

Abstract
Arylmetals are highly valuable carbon nucleophiles that are readily and inexpensively prepared from aryl halides or arenes and widely used on both laboratory and industrial scales to react directly with a wide range of electrophiles. Although C–C bond formation has been a staple of organic synthesis, the direct transfer of primary amino (–NH2) and hydroxyl (–OH) groups to arylmetals in a scalable and environmentally friendly fashion remains a formidable synthetic challenge because of the absence of suitable heteroatom-transfer reagents. Here, we demonstrate the use of bench-stable N–H and N–alkyl oxaziridines derived from readily available terpenoid scaffolds as efficient multifunctional reagents for the direct primary amination and hydroxylation of structurally diverse aryl- and heteroarylmetals. This practical and scalable method provides one-step synthetic access to primary anilines and phenols at low temperature and avoids the use of transition-metal catalysts/ligands/additives, nitrogen-protecting groups, excess reagents and harsh workup conditions.
Graphical Abstract
The direct transfer of primary amino and hydroxyl groups to aryl-metals in a scalable and environmentally friendly fashion remains a formidable synthetic challenge. Here, it is demonstrated that bench-stable N–H and N–alkyl oxaziridines can be used as efficient multifunctional reagents, without deprotonation, for the direct primary amination and hydroxylation of (hetero)arylmetals.
Nitrogen- and oxygen-substituted aromatic rings (that is, anilines and phenols) appear as substructures in a large number of industrially and commercially significant organic compounds such as active pharmaceutical ingredients, agrochemicals and functional materials1,2. For example, primary anilines (that is, Ar–NH2) and phenols (that is, Ar–OH) are used as intermediates or building blocks for the preparation of drug candidates, azo dyes and polyanilines1,2. Today, anilines are mostly prepared via one of the following methods: (1) the reduction of aromatic nitro compounds3; (2) transition-metal-catalysed (Pd, Ni or Cu) cross-coupling of haloarenes and arylboronic acids with ammonia or substituted primary and secondary amines4–7, (3) transition-metal-catalysed (Pd, Ni or Cu) electrophilic amination of various organometallics8–11 (for example, Li, Zn and B), (4) nucleophilic aromatic substitution (SNAr) and nucleophilic substitution of hydrogen in electron-deficient systems12–15 and (5) direct C–H amination16 of aromatic rings. Phenols are often prepared via the following methods: (1) SNAr in heteroaromatic systems1,14,15,17, (2) oxidation of arylboronic acids and derivatives18 and (3) metal-catalysed direct hydroxylation of aromatic rings19,20.
Nearly all the methods that are catalysed or mediated by transition metals and their complexes require forcing conditions (high temperature, high pressure, strong oxidants and so on), which results in limited functional group tolerance. From a practical and environmental point of view, transition-metal-free processes are much preferred, especially in the pharmaceutical industry for which the removal of undesired metal contamination can be expensive21. Therefore, we became intrigued by the possibility of the application of readily available and inexpensive aryl-Grignard22,23 and aryllithium24 reagents (that is, Ar–Mg–X and Ar–Li) that might be directly primary aminated or hydroxylated with bench-stable aminating and hydroxylating reagents. A survey of the literature revealed that the direct primary amination of arylmetal reagents (that is, ArMgX, ArLi) is exceedingly problematic as most hydroxylamine-derived aminating agents (H2N–OR, where OR is a leaving group) undergo rapid proton transfer, and thus consume a total of three equivalents of the precious arylmetal substrate and tend to give a poor yield of the desired primary arylamine on workup (Fig. 1a)25–27. It was also surprising to find that currently there are no general methods/reagents available for the direct hydroxylation of structurally diverse arylmetal reagents that would allow the effi-cient synthesis of phenols under operationally simple and mild reaction conditions.
Figure 1. Current approaches to primary arylamine and phenol synthesis from arylmetals in the absence of transition-metal catalysts, impact of steric hindrance on kinetic acidity and the discovery of multifunctional oxaziridine reagents for heteroatom-transfer reactions.

a–c, Known two-step procedures for the synthesis of primary arylamines (4) from the corresponding arylmetals (2 and 2c) using electrophilic aminating agents (1, 5 and 7). d, Conversion of arylmagnesium halides (2a) into phenols (12) using molecular oxygen in a flow system. e, Non-hindered amines undergo rapid proton exchange with arylmetals. f, Sterically bulky secondary amines, such as 13, do not undergo proton exchange with arylmetals. g, Our hypothesis: a sterically hindered electrophilic N source, such as an N–H oxaziridine, will undergo NH transfer rather than proton exchange when reacted with arylmetals. h,i, Camphor-derived N–Me and N–benzyl oxaziridine (19a (h) and 19b (i), respectively) react with 15 directly to give 2-naphthol (20) on workup. j,k, Camphor- and fenchone-derived N–H oxaziridines (16 (j) and 18 (k), respectively) react with 2-naphthylmagnesium bromide (15) directly at low temperature and under a protective argon atmosphere to afford 2-naphthylamine (17) on simple aqueous workup. Bn, benzyl; TMDEA, tetramethylethylenediamine.
