Scholar]
- 44.Hamilton AL, Eder JP, Pavlick AC, Clark JW, Liebes L, Garcia-Carbonero R, et al. Proteasome inhibition with bortezomib (PS-341): a phase I study with pharmacodynamic end points using a day 1 and day 4 schedule in a 14-day cycle. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(25):6107–16. doi: 10.1200/JCO.2005.01.136. [DOI] [PubMed] [Google Scholar]
- 45.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer research. 2007;67(13):6383–91. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 46.Papadopoulos KP, Burris HA, 3rd, Gordon M, Lee P, Sausville EA, Rosen PJ, et al. A phase I/II study of carfilzomib 2–10-min infusion in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2013;72(4):861–8. doi: 10.1007/s00280-013-2267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer cell. 2013;24(6):791–805. doi: 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.