Over the past two decades, several approaches have been developed for the two-step synthesis of primary arylamines from the corresponding arylmagnesium or aryllithium reagents. In the first approach (Fig. 1a), an electrophilic nitrogen source is reacted with an arylmetal and, after C–N bond formation, the activating group is removed, typically under harsh conditions (that is, strongly acidic hydrolysis at elevated temperatures)28–31. Additionally, the free primary amine has to be liberated from its salt using basic conditions. Clearly, this type of approach prohibits the use of highly functionalized arylmetal reagents or those that have acid- or base-sensitive functionalities. In the second approach (Fig. 1b), an O-alkylhydroxylamine (for example, methoxyamine) is first treated with MeLi and the resulting lithium amide has to be used in large excess for the efficient amination of the arylmetal reagent25–27. Although the arylamine can be obtained in the free-base form right after the aqueous workup, the need to use two separate organometallic reagents (one of them in excess) and the modest overall efficiency/yield are the two obvious drawbacks of this method. Recently, our group and the Morken group pioneered the development of a transformation in which arylboronic acids or borate esters may be converted into the primary arylamines in the absence of transition-metal catalysts; however, elevated temperatures are often required (Fig. 1c)32,33. The direct hydroxylation of arylmagnesium or aryllithium reagents has not been extensively exploited beyond what was shown by Davis and co-workers34,35. One method by the Jamison group stands out (Fig. 1d) as being quite general; however, it uses oxygen (O2) in air at a high pressure (1,720 kPa (250 pounds per square inch (p.s.i.))) and requires a specialized flow-reactor system36.
Given the apparent gaps in existing synthetic methodology, we decided to develop a mild and direct primary amination as well as hydroxylation of structurally diverse and abundant arylmetal reagents (that is, arylmagnesium halides and aryllithium) in which the unprotected primary aniline and phenolic products are isolated readily in their non-ionized (that is, free-base) form. To achieve this goal, suitable bench-stable/highly chemoselective reagents are required that will allow the primary amination and hydroxylation to proceed rapidly at or below ambient temperature and without the need to use excess reagents (that is, ideally a near 1:1 molar ratio of arylmetal and the aminating or hydroxylating reagent). The new method would enable the introduction of valuable unprotected primary amino (–NH2) as well as hydroxyl groups (–OH) into a large variety of functionalized aromatic rings, even at a later stage of a complex synthetic sequence.
When arylmetals react with non-hindered primary and secondary amines, the N–H group undergoes fast proton transfer, which quenches the arylmetals and is a major drawback of the amination approach (Fig. 1e). Highly sterically hindered secondary amines have a reduced kinetic acidity, and thus resist N–H deprotonation even in the presence of excess alkyllithium reagents at elevated temperatures, as shown by Corey and Gross (Fig. 1f; 13→14)37. We surmised that a sterically hindered electrophilic nitrogen source may also resist deprotonation because of the reduced kinetic acidity of the N–H bond and thus avoid the unproductive protonation of the arylmetal reagent.
When looking for alternative reagents to replace O-alkylhydroxyl-amines as aminating agents, we considered sterically hindered N–H oxaziridines (Fig. 1g)38–41. Compared with the widely used and studied N-sulfonyloxaziridines (for example, Davis’ oxaziridines), N–H oxaziridines have been exploited only rarely in synthesis because of their perceived low stability/high reactivity. However, a handful of bench-stable N–H oxaziridines have been reported and used for the amination of alcohols and enolates, albeit in fairly limited ways39,40,42,43. N–H oxaziridines (Fig. 1, 16 and 18) are readily prepared from inexpensive terpenoids (camphor and fench-one) on a multidecagram scale. Preliminary differential scanning calorimetry (DSC) profiles demonstrated an onset for the decomposition at around 100 °C. In an explosive screening, compound 18 gave a maximum pressure-rise rate of 480.06 bar min−1, and therefore it does not pose an explosion hazard with respect to transport classification. However, further safety assessments are currently underway to evaluate the safety profile for these oxaziridine reagents (see DSC data on page 102 of the Supplementary Information). Moreover, density functional calculations suggest that the fench-one-derived N–H oxaziridine has a low kinetic acidity for reactivity with arylmetals (see later results).
Our hypothesis was that N–H oxaziridines, such as 16 and 18, might undergo amination with arylmetal reagents faster than the deprotonation of their N–H functionality. Indeed, we found that camphor-derived N–H oxaziridine 16 was an efficient N–H-transfer agent and reacted smoothly with 2-naphthylmagnesium bromide (15) to afford the corresponding 2-naphthylamine (17) directly in its unprotected free-base form (Fig. 1h). Remarkably, the use of either a large excess of aminating reagent (16) or arylmetal substrate (15) was not required to obtain a synthetically useful isolated yield (50%, 1 mmol scale) in this direct primary amination reaction. The main side product was the protonated aryl-Grignard reagent (that is, naphthalene, 25% isolated yield), which suggests a competing deprotonation of the N–H group. We presumed that this undesired deprotonation pathway could potentially be suppressed by increasing the steric bulk and thus decreasing the kinetic acidity of the N–H oxaziridine. In fact, reaction of the more sterically hindered fenchone-derived N–H oxaziridine (18) with 2-naphthylmagnesium bromide (15) furnished 2-naphthylamine (17) in a significantly improved isolated yield (>80%; Fig. 1i). Apparently the kinetic acidity was, indeed, lower in the case of 18 compared with 16. Remarkably, both 16 and 18 acted as exclusive N-transfer agents—the trace amounts (<1%) of phenols that we detected were presumably the products of air oxidation.
Naturally, it was intriguing to contemplate if direct N–alkyl transfer was also a possibility by utilizing the N–alkyl versions of oxaziridines 16 and 18. To our great surprise, N–Me oxaziridine 19a (the N–Me analogue of 16) did not react at all with 2-naphthylmagnesium bromide (15) at −78 °C; however, at ambient temperature it acted as an effective and exclusive O-transfer reagent and converted 15 into the corresponding phenol 20 in good isolated yield (Fig. 1j). A control experiment in the absence of 19a, but still under argon atmosphere, led to the formation of only trace amounts of 20, which indicates that the origin of the oxygen atom in phenol 20 is the oxaziridine (that is, 19a) and not the adventitious oxygen (O2) from air. It is clear from these results that this reagent scaffold is multifunctional because it can be modified easily to transfer either hetereoatom.
Encouraged by these promising preliminary results, we conducted systematic optimization studies (Supplementary Tables 1 and 2), which concluded that the highest isolated yields are obtained at −78 °C in a toluene/THF mixture when using a 1:1.2 ratio of aryl-metal and aminating agent. With the optimum reaction conditions in hand, the stage was set to explore the scope and limitations of this method by subjecting dozens of substituted arylmetal reagents to primary amination (Tables 1 and 2). The initially tested aryl Grignard reagents represent an extensive sampling of both fused and monocyclic aromatic rings as well as electron-rich and electron-deficient examples (Table 1). Fused aromatic rings (entries 1–5, Table 1) and biaryl systems (entries 11–16, Table 1) underwent smooth primary amination with good-to-excellent isolated yields, except for one instance (entry 4) in which an electronegative fluorine atom was in the para position of the carbon–magnesium bond. Ring-halogenated substrates (entries 30–43, Table 1) furnished the corresponding primary anilines in moderate-to-good yields and clearly illustrate the true complementary nature of this method to transition-metal-catalysed aminations given that halogen atoms are tolerated well.
Table 1.
Direct primary amination of arylmagnesium halide substrates with N–H oxaziridine 18.

|
All the aromatic Grignard reagents (21) were prepared from the corresponding aryl halides using turnings of freshly activated Mg metal and THF as the solvent. The concentration of the arylmetal solution was targeted to be around 0.5 M, but was carefully determined by titration immediately before use. The amination reactions were conducted on a 1 mmol scale at the indicated temperature and considered complete on full consumption of the aminating agent (18) by thin-layer chromatography analysis; a number of experiments showed that 18 undergoes decomposition in the presence of strong metal bases.
Table 2.
Direct primary amination of aromatic and heteroaromatic arylmetal substrates using N–H oxaziridine 18.

|
The aromatic and heteroaromatic metal reagents (25 and 26) were prepared using one of the following methods:
from aryl halides using activated Mg metal;
from aryl halides using the i-PrMgCl·LiCl complex (Knochel’s procedure);
performed with the aryllithium reagent via Li/halogen exchange;
the primary amination was performed with N–H oxaziridine 30;
direct C–H deprotonation with TMPMgCl·LiCl;
Li/Br exchange followed by transmetallation with MgBr2.
When one or more alkyl substituents are located adjacent to the aryl–metal bond (that is, ortho positions), primary aminations proceed only in fair-to-moderate yields (entries 7, 9, 28 and 37, Table 1); however, substrates with O–methoxy substituents (entries 18 and 20, Table 1) furnished the primary arylamines in good isolated yields. It is likely that when both the substrate and the aminating agent are sterically bulky, the rate and efficiency of primary amination are reduced—in these cases elevation of the reaction temperature (–45 °C instead of −78 °C) was necessary to observe synthetically useful reaction times and isolated yields (see specifically entry 37, Table 1). Note that substrates having tertiary amine moieties (–NR2), which are usually quite sensitive to oxidation, furnished the corresponding primary arylamines in good-to-excellent yields (entries 44–46, Table 1). These cases highlight the remarkable chemoselectivity of aminating agent 18.
To illustrate further the unprecedented mildness of our direct primary amination method, we selected aromatic and heteroaromatic substrates that have redox- or hydrolytically sensitive moieties (Table 2), such as a primary alkyl halide (entry 52), isolated and conjugated olefins (entries 47, 48 and 53), a 1,3-diene (entry 49), alkynes (entries 50 and 51), ethers and thioethers (entries 54–60), acetals and ketals (entries 61–63), one or more halogen/pseudo-halogen atoms (entries 64–72) and heterocycles (entries 73–76, 78 and 79). In a few cases (entries 58, 64, 70, 74, 75 and 79) the isolated yields were poor (for example, <30%), which can be attributed to a combination of steric and electronic factors or to the volatility of the product (entry 58). Even bis-metallated arenes can undergo efficient double amination (entry 77). Most of these functionalities are tolerated well under the reaction conditions—especially noteworthy are the high isolated yields of anilines that feature highly acid-sensitive acetal or ketal functionalities (entries 61–63), oxidatively highly sensitive thioether moieties (entries 59 and 60) and bromine atoms, all of which usually lead to side reactions under transition-metal catalysis. This methodology can also be used for the late-stage functionalization of structurally complex and pharmaceutically relevant intermediates (for example, oestradiol derivatives (entries 80 and 8, Table 2), a terpenoid derivative (entry 82) and a carbohydrate derivative (entry 83)).
Given the poor primary amination performance of N–H oxaziridine 18 with sterically hindered substrates, we decided to examine the impact of reducing the steric bulk around the oxaziridine moiety (Table 3). Therefore, we prepared two additional N–H oxaziridines (30 and 31, Table 3) that were, in our judgement, sterically less hindered than N–H oxaziridines 16 and 18 and were expected to improve the isolated yields of primary arylamines for a handful of challenging substrates. Indeed, we found that as the steric bulk of the N–H oxaziridines was reduced, their primary amination performance improved markedly with sterically hindered arylmetals. In contrast, the primary amination of sterically unencumbered aryl-metals (for example, 15 → 23a) became less efficient with a decrease in the steric bulk of the N–H oxaziridines, presumably as a result of easier N–H deprotonation. It appears that N–H oxaziridine 18 can efficiently transfer the primary amino group (–NH2) to most arylmetals. The results in Table 3 indicate that one can readily find a suitable N–H oxaziridine aminating agent for both sterically demanding and less nucleophilic arylmetals.
Table 3.
Direct primary amination of arylmagnesium halides with structurally and sterically different N–H oxaziridines.

| |||
|---|---|---|---|
| (Entry no.) Compound; T(°C), t (h), iσolated yield (%) | |||
 23a |
 23i |
 24b |
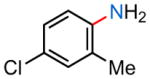 24k |
| (1) 16, −78 °C, 2 h; 50% | (1) 16, −45 °C, 3 h; 26% | (1) 16, −45 °C, 3 h; 26% | (1) 16, −45 °C, 3 h; 57% |
| (2) 18, −78 °C, 2 h; 89% | (2) 18, −45 °C, 3 h; 0% | (2) 18, −45 °C, 3 h; 46% | (2) 18, −45 °C, 3 h; 46% |
| (3) 30, −78 °C, 2 h; 83% | (3) 30, −45 °C, 2 h; 31% | (3) 30, −45 °C, 2 h; 52% | (3) 30, −45 °C, 2 h; 65% |
| (4) 31, −78 °C, 2 h; 46% | (4) 31, −45 °C, 2 h; 0% | (4) 31, −45 °C, 2 h; 65% | (4) 31, −45 °C, 2 h; 9% |
Studies aimed at improving the efficiency of primary amination for sterically hindered (that is, ortho-substituted) arylmetals. Four bench-stable N–H oxaziridines (16, 18, 30 and 31) were evaluated as aminating agents under the indicated reaction conditions.
For the direct hydroxylation of arylmetals, several N–alkyl oxaziridines have been prepared and evaluated (Supplementary Information, pages 7 and 8). N–benzyl oxaziridine 19b was selected as the optimal reagent given its ease of synthesis, bench stability and chemoselectivity. The examples in Table 4 amply illustrate the unprecedented functional group tolerance of this reagent as both oxidatively (entries 9, 17, 19, 20, 21 and 24, Table 4) and hydrolytically (entries 22 and 23, Table 4) sensitive functionalities remained untouched during the hydroxylation process. The phenols themselves are usually highly oxidatively sensitive even without additional electron-donating groups on their aromatic rings1; that most of the phenol products were isolated in good yield attests to the unprecedented mildness of this method.
Table 4.
Direct hydroxylation of arylmagnesium halide substrates with N–benzyl oxaziridine 19b.

|
The N–benzyl derivative of camphor-derived oxaziridine (19b) serves as an exclusive O-transfer agent when reacted with arylmetals. Reactions with aryl-Grignard reagents were conducted between 0 °C and 25 °C followed by mild workup with aqueous NH4Cl solution. r.t., room temperature.
Our hypothesis of low kinetic acidity is based on M06-2X/def2-TZVP density functional calculations in THF continuum solvent for the reaction of 18 with (PhMgBr)2 (modelled as dinuclear bridging and Schlenk-type structures (details are given on page 65 of the Supplementary Information))44. Consistent with this hypothesis is that the calculated pKa for 18, assuming MgBr coordination of the anion in THF solution, the transfer of ~34 protons between (PhMgBr)2 and 18 is thermodynamically favourable (ΔG = −13.4 kcal mol−1), and therefore the selective amination versus proton transfer must be governed by kinetic pathways37.
Several kinetic pathways were readily ruled out. For example, outersphere one-electron transfer from (PhMgBr)2 to 18 generates [(PhMgBr)2]•+ and 18•− with an energy penalty of >80 kcal mol−1 (ref. 45). This large electron-transfer energy is consistent with Woerpel’s observations that Grignard reagents do not react with electrophiles (for example, aldehydes) through one-electron transfer46. The ~30 kcal mol−1 strain energy of the oxaziridine ring and the relatively weak N–O bond could suggest homolytic cleavage of the N–O bond. However, the constrained elongation of the N–O bond to the corresponding 1,3-diradical suggests that the bond energy is ~40 kcal mol−1, which is significantly larger than that of alternative pathways. Isomerization products (for example, amide) were not detected experimentally47.
The most-viable amination pathway identified involves the formation of a (PhMgBr)2–18 complex followed by the transfer of a nucleophilic phenyl group to the nitrogen atom by TS1 (Fig. 2a). This transition state features PhMg coordination to the N–H group, delivery of the phenyl anion to the N–O σ* orbital, which results in bond cleavage, and stabilization of the developing oxygen anion by the second Mg (ref. 48). The ΔG‡ for TS1 is 19.7 kcal mol−1 relative to a (PhMgBr)2–18 complex. Importantly, the ΔG‡ for proton transfer between (PhMgBr)2 and 18 by TS2 (Fig. 2a) is 23.5 kcal mol−1, which is 3.8 kcal mol−1 larger than that for TS1. Using another accurate functional (ωB97X-D/ def2-TZVP), the ΔΔG‡ between TS1 and TS2 is 5.5 kcal mol−1 (ref. 49). This indicates that amination kinetically outcompetes proton transfer, but competitive ΔG‡ values for TS1 and TS2 is consistent with the minor amount of arene product (~10–20%) found. The deuterium-trapping experiments (Fig. 2c) support the hypothesis that, in addition to the predominant N transfer, deprotonation of the aminating agent also occurs to a minor extent at low temperatures. In other words, the active Grignard reagent 21b is quickly consumed at the reaction temperature, and hence no deuterated product (35) was observed. The density functional calculations also show that amination is also significantly favoured for the N–H oxaziridine 18 over hydroxylation via TS3 (Fig. 2a) with ΔG‡ > 30 kcal mol−1. Transition-state calculations for a phenyl-group transfer between (PhMgBr)2 and 19b show that the N versus O selectivity is reversed and hydroxylation has a ~2 kcal mol−1 lower ΔG‡ value (the transition-state structures are given on pages 65–84 of the Supplementary Information).
Figure 2. Three-dimensional representation of competitive amination and proton-transfer transition states, and the proposed mechanism of N transfer and deuterium-trapping experiments.

a, Free-energy barriers and enthalpy barriers (kcal mol−1) (the top row numbers correspond to M06-2X and the bottom row to wB97X-D.) b, Proposed mechanism of the direct primary amination of arylmetals using bench-stable N–H oxaziridines: I, the strained three-membered oxaziridine ring is easy to open and 21a attacks the N–O σ* orbital; II, breakdown of the aminal intermediate to afford the magnesium amide of the arylamine product. c, Deuterium-trapping experiments using a delayed addition (2 h) at −78 °C in THF/toluene and then quenched with NH4Cl (aq). The aryl-Grignard reagent 21b was titrated before use, approximately 0.2 mmol of 36 was present in the aryl-Grignard reagent because of inadvertent/unavoidable protonation during its preparation and the addition of the D2O after only 5 s gave almost identical results.
Conclusion
We have demonstrated that bench-stable N–H and N–alkyl oxaziridines derived from inexpensive terpenoid scaffolds can serve as effective primary aminating and hydroxylating reagents to give rise directly to primary arylamines and phenols from readily available arylmetals in the absence of transition-metal catalysts and under exceedingly mild conditions. This one-step/one-pot method is expected to become widely used in both academic and industrial laboratories as it will provide practical and scalable synthetic access to a vast array of structurally diverse unprotected aromatic amines and hydroxyarenes to be used as building blocks or as value-added compounds. Although this approach is a step in the right direction towards ‘greener’ chemistry, the preparation of the oxaziridine reagents needs further improvements to achieve a higher level of sustainability. Therefore, more-efficient synthetic routes to these reagents are currently being developed.
Supplementary Material
Acknowledgments
L.K. gratefully acknowledges the generous financial support from Rice University, National Institutes of Health (R01 GM-114609-01), National Science Foundation (CAREER: SusChEM CHE-1455335), the Robert A. Welch Foundation (Grant C-1764), Amgen (2014 Young Investigators’ Award) and Biotage (2015 Young Principal Investigator Award), which are greatly appreciated. D.H.E. thanks BYU and the Fulton Supercomputing Lab. We thank S. Sutton and P. Richardson at Pfizer LaJolla for the thorough DSC analysis of the oxaziridine reagents used in this manuscript—the full DSC analysis and interpretation data are included in the Supplementary Information. We also thank E. M. Carreira, A. J. Catino, E.J. Corey, F. A. Davis, A. Ganesan, M. M. Joullie, J. Lopchuk, I. Marek, A. G. Myers, K.C. Nicolaou, J. Njardarson, A. Padwa, R. Sarpong, A. Toro and E. Vedejs for helpful commentary. We dedicate this article to Professor K. C. Nicolaou on the occasion of his 70th birthday.
Footnotes
Author contributions
H.G. and L.K. conceived this work; H.G., Z.Z. and L.K. designed the organic chemistry experiments; H.G., Z.Z. and N.E.B. conducted the organic chemistry experiments and analysed the data; D.-H.K., D.H.E. and L.K. designed the computational studies; D.-H. K., J.C., S.J. and D.H.E. conducted the calculations and analysed the data; D.H.E. and L.K. wrote the manuscript.
Supplementary information and chemical compound information are available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Rappoport Z. The Chemistry of Phenols. John Wiley & Sons; 2004. [Google Scholar]
- 2.Rappoport Z. The Chemistry of Anilines, Parts 1–2. John Wiley & Sons; 2007. [Google Scholar]
- 3.Blaser HU, Steiner H, Studer M. Selective catalytic hydrogenation of functionalized nitroarenes: an update. ChemCatChem. 2009;1:210–221. [Google Scholar]
- 4.Klinkenberg JL, Hartwig JF. Catalytic organometallic reactions of ammonia. Angew Chem Int Ed. 2011;50:86–95. doi: 10.1002/anie.201002354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiao JX, Lam PYS. In: Boronic Acids. 2. Hall DG, editor. Vol. 1. Wiley-VCH; 2011. pp. 315–361. [Google Scholar]
- 6.Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Rational development of practical catalysts for aromatic carbon–nitrogen bond formation. Acc Chem Res. 1998;31:805–818. [Google Scholar]
- 7.Vo GD, Hartwig JF. Palladium-catalyzed coupling of ammonia with aryl chlorides, bromides, iodides, and sulfonates: a general method for the preparation of primary arylamines. J Am Chem Soc. 2009;131:11049–11061. doi: 10.1021/ja903049z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker TJ, Jarvo ER. Umpolung amination: nickel-catalyzed coupling reactions of N,N-dialkyl-N-chloroamines with diorganozinc reagents. J Am Chem Soc. 2009;131:15598–15599. doi: 10.1021/ja907038b. [DOI] [PubMed] [Google Scholar]
- 9.Rucker RP, Whittaker AM, Dang H, Lalic G. Synthesis of hindered anilines: copper-catalyzed electrophilic amination of aryl boronic esters. Angew Chem Int Ed. 2012;51:3953–3956. doi: 10.1002/anie.201200480. [DOI] [PubMed] [Google Scholar]
- 10.Berman AM, Johnson JS. Copper-catalyzed electrophilic amination of diorganozinc reagents. J Am Chem Soc. 2004;126:5680–5681. doi: 10.1021/ja049474e. [DOI] [PubMed] [Google Scholar]
- 11.Barker TJ, Jarvo ER. Developments in transition-metal-catalyzed reactions using electrophilic nitrogen sources. Synthesis. 2011:3954–3964. [Google Scholar]
- 12.Mąkosza M. Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value. Chem Soc Rev. 2010;39:2855–2868. doi: 10.1039/b822559c. [DOI] [PubMed] [Google Scholar]
- 13.Makosza M. Reactions of nucleophiles with nitroarenes: multifacial and versatile electrophiles. Chem Eur J. 2014;20:5536–5545. doi: 10.1002/chem.201400097. [DOI] [PubMed] [Google Scholar]
- 14.Terrier F. Modern Nucleophilic Aromatic Substitution. John Wiley & Sons; 2013. [Google Scholar]
- 15.Alvarez-Builla J, Vaquero JJ, Barlueng J. Modern Heterocyclic Chemistry. John Wiley & Sons; 2011. [Google Scholar]
- 16.Yoo EJ, Ma S, Mei TS, Chan KSL, Yu JQ. Pd-catalyzed intermolecular C–H amination with alkylamines. J Am Chem Soc. 2011;133:7652–7655. doi: 10.1021/ja202563w. [DOI] [PubMed] [Google Scholar]
- 17.Romero NA, Margrey KA, Tay NE, Nicewicz DA. Site-selective arene C–H amination via photoredox catalysis. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 18.Chinnusamy T, Feeney K, Watson CG, Leonori D, Kin Aggarwal V. In: Comprehensive Organic Synthesis. 2. Knockel P, Molander GA, editors. Vol. 7. Elsevier; 2014. pp. 692–718. [Google Scholar]
- 19.Enthaler S, Company A. Palladium-catalyzed hydroxylation and alkoxylation. Chem Soc Rev. 2011;40:4912–4924. doi: 10.1039/c1cs15085e. [DOI] [PubMed] [Google Scholar]
- 20.Alonso DA, Najera C, Pastor IM, Yus M. Transition-metal-catalyzed synthesis of hydroxylated arenes. Chem Eur J. 2010;16:5274–5284. doi: 10.1002/chem.201000470. [DOI] [PubMed] [Google Scholar]
- 21.Garrett CE, Prasad K. The art of meeting palladium specifications in active pharmaceutical ingredients produced by Pd-catalyzed reactions. Adv Synth Catal. 2004;346:889–900. [Google Scholar]
- 22.Knochel P, et al. Highly functionalized organomagnesium reagents prepared through halogen–metal exchange. Angew Chem Int Ed. 2003;42:4302–4320. doi: 10.1002/anie.200300579. [DOI] [PubMed] [Google Scholar]
- 23.Klatt T, Markiewicz JT, Sämann C, Knochel P. Strategies to prepare and use functionalized organometallic reagents. J Org Chem. 2014;79:4253–4269. doi: 10.1021/jo500297r. [DOI] [PubMed] [Google Scholar]
- 24.Rappoport Z, Marek I. The Chemistry of Organomagnesium Compounds. John Wiley & Sons; 2008. [Google Scholar]
- 25.Erdik E. In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Patai S, editor. Wiley; 2009. pp. 303–341. [Google Scholar]
- 26.Corpet M, Gosmini C. Recent advances in electrophilic amination reactions. Synthesis. 2014;46:2258–2271. [Google Scholar]
- 27.Starkov P, Jamison TF, Marek I. Electrophilic amination: the case of nitrenoids. Chem Eur J. 2015;21:5278–5300. doi: 10.1002/chem.201405779. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura M, Suga T, Chiba S, Narasaka K. Synthesis of primary amines by the electrophilic amination of Grignard reagents with 1,3-dioxolan-2-one O-sulfonyloxime. Org Lett. 2004;6:4619–4621. doi: 10.1021/ol0479951. [DOI] [PubMed] [Google Scholar]
- 29.Kitamura M, Chiba S, Narasaka K. Synthesis of primary amines and N-methylamines by the electrophilic amination of Grignard reagents with 2-imidazolidinone O-sulfonyloxime. Bull Chem Soc Jpn. 2003;76:1063–1070. [Google Scholar]
- 30.Tsutsui H, Ichikawa T, Narasaka K. Preparation of primary amines by the alkylation of O-sulfonyloximes of benzophenone derivatives with Grignard reagents. Bull Chem Soc Jpn. 1999;72:1869–1878. [Google Scholar]
- 31.Chiba S, Narasaka K. In: Amino Group Chemistry. Ricci A, editor. Wiley VCH; 2008. pp. 1–54. [Google Scholar]
- 32.Zhu C, Li G, Ess DH, Falck JR, Kürti L. Elusive metal-free primary amination of arylboronic acids: synthetic studies and mechanism by density functional theory. J Am Chem Soc. 2012;134:18253–18256. doi: 10.1021/ja309637r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mlynarski SN, Karns AS, Morken JP. Direct stereospecific amination of alkyl and aryl pinacol boronates. J Am Chem Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis FA, Mancinelli PA, Balasubramanian K, Nadir UK. Coupling and hydroxylation of lithium and Grignard reagents by oxaziridines. J Am Chem Soc. 1979;101:1044–1045. [Google Scholar]
- 35.Davis FA, Wei J, Sheppard AC, Gubernick S. The mechanism of hydroxylation of organometallic reagents by 2-sulfonyloxaziridines. Tetrahedron Lett. 1987;28:5115–5118. [Google Scholar]
- 36.He Z, Jamison TF. Continuous-flow synthesis of functionalized phenols by aerobic oxidation of Grignard reagents. Angew Chem Int Ed. 2014;53:3353–3357. doi: 10.1002/anie.201310572. [DOI] [PubMed] [Google Scholar]
- 37.Corey EJ, Gross AW. Methods for the synthesis of chiral hindered amines. J Org Chem. 1985;50:5391–5393. [Google Scholar]
- 38.Blanc S, Bordogna CAC, Buckley BR, Elsegood MRJ, Page PCB. New stable N–H oxaziridines—synthesis and reactivity. Eur J Org Chem. 2010;2010:882–889. [Google Scholar]
- 39.Page PCB, Limousin C, Murrell VL. Asymmetric electrophilic amination of various carbon nucleophiles with enantiomerically pure chiral N–H oxaziridines derived from camphor and fenchone. J Org Chem. 2002;67:7787–7796. doi: 10.1021/jo020306j. [DOI] [PubMed] [Google Scholar]
- 40.Page PCB, et al. The first stable enantiomerically pure chiral N–H oxaziridines: synthesis and reactivity. J Org Chem. 2000;65:4204–4207. doi: 10.1021/jo000176j. [DOI] [PubMed] [Google Scholar]
- 41.Choong IC, Ellman JA. Synthesis of alkoxylamines by alkoxide amination with 3,3′-di-tert-butyloxaziridine. J Org Chem. 1999;64:6528–6529. doi: 10.1021/jo990490h. [DOI] [PubMed] [Google Scholar]
- 42.Williamson KS, Michaelis DJ, Yoon TP. Advances in the chemistry of oxaziridines. Chem Rev. 2014;114:8016–8036. doi: 10.1021/cr400611n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis FA, Chen B-C, Zhou P. In: Comprehensive Heterocyclic Chemistry III. Katritzky AR, editor. 1A. Elsevier; 2008. pp. 559–622. [Google Scholar]
- 44.Mori T, Kato S. Analytical RISM-MP2 free energy gradient method: application to the Schlenk equilibrium of Grignard reagent. Chem Phys Lett. 2007;437:159–163. [Google Scholar]
- 45.Haines BE, Wiest O. SET-induced biaryl cross-coupling: an SRN1 reaction. J Org Chem. 2014;79:2771–2774. doi: 10.1021/jo500222d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otte DAL, Woerpel KA. Evidence that additions of Grignard reagents to aliphatic aldehydes do not involve single-electron-transfer processes. Org Lett. 2015;17:3906–3909. doi: 10.1021/acs.orglett.5b01893. [DOI] [PubMed] [Google Scholar]
- 47.Aminova RM, Ermakova E. Rearrangements and proton transfer in nitrones by quantum chemistry and molecular dynamics. Chem Phys Lett. 2002;359:184–190. [Google Scholar]
- 48.Jiménez-Osés G, Brockway AJ, Shaw JT, Houk KN. Mechanism of alkoxy groups substitution by Grignard reagents on aromatic rings and experimental verification of theoretical predictions of anomalous reactions. J Am Chem Soc. 2013;135:6633–6642. doi: 10.1021/ja4015937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chai JD, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.